
Abstract
The CAR-HEMATOTOX (CAR-HT) score can assess hematotoxicity risk, but its utility in guiding supportive care remains underexplored. We analyzed 224 multiple myeloma patients treated with BCMA-directed CAR-T therapy (2016–2024), stratified by CAR-HT score (low <2 vs high ≥2). We evaluated the incidence and timing of early immune effector cell-associated hematotoxicity (ICAHT), other cytopenias, transfusion burden, cytopenia-directed interventions (growth factors and stem cell boosts), infections, and survival outcomes. High CAR-HT scores (58%) were associated with more grade 3 ICAHT (45.4% vs 23.3%) and grade 4 ICAHT (5.4% vs 1.2%; p = 0.0069), higher rates of grade ≥3 anemia (43.1% vs 24.4%, p < 0.0001) and thrombocytopenia (50.8% vs 27.9%, p < 0.0001). These patients were 7 times more likely to require red cell and platelet transfusions (p < 0.0001). G-CSF and TPO agonists were variably effective. Stem cell boosts (4%) led to rapid trilineage recovery. Infections occurred more frequently among high CAR-HT patients and those receiving intensive cytopenia-directed interventions. There were no significant differences in progression-free or overall survival between CAR-HT groups. Cytopenia-directed interventions did not affect survival. The CAR-HT score is a practical clinical risk tool that predicts transfusion burden and helps guide supportive care after BCMA CAR-T therapy.
Similar content being viewed by others

Introduction
B-cell maturation antigen (BCMA) directed chimeric antigen receptor T-cell (CAR-T) therapy has transformed outcomes for patients with relapsed and refractory multiple myeloma (MM), resulting in exceptional overall response rates, and with no requirement for maintenance therapy at this time [1,2,3]. However, prolonged cytopenias, now coined immune effector cell-associated hematotoxicity (ICAHT), have emerged as a significant complication of CAR-T therapy [4]. More than 90% of patients experience cytopenias with ≥grade 3 neutropenia in up to 80%, and grade 3 thrombocytopenia and anemia in up to 50% in the first 100 days [1, 2, 5]. Severe cytopenias are associated with higher rates of severe infections, increased non-relapse mortality, and worse overall survival outcomes, highlighting their significant clinical burden [6]. These findings emphasize the importance of optimizing management strategies to mitigate the morbidity and mortality associated with post-CAR-T cytopenia.
The mechanisms and etiology of the various patterns of cytopenias remain an area of investigation, but pre-treatment baseline characteristics are an important contributor, including percentage plasmacytosis at time of lymphodepletion chemotherapy prior to CAR-T administration, prior treatments and transplant [6, 7]. The CAR-HEMATOTOX prognostic score, identifies those at risk for severe cytopenias after CAR-T therapy [7]. The mainstay management strategies are supportive blood product transfusions, antimicrobial therapies, and growth factor administration [8]. For those with refractory cytopenias, administration of a stem cell boost, adapted from the post-autologous stem cell transplant experience, is now a well-recognized management strategy [9,10,11,12,13]. This study builds upon prior validation of the CAR-HEMATOTOX (CAR-HT) score in lymphoma and myeloma by providing a real-world, practice-evolution analysis across multiple CAR-T eras (2016–2024). Our work reflects the dynamic shift in supportive-care approaches, quantifies transfusion burden, details timing of cytopenia-directed interventions, infection outcomes, and offers practical insight into management of hematologic toxicity beyond just guidelines. These real-world data address a gap not captured by earlier validation studies and serve as a framework for optimization of supportive care as CAR-T is implemented earlier in therapy.
Methods
Study design and population
This retrospective study analyzed the medical records of 224 adult (aged ≥18 years) patients with relapsed/refractory multiple myeloma (RRMM) treated with BCMA-targeted CAR-T cell therapy at Mayo Clinic from August 2016 to July 2023. Patients included in the study received either standard-of-care commercial CAR-T products or CAR-T therapy as part of a clinical trial.
Data collection
Data were extracted from electronic medical records and included demographic details (age, sex, race and ethnicity), disease characteristics (duration of MM, cytogenetic risk, prior treatments, and CAR-T product used), laboratory parameters (neutrophil, hemoglobin, and platelet counts), supportive care interventions (G-CSF, thrombopoietin [TPO] agonists, stem cell boosts, and transfusions), infectious complications, and survival outcomes.
Clinically significant anemia, neutropenia, and thrombocytopenia were assessed at the time of lymphodepleting chemotherapy, prior to CAR-T infusion, and defined as hemoglobin <8 g/dL, absolute neutrophil count (ANC) < 1.5 × 10⁹/L, and platelet count <50 × 10⁹/L, respectively. Cytopenias were graded per CTCAE v5.0. The baseline risk for cytopenia was assessed using the CAR-HEMATOTOX (CAR-HT) score, calculated at the start of lymphodepletion chemotherapy. This score incorporates baseline ANC, hemoglobin, platelet count, C-reactive protein (CRP), and ferritin to stratify hematologic vulnerability risk prior to CAR-T, with higher points assigned for more aberrant values. Patients with a CAR-HT score ≥2 were categorized as high-risk, while those with a score <2 were considered low-risk.
