
Abstract
Emerging evidence suggests expression from human endogenous retrovirus (HERV) loci likely contributes to, or is a biomarker of, glioblastoma multiforme (GBM) disease progression. However, the relationship between HERV expression and GBM malignant phenotype is unclear. Applying several in silico analyses based on data from The Cancer Genome Atlas (TCGA), we derived a locus-specific HERV transcriptome for glioma that revealed 211 HERVs significantly dysregulated in the comparisons of GBM vs. normal brain (NB), GBM vs. low-grade glioma (LGG), and LGG vs. NB. Our analysis supported development of a unique HERV scoring algorithm that segregated GBM, LGG, and NB. Interestingly, lower HERV scores showed correlation with lower survival in GBM. However, HERV scores were less robust in predicting LGG survival or LGG progression to GBM. Functional prediction analysis linked the 211 HERV loci with 18 voltage-gated potassium channel genes. The functional link between dysregulated HERVs and specific potassium channel genes may contribute to better understanding of GBM pathogenesis, disease progression, and possibly drug resistance.
Similar content being viewed by others

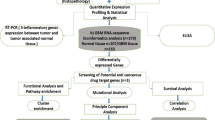

Introduction
Glioma is the umbrella term for various brain tumors that arise from glial cells and encompassing several types of tumors. Gliomas are categorized into four grades: grade 1 and grade 2—low-grade glioma (LGG), grade 3 and grade 4—high-grade glioma (HGG). Median survival decreases with increasing tumor grade from >11 years for LGG and approximately 3 years for grade 3 [1] to a dozen months in glioblastoma multiforme (GBM). The most aggressive subgroup of grade 4 glioma, GBM, is one of the most treatment-resistant cancers and the deadliest in the central nervous system (CNS) [2]. Interestingly, GBM may develop de novo or progress from slow-growing LGG [3]. However, LGG to GBM transition is poorly understood and requires more insight.
Human endogenous retroviruses (HERVs) are relics of ancient retroviral infections accumulated during evolution, and they comprise around 8% of the human genome in approximately 500,000 loci. They have been implicated in normal development, stemness, cancer biology, as well as neurological and neuropsychiatric disorders [4]. Over evolutionary time, most HERVs have become highly mutated and no longer encode functional genes. However, the more recently introduced to the human genome HERV-K family contains intact open reading frames and expresses viral proteins. HERVs loci may also be a natural source of genomic instability and provide a multitude of regulatory DNA sequences.
Emerging data suggests that HERVs may be involved in glioma biology [5,6,7], especially GBM stemness [8]. However, there is a gap in knowledge because of the main focus on HERV expression in GBM, while little data is available for HERVs in LGG. Our study is based on comparisons between GBM, NB, and LGG from The Cancer Genome Atlas (TCGA) RNA-Seq datasets exploring multiple HERV families with state of the art highly accurate locus-specific approach.
Our results show significant locus-specific differences in HERV transcriptomes of GBM, NB, and LGG occurring in multiple genetically distinct HERV families. Principal Component Analysis (PCA) of the most dysregulated 211 HERVs in comparisons of GBM, NB, and LGG segregated all three groups. Interestingly, GBM and LGG were separated along PCA Principal Component 1 (PC1) parameter, thus termed Glioma-specific HERV Score (GH Score). Importantly, lower values of GH Score showed a correlation with lower survival in GBM. However, GH Score was less robust in predicting LGG disease survival and progression to GBM. Analysis of functional significance linked the 211 HERV loci most consistently with 18 voltage-gated potassium channel genes. This finding suggests an interesting new link between dysregulated HERVs and altered transmembrane transport proved to play an important role in GBM biology [9]. Our results may have exciting implications for improved understanding of glioma pathogenesis, progression, and drug resistance, and for identifying locus-specific HERVs with possible diagnostic, prognostic, or therapeutic potential.
Materials and methods
Data
RNA-Seq data were downloaded in BAM format (*.bam) from The Cancer Genome Atlas—TCGA [10] using the Genomic Data Commons (GDC) Data Portal [11, 12]. In total, RNA-Seq from 5 normal brain (NB), and 158 GBM (“Glioblastoma Multiforme” TCGA-GBM project, dbGaP Study Accession: phs000178 [13, 14]), and 511 LGG (“Brain Lower Grade Glioma” TCGA-LGG project, dbGaP Study Accession: phs000178 [15]) were used. Corresponding clinical and demographic metadata were also downloaded. For cases with multiple samples, only the first sample was analyzed. GBM subtype data was obtained from the cBioPortal database [16,17,18].
HERV transcriptome quantification
For HERV transcriptome quantification, we selected Telescope—a computational software tool that provides estimation of HERV expression resolved to specific genomic locations [19]. We followed the analysis pipeline workflow required by Telescope [19]. Briefly, RNA-Seq BAM files were converted to FASTQ using SAMtools (RRID:SCR_002105) version 1.9 [20]. Converted data were aligned to human reference genome Hg38 using Bowtie2 with a very sensitive local alignment search, with fragments having at least 95% or greater sequence identity reported, and up to 100 alignments per read. The Telescope was utilized for HERV quantification using the HERV annotation described in [19].
Functional prediction with Genomic Regions Enrichment of Annotations Tool (GREAT) and GeneMANIA tool
As HERV regions lack biological function annotations, we used the Genomic Regions Enrichment of Annotations Tool (GREAT - version 4.0.4) [21, 22] in order to analyze the functional significance of HERV cis-regulatory regions. GREAT associates each HERV with all genes in whose regulatory domains it lies and analyzes biological function annotations of these associated genes. GREAT web application (available at: http://great.stanford.edu/public/html/) was used, and default settings of GREAT parameters were implemented (version 4.0.4). GeneMANIA tool [23] (available at: https://genemania.org/) was used to help predict the function of gene sets by revealing association networks and interactions.
Pathway analysis
To discover signaling pathways linked to HERV-associated genes (from GREAT analysis described above), we performed pathway analysis. Data were analyzed with the use of QIAGEN IPA (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA, RRID:SCR_008653) [24]. Genes were mapped onto canonical functional pathways determined by Fisher’s exact test. Discovered pathways were ranked based on their log-transformed p-values.
