
Abstract
Despite the success of cancer immunotherapy in treating hematologic malignancies, their efficacy in solid tumors remains limited due to the immunosuppressive tumor microenvironment (TME), which is mainly formed by myeloid-derived suppressor cells (MDSCs). MDSCs not only exert potent immunosuppressive effects that hinder the success of immune checkpoint inhibitors (ICIs) and adaptive cellular therapies, but they also promote tumor advancement through non-immunological pathways, including promoting angiogenesis, driving epithelial-mesenchymal transition (EMT), and contributing to the establishment of pre-metastatic environments. While targeting MDSCs alone or in combination with conventional therapies has shown limited success, emerging evidence suggests that MDSC checkpoint blockade in combination with other immunotherapies holds great promise in overcoming both immunological and non-immunological barriers. In this review, we discussed the dual roles of MDSCs, with a particular emphasis on their underexplored checkpoints blockade strategies. We discussed the rationale behind combination strategies, their potential advantages in overcoming MDSC-mediated immunosuppression, and the challenges associated with their development. Additionally, we highlight future research directions aimed at optimizing combination immunotherapies to enhance cancer therapeutic effectiveness, particularly in solid tumor therapies where MDSCs are highly prevalent.
Similar content being viewed by others
Introduction
Immunotherapy has gained recognition as a highly promising strategy for cancer treatment, with the potential to form the backbone of future therapeutic strategies [1]. Unlike traditional therapies, which directly target tumor cells, immunotherapy leverages the patient’s immune system to identify and destroy cancer cells [2]. Despite significant success in hematologic malignancies, the efficacy of immunotherapies in solid tumors remains limited, largely due to the immunosuppressive nature of the microenvironment. The TME comprises a diverse range of immune cells populations, with MDSCs playing a prominent role in driving immune suppression [3]. MDSCs have gained attention for their role in dampening anti-tumor immunity. These cells, derived from abnormal hematopoiesis, are recruited to the TME in response to cancer, chronic inflammation, and other pathological conditions. Once in the TME, MDSCs suppress both innate and adaptive immune responses, which restricts the efficacy of immunotherapies [4].
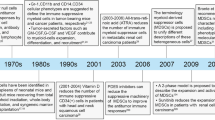
A significant obstacle in cancer treatment is the development of resistance to immune checkpoint inhibitors, such as therapies targeting lymphocyte-activation gene 3 (LAG-3), programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [5]. While checkpoint inhibitors have transformed cancer treatment, a significant percentage of patients are unresponsive. This response is often attributed to the profound immunosuppression exerted by MDSCs within the TME, which can limit the infiltration and activity of effector immune cells [6]. In both mice and humans, MDSCs are categorized into distinct phenotypic subsets. In mice, MDSCs are defined by the expression of Gr-1+ and CD11b+ and are subdivided into two main subsets: polymorphonuclear (PMN)-MDSCs, characterized by CD11b+, Ly6Ghigh, Ly6Clow, CD117+, and monocytic (M)-MDSCs, which express CD11b+, Ly6Chigh, Ly6Glow, these subsets function predominantly to suppress T-cell activation via several pathways. In humans, MDSCs lack Gr-1 expressions and are similarly classified based on CD11b expression. PMN-MDSCs are defined as CD11b+, CD33+, CD14−, CD15+ (or CD66b+), HLA-DR−, while M-MDSCs express CD11b+, CD33+, CD14+, CD15−, HLA-DR−/low. Furthermore, early-stage MDSCs (eMDSCs), a third subset, have been found in humans, characterized by Lin (comprising CD3, CD14, CD15, CD19 and CD56)−, HLA-DR−, CD33+, though their precise function remains under investigation [7]. Despite the critical role MDSCs play within the TME, originality, phenotype, their contributions to tumor progression and therapeutic resistance remain one of the least understood aspects of the immune landscape. In this regard, Nature Reviews Immunology brings together insights from eight leading experts in the field to discuss the critical questions and challenges surrounding research on MDSCs [8].
