
Abstract
Myelodysplastic/myeloproliferative neoplasms (excluding CMML) are rare and heterogenous malignancies with recurrent and overlapping clinicopathological abnormalities. Classification and characterization continue to evolve, but outcomes remain unsatisfactory and allo-HCT is currently the only curative therapy. Experience with these diseases is limited by rarity, accounting for <1% of allo-HCT procedures in Europe annually. Transplant indications have recently been described by this group, however response assessment, post-transplant surveillance and management of poor graft function, splenomegaly and relapse are not well established. To address this gap, the European Society for Blood and Marrow Transplantation (EBMT) Practice Harmonization & Guidelines (PH&G) Committee and the Chronic Malignancies Working Party (CMWP) collaborated to develop a practical and expert consensus-based guideline. Criteria for remission confirmation are proposed, including timing/thresholds for morphological, molecular, cytogenetic and chimerism analysis, aiming to harmonize comparisons in registry data and between international series. Suggestions for managing poor graft function and splenomegaly are designed to broadly align with those for myelofibrosis and related disorders. Relapse post-transplant remains a frequent challenge and the lack of robust evidence, in both the pre-transplant/post-transplant setting, underpins a need for collaborative clinical trials. This document serves as a framework to model post-transplant care, guide future research and address unresolved questions.
Similar content being viewed by others
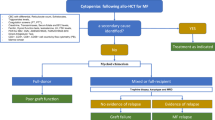
Introduction
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) are rare and heterogeneous clonal myeloid disorders with widely varying prognoses that frequently pose significant clinical management challenges [1]. Despite advances in our understanding of the clonal architecture of these disorders and refined classifications, development of novel, effective therapies across this disease arena has unfortunately met significant barriers [1, 2]. Regarding allogeneic haematopoietic cell transplantation (allo-HCT), contemporary clinical practice focussed on non-chronic myelomonocytic leukemia (CMML) MDS/MPN remains largely heterogeneous across centres as regards disease burden assessment, pre-transplant therapeutic strategies, management of bulky splenomegaly and therapeutic approaches to both poor graft function (PGF) and relapse. Moreover, there are no harmonized definitions of remission and relapse status for these disease entities. Despite remaining a relatively rare indication for allo-HCT in the context of overall global activity, there has been a sustained and upward trend in the numbers of patients being brought forward for allo-HCT, highlighting a need for more standardized approaches globally [3, 4]. Recently the European Society for Blood and Marrow Transplantation (EBMT) published much needed guidance on the role and indications of allo-HCT in ‘non-classical’ MPN and MDS/MPN (non-CMML) [3]. A remaining challenge, however, is the best way to define post allo-HCT remission for this heterogeneous group of disorders, as there are difficulties directly applying previously suggested response criteria developed for the non-transplant setting [5]. Moreover, harmonizing definitions of disease persistence and relapse post-transplant, as well as approaches regarding disease monitoring, management of PGF and therapeutic strategies for relapsed disease are urgently needed. This will provide more accurate assignment of responses for national and international registries and also for future prospective clinical trials. We hereby provide a practical, clinically relevant and up-to-date framework on how best to approach these issues regarding allo-HCT in the ‘non-CMML’ MDS/MPN.
Questions asked/issues to be addressed
Primarily, we consider best practice for which patients with MDS/MPN should be suggested for allo-HCT. Next, we aim to generate pragmatic definitions of disease persistence, remission, progression and relapse post allo-HCT for ‘non CMML’ MDS/MPN. In addition, we address the issues of peri-transplant splenomegaly evaluation and management, management of PGF and suggested strategies for relapse.
Current ‘State of the Art’
Epidemiology and classification
MDS/MPN syndromes are recognized by both the International Consensus Classification (ICC) and the World Health Organization (WHO) classification 5th edition (WHO-5), with some important differences between groups concerning both the recognized entities as well as classification criteria (Table 1,2) [6, 7]. From an epidemiological stance, all MDS/MPN are ‘ultrarare’ diseases (prevalence <1:50,000 persons) and data on epidemiology of MDS/MPNs other than CMML is extremely limited. The entity of atypical CML (ICC nomenclature), termed MDS/MPN with neutrophilia in WHO-5 (from now in the paper referred to as MDS/MPN-N) has a relative incidence of 1–2 cases for every 100 patients with BCR::ABL1-positive CML. It presents in elderly patients with a reported median age at diagnosis of 70–74 years and a male preponderance [8]. MDS/MPN with SF3B1 and thrombocytosis (MDS/MPN-SF3B1-T) constitutes < 1% of new MDS or MDS/MPN cases and the median age for presentation usually ranges from 71 to 75 years [9,10,11]. There is no robust epidemiological data for MDS/MPN-not otherwise specified (NOS); the median age at diagnosis is approximately 70 years, and male patients are more frequently affected [12]. Importantly, based on the mutational profile, specific subcategories of MDS/MPN-NOS can be identified, i.e. “CMML-like”, “aCML-like”, “MDS/MPN-SF3B1-T-like”, and MDS/MPN with mutated TP53. Such molecular classifiers with the MDS/MPN-NOS group can facilitate estimation of patient outcomes and can aid in the clinical decision making process [2, 13, 14].