Statistical analyses
Differential expression analysis of HERV transcriptomic data for 3 comparisons among GBM, LGG, and NB (GBM vs. NB, GBM vs. LGG, and LGG vs. NB) was performed based on the negative binomial distribution and variance stabilization transformation using the DESeq2 (RRID:SCR_000154) R package [25]. For multiple comparisons justification adjusted p-values based on the Benjamini-Hochberg false discovery rates were calculated. For the 211 top differentially expressed HERVs, which were defined as p-value < 0.01 and the absolute value of log2 fold-change > 3, principal component analysis (PCA) was performed for variable reduction. Based on the principal component 1 (PC1) in the PCA, we developed “Glioma-specific HERV Score (GH Score).” Moreover, we tested whether the 211 HERVs could distinguish LGG with different therapy outcomes [Complete Remission/Response (CR/R) and Progressive Disease (PD)] using the PCA method. In addition, we were interested in testing if GH Score can predict survival status for GBM and LGG patients. Survival curves were generated based on the Kaplan–Meier method for the 3 groups (low, medium, and high) of patients based on the GH Scores.
Survival analysis was performed using the log-rank test and Cox proportional hazards regression. The Cox models with and without adjusting for age at diagnosis were performed. Data were not randomized into groups because this was deemed irrelevant to this study. Experimenters were not blinded.
Data visualization
Visualization of the 211 HERVs on chromosomes was performed with PhenoGram [26] (https://visualization.ritchielab.org/phenograms/plot).
Results
Dysregulation of HERV transcripts in gliomas follows a consistent downregulation pattern and involves multiple HERV families
To fully characterize glioma HERV transcriptome, we implemented Telescope-based [19] locus-specific bioinformatics pipeline to compare GBM (glioblastoma multiforme), LGG (low-grade glioma), and NB (normal brain). A total of 1682 HERVs were found differentially expressed (DE) in at least one of the comparisons: GBM vs. NB, GBM vs. LGG, LGG vs. NB (Fig. 1A). In GBM vs. NB comparison, 712 (16%, orange) of differentially expressed HERVs were found, with more HERVs being downregulated (536, green) than upregulated (176, red). Comparison of GBM vs. LGG showed 1271 (21%, orange) differentially expressed HERVs with 707 HERVs downregulated (green) and 564 upregulated (red). When LGG vs. NB data were compared, a much lower number of HERVs was significantly differentially expressed (246, 7%, orange) with 170 of them being downregulated (green) and 76 upregulated (red). Importantly, there were more differentially expressed HERVs in the comparison between GBM vs. LGG (1271, 21%) than in comparisons between GBM vs. NB (712, 16%), and between LGG vs. NB (246, 7%). This finding indicates that with respect to HERV expression, GBM and LGG differ more from each other than each of them differs from NB. Detailed intersections for GBM, LGG, and NB comparisons are shown in Suppl. Fig. 1B. p-values and log2-transformed fold changes of HERV expression comparisons among the 3 brain tissue types are shown in the volcano plots (Fig. 1B). Notably, the p-values of comparing HERVs expression levels between GBM and LGG were very small (29 HERVs with p < 1 × 10−70). Differentially expressed HERVs were detected across all chromosomes (Manhattan plots shown in Suppl. Fig. 1A). Differentially expressed HERVs occurred in a variety of HERV superfamilies (ERV1, ERVL, ERVK) and families (Suppl. Fig. 1C), that are particularly enriched in stemness-associated HERV-H family members [27].

Comparisons of HERV transcription levels in GBM, LGG, and NB shown as circular diagrams (A) and volcano plots (B) for three comparisons (GBM vs. NB, GBM vs. LGG, and LGG vs. NB) with numbers and % of significantly differentially expressed HERVs indicated. Significance criteria: adj p-value < 0.05 and |log2 fold-change| > 1.
211 highly dysregulated HERVs differentiate between GBM, LGG, and NB
Out of 1682 differentially expressed HERVs found in GBM, LGG, and NB comparisons, 211 were highly dysregulated (p-value < 0.01 and absolute value of log2 fold-change >3, Suppl. Table 1) and sufficient to differentiate GBM, LGG, and NB as determined by the Principal Component Analysis (PCA) (Fig. 2A). As PC1 in the PCA distinguished GBM and LGG, we termed it “Glioma-specific HERV Score (GH Score).” The ability of this specific group of 211 HERVs to discriminate between GBM, LGG, and NB may suggest their potential importance in glioma tumor biology. Overall, among these 211 HERVs, total number of downregulated HERVs was higher than upregulated HERVs (Fig. 2B)—157 HERVs were downregulated, while 76 HERVs were upregulated in at least one of the comparisons (GBM vs. NB, GBM vs. LGG, LGG vs. NB). In GBM vs. NB comparison, 111 HERVs were downregulated and 31 upregulated (Fig. 2B). For GBM vs. LGG, 40 HERVs were downregulated, and 28 were upregulated. However, for LGG vs. NB only 6 HERVs were downregulated, while 17 were upregulated (Fig. 2B). Additionally, the 211 HERVs belong to different HERV superfamilies (ERV1, ERVL, ERVK) and families (marked in different colors, Fig. 2C), with the most represented HERV-H family members (light blue) known to be associated with stemness [27]. Moreover, the 211 dysregulated HERVs were detected on all chromosomes, with the exception of Y chromosome, which did not show any dysregulated HERV loci (Suppl. Fig. 2A). Chromosomes with the highest numbers of the 211 dysregulated HERVs were chromosomes: 7 (20 HERVs—15 upregulated and 5 downregulated), 1 (18 HERVs—2 upregulated and 16 downregulated), 2 (18 HERVs—5 upregulated and 13 downregulated), and 12 (18 HERVs—7 upregulated and 11 downregulated). All other chromosomes had 14 or less dysregulated HERVs. In respect to HERV superfamilies, chromosome 7 had 14 ERV1 family members and 6 ERVL family members, chromosome 1: 2 ERVK, 5 ERVL, 11 ERV1, chromosome 2: 7 ERVL, 11 ERV1, and chromosome 12: 1 ERVK, 4 ERVL, 13 ERV1.