The hematotoxicity observed after CAR-T was further stratified using the Immune Effector Cell–Associated Hematotoxicity (ICAHT) grading system. ICAHT grade was determined based on the depth and duration of neutropenia, anemia, and thrombocytopenia following CAR-T infusion.
Recovery from cytopenias was defined as follows: neutrophil recovery: ANC > 1000 cells/µL, platelet recovery: >50 × 10⁹/L (with subset analysis for recovery >20 × 10⁹/L), hemoglobin recovery: >8 g/dL. Each threshold had to be sustained for at least three consecutive days to be considered recovered.
Supportive care and cytopenia-directed interventions were recorded. Autologous stem cell boosts were offered to patients with persistent or refractory cytopenias when an eligible product was available based on clinician discretion. CAR-T–related toxicities, infectious complications, response to therapy, and clinical outcomes were also documented.
Statistical analysis
All statistical analyses were performed using the JMP statistical software package (SAS Institute, Cary, NC, USA). Descriptive statistics were used to summarize patient demographics, clinical characteristics at baseline and prior to CAR-T infusion, pre-lymphodepleting laboratory values, and the type, depth, and duration of post–CAR-T cytopenias, as well as interventions used to manage cytopenias.
Time to hematologic recovery for neutrophils, platelets, and hemoglobin was analyzed using Kaplan-Meier survival methods, with recovery defined as sustained counts above pre-specified thresholds. Patients who did not achieve hematologic recovery were censored earlier either at the last follow-up date (if in remission) or the date of disease relapse, whichever occurred first. Log-rank tests were used to compare time-to-recovery distributions between groups.
Univariate analyses were also conducted to explore potential prognostic factors associated with hematologic recovery and survival outcomes. A p-value < 0.05 was considered statistically significant for all analyses.
To evaluate the independent impact of clinical and treatment-related variables on hematologic recovery, we performed multivariable Cox proportional hazards modeling, adjusting for relevant covariates including CAR-HT score and type of cytopenia-directed intervention.
Ethical considerations
This retrospective study was approved by the Mayo Clinic Institutional Review Board and conducted in accordance with the Declaration of Helsinki. Given the retrospective design and use of existing clinical data, the requirement for informed consent was formally waived by the Institutional Review Board.
Results
Cohort demographics and baseline characteristics
The cohort included 224 patients; 38.4% had a low CAR-HT score, 58.0% had high CAR-HT score, and 8 patients were not classifiable because ferritin or CRP were missing. Baseline demographics and clinical characteristics, stratified by CAR-HT score, are described in Table 1, and CRS, ICANS and infections in Table 2. Prior to the administration of LD-chemotherapy, high CAR-HT patients experienced higher rates of grade 3 or higher anemia (24.6 vs 1.2%), neutropenia (17.7 vs 1.2%) and thrombocytopenia (20 vs 0%) than low CAR-HT patients. After CAR-T administration, high CAR-HT patients had significantly higher odds of CRS, 2.83 (95% CI: 1.66–4.82, p = 0.0001) compared to low CAR-HT patients. There was no difference in the occurrence of ICANS. High CAR-HT scores were associated with increased risk of infectious complications, OR 2.06 (95% CI: 1.15–4.17, p = 0.016) and 4-fold higher odds of developing MAS/HLH (13.8%, n = 18) compared to those with low CAR-HT score (3.5%, n = 3); p = 0.01 (95% CI 1.24, 15.2).
Characterization of cytopenias after CAR-T
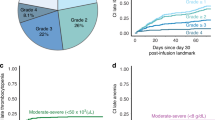
Table 3 characterizes ICAHT, anemia and thrombocytopenia occurring in patients after LD chemotherapy stratified by the CAR-HT score. Of the high CAR-T group, 16.9% (n = 22) had preexisting cytopenias compared to 1.2% (n = 1) of the low CAR-HT patients; Fig. 1a shows the distribution of unilineage-, bi- and pancytopenia occurring after BCMA-CART among the patients without preexisting cytopenias by CAR-HT status.

a Mosaic plot showing post–CAR-T cytopenia patterns stratified by CAR-HEMATOTOX (CAR-HT) score. b Sankey diagrams depicting changes in neutropenia grade at baseline, prior to pre–lymphodepleting chemotherapy to after CAR-T administration (ICAHT).
We examined how neutropenia grades changed from the pre-lymphodepletion period to the onset of ICAHT, stratified by the CAR-HT group. Figure 1b presents these transitions using Sankey plots, which visually track how patients’ neutropenia grades shifted across timepoints within the high and low CAR-HT groups. We found no statistically significant associated between the pre-LD neutropenia grade and subsequent ICAHT severity within either group (p = 0.11 high CAR-HT, p = 0.37 low CAR-HT). ICAHT severity was significantly associated with CAR-HT score (p = 0.0036). High CAR-HT patients had higher rates of severe ICAHT (Grade 3: 45.4%, Grade 4: 5.4%) compared to low CAR-HT patients (23.3% and 1.2%, respectively) and grade 0 ICAHT was more frequent in the low CAR-HT group (40.7% vs. 16.9%). Thirty-four patients had preexisting grade 3-4 anemia when receiving their LD chemo, and a further 80 patients experienced onset of anemia after CAR-T: 24.4% (n = 21) low CAR-HT patients, and 43.1% (n = 56) of the high CAR-HT patients. Twenty-eight patients (12.5%) had pre-existing grade 3 thrombocytopenia prior to LD chemo, and 41.5% (n = 93) developed grade 3 and higher thrombocytopenia after their LD chemotherapy. Table 3 categorizes the type and time to grade 3 and higher cytopenias occurring in patients by the CAR-HT score.