Insights into molecular and cytogenetic landscape
Only a minority of MDS/MPN clones harbor cytogenetic abnormalities, and where identified are typically not disease entity defining, with few complex or monosomal profiles [2, 15]. Conversely, a large majority of cases demonstrate somatic mutations by NGS testing, either in marrow or peripheral blood [2]. The most commonly mutated gene groups in these disorders affect epigenetic modifiers (SETBP1, TET2, ASXL1, DNMT3A), splicing factors (SF3B1, U2AF1, SRSF2) and cell signaling molecules (JAK2, CALR, MPL, NRAS/KRAS) [16]. The composition of molecular lesions may have specific phenotypic manifestations, such as proliferative disease in those with predominantly cell-signaling mutations, or cytopenia/dysplasia in those with splicing mutations. The combination of mutations in SETBP1 and ASXL1 is overrepresented in MDS/MPN-N. SF3B1 is present by definition in MDS/MPN-SF3B1-T, but additional mutations such as JAK2 are frequently seen. The ICC recognizes a related group of MDS/MPN with ring sideroblasts and thrombocytosis, not otherwise specified (MDS/MPN-RS-T-NOS). MDS/MPN-NOS, akin to its clinical manifestations, remains the most heterogenous entity by molecular profiling, however as testing becomes more widespread it is now beginning to be stratified more clearly. Risk stratification in MDS/MPN-N is typically performed using the ‘Mayo Clinic’ model, which evaluates age, hemoglobin and TET2 mutation status to identify a ‘low’ and ‘high-risk’ group, however the predicted difference in median overall survival (OS) is small at 18 and 7 months respectively, underlining the overall poor prognosis [8, 17]. MDS/MPN-SF3B1-T, in contrast, typically follows a relatively indolent course, which is reflected in the very small number of allografts performed for this disease entity in recent EBMT registry data [3]. No harmonized risk stratification covers the heterogeneity of MDS/MPN-NOS, but those used in CMML (CPSS-Mol) and MDS (IPSS-M) may be applicable [18, 19]. Of note, considering CMML, there is the recently suggested international CMML Prognostic Scoring System (iCPSS), a novel prognostic model incorporating hematological parameters, cytogenetic anomalies, and evaluation of mutations in 10 genes (ASXL1, DNMT3A, EZH2, NRAS, RUNX1, SETBP1, STAG2, TET2, TP53, U2AF1), which has been shown to predict both OS and relapse risk post allo-HCT for CMML, and may have applicability in the MDS/MPN-NOS group [20]. The prognostic impact of typically high-risk mutations in myeloid malignancies such as TP53 are not specifically clarified in these ‘non-CMML’ disease subtypes, but lessons from other acute and chronic myeloid diseases are likely to be applicable.
Overall current position
To advance the care of patients with MDS/MPN in the molecular era, harmonized registry-based collaborations and multicentre prospective studies are urgently needed. Such cooperative efforts would provide more robust data on disease incidence and phenotype, refine prognostic models, and validate thresholds for intervention. Importantly, they could also establish standardized eligibility criteria and optimize timing of transplantation. By aligning practices across centers, we can strengthen surveillance strategies, enhance the evidence base guiding post-transplant care, and create the foundation for well-designed prospective trials in both transplant and non-transplant settings. While the rarity of these disorders poses significant challenges, coordinated international efforts remain essential to generate the evidence required to improve patient outcomes.
Methodology
This paper was generated under the auspices of the methodology of the Practice Harmonization and Guidelines committee of the EBMT [21]. The EBMT is a non-profit, scientific society representing more than 700 transplant and cellular therapy centres mainly in Europe but also wider afield. A two day meeting was held in Berlin for in-depth discussion of the topics under evaluation. International experts from Europe, the Americas and Australia were additionally invited to develop the manuscript. Over the course of two months in advance of the two-day hybrid workshop, preparatory reviews through virtual communication were carried out by the group, which comprised key opinion leaders in the fields of MDS/MPN, haematopathology and/or allo-HCT. Relevant literature in the PubMed database was examined by subgroups of 4-5 experts, who then generated summative appraisals. These evidence reviews were then discussed in a hybrid workshop based in Berlin, Germany, in September 2025. A draft paper was subsequently generated and circulated to the group for final revision. As there is a lack of evidence from randomized trials and a paucity of data for this topic generally, it was not possible to grade these recommendations and the views expressed are therefore the expert consensus of all the authors. No external or industry funding was utilized to complete this project.
Workshop recommendations
Which patients with MDS/MPN are transplant candidates?
Allo-HCT remains the only therapeutic strategy with curative potential for patients with MDS/MPN. Current evidence, mainly derived from retrospective series, transplant registry analyses, and extrapolation from studies in MDS, 'classical' CMML, and myelofibrosis (MF), supports its use in selected patients. However, as discussed above, the rarity and biological heterogeneity of these entities, limit the strength of current recommendations [3]. In MDS-MPN-N, prognosis is generally poor with conventional approaches, and in our view early allo-HCT should be considered for all eligible patients [8, 22] Conversely, MDS/MPN-SF3B1-T usually follows an indolent course, as do patients with the related ICC entity MDS/MPN-RS-T-NOS; patients with excess blasts, often with a monosomal and complex karyotype, have a more aggressive course, but are excluded from this category and are classified as MDS/MPN-NOS [23]. Allo-HCT hence is normally reserved for so-called ‘high-risk candidates’ (e.g., refractory anemia/ transfusion dependence, adverse cytogenetics, and/or co-existing ASXL1 or SETBP1 mutations), with a timely donor search initiated when appropriate [23]. For MDS/MPN-NOS, where median survival is frequently short (12–33 months), transplantation is recommended by this working group in all eligible candidates, particularly in those patients with transfusion dependency, aggressive clinical behavior/rapid progression, or high-risk biological features, in line with the general principles established for CMML and ‘non-CMML’ overlap syndrome allo-HCT by expert EBMT groups [3, 24]. Eligibility for allo-HCT must be assessed by integrating standard patient-related factors (age, comorbidities, performance status, frailty index) with disease-specific parameters such as proliferative burden, cytogenetic and molecular profile and dynamic evolution. Donor availability, conditioning intensity and GVHD prophylaxis strategies remain crucial determinants of outcome [3].