A Principal component analysis (PCA) based on differentially transcribed HERVs between GBM, LGG, and NB at significance criteria: adj p-value < 0.01 and |log2 fold-change| > 3, resulting in a set of highly dysregulated 211 HERV transcripts. B Intersections for the 211 highly dysregulated HERVs (adj p-value < 0.01 and |log2FC| > 3). C Differentially transcribed HERV families of the highly dysregulated 211 HERVs (adj p < 0.01 and |log2FC| > 3). Black top numbers over bars show what % of ERV1, ERVL, and ERVK superfamilies differentially transcribed HERV members constitute. Colorful numbers over bars show what % of each HERV family differentially transcribed members constitute.
The 211 HERVs are associated with survival in GBM patients
We further explored whether the GH Score can predict the survival status of GBM patients. As shown in Fig. 3A, the GH Score was significantly associated with survival for GBM patients (log-rank test p = 0.024). We found that GBM patients with low GH score (red) had low survival. This correlation was also confirmed using Cox analysis adjusting for gender, race, and age at diagnosis (Suppl. Table 2). Among the top 20 HERVs, that most significantly affected GH Score value (Fig. 3B), 17 were upregulated in at least one of the comparisons among GBM, LGG, and NB. Most importantly, these 17 HERVs contributed to lower values of the GH Score (Fig. 3B, negative PC1/GH Score loading value—indicates inverse correlation with GH Score). This finding indicates that despite the fact that the majority of HERVs in multiple comparisons among GBM, LGG, and NB are downregulated (Figs. 1, 2B, Suppl. Fig. 1B), the upregulated HERVs may also contribute to GBM tumor growth and could impact patients’ survival. Moreover, additional HERV-based PCA based on GBM subtypes discriminated subtype characterized by the best survival rate—G-CIMP (Glioma CpG Island Methylator Phenotype) from other subtypes (blue, Suppl. Fig. 2B–D). PCA was performed for 5 subtypes present in GBM dataset [mesenchymal, classical, proneural, neural, G-CIMP (Suppl. Fig. 2B)]. The best separation was achieved for subtype characterized by exceptionally better survival rate—G-CIMP (blue, Suppl. Fig. 2C, D), while separation among other subtypes was less robust.

A Results of Kaplan–Meier survival analysis—3 groups created based on GH Score quantile values (Q1 = 3.06, Q2 = 3.97). B Visualization of individual HERV contributions to Principal Component 1 (PC1/GH Score) and Principal Component 2 (PC2) and table of the top 20 HERVs affecting GH Score (PC1) values (HERVs in red, not underlined—upregulated in one or more comparisons, HERVs underlined—upregulated in one comparison and downregulated in another comparison, HERVs in black, not underlined—downregulated in one or more comparisons).
We followed with the exploration of potential GH Score prognostic capability also in LGG (Fig. 4A). Similar to GBM, we compared the survival of individuals with low (red), medium (green), and high (blue) GH Score values. The results showed that, in contrast to GBM, GH Score was not significantly associated with survival for LGG patients (log-rank test p = 0.870). In addition, the 211 HERVs could not discriminate LGG patients with different therapy outcomes [CR/R (Complete Remission/Response, light green) vs. PD (Progressive Disease, orange), Fig. 4B].

A Results of Kaplan–Meier survival analysis—3 groups created based on GH Score quantile values (Q1 = 3.06, Q2 = 3.97), log-rank test p = 0.870. B PCA comparing “primary therapy outcome” parameter in LGG: “Complete Remission/Response” (CR/R, light green) vs. “Progressive Disease” (PD, orange).
Functional prediction links the 211 HERVs with voltage-gated potassium channels
Functional prediction showed the 211 (211-6) HERVs to be associated with a total of 307 genes. 6 HERVs were not associated with any genes. Out of the 211-6 HERVs, 43 HERVs were associated with 1 gene and 162 HERVs were associated with 2 genes (Fig. 5A). The distances of 4 HERVs to the transcription start site (TSS) were less than 5 kb, 88 HERVs were between 5 and 50 kb, while 220 HERVs were between 50 and 500 kb, and 55 HERVs were more than 500 kb apart from TSS (Fig. 5B, C). Based on the 307 associated genes, Gene Ontology (GO) Cellular Component analysis revealed the possible involvement of HERVs in regulating genes encoding “voltage-gated potassium channel complex” and its parent terms: “potassium channel complex,” “cation channel,” “ion channel complex,” “transmembrane transporter complex,” and “transporter complex” (Fig. 5D, Suppl. Table 3). GREAT analysis was also performed for up- and downregulated portions of the 211 HERVs. It revealed that downregulated HERVs are mostly responsible for association with potassium channels genes, as they are associated with Potassium Channels GO terms (Fig. 5D lower panel). Upregulated portion of the 211 HERVs did not result in any GO Cellular Component terms. Consistently, within 22 specific HERVs associated with Potassium Channels genes, 19 HERVs were downregulated in at least one of the comparisons between GBM vs. NB, GBM vs. LGG, and LGG vs. NB (downregulated HERVs marked in green, Suppl. Table 3). Only 3 HERVs were upregulated in at least one comparison (upregulated HERVs marked in red, Suppl. Table 3). Interestingly, correlation between expression of these 22 HERVs and associated 18 potassium channels genes is mostly positive in GBM and there is no correlation in LGG (Suppl. Table 3).

A–D Visualization of GREAT tool analysis. Graphs displaying statistics about the association of HERVs to the TSS of all genes putatively regulated by HERVs. A–C y-axis in percentages, numbers above bars in the graph show absolute number of items being counted. A shows how many genes each HERV is assigned as putatively regulating. B, C The distance to TSS graphs showing the distance between HERVs and their putatively regulated genes. The distances are divided into four separate groups: one from 0 to 5 kb, another from 5 kb to 50 kb, a third from 50 kb to 500 kb, and a group of all associations over 500 kb. B Associations precisely at 0 (i.e., on the TSS) are split between the [−5 kb, 0] and [0, 5 kb] groups. HERV-gene associations grouped by both distance and gene orientation (so an association of HERV that is 10 kb upstream of its predicted target gene is counted in a separate group from another HERV that is 10 kb downstream of its predicted target gene). C Only the distance of HERVs to TSS is shown. D GO Cellular Component summary for the 211 HERVs (upper panel) and for downregulated portion of the 211 HERVs (lower panel). E Top 5 canonical pathways from pathway analysis of 307 human genes associated with the 211 highly dysregulated HERVs from GREAT analysis (A).