Supportive interventions
Patients with high CAR-HT scores were twice as likely to receive red cell transfusions and almost 7 times more likely to receive both red cell and platelet transfusions than patients with low CAR-HT score (p < 0.0001) as shown in Fig. 2a. Figure 2b illustrates the patient level transfusion burden stratified by CAR-HT score over 6 months after CAR-T therapy; patients with high CAR-HT scores had a greater transfusion burden, with a median of 1 PRBC transfusion (range 0–38; n = 63) compared to a median of 0 in the low CAR-HT group (range 0–5; n = 55; p < 0.0001). Platelet transfusion requirements over 6 months were markedly skewed in both high and low CAR-HT groups. Among patients with high CAR-HT scores, the median number of transfusions was 0, but the mean was 6.0 (range 0–51), reflecting a substantial transfusion burden in a subset of patients. In contrast, the low CAR-HT group had a mean of 0.64 transfusions (range 0–12), median of 0; p < 0.0001). The distribution of platelet transfusions among patients with high CAR-HT scores was right-skewed, with a median of 0 but a mean of 6.0 transfusions over 6 months. Although the majority did not require transfusions, a notable subgroup had high transfusion needs, with a maximum of 51 platelet transfusions. Most low CAR-HT patients required no transfusions, underscoring the discriminative utility of the CAR-HT score in forecasting transfusion burden. The transfusion requirement by low and high CAR-HT score for each of the additional cytopenia- directed interventions (GCSF, TPO agonist, GCSF and TPO agonist and stem cell boost), is shown in Fig. 2c, d. Patients who required G-CSF and TPO combination and stem cell boosts required highest level of transfusion dependence consistent with the expected degree of marrow suppression in the high CAR-HT patients (p < 0.0001). In the low CAR-HT group, patients requiring GCSF and TPO agonist required significant dual transfusion dependence (p < 0.0001).

a Stacked bar plot showing transfusion type by CAR-HEMATOTOX (CAR-HT) score. b Beeswarm plot displaying individual patient-level transfusion burden (red cell and platelet units) over 6 months following CAR-T therapy, stratified by CAR-HT score. c Transfusion burden by cytopenia-directed intervention in patients with low CAR-HT scores. d Transfusion burden by cytopenia-directed intervention in patients with high CAR-HT scores.
Cumulative incidence functions were performed to evaluate the overall time to recovery of neutrophils >500 U/L, platelets >50 and hemoglobin >8 g/dl by the cytopenia-directed intervention, as shown in Fig. 3. Patients who had no cytopenia-directed intervention demonstrated fastest time of neutrophil recovery, median 10 days (95% confidence interval [CI], 6–14). As anticipated, the use of supportive interventions such as G-CSF + TPO agonist, TPO agonist alone, or stem cell boost was associated with more profound cytopenias in these groups (median recovery: 115 (95% CI, 17–115), 71 (95% CI, 24–151), and 78 (95% CI, 24–151) days, respectively; p = 0.021). The time to recovery of Hb > 8 g/dl was longest in the G-CSF + TPO agonist group, at 126 days (95% CI, 68–184) followed by stem cell boost group, median 100 days (95% CI, 11-NR), then the GCSF group median 80.5 days (95% CI, 29–132). The no intervention group recovered Hb median 3 days after LD chemo (95% CI, 1–12 days), p = 0.0017. Time to platelets > 50 was shortest in the no intervention group (25 days, 95% CI, 7–61), and the longest time was the stem cell boost cohort (214 days, 95% CI, 78–244), followed by the TPO agonist group (138, 95% CI, 44–170); p = 0.014.

Cumulative incidence plots showing time to recovery of (a) absolute neutrophil count (ANC > 500/μL), (b) hemoglobin ≥8 g/dL, and (c) platelet count >50 × 10⁹/L, stratified by cytopenia-directed intervention.
Administration of growth factors and stem cell boost
Excluding the 9 patients who received stem cell boost, 56 patients received G-CSF for ANC < 0.5 × 109/L and 18 patients for ANC < 1.0 × 109/L. Time to G-CSF initiation was 12 days (6–47) and 13 days (5–146) in the patients in the high CAR-HT group tended to require longer time to ANC > 0.5 median 28.5 (9–39) days, compared to low CAR-HT 7.5 days (3–36); but this was not statistically significant (p = 0.07). In the analysis of time to ANC > 1.0 after G-CSF, this was statistically significant longer for the high CAR-HT group, 49 days (95% CI, 32–81) compared to low CAR-HT group 15 days (95% CI, 3–49) p = 0.05. (Fig. 4a, b)

a, b Cumulative incidence plots showing time to absolute neutrophil count (ANC) recovery >500/μL and >1000/μL after G-CSF administration, stratified by CAR-HEMATOTOX (CAR-HT) score. c Swimmer plot displaying individual time to ANC, hemoglobin, and platelet recovery following stem cell boost. Each line represents one patient, with recovery marked by colored dots and censoring indicated by black crosses.
