
Abstract
T-cell lymphoblastic lymphoma (T-LBL) and T-cell acute lymphoblastic leukemia (T-ALL) originate from thymic T-cell precursors, with ongoing debate on whether they are variants of the same disease or distinct entities. For 211 patients, including pediatric and adult T-ALL and T-LBL cases, targeted next-generation sequencing and SNP-arrays were performed, and single-nucleotide variants, indels and copy-number variants (CNVs) were analyzed. We aimed to assess genetic differences between T-ALL and T-LBL across age. Generally, mutational landscape analysis identified mutated PHF6 being associated with higher, NOTCH1 with lower age at diagnosis for both T-LBL and T-ALL. Association of CNVs with higher age was evident for T-ALL, but not T-LBL. Analysis of clonal evolution revealed that CNVs – especially deletions and LOH in chromosome 9 (LOH_in_9p) – were observed as first mutational event in both pediatric T-ALL and T-LBL. The sequence of genetic events, starting with LOH_in_9p followed by mutations in NOTCH1, was significantly more frequent in pediatric T-ALL and T-LBL. Detailed evaluation of the patients’ individual clonal evolution indicated that the proportion of malignant cells without NOTCHMT determines the risk of relapse (hazard ratio 1.032, p = 4.65*10−5). In T-ALL, aside from MRD, validated molecular markers for risk-group stratification remain limited. Our data suggest that molecular metrics analogous to those in T-LBL may help refining risk stratification in T-ALL as well.

Similar content being viewed by others

T-cell lymphoblastic lymphoma (T-LBL) and T-cell acute lymphoblastic leukemia (T-ALL) arise from malignant transformation of immature T-cell precursors. They are clinically distinguished by the extent of bone marrow (BM) infiltration at diagnosis, with T-ALL defined by ≥25% BM involvement. T-ALL represents 15% of childhood ALL cases, with current treatment protocols achieving an event-free survival (EFS) of ~85%, but relapse EFS is only ~20%. In adults, T-ALL comprises ~25% of ALL cases with a median age of 25–30 years and overall survival rates of 40–50%, again with poor outcomes at relapse [1, 2].
T-LBL is the second most common Non-Hodgkin lymphoma (NHL) subtype in children and adolescents, with a median onset age of 8.8 years and EFS rates of ~80%. Relapsed pediatric T-LBL patients have a poor prognosis, with survival rates of 10–30% [3]. In adults, T-LBL accounts for 90% of all LBL cases, with survival of 50–60% and dismal outcomes after relapse [4]. The World Health Organization classifies T-LBL and T-ALL as a single entity due to overlapping clinical and biological features, yet recent studies have revealed molecular differences [5], including TRB::NOTCH1 fusions that are restricted to pediatric T-LBL and associated with high relapse risk [6].
Clonal evolution analyses allow assessment of tumor progression over time. However, these approaches are not widely used clinically, as automated reconstruction tools often generate unreliable results [7] and single-cell DNA sequencing still faces technical and analytical barriers. Existing studies primarily examine single-nucleotide variants (SNVs) and indels while modeling their underlying copy number. Integrated analyses combining SNVs, indels, and copy number variants (CNVs) remain limited. Here, we use next-generation sequencing (NGS) and SNP-array data to characterize molecular genetic differences between pediatric and adult T-LBL and T-ALL and evaluate their clonal evolution.
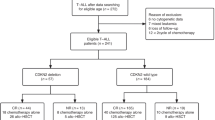
Clinical characteristics of the four subgroups, pediatric T-LBL, pediatric T-ALL, adult T-LBL and adult T-ALL, are summarized in Supplementary Table 1. Pediatric T-LBL samples were obtained from reference-reviewed cases from the NHL-BFM Registry 2012 and the LBL-2018 clinical trial, depending on material availability. Pediatric T-ALL cases represent relapses with 1:2 matched controls from the AIEOP-BFM ALL-2009 study [8].
We analyzed SNVs and indels in 211 age-stratified T-ALL and T-LBL cases using a 52-gene NGS panel and appreci8 for variant calling (Supplementary Table 2, Supplementary Methods). The total mutation burden did not differ significantly between subgroups (T-ALL: pediatric x̅ = 3.08 adult x̅ = 3.32; T-LBL pediatric x̅ = 3.13 adult x̅ = 3.28). Pathway-based grouping revealed distinct age- and entity-specific patterns, although NOTCH1 remained the most frequently mutated gene in all groups (Fig. 1A, Supplementary Data 1; detection threshold 1% in Supplementary Fig. 1).