Given the central role of MDSCs in facilitating immune-mediated tumor evasion, novel strategies aimed at targeting MDSC-mediated immunosuppression are gaining concern. Checkpoint blockade of MDSCs in combination with existing immunotherapies has emerged as a potential approach to enhance anti-tumor immunity [9]. Combination immunotherapy may increase the recognition and elimination of tumor cells by the immune system, improve patient response rates, and overcome resistance to current treatments. Furthermore, the exploration of prognostic biomarkers for immunotherapy response, particularly those related to MDSC function, could guide personalized treatment decisions and improve clinical outcomes. In this review, we have explored the mechanisms of MDSC-induced immune suppression and the potential of targeting MDSC checkpoints as a strategy to enhance the efficacy of immunotherapy. By investigating combination therapies that simultaneously target MDSCs and reinvigorate immune responses, we aim to highlight promising avenues for overcoming immunosuppression and improving cancer treatment outcomes.
MDSCs as pillars of immunosuppressive network
MDSCs are critical components of the tumor microenvironment and play a pivotal role in the failure of various immunotherapeutic approaches, including ICIs, CAR-T, CAR-NK, CAR-NKT, CAR-macrophage (CAR-M), oncolytic virus, and cancer vaccine [10].
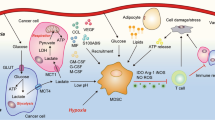
MDSCs exert their immunosuppressive effects through multiple mechanisms. One significant mechanism involves the production of immunosuppressive factors, with MDSCs secreting high levels of arginase-1 (Arg-1) [11], inducible nitric oxide synthase (iNOS) [12], and reactive oxygen species (ROS) [13], all of which play key roles in dampening T cell activity. Arg-1 depletes l-arginine, an amino acid crucial for T cell proliferation and function [14]. iNOS and ROS induce oxidative stress, leading to T cell apoptosis and the inhibition of T cell receptor (TCR) signaling [15]. Furthermore, MDSC is an important cell that secretes TGF-β and interleukin-10 (IL-10) [16] cytokines that suppress effector T cells and promote the expansion of regulatory T cells (Tregs), further enhancing immunosuppression [17]. In addition to the secretion of immunosuppressive factors, MDSCs engage in direct interactions with T cells, preventing their activation and proliferation [18]. Through the expression of inhibitory ligands such as PD-L1, MDSCs inhibit T cell function by binding to PD-1 on T cells, contributing to immune evasion [19]. In addition, MDSCs have the capability to activate the T cells to differentiate into Tregs, a process that further suppresses immune responses within the TME. Moreover, MDSCs alter the metabolic landscape of the TME by promoting hypoxia and nutrient depletion [20]. By consuming large amounts of L-arginine and cysteine, MDSCs deprive T cells of essential nutrients, impairing their function [21]. Hypoxia-inducible factors (HIFs) also regulate the metabolic reprogramming of MDSCs, enhancing their suppressive functions under the hypoxic conditions that are common in solid tumors [22]. MDSCs also actively recruit other immunosuppressive cells, such as Tregs and tumor-associated macrophages (TAMs), which amplify the immunosuppressive network in the TME [23]. The interaction between MDSCs and TAMs is particularly significant, as it drives the polarization of macrophages toward an anti-inflammatory M2 phenotype, supporting tumor growth while suppressing cytotoxic immune responses. These cells significantly influence immune suppression in cancer, contributing to tumor vascular development, therapy resistance, and increased metastasis, all of which are well supported by existing evidences.
It is now recognized that M-MDSCs and PMN-MDSCs employ distinct strategies to achieve this suppression. M-MDSCs primarily use pathways that do not necessarily require direct cell–cell contact to inhibit effector T cell responses, achieving this through the production of nitric oxide and anti-inflammatory cytokines [24]. In contrast, PMN-MDSCs can predominantly suppress immune reactions in an antigen-dependent manner, facilitating T cell tolerance specific to antigens. This ability relies on the production of free radicals, including ROS [25]. One of the significant reactive species produced by MDSCs is peroxynitrite (PNT), formed when superoxide and nitric oxide react. PNT directly suppresses T cell functionality by nitrating their T cell receptors (TCRs), diminishing their responsiveness to MHC-antigen complexes [26]. Additionally, peroxynitrite impedes T cell migration by nitrating TCRs, which hinders the binding of epitope-specific amino acids to MHC molecules on tumor cells [27]. By disrupting these immunosuppressive capabilities of MDSCs, we can foster a more favorable microenvironment for effective immunotherapies, paving the path to novel and effective cancer treatment strategies.