To assess the safety of G-CSF administration following CAR-T therapy, we examined the association between G-CSF use and the incidence of any grade of CRS and by CAR-HT score. Among those with high CAR-HT scores, CRS occurred in 92% (n = 46) in those who received G-CSF, compared to 82.5% (n = 66) in those who did not receive G-CSF; however, this difference was not statistically significant (p = 0.13). Similarly, in the low CAR-HT group, CRS incidence was 79.2% (n = 19) in the G-CSF group and 64.5% in the non–G-CSF group (p = 0.19).
Of the 121 patients with at least grade 3 thrombocytopenia (93 with new onset, 28 with preexisting) after CAR-T, only 7.4% (n = 9, 8 high CAR-HT and 1 low CAR-HT) were treated with TPO agonist at median 129 days (range 49-303) from LD chemotherapy. Among the 6 patients who received TPO agonist alone without a stem cell boost (5 with high CAR-HT scores, 1 with low), the median time to sustained platelet recovery ( ≥ 50 × 10⁹/L) could not be estimated, as 4 patients had not reached this threshold by the end of the observation period. All patients who failed to respond to TPO agonists, were found to have progression of their myeloma on further evaluation.
A total of nine patients received a stem cell boost following CAR-T therapy (8 with high CAR-HT scores, 1 with low CAR-HT) at median 122 (51–209) days after administration of LD chemotherapy. The median dose of stem cells was 3.72 × 106 (2.4–6.98). Indications included pancytopenia in seven patients, isolated thrombocytopenia in one, and combined anemia with thrombocytopenia in one. None of these patients had evidence of evolution to myeloid neoplasm at the time of stem cell boost. The median time to hematologic recovery following boost was 21 days for ANC > 1000/μL (95% CI, 6–45), 43 days for hemoglobin >8 g/dL (95% CI, 6–70), and 21.5 days for platelets >50 × 10⁹/L (95% CI, 6–46.5). One patient was censored in the hemoglobin recovery analysis due to disease progression, while all patients achieved neutrophil and platelet recovery. Three patients continued to receive G-CSF and one, G-CSF and TPO agonist, after the boost was administered until adequate recovery. The swimmers plot in Fig. 4c outlines the recovery for each patient.
Infections
Among the 78 patients who developed infections in the first year following CAR-T therapy, bacterial infections were the most common, occurring in 43.6% (n = 34), followed closely by viral infections in 41.0% (n = 32), with cytomegalovirus (CMV) accounting for 70.6% (n = 24) of these cases. With the exception of one case of CMV retinitis, all infections were limited to CMV viremia without end-organ disease. By institutional guidelines, CMV is monitored during the first 30 days after CAR-T administration, and monthly for the first year. If there is low level CMV viremia by PCR, weekly checks are conducted until negative. For high level viremia > 100 IU/mL, treatment with valganciclovir is initiated. Fungal infections were observed in 6.4% (n = 5), and 8.9% (n = 7) of patients experienced both viral and bacterial infections, with CMV contributing to 20% (n = 6) of the viral co-infections. Among the 224 patients evaluated, infectious complications occurred more frequently in the high CAR-HT group compared to the low CAR-HT group (54/130 [41.5%] vs. 22/86 [25.6%]), with the remaining 1/8 (12.5%) occurring in patients with missing CAR-HT scores. (p = 0.022), indicating a meaningful association between CAR-HT risk and infectious complications following CAR-T therapy. As expected, the higher infection rates among those requiring G-CSF [G-CSF alone (35.2%, n = 25), G + TPO (33.3%, n = 1)] or stem cell boost (77.8%, n = 7) compared to those managed without cytopenia-directed intervention (24%, n = 19) or transfusion support only (39%, n = 23) (p = 0.022) likely reflect more severe and prolonged neutropenia necessitating these interventions.
Evolution to myelodysplastic syndrome
Four patients developed confirmed evolution to myelodysplastic syndrome/myeloid neoplasm without relapse at median 11 months (range 4–18) after administration of LD chemotherapy. All these patients received prior ASCT. These patients required only transfusion support in the first three months, without need for growth factor support or stem cell boost administration. None of the patients were evaluated for clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenia of undetermined significance (CCUS) prior to receiving their CAR-T therapy.