Pre-transplant therapy is not standardized across centres. Hydroxyurea or interferon are useful for cytoreduction in certain proliferative cases, while hypomethylating agents (HMA) or induction chemotherapy are generally reserved for patients with higher blast counts or AML transformation but robust data on treatment choice is limited [25]. Data on ruxolitinib, alone or in combination with azacitidine, are promising but overall remain investigational [26, 27]. The primary goal of pre-transplant therapy is transient ‘disease control’ in hyperproliferative cases and patient optimization, rather than an aim for deep remission, but we need to be cognizant that some patients initially considered transplant-eligible may fail to get to transplant if they experience therapy-related complications or disease progression during this period. Conditioning regimens should be individualized as per patient phenotype. In general, myeloablative conditioning (MAC) is preferred in younger, fit patients, especially with proliferative disease, while reduced-intensity regimens (RIC) are appropriate for older or comorbid patients. In summary, allo-HCT should be offered to patients with high-risk or clinically aggressive forms of ‘nonclassical’ MDS/MPN, while a watch-and-wait or non-transplant approaches remain appropriate for the indolent subtypes.
Summary
-
Allo-HCT should be offered to patients with high-risk (i.e. all eligible patients with MDS/MPN-N and MDS/MPN-NOS) or clinically aggressive forms of nonclassical MDS/MPN (i.e. patients with significant disease activity or evidence of disease evolution), while a watch-and-wait or non-transplant approach remains appropriate for indolent subtypes.
-
Early transplant referral is essential for potential transplant candidates based on patient and disease features without delaying for disease-modifying treatment and facilitating early donor identification.
-
There are no current robust data to guide on choice and indication of pre-transplant therapy, whether treatment prior to transplant is necessary and the decision should be made based on patient- and disease characteristics. Where possible patients should be enrolled on applicable clinical trials to help clarify this knowledge gap.
Challenges posed by defining remission and relapse post Allo-HCT in MDS/MPN
The inherent hematological, histological, and molecular heterogeneity, together with the need to assess both dysplastic and proliferative components of MDS/MPN, remains a major challenge. More than a decade ago, an international consortium proposed pragmatic response criteria based on blood counts, blast percentage, fibrosis, splenomegaly, and constitutional symptoms that at the time of writing are currently undergoing revision [5]. Yet, no standardized post allo-HCT definition of remission or relapse specific to these entities exists and as allo-HCT is performed with curative intent, the categorization of response is of particular importance. Given the relative rarity of these disorders, this is not surprising, and there remains a lack of well-annotated cohorts which include sequential marrow morphology with molecular and chimerism assessment post allo-HCT. As seen in the context of MF, we would expect variable kinetics in the resolution of splenomegaly, cytopenias, fibrosis, abnormal marrow architecture, and measurable residual disease (MRD) clearance [28]. Moreover, in the context of MDS/MPN, there is complexity related to the definition of MRD and how this should be evaluated. Here, we have focussed on ‘molecular’ MRD rather than immunophenotypic-based MRD as this modality continues to undergo sustained development across myeloid malignancies. Clear and pragmatic definitions of persistent/ progressive disease, achievement of complete remission, and relapse are essential for the transplant community for optimization of patient management and registry harmonization. Relapse remains a leading cause of transplant failure as highlighted by several studies. Desai et al. reported a 2-year cumulative incidence of relapse (CIR) of 17.5% in 75 patients with MDS/MPN undergoing allo-HCT in a single centre [29]. Onida et al., harnessing EBMT registry data, reported a significantly higher 40% CIR at 5 years in 42 patients with MDS-MPN-N who underwent allo-HCT [30]. A similar CIR of 40.4% was reported very recently by Itonaga et al. from the Japanese Society for Transplantation and Cellular Therapy on 74 patients MDS-MPN-N who underwent allo-HCT [31]. Kurosawa et al. described a nationwide series of 86 MDS/MPN-NOS allo-HCT cases, with a 3-year CIR of 23.7% [32]. The absence of standardized response criteria in these single center or registry-based studies unfortunately limits direct comparisons. Post-transplant complications such as PGF or slow to resolve fibrosis can clearly overlap/ mimic pending or overt relapse, underscoring the need for integrated approaches combining assessment of morphology, donor chimerism, and MRD monitoring. For example, EBMT guidelines in MF recommend sensitive MRD assays with minimum detection limits of 0.01–1%, including utilization of either droplet digital (dd) or quantitative PCR (qPCR), but we have no such robust data in the ‘non-CMML’ MDS/MPN setting with limited data on sensitivity thresholds [3, 33]. A combinatorial approach evaluating both donor chimerism and sensitive MRD appears essential for assessing disease remission/ relapse in myeloid disorders in general and can facilitate pre-emptive intervention as required [34, 35]. Standardizing these strategies across centres with agreed timings in the post-transplant setting would provide a much-needed evidence base.