A Variants detected by NGS (detection threshold 10%). Mutated genes are categorized by affected pathway. Pediatric and adult cases are distinguished. For pediatric cases, information on follow-up and time to relapse is provided, respectively. B Age-dependent distribution of the 22 genes with highest mutation frequencies in T-ALL and T-LBL. An ‘x’ marks a gene being mutated in one patient. Colors define the mutation density normalized for baseline age distribution, additionally depicted as grey curve on top of the plot (light grey: all patients, dark grey: relapsed patients). All genes mutated in at least 5% of T-ALL or T-LBL patients are included. Adjusted p-values (Wilcoxon Rank-Sum test) are reported if a gene is mutated in ≥5% of the patients per subgroup. C Cumulative frequencies of CNVs detected by SNP arrays. CNVs are distinguished by their precise copy number: CN = 0 and CN = 1 (red: deletion; del), CN = 2 (gold: loss of heterozygosity; LOH), CN = 3 and CN = 4 (blue: amplification; amp). Results for the subgroups T-ALL pediatric, T-ALL adult, T-LBL pediatric and T-LBL adult are displayed. Only primary tumor samples are included. Data are scaled for a maximum frequency of 50%. D Age-dependent distribution of the top-20 mutated chromosomes in T-ALL and T-LBL. An ‘x’ marks a chromosome being mutated in one patient. Colors define the mutation density normalized for baseline age distribution, additionally depicted as gray curve on top of the plot (light grey: all patients, dark grey: relapsed patients). All chromosomes mutated in at least 5% of T-ALL or T-LBL patients are included. Adjusted p values (Wilcoxon Rank-Sum test) are reported if a gene is mutated in ≥5% of the patients per subgroup.
Alterations in RAS, JAK-STAT, and PI3K signaling varied between cohorts. PI3K-pathway mutations (PIK3CA, PTEN, FBXW7) were enriched in pediatric cases, particularly in T-LBL. JAK1MT, SUZ12MT and DNM2MT showed no significant age associations, but JAK1MT occurred only in adult T-LBL (9%), SUZ12MT only in adult T-ALL (10%), and DNM2MT was more common in adult T-LBL (17% vs. 3%). PHF6MT were significantly more frequent in adult compared to pediatric T-LBL (34% vs. 13%; padj = 0.0410). In pediatric T-LBL, PHF6MT occurred exclusively in non-relapsed cases. In T-ALL, PHF6MT were more common in adults, but age-adjusted analyses showed no significant differences (adult 37% vs pediatric 19%; Fig. 1A, B; Supplementary Figs. 2, 3).
NOTCH1MT were more prevalent in pediatric than adult patients (T-ALL: 78% vs. 61%; T-LBL: 60% vs. 49%). In pediatric T-LBL, NOTCH1MT were enriched in non-relapsed cases (73% vs. 30%). Furthermore, NOTCH1 status was associated with time to relapse (p = 0.0256; Supplementary Fig. 4). Most NOTCH1 hotspot mutations affected the intracellular and HD domains, whereas truncating variants were confined to the PEST domain, consistent with constitutive activation [9] (Supplementary Fig. 5).
Exploratory analyses of unadjusted p-values revealed mutual exclusivity of NOTCH1 and PTEN in pediatric T-ALL (odds ratio OR = 0.06; p = 0.0194) and pediatric T-LBL (OR = 0.2; p = 0.0193). This pattern persisted in non-relapsed pediatric T-ALL (OR = 0.05; p = 0.0209) but not in relapsed cases. Pediatric T-LBL additionally showed exclusivity of NOTCH1 with PIK3R1, PIK3CA, and SETD1B. In adult T-LBL, NOTCH1 and NOTCH3 were mutually exclusive (OR = 0; p = 0.0046) (Supplementary Figs. 6, 7). Because pediatric samples were enriched for relapse events due to case–control design, relapse proportions do not reflect population incidence.
Overall, the mutational landscape of T-LBL and T-ALL was highly similar, consistent with prior studies [5]. Both share common driver pathways, including NOTCH1MT, FBXW7MT and PTENMT. PHF6 was the second most frequently mutated gene in our cohort (T-LBL 20%, T-ALL 29%), comparable to reported frequencies [5, 10]. In pediatric T-LBL, PHF6MT were confined to non-relapsed patients, aligning with favorable associations in adult T-LBL [11], whereas in pediatric T-ALL PHF6MT have been linked to inferior outcome [12], suggesting disease- and age-specific functional contexts.