Survival
The progression-free survival (PFS) for the high CAR-HT group was 6.9 months (95% CI, 5.2–8.5), low CAR-HT group 8.3 months (95% CI, 5.9–11.3) and missing CAR-HT group 3.84 months (95% CI, 0.98–11); with no difference in survival between the groups (log-Rank p = 0.09) [Fig. 5]. We performed two multivariable Cox proportional models. The first tested whether specific cytopenia features predicted PFS. It included duration of neutropenia <500/μL, duration of thrombocytopenia <20 × 10⁹/L and hemoglobin<8 g/dl, CAR-HT and high-risk cytogenetics. Only duration of hemoglobin below 8 g/dL showed an independent association with progression-free survival (HR 1.0039 per day, 95 percent CI 1.001–1.0066, p 0.006). Duration of neutropenia, thrombocytopenia, CAR-HT score, and high-risk cytogenetics were not associated, and overall, the model was not significant (p value = 0.106) (Supplemental data). The second model was used to evaluate the impact of cytopenia-directed interventions (no intervention, transfusion only, G-CSF only, TPO agonist only, G-CSF plus TPO agonist, or stem cell boost) on progression-free survival (PFS) stratified by CAR-HT score; and across all CAR-HT strata, none of the interventions were associated with a statistically significant difference in PFS (high CAR = HT overall model p = 0.82, low CAR-HT, overall model p = 0.71) and among those with missing CAR-HT, the small sample size limited interpretability (Supplemental data).

Kaplan–Meier curves showing (a) progression-free survival (PFS) and (b) overall survival (OS) stratified by CAR-HT score (high, low, or missing).
The overall survival similarly did not differ between the high, low or missing CAR-HT groups. Median overall survival was 12.1 months in the high CAR-HT group (95% CI, 6.48–19.0), 15.0 months in the low CAR-HT group (95% CI, 6.4–20.0), and 16.5 months in those with missing CAR-HT scores (95% CI, 9.5–45.8) [(Log-rank p = 0.1794]. With multivariable Cox proportional hazards modeling, there was no additional impact of the particular cytopenia-directed intervention (overall model effects not shown; High CAR-HT p = 0.21, low CAR-HT p = 0.62) or number of prior lines of therapy on overall survival (overall model effects not shown; High CAR-HT p = 0.35, low CAR-HT p = 0.73)
Survival analysis by CAR-T product and CAR-HT score
When analyzed by CAR T product, median progression-free survival was 7.1 months for idecabtagene vicleucel, 8.8 months for ciltacabtagene autoleucel, and 6.2 months for other BCMA products, with no statistically significant difference across groups by log-rank testing (p = 0.33). However, when outcomes were stratified by CAR-HT score, the pattern shifted. For patients with low CAR-HT score, patients treated with idecabtagene vicleucel demonstrated longer PFS than ciltacabtagene autoleucel (median 13.1 vs 7.9 p < 0.001) [Supplemental data]. For those with high CAR-HT score, ciltacabtagene autoleucel demonstrated the survival advantage with median PFS 9.1 (95%CI 3.7–17.1) versus median PFS 5.49 months (95% CI 3.25–7.49) for idecabtagene vicleucel, and 12.2 (95%CI 1.9–18.1) months for other BCMA CART products [Log rank p = 0.021] [Supplemental data].
Discussion
The CAR-HT score emerged as a powerful risk stratification tool in this cohort [7]. High CAR-HT patients had earlier onset of anemia and thrombocytopenia, more frequently experienced ICAHT and demonstrated higher prevalence of pancytopenia reflecting the predictive value of the score [7, 14]. CAR-HT score was predictive of ICAHT after LD chemo more so than preexisting neutropenia alone [7]. High CAR-HT patients required significantly more transfusion support, which offers practical information for clinicians counseling patients about expected supportive care needs after CAR-T. Moreover, the magnitude of transfusion dependence in high CAR-HT patients suggests a role for earlier risk adapted strategies to reduce the clinical and resource burden associated with prolonged hematotoxicity [15].
Prior studies established the predictive value of CAR-HT for cytopenias [7]. This analysis extends its utility by defining the depth, timing and trajectory for the hematologic toxicity, characterization of transfusion burden and timing of interventions across over a decade of CAR-T practice. These data highlight how supportive-care intensity evolves as institutional familiarity increases, providing context for designing risk-adapted supportive care practices. This cohort also provides one of the most detailed real-world descriptions of longitudinal transfusion burden and infection patterns following BCMA CAR-T.
TPO agonists were used in relatively few patients and generally late in the course. Most patients who received TPO agonist alone did not achieve platelet recovery and were later found to have myeloma progression. This aligns with larger retrospective data showing the limited benefit of TPO agonists after CAR-T [16]. These findings support reassessment for occult myeloma progression in patients with isolated refractory thrombocytopenia [16]. G-CSF was administered to mitigate the duration of ICAHT and high CAR-HT patients consistently required longer time to ANC recovery >1000/μL despite its use. Reports including lymphoma patients have reported increased risk of grade ≥2 CRS with G-CSF [17], but in our cohort the same did not apply. Use was conservative and often delayed until the highest risk of CRS had passed [18]. This reflects earlier treatment practices, since we have now shifted towards earlier proactive G-CSF use for severe neutropenia, after day 14 [18]. Stem cell boosts were effective [10, 13], even though only 4% of patients required them. Recovery was reliable across all lineages except for one patient who relapsed one month after the boost. Some patients received growth factors concurrently, reflecting the ongoing supportive measures necessary to support patients through hematopoietic recovery.