Disease persistence, remission status and relapse
As introduced above, it is clear that the international consensus group 2015 criteria generated for the non-transplant setting cannot be fully transcribed for post-transplant remission and relapse status as they do not include transplant-specific issues such as chimerism, poor graft function and dynamic assessment, yet there are pivotal aspects from this expert group consensus that we should take into consideration [5]. Table 3 summarizes the suggested original criteria for measurement of treatment response in adult MDS/MPN patients in the non-transplant setting by Savona and colleagues. Of note, suggested response criteria encompassed both CMML and ‘non-CMML’ entities and an estimation of clinical benefit to treatment in the absence of progression or achievement of a complete or partial response. The authors recognized the frequent scenario of potential dyssynchronous responses e.g. in the more proliferative entities should we focus on marrow responses and prioritize spleen response criteria whereas in the more dysplastic cases should we focus on marrow responses paralleled with an emphasis on improvement in cytopenia? Additional candidate metrics for response assessment included symptom burden assessed by symptom assessment tools with inclusion of a provisional category ‘CR with resolution of symptoms’, noting that achievement of CR did not require symptom resolution. Regarding marrow fibrosis assessment, the authors also recognized the dynamic nature of fibrosis resolution and suggested that where baseline fibrosis has been detected, a minimum of two marrows should be performed to confirm resolution of fibrosis with a minimum interval period of 2-months. Additionally, it should be noted that the morphology (and blood count parameters, i.e. specific pattern of cytosis and cytopenia seen originally) of disease at relapse may differ from the original disease, which complicates morphological evaluation of a potential disease recurrence. Documented improvements in blood counts should be maintained for a minimum of 8 weeks as regards hematological responses and the authors chose the transfusion-related endpoint of achievement of transfusion independence (TI) only. The authors also brought together potential criteria for progressive disease, but we should note that given the era, there was less emphasis on molecular annotation coupled with prognostication/ progression assessment, with recognition of the complexities of clonal and sub clonal dynamics and variant allelic frequency (VAF) assessment. This should be re-appraised in the current landscape and the group are working on updated guidance.
Defining disease remission, relapse and persistence/progression post allo-HCT
Below we suggest a pragmatic set of criteria for defining remission post allo-HCT for the following entities: MDS-MPN-N, MDS/MPN-SF3B1-T and MDS/MPN-NOS (Table 4). We have considered the current ICC/WHO diagnostic criteria and subsequently applied suggested criteria for defining (i) complete remission, (ii) complete remission with presence of poor graft function, (iii) persistent/ progressive disease, (iv) relapse, in the post allo-HCT setting. We need to recognize that disease heterogeneity and variable recovery post allo-HCT may mean that in the majority of cases it will be essential to comprehensively and dynamically assess the patient at sequential time points (at least 2) dependent on the disease phenotype and baseline characteristics such as the presence of established fibrosis, which we know may have variable resolution. Akin to primary myelofibrosis (PMF), we predict there could be variable kinetics regarding clearance of disease-associated mutations, coupled with wide variability in the sensitivity of the testing platform, and this data needs prospective collation. Another aspect that is worth recognizing is the plasticity of hematologic disease that may occur in the post-allo-HCT state i.e. disease may change form based on sub-clonal evolution to manifest as phenotypes not present originally (eosinophilia, basophilia, different blast populations etc.) which complicates morphologic evaluation of potential disease recurrence.
Post-transplant monitoring in MDS/MPN: molecular and chimerism analysis
Post-transplant surveillance in MDS/MPN should follow a risk-adapted framework that combines serial chimerism analysis and molecular MRD assessment. When feasible, trackable disease-related mutations identified at diagnosis could be used for longitudinal molecular MRD monitoring in line with previous reports in MPN [3, 33, 36]. For other somatic mutations, NGS could be applied, in line with previous findings and consensus from MDS and CMML, but this remains investigational [23, 37, 38]. Chimerism testing, preferably with sensitive platforms such as Short Tandem Repeat (STR)-PCR, qPCR, or sensitive NGS-based approaches, should be assessed at the same time points. [3, 33, 34, 39]. Lineage-specific analysis, specifically T-cell chimerism, may provide additional predictive value. Suggested guidance for MRD and chimerism analysis are shown below.
-
Serial chimerism evaluation is required e.g. the following time points in the first year: day+30, day +90, day +180, +270 and 12-months and further as clinically indicated. Some centres may perform later chimerism time points or more frequent monitoring if chimerism remains mixed.
-
Molecular MRD monitoring is suggested at day+30, day+90, day+180, day+270 and then at 12 months and further as clinically indicated (depending on disease subtype and risk profile, clinical presentation, GVHD occurrence, and hematological status) post-transplant.
Conversion from full to mixed chimerism or persistent mixed chimerism, which may occur in parallel with dynamic rises in molecular MRD should be considered actionable trigger warnings for closer monitoring and intervention if required. This may involve standard approaches in such a setting such as tapering immunosuppression therapy (IST) if applicable and adoptive immunotherapeutic approaches with donor lymphocyte infusions (DLI). In the case of socioeconomic challenges, monitoring should be adapted to available resources. A suggested schema for the tests and timepoints required for determining remission, persistence/progression and relapse is outlined in (Table 5).
Management of splenomegaly in MDS/MPN pre- and post allo-HCT
Based on current evidence in PMF, significant splenic enlargement prior to allo-HCT (arbitrarily defined as ≥5 cm below the left costal margin) may be associated with an increased risk of graft failure, delayed engraftment and PGF [33, 40]. Therefore, in our opinion for MDS/MPN associated by clinically significant splenomegaly, all reasonable efforts should be made to reduce spleen volume prior to transplantation, although the data is lacking when considering medical therapies, splenic irradiation or splenectomy. Considerations to pre-transplant spleen management have already been addressed in the paper by Polverelli et al. and readers are recommended to evaluate/ manage patients in a similar fashion if appropriate [3].