GREAT analysis was additionally performed for different HERV superfamilies and families among the 211 HERVs but did not result in any GO Cellular Component terms. HERVs associated with potassium channels genes belong to various superfamilies and families (Suppl. Table 3). Among 22 HERVs associated with potassium channels genes, 14 HERVs belong to ERV1 superfamily, 7 to ERVL, and 1 to ERVK. Five of them belong to HERVH family, 2 to ERVLE, 2 to HERVL18, and 2 to MER61 family. Remaining 11 HERVs are single representatives of various other families.
The association between the 211 HERVs and potassium channels genes was also supported by pathway analysis of the 307 genes, in which “Potassium Channels” was the second top identified pathway (Fig. 5E). The top pathway identified was “CREB Signaling in Neurons.” The top 5 pathways also included: “GABAergic Receptor Signaling Pathway,” “Class A/1 (Rhodopsin like receptors),” and “S100 Family Signaling Pathway.” These terms are consistent with published data showing crucial contributions of potassium channels in GBM [9]. Additionally, all top 5 pathways are described in the literature as important players in GBM [28,29,30,31].
GeneMANIA analysis performed for 18 potassium channel genes associated with the 211 HERVs by GREAT analysis revealed the top association networks as “Physical Interactions”, “Shared protein domains”, “Co-expression”, and “Co-localization” (Suppl. Fig. 3). Similarly, GenMANIA analysis performed for each of the top 5 pathways from pathway analysis, revealed “Physical Interactions” to be the top association network also for “CREB Signaling in Neurons” (Suppl. Fig. 4), “GABAergic Receptor Signaling Pathway” (Suppl. Fig. 6), and “Class A/1 (Rhodopsin-like receptors)” (Suppl. Fig. 7). While “Shared protein domains” was the top association network for “Potassium Channels” (Suppl. Fig. 5) and “S100 Family Signaling Pathway” (Suppl. Fig. 8).
Discussion
To address the gap of scarce information on HERVs transcriptome in gliomas, we implemented in silico locus-specific approach in a unique setting of comparing not only GBM and NB but also LGG datasets. We characterized HERV transcriptomes of GBM, LGG, and NB, which enabled us to develop unique GH Score with prognostic capabilities in GBM. We also discovered that prognostic HERVs are functionally linked with voltage-gated potassium channels.
For HERV transcriptome quantification, we selected Telescope, as it provides an estimation of HERV expression resolved to specific genomic locations [19]. Additionally, Telescope has the greatest resolution and accuracy compared to other approaches (unique counts, best counts, RepEnrich, TEtranscripts, RSEM, SalmonTE) [19]. Another tool—GREAT, was selected to perform functional prediction because, compared to other methods, it takes into account not only binding sites proximal to genes but also properly incorporates distal binding sites [22].
We show dysregulation of HERVs in GBM (Fig. 1A, B) vs. NB, which agrees with findings in other cancers compared to normal tissues [32,33,34]. Interestingly, when it comes to HERV transcriptome, GBM and LGG differ more from each other than each of them differs from NB. GBM differs from LGG with 21% differentially expressed HERVs and from NB with 16% DE HERVs (Fig. 1A), while LGG differs from NB with 7% differentially expressed HERVs. This finding may be explained by the fact that majority of glioblastomas (~90%) develop de novo without evidence of a less malignant precursor lesion [35], while only ~10% rise from LGG. Our data suggest that although LGG can progress to GBM, they are not always different stages of the same disease, but rather separate diseases. The drawback of our study is limited number of NB samples [5], which should be addressed in future analyses.
In all comparisons among GBM, LGG, and NB we see more downregulated than upregulated HERVs (Fig. 1), which aligns with the data from breast and colon cancers [32]. Pointing out HERVs downregulation in GBM is important since previous reports focused mostly on HERVs upregulation. Downregulation of HERVs in cancer may not only constitute a diagnostic marker but also therapy target. Reversal of HERV silencing can trigger the utilization of immune antiviral response against HERVs (viral mimicry) in cancer treatment [36]. This could be used, for example, to circumvent drug resistance and reduce immune evasion. Such effects could be achieved, for example, by the use of EZH2 inhibitors [37] and histone deacetylase (HDACs) inhibitors [38]. In fact, it has been shown that demethylation agents such as 5-azacytidine (5-Aza) provided a survival benefit as a single agent and synergize with temozolomide in IDH1-mutant glioma models [39].
Principal Component Analysis (PCA) based on the 211 most dysregulated HERVs distinguished GBM, LGG, and NB (Fig. 2A) and resulted in development of GH Score that differentiated GBM from LGG. Majority of the 211 HERVs were downregulated (Fig. 2B) and showed dysregulation in a variety of superfamilies and families (Fig. 2C), in addition to the previously reported HERV-K family [7, 8, 40]. Our approach revealed high dysregulation of stemness-associated HERV-H family [27] that contributes to various cancers [41], and could be potential candidate for further studies in GBM. From other dysregulated families, HERVL family is associated with transcription activation of early embryonic genes and totipotency [42]. HERV9 family is involved in cancer as enhancers [38] and through RNA products [43].