The question of whether all BCMA CAR-T candidates should undergo pre-collection of stem cells, regardless of intent to transplant, remains unresolved. Although our center routines collects sufficient for two transplants, typically 6 × 10⁶ CD34⁺ cells/kg, only a small proportion ultimately required a boost, and early intervention strategies may mitigate this need. Prior studies have highlighted the cost and low utilization rate of cryopreserved stem cells [19,20,21].
We did not observe a significant difference in PFS or OS by CAR-HT score [7] and none of the cytopenia-directed interventions influenced survival outcomes. This differs from findings in lymphoma cohorts such as the population based validation by de Boi et al. [22]. This discrepancy likely reflects key differences in disease biology and treatment context. Although some studies link prolonged hematologic toxicity to inferior outcomes, robust survival associations with the CAR-HT score in BCMA CAR-T myeloma remain limited. For instance, Li H et al. reported shortened PFS and OS in myeloma patients with prolonged severe cytopenias [23]. In contrast, Rejeski K et al. validated the CAR-HT score in myeloma but did not report strong independent associations with PFS/OS [7]. Further Avigan et al. compared Duffy null vs Duffy non-null myeloma and demonstrated similar ANC prior to lymphodepletion, comparable G-CSF support in the first 100 days after BCMA CAR-T and despite delayed neutrophil recovery in the Duffy-null group, there was no adverse impact on survival [24]. These results reinforce that CAR-HT score is a predictive biomarker of post CAR-T hematologic toxicity but not as a prognostic marker of survival [10, 17, 25].
In exploratory analyses, we evaluated if PFS differed by infused CAR-T product, stratified by CAR-HT score. Though we found differences in PFS between the product by CAR-HT score, this signal is likely not reliable and we have interpreted these differences with caution given the relatively small sample size and influence of unmeasured confounders. The variation across strata likely reflect case mix and disease characteristics within each subgroup rather than a true biologic association between CAR HT score and product efficacy. Although retrospective series have described differential toxicity and survival profiles between idecabtagene vicleucel and ciltacabtagene autoleucel, there are no published data demonstrating a statistically validated interaction between CAR-HT score and PFS by CAR-T product in multiple myeloma.
CHIP status was not available for this cohort. CHIP is common in 30-50% of CAR-T recipients, may be associated with increased inflammatory toxicity, and in some series, delayed hematopoietic recovery or later myeloid neoplasms [26,27,28]. However, the relationship to early post CAR-T cytopenias is inconsistent [26, 28, 29]. Universal CHIP screening prior to lymphodepletion is therefore not supported at this time, and targeted evaluation should be performed for prolonged and unexplained cytopenias. Prospective studies incorporating baseline myeloid sequencing will be essential to define how clonal hematopoiesis influences hematologic toxicity and long-term marrow fitness after CAR-T.
Our study has limitations. Patients were heavily pretreated and may not reflect marrow reserve in contemporary second line Cilta-cel recipients [1]. Growth factors and stem cell boosts are now used much earlier than earlier treatment eras represented in this cohort. In our study, the median time to stem cell boost was 122 days, similar to the 116 day median reported by Mohan et al. [13]. By contrast, Davis et al., reported a much shorter median time of 53 days for patients between 2020 and 2023, likely reflecting increased clinical familiarity with this strategy and evolving supportive care practices [10].
This study was not designed to propose a new therapeutic intervention but to describe the depth and duration and management of cytopenias occurring after BCMA CAR-T. These data offer clinicians, particularly community hematologists who assume joint care after the post infusion period, a real-world view of the expected count recovery and sequence of available interventions. By outlining the full spectrum of cytopenias, this work supports coordinated care between CAR-T centers and local practices, and provides a practical framework for anticipating recovery, managing prolonged cytopenias and counseling patients as CAR-T moves into earlier treatment lines.
Our findings further support the integration of CAR-HT into routine clinical practice as practical, accurate biomarker for hematotoxicity risk. High-risk patients may benefit from earlier G-CSF initiation, closer monitoring and timely evaluation for progression when thrombocytopenia persists. Stem cell boosts remains an effective option for selective patients with multilineage cytopenias. Strategies targeting the biologic drivers of cytopenias and prophylactic measures based on CAR-HT risk will be important areas of future study [5].
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. All relevant data supporting the findings of this study will be made available to qualified researchers for non-commercial use, in a manner that does not compromise participant confidentiality.
References
San-Miguel J, Dhakal B, Yong K, Spencer A, Anguille S, Mateos M-V, et al. Cilta-cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. New England Journal of Medicine. 2023;389:335–47.
Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. New England Journal of Medicine. 2023;388:1002–14.
Delforge M, Shah N, Rodríguez-Otero P, Braverman J, Dhanda D, Shi L, et al. Updated Health-Related Quality of Life Results from the KarMMa Clinical Study in Patients with Relapsed and Refractory Multiple Myeloma Treated with the B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy Idecabtagene Vicleucel (ide-cel, bb2121). Blood. 2021;138:2835.
Jain T, Olson TS, Locke FL. How I treat cytopenias after CAR T-cell therapy. Blood. 2023;141:2460–9.