In PMF, progressive reduction of splenomegaly during the first 12 months post-transplant has been reported, paralleling the resolution of bone marrow fibrosis [41, 42]. Nonetheless, spleen size reduction post allo-HCT is variable; some patients may even require more than 36 months for complete resolution [41]. Conversely, persistent splenomegaly post-transplant, particularly when associated with cytopenias, may signal residual or progressive disease. Whether this is the case in MDS/MPN remains unknown. Anecdotal case reports support the feasibility of performing post-allo-HCT splenectomy in selected patients with prolonged and significant PGF, but this remains investigational and there is no data to support such an approach specifically in MDS/MPN [43]. Whilst we cannot provide definitive recommendations for splenomegaly management in the MDS/MPN due to a lack of evidence, a pragmatic approach based on our recommendations in PMF may be appropriate [44].
-
Comprehensive pre- and post-transplant spleen assessment should be conducted using physical examination (documented as centimeters below the left costal margin), ultrasound (as the most validated and optimal modality for assessing splenomegaly) or CT or MRI, where appropriate for detailed imaging.
-
In the absence of cytopaenias and with complete donor chimerism, post-transplant spleen size reduction may be a gradual, prolonged process and persistent splenomegaly should not, by itself, be considered a sign of disease persistence. Complete normalization may take 12 months, or even up to 36 months after transplantation so dynamic assessment is mandated.
-
If persistent splenomegaly is accompanied by cytopenias, a thorough differential diagnosis should be performed to evaluate for graft failure (standard definition applies), PGF, disease relapse or progression and other secondary causes
Post-transplant management of poor graft function in MDS/MPN
Disease-specific high-quality data to guide post-transplant management of PGF post allo-HCT in MDS/MPN is lacking thus making it difficult not only to define the magnitude of the problem but also consequent management. As a result, inspiration from other more common myeloid malignancies undergoing allo-HCT with a high incidence of PGF, such as PMF, which shares some clinico-pathological features, could be considered, albeit we recognize the limitations of such an approach. Conventional definitions vary but a clinically relevant definition of PGF is the presence of mild/moderate cytopenias in at least two haematopoietic lines (ANC ⩽1.5 × 109/L, platelet count ⩽30 × 109/L, Hb ⩽85 g/L) lasting for more than two consecutive weeks following documented engraftment, beyond day +14 and in the presence of full donor chimerism [45, 46]. This is in the absence of severe acute or chronic GVHD, relapse, and either drug- or CMV reactivation-related myelosuppression. There is no data to inform how long PGF can persist post-transplant across these heterogenous disorders and risk factors to take into consideration include the presence of pre-transplant splenomegaly and persistence post-, HLA-senitization and iron overload etc [33]. Standard approaches to PGF should be employed:
-
1)
Utilization of growth factor support: GCSF and recombinant erythropoietin stimulating agents as required. There is insufficient evidence for routine use of thrombopoietin receptor agonists such as eltrombopag or romiplostim, which remain experimental and further data beyond phase 1/2 studies are needed [47].
-
2)
Appropriate infectious prophylaxis dependent on neutrophil and lymphocyte subset recovery.
-
3)
A CD34+ selected stem cell boost can be considered if PGF fails to respond to these supportive measures and is prolonged. Timing and GVHD risk assessment requires close consideration.
-
4)
Rarely, consideration to a second allo-HCT may be required.
Management of relapse post allo-HCT
A lack of standard therapies for MDS/MPN as discussed above makes the management of post-transplant relapse a particular challenge [1]. Obviously, the enhancement of a graft versus disease effect by tapering immunosuppressive therapy and/or considering DLI is an obvious choice for patients without current GvHD. Consideration must be given to the timing of relapse (early versus late) and whether this is molecular, cytogenetic or morphologic relapse. There is, however, no evidence to guide DLI use post allo-HCT for MDS/MPN. Real world experience, suggests response to DLI in post-transplant relapse of MDS/MPN could be directed by phenotype: DLI alone may be preferred in ‘MPN-like disease’ phases, whereas in more dysplastic disease phases, the combination of DLI with HMA may be preferred [48,49,50,51]. The schedule and sequencing of such approaches are frequently centre dependent and there is an urgent need for more data.
HMA plus venetoclax has shown encouraging results as a pre-transplant bridging regimen in MDS/AML and is being investigated in the post-transplant setting across a range of myeloid malignancies [52, 53]. However, only single patients with MDS/MPN were included in these studies hence wider applicability overall of such an approach remains uncertain. Whether a second transplant in suitable candidates yields superior results compared to a DLI or DLI/HMA based approach is highly controversial in myeloid neoplasms in general [54]. In MDS/MPN, a second transplant should be considered for primary or secondary graft failure in fit patients, but whether a patient responding to relapse treatment with hematopoietic recovery should be offered a second transplant remains debatable and should consider variables with regard to fitness, time of relapse, presence of active GVHD, donor availability and patient preference.
A number of novel agents have been proposed as candidates for post-transplant relapse strategies. Dependent on phenotype, therapeutics like conventional cytoreductives, anemia-directed agents, JAK-inhibitors or imetelstat may improve anemia and/or symptoms but may be difficult to obtain dependent on reimbursement. Agents such as conventional cytoreductive agents (e.g. interferon and hydroxycarbamide) and JAK-inhibitors alone will be insufficient to suppress the malignant clone and revert chimerism to a clinically relevant degree and are not curative in intent [55,56,57,58]. Targeting IDH1 or IDH2 mutations, if present, may be a more promising approach in this respect, albeit the overall incidence across these disease entities is low [59].