Developed GH Score correlated with GBM survival. Interestingly, majority of the 211 HERVs that were the basis for the GH Score were downregulated (Fig. 2B). However, among the top 20 HERVs, that most significantly affected GH Score values, 17 HERVs were upregulated (Fig. 3B). Data showed that the higher the 17 HERVs upregulation was, the lower GH Score and survival in GBM. This indicates that the upregulated HERVs may be crucial in GBM survival. Interestingly, the prognostic set of 211 HERVs did not include HML6_19q13.43b, which encodes the envelope transcript (ERVK3-1) previously reported as negative prognosticator in GBM [6]. However, upregulated HML6_3p21.31b—the top HERV that most significantly affected GH Score value corresponding with worse GBM survival, could work in a similar manner. It would be important in the future to verify its transcripts. Similarly, it should be verified for HML6_8q12.1. Second top HERV—HERVH_1p31.3d was found to be significantly associated with worse esophageal adenocarcinoma prognosis [44]. In colorectal cancer, this HERVH_1p31.3d expression (among other HERVH) predicts poorer survival and can promote tumorigenesis by stimulation of BRD4-dependent transcription [45]. Our top 20 set of HERVs contributing to GH score contains 3 HERVH loci (HERVH_1p31.3d, HERVH_12q21.2a, HERVH_6q12f), and all of them are upregulated. Interestingly, it has been shown that ARID1A loss derepresses a group of HERVs including HERVH_1p31.3d, but not HERVH_6q12f [45], which could be a part of a different mechanism. Top third upregulated HERV9_11q22.3 is located upstream of FDX1 gene and can play enhancer role, as it was previously shown for HERV9 family [38]. It may be important as FDX1 expression is a prognostic biomarker in GBM and promotes progression via immune response regulation [46]. Among other top 20 HERVs, MER4B was also shown to be in top upregulated HERVs in prostate cancer and colon cancer, but in different genomic locations [32]. ERVLB4 were shown to be top-upregulated HERVs in prostate cancer and both up- and downregulated in different locations in breast and colon cancers [32]. MER4 in a different location (MER4 6p22-3c) than identified in our data (MER4_7p11.2f, MER4_7p11.2d) was shown to correlate with better efficacy of immunotherapy in non-small cell lung cancer [47]. However, interestingly, two upregulated MER4 elements (MER4_7p11.2f, MER4_7p11.2d) are flanking GBM characteristic EGFR amplification region and could possibly be connected to genome instability via non-allelic homologous recombination [48]. As MER61 family is enriched in p53 binding sites [49, 50], it may be worth verifying in the future if upregulated MER61_18q12.3a and MER61_7p15.3 contain such site. Identified upregulation of HERVL_5q12.3 corresponds with significant upregulation of the exact same loci in breast cancer [32]. HUERSP1 and HUERSP2 are not described in the literature, but GREAT associates them with gene regulation. Downregulated HUERSP1_14q23.1 is associated with JKAMP and RTN1. Interestingly, RTN1 has survival predictive properties in GBM with lower expression in aggressive phenotype [51]. Upregulated HUERSP HERVs are associated with TMEM182 (HUERSP1_2q12.1), as well as KCNK10 and GPR65 (HUERSP2_14q31.3). While TMEM182 is a new target in GBM, KCNK10 belongs to potassium channels and corresponds to our GREAT analysis results. GPR65 is overexpressed in GBM with high expression predicting poor clinical outcome [52]. In the top 20 GH Score-associated HERVs we see two ERV316A3 elements—one upregulated and one downregulated, which corresponds with literature showing up- and downregulation of these elements in different GBM cell lines [6]. Dysregulated (up- and downregulated in different comparisons) HERVL_15q11.2 and upregulated HERVS71_18q21.1, to our knowledge, are new targets in the cancer field.
The association of the GH Score with survival in GBM could be connected to different GBM subtypes, as HERV-based analysis of GBM dataset shows the best separation of G-CIMP subtype (Suppl. Fig. 2B–D) characterized by favorable survival rate (Suppl. Fig. 2C). As G-CIMP is defined by genome-wide hypermethylation of CpG (cytosine-phosphate-guanine) islands, it could be responsible for HERVs downregulation. Literature shows prognostic G-CIMP value not only in GBM but also in LGG [53]. However, in our results, we observed no correlation between GH Score and survival or therapy outcomes in LGG (Fig. 4A, B). Our results showing that GH Score correlates with survival for GBM patients, but not for LGG patients, may be caused by the fact that majority of the 211 HERVs belongs to group that is dysregulated in GBM vs. NB comparison (31 upregulated, 111 downregulated HERVs, Fig. 2B). Smaller portion of the 211 HERVs is dysregulated in GBM vs. LGG (28 upregulated, 40 downregulated), and the smallest in LGG vs. NB (17 upregulated, 6 downregulated). Approx. 40% of LGG advance to GBM [54] and LGG to GBM transition is poorly understood. Future experiments focused on HERVs in LGG are needed to answer the question if HERVs could serve as prognostic markers of LGG to GBM progression.
Our results link the 211 HERVs with regulation of genes encoding voltage-gated potassium channels (Fig. 5D, E, Suppl. Table 3). Most of the 18 potassium channels genes are associated with HERVs from HERVH, ERVLE, HERVL18, and MER61 families. HERVH family is associated with stemness [27] and contributes to various cancers [41]. MER61 family is enriched in p53 binding sites [50]. Functions of ERVL and HERVL18 families are less explored.
Interestingly, literature shows potassium channels involvement in GBM biology [9]. Potassium channels play a crucial role in GBM proliferation, apoptosis, cell cycle, migration, invasion, metastasis, and drug resistance. In addition, potassium channels have been identified as important contributors to glioblastoma stem cells (GSC) biology [55]. The vast involvement of potassium channels in cancer biology positions them as therapeutic targets [56] including GBM treatment [57]. In particular, KCNB1 could be more important than others, as it is involved in sensitivity to chemotherapy in GBM [58] and cancer stem cells biology [59]. GRIN2B gene is confirmed to play a role in glioblastoma stemness [60], while KCNB1 [59] and KCNK1 [61] are described as cancer therapeutic targets. We are also introducing novel transmembrane transporter genes in the glioma landscape—that were described to be important in other cancers but, to our knowledge, were not reported in GBM: KCNJ2 [62], KCNMB4 [63], KCNV1 [64], and SCRIB [65]. Our results also point out an emerging target in the cancer research field—triadin (TRDN), which is a part of calcium release complex [66]. Pathway analysis additionally identified “CREB signaling in neurons” with proto-oncogenic [67, 68] and temozolomide-resistance properties in GBM [28]. All top 5 identified pathways are important players in GBM [9, 28,29,30,31]. GABAergic signaling pathway [29] and rhodopsin-like receptors [31] are described as cancer therapeutic targets, while S100 family signature is associated with GBM survival and prognosis [30]. Our study focuses on HERV cis-regulatory function of genes, so it does not include RNA or protein expression and interactions, which are subject of other studies in the field. Presented in silico approach brings interesting HERV-gene associations that should be further verified in vitro.