Palacios-Berraquero ML, Rodriguez-Marquez P, Calleja-Cervantes ME, Berastegui N, Zabaleta A, Burgos L, et al. Molecular mechanisms promoting long-term cytopenia after BCMA CAR-T therapy in multiple myeloma. Blood Adv. 2024;8:5479–92.
Cheng H, Shao L, Wang D, Chen Y, Sun Y, Chen Z, et al. Comprehensive characterization of cytopenia after chimeric antigen receptor-T cell infusion in patients with relapsed or refractory multiple myeloma. Cytotherapy. 2025;27:16–24.
Rejeski K, Hansen DK, Bansal R, Sesques P, Ailawadhi S, Logue JM, et al. The CAR-HEMATOTOX score as a prognostic model of toxicity and response in patients receiving BCMA-directed CAR-T for relapsed/refractory multiple myeloma. Journal of Hematology & Oncology. 2023;16:88.
Ma S, Li H, Zhou D, Zhang X, Shi M, Cao J, et al. Associations of granulocyte colony-stimulating factor with toxicities and efficacy of chimeric antigen receptor T-cell therapy in relapsed or refractory multiple myeloma. Cytotherapy. 2023;25:653–8.
Cook J, Gonsalves WI, Gertz MA, Visram A, Warsame R, Lacy MQ, et al. Success of the autologous stem cell boost after autologous graft failure in multiple myeloma and AL amyloidosis. Bone Marrow Transplant. 2022;57:1007–9.
Davis JA, Sborov DW, Wesson W, Julian K, Abdallah AO, McGuirk JP, et al. Efficacy and Safety of CD34+ Stem Cell Boost for Delayed Hematopoietic Recovery After BCMA Directed CAR T-cell Therapy. Transplant Cell Ther. 2023;29:567–71.
Bhutani M. Boosting CAR T cytopenias with backup autologous stem cells. Transplantation and Cellular Therapy. 2023;29:535–6.
Yadav K, Patel TH, Bachu R, Esselmann J, Rein LE, Szabo A, et al. Application of Stem Cell Boost for Immune Effector Cell Associated Hematotoxicity (ICAHT) Following BCMA Directed Chimeric Antigen Receptor (CAR) T-Cell Therapy for Multiple Myeloma (MM). Blood. 2024;144:254.
Mohan M, Szabo A, Patwari A, Esselmann J, Patel T, Bachu R, et al. Autologous stem cell boost improves persistent immune effector cell associated hematotoxicity following BCMA directed chimeric antigen receptor T (CAR T) cell therapy in multiple myeloma. Bone Marrow Transplantation. 2024;59:647–52.
Palacios-Berraquero ML, Rodriguez-Marquez P, Calleja-Cervantes ME, Berastegui N, Zabaleta A, Burgos L, et al. Molecular mechanisms promoting long-term cytopenia after BCMA CAR-T therapy in multiple myeloma. Blood Advances. 2024;8:5479–92.
Jacobs JW, Diaz M, Arevalo Salazar DE, Tang A, Stephens LD, Booth GS, et al. United States blood pricing: A cross-sectional analysis of charges and reimbursement at 200 US hospitals. American Journal of Hematology. 2023;98:E179–E82.
Waddell D, Collins J, Sadrameli S. Utility of Thrombopoietin Receptor Agonists for Prolonged Thrombocytopenia After Chimeric Antigen Receptor T-cell Therapy. Transplantation and Cellular Therapy. 2025;31:238.e1–.e12.
Cao CL, Martinez A, Dains J. Growth Factor in the Setting of CAR T-Cell Therapy: To Use or Not to Use. J Adv Pract Oncol. 2024;15:253–64.
Lin Y, Qiu L, Usmani S, Joo CW, Costa L, Derman B, et al. Consensus guidelines and recommendations for the management and response assessment of chimeric antigen receptor T-cell therapy in clinical practice for relapsed and refractory multiple myeloma: a report from the International Myeloma Working Group Immunotherapy Committee. The Lancet Oncology. 2024;25:e374–e87.
Ahmed N, Li L, Rojas P, Covut F, Reese-Koc J, Kolk M, et al. Significant costs and low utilization of stored peripheral blood stem cells for salvage autologous transplant in multiple myeloma patients including those meeting mSMART criteria. Bone Marrow Transplant. 2021;56:1458–61.
Krummradt F, Sauer S, Pavel P, Klein EM, Schmitt A, Kriegsmann M, et al. Storage, Utilization, and Disposal of Hematopoietic Stem Cell Products in Patients with Multiple Myeloma. Biol Blood Marrow Transplant. 2020;26:1589–96.
Chhabra S, Thapa B, Szabo A, Konings S, D’Souza A, Dhakal B, et al. Utilization and Cost Implications of Hematopoietic Progenitor Cells Stored for a Future Salvage Autologous Transplantation or Stem Cell Boost in Myeloma Patients. Biol Blood Marrow Transplant. 2020;26:2011–7.
de Boer JW, Keijzer K, van Dorp S, Mutsaers PGNJ, Niezink AGH, van Doesum JA, et al. Population-based validation of the CAR-HEMATOTOX for hematotoxicity, infections, and survival after CART in R/R LBCL. Blood Advances. 2025;9:5641–50.