MDSCs contribute in cancer resistance to immunotherapies
In the last few years, the crucial role of MDSCs in mediating resistance to immunotherapy has become increasingly evident. This discovery underscores the complex interactions among malignant neoplasms, MDSCs, various immune cell types, fibroblasts, and tumor vasculature [28]. MDSCs significantly contribute to immunotherapeutic resistance by suppressing adaptive immunity and fostering angiogenesis through the secretion of pro-angiogenic factors such as VEGFA and immunosuppressive cytokines like TGF-β [29]. Various researches on MDSCs accumulate in various malignancies, including breast, lung, and prostate cancers, where they disrupt anti-tumor immune responses and hinder the therapeutic effects of ICIs. It has been reported that, as levels of circulating MDSCs rises, resistance to immune checkpoint inhibitors is likely to increase [30]. These cells function as critical protectors of TME in concert with M2-polarized TAMs to shield tumors from the host immune response and therapeutic interventions. Numerous studies have established a strong correlation between both intratumoral and circulating MDSCs and resistance to immunotherapy across various cancer models. Upon their recruitment into the TME, MDSCs are activated by chemokines secreted by cancer cells, leading to T-cell immunosuppression via STAT3-dependent pathways. This process involves the activation of arginase and iNOS, depleting critical nutrients such as arginine necessary for T-cell function [13]. Thus, targeting MDSCs has emerged as a promising therapeutic strategy to overcome treatment resistance. Finally, advancing cancer immunotherapy requires addressing MDSC-driven immunosuppression, which hinders the efficacy of ICB and adaptive cellular therapies (Fig. 1). Recent insights into MDSC biology highlight their dynamic plasticity, metabolic regulation and their suppression of diverse immune populations. Promising strategies include targeting novel checkpoints leveraging metabolic inhibitors, and exploring combination therapies that simultaneously target MDSCs and enhance effector immune cells function. Advances in single-cell transcriptomics and predictive biomarkers further enable personalized and effective treatments. This integrative approach is critical for overcoming immunotherapeutic resistance and improving clinical outcomes.

In the tumor microenvironment (TME), MDSCs inhibit the key functions of effector immune cells, impairing TME responsiveness and contributing to immunotherapy resistance in tumors (lower right panel). Conversely, following MDSC functional blockade, this renders previously immunotherapy-resistant tumors sensitive to immunotherapeutic interventions (upper right panel).
MDSC checkpoint blockade is the new avenue for solving the cancer immunotherapeutic resistance
MDSC checkpoint blockade is at the forefront of modern immunotherapy. These checkpoints act as modulators of MDSC-driven immune suppression, offering promising avenues to enhance therapeutic efficacy against cancer [4]. These checkpoints consist of inhibitory or activating pathways and molecules predominantly expressed by MDSCs, which govern their immunosuppressive behavior.
Inflammatory cytokines and mediators, including TNF-α, IL-1β, PGE2, IL-6, and VEGF, TGF-β1, are among the tumor-associated factors that promote MDSC proliferation, expansion, and differentiation [31]. The balance between immunological activation and suppression can be significantly influenced by these checkpoints. Various checkpoints have been investigated in relation to MDSCs, but several key targets for reprogramming MDSCs include Cyclooxygenases, HIF-1α TNF-α, PD-L1, STAT3, C/EBPβ, c-Rel, PGE2, CCR2, PI3K, CHOP, TIPE2, CD300ld, FATP2, HIF-1α, Alox12/15, S100A8/A9, Dkk1, PLCγ 2, TRAIL-Rs, β2AR, LILRB, SLFN4, PERK, ASAH2, Tet2, CD45 phosphatase, RORC1, VISTA, SIRT 1, AMPK alpha 1, Retinoblastoma Gene 1, and Prokineticin 2 (BV8). These MDSC checkpoints orchestrate a highly efficient immunosuppressive network within tumors. Targeting these pathways offers a promising strategy to reprogram MDSCs, thereby reducing their immunosuppressive capacity and enhancing the efficacy of immunotherapies. By disrupting MDSC-mediated suppression, immune checkpoint inhibitors, CAR-T cells, CAR-Macrophages, and other adaptive therapies may achieve greater clinical success, especially in overcoming resistance in solid tumors. In Table 1, we summarized key MDSC-associated checkpoints, detailing the signaling pathways involved, mechanisms by which these checkpoints mediate immunosuppression, and potential drug candidates targeting these pathways. Some of these candidates are already FDA-approved for clinical use, while others are currently under investigation in clinical trials (Table 2).