Interestingly, association with potassium channels genes relies mostly on downregulated HERVs (Fig. 5D, Suppl. Table 3), underlining the need for more research focusing on HERVs downregulation. As mechanisms of gene regulation by HERVs are not well described, more research is needed to decipher correlations between HERVs and potassium channels genes. Insight is also needed into correlations between expression of 22 HERVs and associated 18 potassium channels genes that are mostly positive in GBM with no correlation in LGG. This result is especially compelling, as it aligns with our other data showing that LGG is very different from GBM and suggests different roles of HERVs and ion channel genes in GBM and LGG. Although most literature data links upregulation of ion channels genes with more aggressive phenotype, there is some indication that also their downregulation can play a role, especially in different parts of the GBM tumor [69]. From the group of downregulated HERVs associated with potassium channels genes HERVFC1 is described in ovarian cancer. Inhibition of histone H3K9 methylation increases the transcripts of HERV-Fc1, activating viral defense response and induction of cell death [70]. In our data, expression of different HERVH family members was not coordinated—some were upregulated, some downregulated similar to the literature data on esophageal adenocarcinoma [44] and colorectal cancer [45].
Our results are based on bulk RNA-Seq data, which limits us from making conclusions about which cells exactly are the source of observed HERV dysregulation. Additionally, literature data on the dysregulation of transposable elements with a single-cell approach is scarce. However, it suggests that HERV dysregulation in GBM comes mainly from neoplastic cells and corresponds with bulk RNA-Seq data [71]. It has been shown that HERV-K (HML-2) contributes to a stem cell niche in GBM [8]. Although we did not see HML2 dysregulation in our analysis, it may be due to the bulk RNA-Seq approach not being sensitive enough to detect the low number of stem cells in niches. However, we see that the most dysregulated HERVs in our dataset, especially in GBM vs. NB comparison, is stemness-associated HERV-H family [27]. Moreover, our analysis shows upregulated HERVH_12p13.1b to be associated with GRIN2B gene, which is confirmed to play a role in glioblastoma stemness [60]. In addition, potassium channels have been identified as important contributors to glioblastoma stem cells (GSC) biology [55].
Multidimensional character of HERVs involvement in human cells biology also translates into HERVs contribution to gliomas. Analysis of genes associated with HERVs and already known to be contributing to GBM, LGG, and other cancers may suggest HERVs may be used as markers in diagnostic, and prognostic tools. Moreover, HERVs unlock a new level of gene regulation that may be exploited in future therapy strategies. On the other hand, genes not described so far as GBM contributors, identified in our analysis bring interesting study material. Further studies are needed to decipher if HERVs dysregulation is the cause of glioma malignancy or is mirroring other changes and can be used as their markers. Differential expression of HERVs in glioma/GBM may provide insights into gliomagenesis, progression, stemness, and therapy resistance, as well as HERVs signatures in glioma/GBM diagnostics. Development of HERV locus-specific research will help decipher the interplay of HERVs, genes, and their products adding significant knowledge to the biology of brain cancer.
Data availability
Data are available in figures and supplementary materials. Raw data used for this study were obtained from The Cancer Genome Atlas—TCGA: “Glioblastoma Multiforme” TCGA-GBM project (dbGaP Study Accession: phs000178) and “Brain Lower Grade Glioma” TCGA-LGG project (dbGaP Study Accession: phs000178). Information about TCGA can be found at https://www.cancer.gov/tcga.
References
Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–89.
Ostrom QT, Cote DJ, Ascha M, Kruchko C, Barnholtz-Sloan JS. Adult glioma incidence and survival by race or ethnicity in the United States from 2000 to 2014. JAMA Oncol. 2018;4:1254–62.
Jaeckle KA, Decker PA, Ballman KV, Flynn PJ, Giannini C, Scheithauer BW, et al. Transformation of low grade glioma and correlation with outcome: an NCCTG database analysis. J Neurooncol. 2011;104:253–9.
Mao J, Zhang Q, Cong YS. Human endogenous retroviruses in development and disease. Comput Struct Biotechnol J. 2021;19:5978–86.
Shah AH, Gilbert M, Ivan ME, Komotar RJ, Heiss J, Nath A. The role of human endogenous retroviruses in gliomas: from etiological perspectives and therapeutic implications. Neuro Oncol. 2021;23:1647–55.
Shah AH, Govindarajan V, Doucet-O’Hare TT, Rivas S, Ampie L, DeMarino C, et al. Differential expression of an endogenous retroviral element [HERV-K(HML-6)] is associated with reduced survival in glioblastoma patients. Sci Rep. 2022;12:6902.
Kessler AF, Wiesner M, Denner J, Kammerer U, Vince GH, Linsenmann T, et al. Expression-analysis of the human endogenous retrovirus HERV-K in human astrocytic tumors. BMC Res Notes. 2014;7:159.
Shah AH, Rivas SR, Doucet-O’Hare TT, Govindarajan V, DeMarino C, Wang T, et al. Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J Clin Invest. 2023;133:e167929.
Younes S, Mourad N, Salla M, Rahal M, Hammoudi Halat D. Potassium ion channels in glioma: from basic knowledge into therapeutic applications. Membranes. 2023;13:434.
Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19:A68–77.
Heath AP, Ferretti V, Agrawal S, An M, Angelakos JC, Arya R, et al. The NCI Genomic Data Commons. Nat Genet. 2021;53:257–62.
Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375:1109–12.
Scarpace L, Mikkelsen T, Cha S, Rao S, Tekchandani S, Gutman D, et al. TCGA-GBM project “Glioblastoma Multiforme.” The Cancer Genome Atlas Glioblastoma Multiforme Collection (TCGA-GBM) 2016.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77.