Unanswered questions and further research to do in the field
International collaboration is necessary to expand the evidence base in these rare entities. By harmonizing response criteria, the quality of data captured by registries will be bolstered, especially as generating high-quality clinical trial data in this field is significantly challenging. As methods for MRD monitoring evolve, establishing which methods and at what time-points to use post-transplant requires further investigation, with a focus on developing monitoring programmes that can reliably predict relapse and allow for timely preventative intervention. New insights into the molecular heterogeneity of MDS/MPN are refining how these disorders are diagnosed and efforts to further characterize clonal and sub-clonal dynamics in the post-transplant setting may allow a deeper understanding of both the disease process and mechanisms of post-transplant relapse.
Serial chimerism monitoring remains a powerful tool and can guide meaningful interventions, however the methods and timing of chimerism analysis are heterogenous across transplant centres. Further clarification of the benefit of highly sensitive chimerism methods and the potential roles of compartment-specific analysis is a subject that may strengthen the consensus recommendations on post-transplant monitoring, and with sufficient quality registry data, could be explored in a collaborative manner. The use of pre-emptive DLI is supported by experience in other myeloid malignancies but the role of prophylactic DLI for MDS/MPN requires further evidence. DLI as part of a therapeutic response to disease relapse is frequently employed, however results from such an approach should be collated and interrogated to establish when and in whom it is most likely to provide benefit.
The pre-transplant management of MDS/MPN is an area of unmet clinical need and therefore future developments in the non-transplant setting are likely to have implications for transplant outcomes and potentially be utilized for peri-transplant optimization, post-transplant maintenance and after relapse.
Finally, specific prognostication models for MDS/MPN undergoing allo-HCT are lacking, and further robust and harmonized retrospective registry data may help to design tools to help guide transplant physicians on estimates of relapse and mortality. The remission criteria here proposed are designed to allow for more comprehensive comparisons between patient populations, which in single series are limited by a low disease incidence and heretofore variably defined criteria.
References
Tremblay D, Hasserjian RP, Rampal RK. Myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: a practical guide to diagnosis and management. Leukemia. 2025;39:1311–24.
Palomo L, Meggendorfer M, Hutter S, Twardziok S, Ademà V, Fuhrmann I, et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood. 2020;136:1851–62.
Polverelli N, Hernández-Boluda JC, Onida F, Gurnari C, Raj K, Czerw T, et al. Role of allo-HCT in “nonclassical” MPNs and MDS/MPNs: recommendations from the PH&G Committee and the CMWP of the EBMT. Blood. 2025;145:2561–73.
Greco R, Ruggeri A, McLornan DP, Snowden JA, Alexander T, Angelucci E, et al. Indications for haematopoietic cell transplantation and CAR-T for haematological diseases, solid tumours and immune disorders: 2025 EBMT practice recommendations. Bone Marrow Transpl 2025. https://doi.org/10.1038/s41409-025-02701-3.
Savona MR, Malcovati L, Komrokji R, Tiu RV, Mughal TI, Orazi A, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125:1857–65.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka H-M, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19.
Szuber N, Orazi A, Tefferi A. Chronic neutrophilic leukemia and atypical chronic myeloid leukemia: 2024 update on diagnosis, genetics, risk stratification, and management. Am J Hematol. 2024;99:1360–87.
Patnaik MM, Lasho TL. Genomics of myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes. Hematol Am Soc Hematol Educ Program. 2020;2020:450–9.
Patnaik MM, Lasho T. Myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: a focused review. Hematol Am Soc Hematol Educ Program. 2020;2020:460–4.
Patnaik MM, Tefferi A. Myelodysplastic syndromes with ring sideroblasts (MDS-RS) and MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T) - ‘2021 update on diagnosis, risk-stratification, and management. Am J Hematol. 2021;96:379–94.
Mangaonkar AA, Swoboda DM, Coltro G, Lasho TL, Novotny PJ, Pophali P, et al. Clinicopathologic characteristics, prognostication and treatment outcomes for myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U): Mayo Clinic-Moffitt Cancer Center study of 135 consecutive patients. Leukemia. 2020;34:656–61.
Mangaonkar AA, Swoboda DM, Lasho TL, Finke C, Ketterling RP, Reichard KK, et al. Genomic stratification of myelodysplastic/myeloproliferative neoplasms, unclassifiable: Sorting through the unsorted. Leukemia. 2021;35:3329–33.
Patnaik MM, Tefferi A. Atypical chronic myeloid leukemia and myelodysplastic/myeloproliferative neoplasm, not otherwise specified: 2023 update on diagnosis, risk stratification, and management. Am J Hematol. 2023;98:681–9.
Palomo L, Acha P, Solé F. Genetic Aspects of Myelodysplastic/Myeloproliferative Neoplasms. Cancers. 2021;13:2120.
Meggendorfer M, Jeromin S, Haferlach C, Kern W, Haferlach T. The mutational landscape of 18 investigated genes clearly separates four subtypes of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2018;103(5):e192–e195. https://doi.org/10.3324/haematol.2017.183160.
Patnaik MM, Barraco D, Lasho TL, Finke CM, Reichard K, Hoversten KP, et al. Targeted next generation sequencing and identification of risk factors in W orld H ealth O rganization defined atypical chronic myeloid leukemia. Am J Hematol. 2017;92:542–8.
Elena C, Gallì A, Such E, Meggendorfer M, Germing U, Rizzo E, et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood. 2016;128:1408–17.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid 2022; 1. https://doi.org/10.1056/EVIDoa2200008.
Lanino L, Hunter AM, Gagelmann N, Robin M, Sala C, Dall’Olio D, et al. A Molecular-Based Ecosystem to Improve Personalized Medicine in Patients with Chronic Myelomonocytic Leukemia (CMML). Blood. 2024;144:1003–1003.