Considering CNVs, mutational burden was comparable across the four subgroups, with mean counts of x̅ = 2.25 (sd = 2.30) in pediatric and x̅ = 4.34 (sd = 4.04) in adult T-ALL, and x̅ = 4.12 (sd = 3.68) in pediatric and x̅ = 5.66 (sd = 7.46) in adult T-LBL. Outcome-based comparisons in pediatric cases revealed no clear differences (pediatric T-ALL: non-relapsed x̅ = 2.71 (sd = 2.65) vs. relapsed x̅ = 1.33 (sd = 0.89); pediatric T-LBL: x̅ = 3.68 (sd = 3.83) vs. x̅ = 4.68 (sd = 3.50); Supplementary Data 2, Supplementary Methods).
CNVs affecting chr9p were the most abundant alterations across all subgroups. Deletions were more frequent in pediatric than adult patients (weighted frequencies: pediatric T-ALL 35% vs. adult 20%; pediatric T-LBL 32% vs. adult 21%). A distinguishing feature between pediatric T-LBL and T-ALL was the enrichment of dup20 in pediatric T-LBL, particularly in relapsed samples: chr20 duplications occurred in 7% of primary non-relapsed, but in 25% of primary relapsed, 27% of first-relapse, and 36% of second/third-relapse pediatric T-LBL cases. Conversely, chr9p duplications were more common in non-relapsed pediatric T-LBL (24% vs 5%). A slight enrichment of chr6q deletions was also observed in relapsed pediatric T-LBL (6% vs. 3%; Fig. 1C; Supplementary Fig. 8).
In T-ALL, age-dependent, cut-off–free analysis demonstrated significant associations between increasing age and CNVs in multiple chromosomes, including chr19 (padj = 0.0166), chr17 (padj = 0.0454), and chr12 (padj = 0.0398), whereas chr14 showed an inverse pattern (Fig. 1D). No comparable age-associated accumulation was observed in T-LBL. Deletions or LOH involving chr9 showed the highest density between 7 and 20 years, although this trend was not statistically significant.
Our findings indicate a clear age-related increase in CNV burden in T-ALL but not in T-LBL. CNVs involving chromosome 9, particularly deletions of chr9p, are well-characterized drivers in both entities. The CDKN2A/2B locus on 9p is frequently lost [5, 10].
For 149 patients, we integrated SNVs, indels and CNVs by cancer cell fraction and reconstructed clonal evolution [13] (details in Supplementary Methods). In pediatric T-ALL, NOTCH1MT frequently co-occurred with chr9p deletions (38.7%) and LOH (35.5%), whereas these combinations were less common in pediatric T-LBL (deletions 15%, LOH 19%). In pediatric T-LBL, NOTCH1MT strongly associated with chr9p CNVs, particularly in non-relapsed patients (Supplementary Figs. 9A, B, 10A, B). Variant-ordering analyses showed that clones harboring NOTCH1MT, and less frequently FBXW7MT, typically arose as descendants of chr9p deletion/LOH events. CNVs preceded small variants in both entities, except for chr9p duplications, which occasionally occurred as descendants of NOTCH1MT or FBXW7MT in non-relapsed pediatric T-LBL (Supplementary Figs. 9C, D, 10C, D).
Analysis of conserved evolutionary trajectories confirmed that CNVs in chr9p represent early founder events. In pediatric T-ALL, LOH_in_9p→NOTCH1 (padj = 0.0152) and LOH_in_9p→BCL11B→NOTCH1 (padj = 0.0046) were significantly enriched patterns. In pediatric T-LBL, highly significant trajectories included LOH_in_9p→NOTCH1 (padj = 0.0016), LOH_in_9p→NOTCH1→FBXW7 (padj = 0.0006) and LOH_in_9p→USP7→FBXW7 (padj = 0.0006). Only non-relapsed pediatric T-LBL showed successive CNVs (dup_in_9p), although not significantly (Fig. 2A, B; Supplementary Fig. 10E, F). Weighted nested-level analysis substantiated early occurrence of chr9 deletions/LOH in both diseases, preceding the accumulation of additional genomic alterations (pediatric T-ALL: nested-leveltree = 3.35, nested-levelchr9del/LOH = 1.04; pediatric T-LBL: nested-leveltree = 4.31, nested-levelchr9del/LOH = 1.13).