Pedano N, Flanders AE, Scarpace L, Mikkelsen T, Eschbacher JM, Hermes B, et al. “Brain Lower Grade Glioma” TCGA-LGG project. The Cancer Genome Atlas Low Grade Glioma Collection (TCGA-LGG)]. 2016.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023;83:3861–67.
Bendall ML, de Mulder M, Iniguez LP, Lecanda-Sanchez A, Perez-Losada M, Ostrowski MA, et al. Telescope: characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput Biol. 2019;15:e1006453.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Tanigawa Y, Dyer ES, Bejerano G. WhichTF is functionally important in your open chromatin data? PLoS Comput Biol. 2022;18:e1010378.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20.
Krämer A, Green J, Pollard Jr, J., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–30..
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Wolfe D, Dudek S, Ritchie MD, Pendergrass SA. Visualizing genomic information across chromosomes with PhenoGram. BioData Min. 2013;6:18.
Sexton CE, Tillett RL, Han MV. The essential but enigmatic regulatory role of HERVH in pluripotency. Trends Genet. 2022;38:12–21.
Jiang LY, Wang GH, Xu JJ, Li XL, Lin XY, Fang X, et al. CREB-induced LINC00473 promotes chemoresistance to TMZ in glioblastoma by regulating O6-methylguanine-DNA-methyltransferase expression via CEBPalpha binding. Neuropharmacology. 2024;243:109790.
Yang Y, Ren L, Li W, Zhang Y, Zhang S, Ge B, et al. GABAergic signaling as a potential therapeutic target in cancers. Biomed Pharmacother. 2023;161:114410.
Wang H, Mao X, Ye L, Cheng H, Dai X. The role of the S100 protein family in glioma. J Cancer. 2022;13:3022–30.
Byrne KF, Pal A, Curtin JF, Stephens JC, Kinsella GK. G-protein-coupled receptors as therapeutic targets for glioblastoma. Drug Discov Today. 2021;26:2858–70.
Steiner MC, Marston JL, Iniguez LP, Bendall ML, Chiappinelli KB, Nixon DF, et al. Locus-specific characterization of human endogenous retrovirus expression in prostate, breast, and colon cancers. Cancer Res. 2021;81:3449–60.
Kolbe AR, Bendall ML, Pearson AT, Paul D, Nixon DF, Perez-Losada M, et al. Human endogenous retrovirus expression is associated with head and neck cancer and differential survival. Viruses. 2020;12:956.
Chang YS, Hsu MH, Chung CC, Chen HD, Tu SJ, Lee YT, et al. Comprehensive analysis and drug modulation of human endogenous retrovirus in hepatocellular carcinomas. Cancers. 2023;15:3664.
Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19:764–72.
Kitsou K, Lagiou P, Magiorkinis G. Human endogenous retroviruses in cancer: oncogenesis mechanisms and clinical implications. J Med Virol. 2023;95:e28350.
Hirukawa A, Singh S, Wang J, Rennhack JP, Swiatnicki M, Sanguin-Gendreau V, et al. Reduction of global H3K27me3 enhances HER2/ErbB2 targeted therapy. Cell Rep. 2019;29:249–57.e8.
Beyer U, Kronung SK, Leha A, Walter L, Dobbelstein M. Comprehensive identification of genes driven by ERV9-LTRs reveals TNFRSF10B as a re-activatable mediator of testicular cancer cell death. Cell Death Differ. 2016;23:64–75.
Yamashita AS, da Costa Rosa M, Borodovsky A, Festuccia WT, Chan T, Riggins GJ. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. 2019;21:189–200.
Misra A, Chosdol K, Sarkar C, Mahapatra AK, Sinha S. Alteration of a sequence with homology to human endogenous retrovirus (HERV-K) in primary human glioma: implications for viral repeat mediated rearrangement. Mutat Res. 2001;484:53–9.
Stricker E, Peckham-Gregory EC, Scheurer ME. HERVs and cancer—a comprehensive review of the relationship of human endogenous retroviruses and human cancers. Biomedicines. 2023;11:936.
Xiang Y, Liang H. The regulation and functions of endogenous retrovirus in embryo development and stem cell differentiation. Stem Cells Int. 2021;2021:6660936.
Xu L, Elkahloun AG, Candotti F, Grajkowski A, Beaucage SL, Petricoin EF, et al. A novel function of RNAs arising from the long terminal repeat of human endogenous retrovirus 9 in cell cycle arrest. J Virol. 2013;87:25–36.
Kazachenka A, Loong JH, Attig J, Young GR, Ganguli P, Devonshire G, et al. The transcriptional landscape of endogenous retroelements delineates esophageal adenocarcinoma subtypes. NAR Cancer. 2023;5:zcad040.
Yu C, Lei X, Chen F, Mao S, Lv L, Liu H, et al. ARID1A loss derepresses a group of human endogenous retrovirus-H loci to modulate BRD4-dependent transcription. Nat Commun. 2022;13:3501.
Zhang G, Shen L, Li Z, Zhao Y. FDX1 serves as a prognostic biomarker and promotes glioma progression by regulating the immune response. Aging. 2023;15:4963–85.
Lecuelle J, Favier L, Fraisse C, Lagrange A, Kaderbhai C, Boidot R, et al. MER4 endogenous retrovirus correlated with better efficacy of anti-PD1/PD-L1 therapy in non-small cell lung cancer. J Immunother Cancer. 2022;10:e004241.
Campbell IM, Gambin T, Dittwald P, Beck CR, Shuvarikov A, Hixson P, et al. Human endogenous retroviral elements promote genome instability via non-allelic homologous recombination. BMC Biol. 2014;12:74.
Su D, Wang X, Campbell MR, Song L, Safi A, Crawford GE, et al. Interactions of chromatin context, binding site sequence content, and sequence evolution in stress-induced p53 occupancy and transactivation. PLoS Genet. 2015;11:e1004885.
Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci USA. 2007;104:18613–8.
Guo XX, Su J, He XF. A 4-gene panel predicting the survival of patients with glioblastoma. J Cell Biochem. 2019;120:16037–43.