Li H, Zhao L, Sun Z, Yao Y, Li L, Wang J, et al. Prolonged hematological toxicity in patients receiving BCMA/CD19 CAR-T-cell therapy for relapsed or refractory multiple myeloma. Front Immunol. 2022;13:1019548.
Avigan ZM, Bodnar S, Pan D, Catlett J, Richter J, Sanchez LJ, et al. Delayed neutrophil recovery following BCMA CAR-T therapy in Duffy-null myeloma does not impact severe infections or survival. Blood Adv. 2025;9:202–6.
Rejeski K, Subklewe M, Aljurf M, Bachy E, Balduzzi A, Barba P, et al. Immune effector cell–associated hematotoxicity: EHA/EBMT consensus grading and best practice recommendations. Blood. 2023;142:865–77.
Miller PG, Sperling AS, Brea EJ, Leick MB, Fell GG, Jan M, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 2021;5:2982–6.
Teipel R, Kroschinsky F, Kramer M, Kretschmann T, Egger-Heidrich K, Krüger T, et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Advances. 2022;6:1941–6.
Goldsmith SR, Shouse G, Wong FL, Bosworth A, Iukuridze A, Chen S, et al. Clonal Hematopoiesis is Associated With Severe Cytokine Release Syndrome in Patients Treated With Chimeric Antigen Receptor T-Cell (CART) Therapy. Transplant Cell Ther. 2024;30:927.e1–.e9.
Teipel R, Kroschinsky F, Kramer M, Kretschmann T, Egger-Heidrich K, Krüger T, et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Adv. 2022;6:1941–6.
Author information
Authors and Affiliations
Contributions
JC and SG contributed equally to the design and writing of the manuscript. YL provided overall study oversight and contributed to manuscript development. JC, SG, NA, MR, CG, LSN, RB, and AC were responsible for data collection and organization. ADMSC, RW, DD, WG, TK, MG, SH, NL, PK, LH, AF, MH, LB, UY, EWN, SC, RF, SA, RP, and SK contributed to data interpretation, critical review of the manuscript, and provided important intellectual content. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
RP: advisory board role for Sanofi Aventis and Astra Zeneca, research funding from Bristol Myers Squibb Foundation and GlaxoSmithKline; SC: honoraria from Sanofi, Ascentage Pharma, Sobi, Legend Biotech, Pfizer, BMS and research funding from Johnson & Johnson, Takeda, C4 Therapeutics, Cynata Therapeutics, Cullinan Therapeutics, Abbvie, Ascentage Pharma, AstraZeneca; MG: personal fees from Ionis/Akcea, honorarium from Alnylym, personal fees from Prothena, personal fees from Sanofi, personal fees from Janssen, personal fees for Data Safety Monitoring board from Abbvie & Arcellex, fees from Johnson & Johnson, Honoraria for Astra Zeneca, Medscape, Dava Oncology. Alexion; PK: Honoraria: Pharmacyclics, Sanofi, BeiGene, MustangBio, AstraZeneca, AbbVie. Consulting or Advisory Role: Sanofi. Research Funding: Amgen, Takeda, Sanofi, AbbVie, GlaxoSmithKline, Sorrento Therapeutics, Karyopharm Therapeutics, Regeneron, Ichnos Sciences, Bristol Myers Squibb/Celgene; PLB: Consultant: Oncopeptidfes. Salarius, Radmetrix, Omeros, CellCentric, AbbVie, Pfizer; SK: Consulting or Advisory Role: Takeda, Janssen Oncology, Genentech/Rocher, Abbvie, BMS/Celgene, Pfizer, Regeneron, Sanofi, K36 Therapeutics; travel, accomodation and expenses: Abbvie, pfizer; Research funding: Takeda, Abbvie, Novartis, Sanofi, Janssen Oncology, MedImmune, Roche/Genentech, CARsgen Therapeutics, Allogene Therapeutics, GSK, Regeneron, BMS/Celgene; SA: Consulting or Advisory Role: Takeda, BeiGene, GlaxoSmithKline, Sanofi, Pharmacyclics, BMS, Amgen, Janssen, Regeneron, Cellectar. Research Funding: Pharmacyclics, Janssen Biotech, Cellectar, BMS, Amgen, GlaxoSmithKline, AbbVie, Ascentage Pharma, Sanofi; RF: consultancy for AbbVie, Adaptive, Amgen, Apple, BMS/Celgene, GSK, Janssen, Karyopharm, Pfizer, RA Capital, Regeneron, Sanofi. Scientific advisory board: Caris Life Sciences. Board of directors: Antengene. Patent for FISH in multiple myeloma; YL: advisory board role for Janssen, Sanofi, BMS, Regeneron, Genentech, Tesserae, Legend, NexT Therapeutics, steering committee for Janssen, Kite/Gilead, research funding from Janssen, BMS, scientific advisory board for Nonimmune, Caribou and sata safety monitor board for Pfizer. Others: No conflicts of interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cook, J., Gupta, S., Abdallah, N. et al. Management of hematological toxicities after BCMA-directed CAR-T cell therapy. Blood Cancer J. 16, 49 (2026). https://doi.org/10.1038/s41408-026-01455-5
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-026-01455-5