A, B To determine significant associations in the order of variants, conserved evolutionary trajectories are analyzed. Patterns of length 2 and 3 are considered. Trajectories observed in 10% (2 levels) and 5% (3 levels) of samples in the corresponding subgroups are included. Significant adjusted p-values are marked (*<0.05, **<0.01, ***<0.001). Data shows an increased pattern of CNVs – especially deletions and LOH affecting chr9p – preceding small variants. A T-ALL pediatric. B T-LBL pediatric. C–F Dolphin plots show the development of cancer cell fraction (CCF) over time. Respectively, shark plots are a simplification of the underlying phylogenetic tree, in the lower left corner. Vertical lines mark the time points at which samples were taken. Numbers specify the days passed since initial diagnosis. If >1 variant per gene is detected for a patient, information on the precise change on protein level is provided. C Patient with 3 relapses. D Patient with 2 relapses. E Patient with 1 relapse. F Patient without relapse. Despite just one time point being available, sequencing data allows to clearly identify two branches co-existing at primary time point: three frame-shift variants affecting PTEN are detected. Lying within a range of 20 bp on the genome, the variants can be clearly identified to affect different alleles. As no duplication of chr10 can be observed, these variants have to emerge on two or three different branches. However, considering CCFs, PTEN p.Q235fs and p.R233fs jointly developing on one branch, leading to bi-allelic drop-out, and PTEN p.N228fs developing on a parallel branch, is the only possible explanation for the observed data.
Individual clonal reconstructions by clevRvis [14] further illustrated these dynamics (Fig. 2C–F; Supplementary Data 3, Supplementary Figs. 12–24). UPN211 showed progressive branching from diagnosis to third relapse, with early CNVs followed by increasing mutational load and later emergence of highly branched TP53/PTEN-altered clones. UPN190 displayed LOH at high frequency already present in germline (Supplementary Fig. 11), two NOTCH1-marked branches that regressed, and later CNV-rich subclones at relapse 2. UPN193 exhibited a linear pattern with sequential CNVs followed by small variants, including a relapse-specific KMT2DMT (p.R5086*). In contrast, relapse-free UPN142 showed two branches with diverse PTEN variants descending from an early NOTCH1MT founder clone.
Across patients, NOTCH1MT frequently appeared as early founding events associated with favorable outcome, consistent with published data [5, 6, 13]. Cox regression–evaluating the difference between the frequency of the largest clone and clones with NOTCH1MT–showed that increasing proportions of tumor cells lacking NOTCH1MT significantly elevated relapse risk (hazard ratio HR = 1.032; 95%-CI = [1.016; 1.048]; p = 4.65 × 10⁻⁵). Thus, an increase of tumor cells with NOTCH1WT by 1% may increase the risk of relapse by 3.2%. Re-analysis of five published relapsed pediatric T-LBL cases showed that although 3/5 harbored NOTCH1MT at diagnosis, all retained subclonal NOTCH1-negative branches (5–25%) [15].
Together, these results suggest that the timing and clonal position of NOTCH1MT, rather than its mere presence, influences relapse risk. Early NOTCH1MT may initiate disease but do not appear to drive relapse, whereas subclones lacking NOTCH1MT may underlie treatment resistance. Integrating SNVs, CNVs, and clonal architectures highlights molecular distinctions between T-ALL and T-LBL and supports incorporating early chr9p CNV/LOH events and NOTCH1 status into future risk stratification, particularly in T-ALL where validated molecular markers remain limited.
Data availability
NGS data are available at SRA under accession number PRJNA1216741. SNP-array data are available at GEO under accession number GSE288263. Information on all detected small variants and CNVs and reconstructed clonal evolution can be found in Supplementary Data 1–3. Code is available at https://github.com/sandmanns/ce_tlbl_tall.
References
Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–84.
Neumann M, Beder T, Bastian L, Hänzelmann S, Bultmann M, Wolgast N, et al. Molecular subgroups of T-cell acute lymphoblastic leukemia in adults treated according to pediatric-based GMALL protocols. Leukemia. 2024;38:1213–22.
Landmann E, Burkhardt B, Zimmermann M, Meyer U, Woessmann W, Klapper W, et al. Results and conclusions of the European Intergroup EURO-LB02 trial in children and adolescents with lymphoblastic lymphoma. Haematologica. 2017;102:2086–96.
Lepretre S, Graux C, Touzart A, Macintyre E, Boissel N. Adult T-type lymphoblastic lymphoma: treatment advances and prognostic indicators. Exp Hematol. 2017;51:7–16.