Wang HX, Chen YH, Zhou JX, Hu XY, Tan C, Yan Y, et al. Overexpression of G-protein-coupled receptors 65 in glioblastoma predicts poor patient prognosis. Clin Neurol Neurosurg. 2018;164:132–7.
Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 2018;20:608–20.
Nakasu S, Nakasu Y. Malignant progression of diffuse low-grade gliomas: a systematic review and meta-analysis on incidence and related factors. Neurol Med Chir. 2022;62:177–85.
Cheng Q, Chen A, Du Q, Liao Q, Shuai Z, Chen C, et al. Novel insights into ion channels in cancer stem cells (Review). Int J Oncol. 2018;53:1435–41.
Zuniga L, Cayo A, Gonzalez W, Vilos C, Zuniga R. Potassium channels as a target for cancer therapy: current perspectives. Onco Targets Ther. 2022;15:783–97.
Ru Q, Li WL, Xiong Q, Chen L, Tian X, Li CY. Voltage-gated potassium channel blocker 4-aminopyridine induces glioma cell apoptosis by reducing expression of microRNA-10b-5p. Mol Biol Cell. 2018;29:1125–36.
Wang HY, Li JY, Liu X, Yan XY, Wang W, Wu F, et al. A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget. 2016;7:74895–903.
Shiozaki A, Konishi T, Kosuga T, Kudou M, Kurashima K, Inoue H, et al. Roles of voltage‑gated potassium channels in the maintenance of pancreatic cancer stem cells. Int J Oncol. 2021;59:76.
Kaluzinska-Kolat Z, Kolat D, Kosla K, Pluciennik E, Bednarek AK. Delineating the glioblastoma stemness by genes involved in cytoskeletal rearrangements and metabolic alterations. World J Stem Cells. 2023;15:302–22.
Sun X, Li Y, Lan H, Jiang T, Wan X, Cheng Y. Identification of KCNK1 as a potential prognostic biomarker and therapeutic target of breast cancer. Pathol Res Pract. 2023;241:154286.
Chen S, Huang M, Hu X. Interference with KCNJ2 inhibits proliferation, migration and EMT progression of apillary thyroid carcinoma cells by upregulating GNG2 expression. Mol Med Rep. 2021;24:622.
Yang J, Annala M, Ji P, Wang G, Zheng H, Codgell D, et al. Recurrent LRP1-SNRNP25 and KCNMB4-CCND3 fusion genes promote tumor cell motility in human osteosarcoma. J Hematol Oncol. 2014;7:76.
Li XX, Wang LJ, Hou J, Liu HY, Wang R, Wang C, et al. Identification of long noncoding RNAs as predictors of survival in triple-negative breast cancer based on network analysis. Biomed Res Int. 2020;2020:8970340.
Shen H, Huang C, Wu J, Li J, Hu T, Wang Z, et al. SCRIB promotes proliferation and metastasis by targeting Hippo/YAP signalling in colorectal cancer. Front Cell Dev Biol. 2021;9:656359.
Zheng S, Wang X, Zhao D, Liu H, Hu Y. Calcium homeostasis and cancer: insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023;33:312–23.
Tan X, Wang S, Zhu L, Wu C, Yin B, Zhao J, et al. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc Natl Acad Sci USA. 2012;109:15805–10.
Daniel P, Filiz G, Brown DV, Hollande F, Gonzales M, D’Abaco G, et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis. 2014;3:e108.
Soeung V, Puchalski RB, Noebels JL. The complex molecular epileptogenesis landscape of glioblastoma. Cell Rep Med. 2024;5:101691.
Liu M, Thomas SL, DeWitt AK, Zhou W, Madaj ZB, Ohtani H, et al. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 2018;78:5754–66.
Bonte PE, Arribas YA, Merlotti A, Carrascal M, Zhang JV, Zueva E, et al. Single-cell RNA-seq-based proteogenomics identifies glioblastoma-specific transposable elements encoding HLA-I-presented peptides. Cell Rep. 2022;39:110916.
Acknowledgements
The results published here are in whole or part based upon data generated by The TCGA Research Network: https://www.cancer.gov/tcga. We thank Dr. John West (Louisiana State University Health Sciences Center, New Orleans, LA, USA) for inspiring ideas on experimental design and preparation of this manuscript. We are also grateful to Dr. Adam Lassak (Louisiana State University Health Sciences Center, New Orleans, LA, USA) for involvement in establishing Telescope pipeline.
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number P20GM121288. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
HM was responsible for data curation, formal analysis, investigation, methodology, visualization, writing—review and editing. HYL was responsible for conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, resources, writing—review and editing. MB was responsible for data curation, investigation, methodology, writing—review and editing, resources. WG was responsible for data curation, investigation, methodology, writing—review and editing. DPV was responsible for project administration, writing—review and editing. CH was responsible for investigation, visualization, methodology, writing—review and editing. DO was responsible for investigation and visualization. FP was responsible for writing—review and editing. ZM was responsible for writing—review and editing. VPB was responsible for conceptual contributions, data interpretation, discussion, and presentation, writing—review and editing. EKF was responsible for conceptualization, methodology, writing—review and editing. KR was responsible for conceptualization, data curation, investigation, methodology, project administration, writing—review and editing, resources, supervision. MR was responsible for conceptualization, data curation, investigation, visualization, methodology, writing—original draft, project administration, writing—review and editing, resources, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Patient consent and approval—not applicable, as this work utilized secondary data analysis based on data from The Cancer Genome Atlas (TCGA): “Glioblastoma Multiforme” TCGA-GBM project (dbGaP Study Accession: phs000178) and “Brain Lower Grade Glioma” TCGA-LGG project (dbGaP Study Accession: phs000178). Information about TCGA can be found at https://www.cancer.gov/tcga.
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Unaffiliated: Wojciech Gałan.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mazumder, H., Lin, HY., Baddoo, M. et al. Human endogenous retroviruses (HERVs) associated with glioblastoma risk and prognosis. Cancer Gene Ther 32, 622–632 (2025). https://doi.org/10.1038/s41417-024-00868-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41417-024-00868-3