Yakoub-Agha I, Greco R, Onida F, De La Cámara R, Ciceri F, Corbacioglu S, et al. Practice harmonization workshops of EBMT: an expert-based approach to generate practical and contemporary guidelines within the arena of hematopoietic cell transplantation and cellular therapy. Bone Marrow Transpl. 2023;58:696–700.
Patterson-Fortin J, Moliterno AR. Molecular Pathogenesis of Myeloproliferative Neoplasms: Influence of Age and Gender. Curr Hematol Malig Rep. 2017;12:424–31.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am J Hematol. 2016;91:492–8.
Onida F, Gagelmann N, Chalandon Y, Kobbe G, Robin M, Symeonidis A, et al. Management of adult patients with CMML undergoing allo-HCT: recommendations from the EBMT PH&G Committee. Blood. 2024;143:2227–44.
Tong X, Li J, Zhou Z, Zheng D, Liu J, Su C. Efficacy and side-effects of decitabine in treatment of atypical chronic myeloid leukemia. Leuk Lymphoma. 2015;56:1911–3.
Dao K-HT, Gotlib J, Deininger MMN, Oh ST, Cortes JE, Collins RH, et al. Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia. J Clin Oncol. 2020;38:1006–18.
Assi R, Kantarjian HM, Garcia-Manero G, Cortes JE, Pemmaraju N, Wang X, et al. A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am J Hematol. 2018;93:277–85.
Gupta S, Courville EL. Bone marrow findings post allogeneic transplant for myeloproliferative neoplasms and chronic myelomonocytic leukemia with increased fibrosis. Lab Med. 2024;55:602–8.
Desai N, Rodriguez-Rodriguez S, Chen C, Moya TA, Al-Shaibani E, Novitzky-Basso I, et al. Outcomes of Allogeneic Hematopoietic Stem Cell Transplantation for Myelodysplastic/Myeloproliferative Overlap Neoplasms. Eur J Haematol. 2025;115:142–52.
Onida F, De Wreede LC, Van Biezen A, Eikema D-J, Byrne JL, Iori AP, et al. Allogeneic stem cell transplantation in patients with atypical chronic myeloid leukaemia: a retrospective study from the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Br J Haematol. 2017;177:759–65.
Itonaga H, Miyazaki Y, Kondo T, Shimazu Y, Aoki J, Kurosawa S, et al. Long-Term Survival After Allogeneic Hematopoietic Stem Cell Transplantation for BCR::ABL1 -Negative Atypical Chronic Myeloid Leukemia: A Nationwide Retrospective Study by Adult CML / MPN and MDS Working Groups of the Japanese Society for Transplantation and Cellular Therapy. Am J Hematol. 2025;100:917–21.
Kurosawa S, Shimomura Y, Tachibana T, Ishiyama K, Ota S, Kobayashi T, et al. Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in Patients with Myelodysplastic/Myeloproliferative Neoplasms-Unclassifiable: A Retrospective Nationwide Study of the Japan Society for Hematopoietic Cell Transplantation. Biol Blood Marrow Transpl. 2020;26:1607–11.
McLornan DP, Hernandez-Boluda JC, Czerw T, Cross N, Joachim Deeg H, Ditschkowski M, et al. Allogeneic haematopoietic cell transplantation for myelofibrosis: proposed definitions and management strategies for graft failure, poor graft function and relapse: best practice recommendations of the EBMT Chronic Malignancies Working Party. Leukemia. 2021;35:2445–59.
Puzo CJ, Tormey CA, Rinder HM, Siddon AJ. Optimizing Donor Chimerism Threshold for Next-Generation Sequencing Monitoring of Measurable Residual Disease Post-Allogeneic Stem Cell Transplantation for Myeloid Neoplasms. Transpl Cell Ther. 2023;29:459.e1–459.e4.
Tobiasson M, Pandzic T, Illman J, Nilsson L, Weström S, Ejerblad E, et al. Patient-Specific Measurable Residual Disease Markers Predict Outcome in Patients With Myelodysplastic Syndrome and Related Diseases After Hematopoietic Stem-Cell Transplantation. J Clin Oncol. 2024;42:1378–90.
Gagelmann N, Quarder M, Badbaran A, Rathje K, Janson D, Lück C, et al. Clearance of Driver Mutations after Transplantation for Myelofibrosis. N Engl J Med. 2025;392:150–60.
Gurnari C, Robin M, Adès L, Aljurf M, Almeida A, Duarte FB, et al. Clinical-genomic profiling of MDS to inform allo-HCT: recommendations from an international panel on behalf of the EBMT. Blood. 2025;145:1987–2001.
Duncavage EJ, Jacoby MA, Chang GS, Miller CA, Edwin N, Shao J, et al. Mutation Clearance after Transplantation for Myelodysplastic Syndrome. N Engl J Med. 2018;379:1028–41.
Sureda A, Carpenter PA, Bacigalupo A, Bhatt VR, De La Fuente J, Ho A, et al. Harmonizing definitions for hematopoietic recovery, graft rejection, graft failure, poor graft function, and donor chimerism in allogeneic hematopoietic cell transplantation: a report on behalf of the EBMT, ASTCT, CIBMTR, and APBMT. Bone Marrow Transpl. 2024;59:832–7.
Kröger N, Wolschke C, Gagelmann N. How I treat transplant-eligible patients with myelofibrosis. Blood. 2023;142:1683–96.
Ciurea SO, Sadegi B, Wilbur A, Alagiozian-Angelova V, Gaitonde S, Dobogai LC, et al. Effects of extensive splenomegaly in patients with myelofibrosis undergoing a reduced intensity allogeneic stem cell transplantation. Br J Haematol. 2008;141:80–83.