Bontoux C, Simonin M, Garnier N, Lhermitte L, Touzart A, Andrieu G, et al. Oncogenetic landscape of T-cell lymphoblastic lymphomas compared to T-cell acute lymphoblastic leukemia. Modern Pathol. 2022;35:1227–35.
Te Vrugt M, Wessolowski JS, Randau G, Alfert A, Müller S, Scholten K, et al. Pediatric T-cell lymphoblastic lymphoma but not leukemia harbor TRB::NOTCH1 fusions with unfavorable outcome. Blood. 2024;144:1412–7.
Sandmann S, Richter S, Jiang X, Varghese J. Reconstructing clonal evolution-a systematic evaluation of current bioinformatics approaches. Int J Environ Res Public Health. 2023;20:5128.
Rizzari C, Möricke A, Valsecchi MG, Conter V, Zimmermann M, Silvestri D, et al. Incidence and characteristics of hypersensitivity reactions to PEG-asparaginase observed in 6136 children with acute lymphoblastic leukemia enrolled in the AIEOP-BFM ALL 2009 study protocol. Hemasphere. 2023;7:e893.
Weng, Ferrando AP, Lee AA, Morris W, JPt, Silverman LB, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71.
Pölönen P, Di Giacomo D, Seffernick AE, Elsayed A, Kimura S, Benini F, et al. The genomic basis of childhood T-lineage acute lymphoblastic leukaemia. Nature. 2024;632:1082–91.
Li Z, Song Y, Zhang Y, Li C, Wang Y, Xue W, et al. Genomic and outcome analysis of adult T-cell lymphoblastic lymphoma. Haematologica. 2020;105:e107.
Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89.
Sandmann S, Behrens YL, Davenport C, Thol F, Heuser M, Dörfel D, et al. Clonal evolution at first sight: a combined visualization of diverse diagnostic methods improves understanding of leukemic progression. Front Oncol. 2022;12:888114.
Sandmann S, Inserte C, Varghese J. clevRvis: visualization techniques for clonal evolution. GigaScience. 2023;12:giad020.
Khanam T, Sandmann S, Seggewiss J, Ruether C, Zimmermann M, Norvil AB, et al. Integrative genomic analysis of pediatric T-cell lymphoblastic lymphoma reveals candidates of clinical significance. Blood. 2021;137:2347–59.
Acknowledgements
Our work was supported by Deutsche Kinderkrebsstiftung (DKS 2014.11, DKS 2016.24 to BB), Deutsche Krebshilfe (reference 70112964, LBL 2018 trial to BB), German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), BMBF, BMG, MWK BW, German Childhood Cancer Foundation (DKS), RTL television, BILD hilft e.V. ("Ein Herz für Kinder") and Scheu family donation. Furthermore, we acknowledge support by the Open‑Access Publication Fund of the Medical Faculty of the Otto-von-Guericke-University Magdeburg.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors read and approved the manuscript. Study concepts: GR, BB, MB, MN, MtV, SS. Data acquisition: AM, CE, GR, MB, BB, MN. Quality control of data and algorithms: SS, TB, TL, JV, JMH. Statistical analysis: SS, MZ, TB, TL, JV. Manuscript preparation: SS, MtV, BB, GR, MH, MB, MN, JMH, SM, AA. Manuscript editing: All authors contributed equally. Manuscript review: TB, MN, SM, CE, CR, AM, JMH, MZ, MB, BB, JV. Correspondence to SS or BB.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The ethical approval for adults was granted under the reference number D 466/21 on March 31, 2021, by the Ethics Committee of the Medical Faculty at Christian-Albrechts-University (CAU) Kiel. The LBL Registry was approved by the Ethics Committee of the Faculty of Medicine at Justus Liebig University Giessen under the reference number 100/02. The NHL-BFM Registry 2012 received approval from the Ethics Committee of the Medical Association of Westphalia-Lippe and the University of Münster, with the reference number 2012-206-f-S. Similarly, the trial LBL 2018 was approved by the same ethics committee under the reference number 2019-037-f-A. The AIEOP-BFM ALL 2009 study was reviewed by the Ethics Committee of the University Medical Center Schleswig-Holstein. All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sandmann, S., te Vrugt, M., Randau, G. et al. Molecular variants, clonal evolution and clinical relevance in pediatric and adult T-cell lymphoblastic neoplasia. Blood Cancer J. 16, 57 (2026). https://doi.org/10.1038/s41408-026-01488-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-026-01488-w