McLornan DP, Yakoub-Agha I, Robin M, Chalandon Y, Harrison CN, Kroger N. State-of-the-art review: allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica. 2019. https://doi.org/10.3324/haematol.2018.206151.
Robin M, Espérou H, de Latour RP, Petropoulou AD, Xhaard A, Ribaud P, et al. Splenectomy after allogeneic haematopoietic stem cell transplantation in patients with primary myelofibrosis. Br J Haematol. 2010;150:721–4.
Polverelli N, Hernández-Boluda JC, Czerw T, Barbui T, D’Adda M, Deeg HJ, et al. Splenomegaly in patients with primary or secondary myelofibrosis who are candidates for allogeneic hematopoietic cell transplantation: a Position Paper on behalf of the Chronic Malignancies Working Party of the EBMT. Lancet Haematol. 2023;10:e59–e70.
Stasia A, Ghiso A, Galaverna F, Raiola AM, Gualandi F, Luchetti S, et al. CD34 Selected Cells for the Treatment of Poor Graft Function after Allogeneic Stem Cell Transplantation. Biol Blood Marrow Transpl. 2014;20:1440–3.
Klyuchnikov E, El-Cheikh J, Sputtek A, Lioznov M, Calmels B, Furst S, et al. CD34+-Selected Stem Cell Boost without Further Conditioning for Poor Graft Function after Allogeneic Stem Cell Transplantation in Patients with Hematological Malignancies. Biol Blood Marrow Transpl. 2014;20:382–6.
Peffault de Latour R, Chevret S, Ruggeri AL, Suarez F, Souchet L, Michonneau D, et al. Romiplostim in patients undergoing hematopoietic stem cell transplantation: results of a phase 1/2 multicenter trial. Blood. 2020;135:227–9.
Rampotas A, Sockel K, Panitsas F, Theuser C, Bornhauser M, Hernani R, et al. Adoptive Immunotherapy via Donor Lymphocyte Infusions following Allogeneic Hematopoietic Stem Cell Transplantation for Myelofibrosis: A Real-World, Retrospective Multicenter Study. Transpl Cell Ther. 2023;29:687.e1–687.e7.
Gagelmann N, Wolschke C, Badbaran A, Janson D, Berger C, Klyuchnikov E, et al. Donor Lymphocyte Infusion and Molecular Monitoring for Relapsed Myelofibrosis After Hematopoietic Cell Transplantation. HemaSphere. 2023;7:e921.
Pagliuca S, Schmid C, Santoro N, Simonetta F, Battipaglia G, Guillaume T, et al. Donor lymphocyte infusion after allogeneic haematopoietic cell transplantation for haematological malignancies: basic considerations and best practice recommendations from the EBMT. Lancet Haematol. 2024;11:e448–e458.
Kröger N, Bacigalupo A, Barbui T, Ditschkowski M, Gagelmann N, Griesshammer M, et al. Indication and management of allogeneic haematopoietic stem-cell transplantation in myelofibrosis: updated recommendations by the EBMT/ELN International Working Group. Lancet Haematol. 2024;11:e62–e74.
Zugasti I, Lopez-Guerra M, Castaño-Díez S, Esteban D, Avendaño A, Pomares H, et al. Hypomethylating agents plus venetoclax for high-risk MDS and CMML as bridge therapy to transplant: a GESMD study. Exp Hematol Oncol. 2025;14:61.
Schuler E, Wagner-Drouet E-M, Ajib S, Bug G, Crysandt M, Dressler S, et al. Treatment of myeloid malignancies relapsing after allogeneic hematopoietic stem cell transplantation with venetoclax and hypomethylating agents—a retrospective multicenter analysis on behalf of the German Cooperative Transplant Study Group. Ann Hematol. 2021;100:959–68.
Robin M, Scheid C. Second allogeneic stem-cell transplantation or donor lymphocyte infusion in patients who relapse after transplantation? Lancet Haematol. 2024;11:e642–e643.
Renneville A, Patnaik MM, Chan O, Padron E, Solary E. Increasing recognition and emerging therapies argue for dedicated clinical trials in chronic myelomonocytic leukemia. Leukemia. 2021;35:2739–51.
McLornan DP, Harrison CN. Guidance on changing therapy choice in myelofibrosis. Blood Adv. 2020;4:607–10.
McLornan DP, Pope JE, Gotlib J, Harrison CN. Current and future status of JAK inhibitors. Lancet Lond Engl. 2021;398:803–16.
Gerds AT, Vannucchi AM, Passamonti F, Kremyanskaya M, Gotlib JR, Palmer JM, et al. A Phase 2 Study of Luspatercept in Patients with Myelofibrosis-Associated Anemia. Blood. 2019;134:557–557.
Chifotides HT, Bose P, Verstovsek S. Givinostat: an emerging treatment for polycythemia vera. Expert Opin Investig Drugs. 2020;29:525–36.
Author information
Authors and Affiliations
Contributions
CA, KR and DPM chaired the workshop and formulated the manuscript design under the direction of the EBMT PH&G Committee members FO, AR, ISO and IYA. The remaining authors contributed written sections of work in advance of the workshop, engaged in the workshop discussion and provided feedback on the collated manuscript, which were then edited by CA and DPM before final approval by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests with regards to this consensus guideline.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Armstrong, C., Raj, K., Battipaglia, G. et al. Defining remission and relapse after allogeneic hematopoietic cell transplantation in myelodysplastic/myeloproliferative neoplasms and optimization of transplantation outcomes: Recommendations from the EBMT practice harmonisation and guidelines committee. Bone Marrow Transplant 61, 591–600 (2026). https://doi.org/10.1038/s41409-026-02843-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41409-026-02843-y