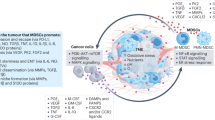
In this review, we also categorize MDSC checkpoints into seven key areas: amino acid metabolism (CHOP, PERK); lipid metabolism (Alox12/15, FATP2, ASAH2, SLFN4, COX, PGE2); mitochondrial metabolism and bioenergetics (AMPKα1, PI3K, SIRT1); hypoxia and oxidative stress (HIF-1α, DKK1); epigenetic and transcriptional regulation (C/EBPβ, STAT3, c-Rel, Tet2, Retinoblastoma Gene 1); membrane receptor signaling (CD74, TRAIL-R, LILRB, PD-L1, VISTA, CD300ld, BV8 receptor); TLR/NF-κB and MAPK signaling (S100A8/A9, TIPE2, CD45 phosphatase, TNF-α, PLCγ2). This categorization highlights how MDSCs leverage diverse metabolic pathways and signaling mechanisms to promote immune suppression (Fig. 2). We propose that targeting these distinct metabolic checkpoints may offer novel therapeutic approaches for overcoming cancer resistance to immunotherapy.

MDSC checkpoints pair and bind with their ligands on the cell membrane (top), and this activates key signaling pathways leading to regulating MDSC expansion, survival, and immunosuppressive activity (top).
Targeting checkpoints involved in lipid metabolism
FATP2
The trafficking of lipids across cellular membranes is mediated by various proteins, fatty acid transport protein (FATP), CD206, and CD36. Among these, the FATP family, consisting of FATP1 through FATP6, functions both as acyl-CoA synthetases (ACS) and as long-chain FA transporters [32]. In cancer, polyunsaturated fatty acids (PUFAs) have been shown to disrupt normal myelopoiesis in the bone marrow of tumor-bearing mice, leading to an increased accumulation and enhanced activity of PMN-MDSCs via the JAK/STAT3 signaling pathway [33]. This PUFA-driven effect on MDSCs was significantly reduced when STAT3 phosphorylation was inhibited by the JAK inhibitor JSI-124. FATP2 is selectively upregulated in both murine and human G-MDSCs, driven by GM-CSF through STAT5 activation [34]. FATP2-mediated immunosuppression involves the assimilation of arachidonic acid, a precursor for PGE2 production. Deletion of FATP2 eliminates the suppressive function of PMN-MDSCs, slowing tumor progression without affecting CD8+ T cell activity or the function of other immunosuppressive cells such as tumor-associated macrophages and monocytic MDSCs [35]. In addition, signaling via the β2-adrenergic receptor enhances fatty acid oxidation (FAO) and increases the expression of the FA transporter CPT1A in MDSCs, further promoting their immunosuppressive function [36]. This signaling pathway activates the arachidonic acid cycle, increasing PGE2 release and driving further immunosuppression [37]. PGE2 produced by MDSCs can elevate PD-L1 expression in both intratumoral MDSCs and TAMs, contributing to the differentiation of IL-10-producing T cells into dysfunctional, and exhausted T cells [38].
Targeting the COX2/mPGES1/PGE2 signaling pathway by blocking FATP2 offers a promising strategy to mitigate PD-L1-mediated immune suppression. These evidences suggests that FATP2 is a pivotal regulator of MDSC function, making it a promising therapeutic target. By inhibiting FATP2, it is possible to reduce MDSC-mediated immune suppression, potentially enhancing the efficacy of cancer immunotherapy. Furthermore, combining FATP2 inhibition with checkpoint blockade therapies such as anti-PD1, as well as adoptive cell therapies like CAR-T, CAR-NK, and CAR-M therapies, could further enhance anti-tumor immune responses. Future research should aim to elucidate the molecular mechanisms underlying FATP2’s interaction with CD36 and FA-binding proteins in MDSC lipid uptake, as well as to evaluate the therapeutic efficacy and safety of targeting FATP2 in combination with other immunotherapies.
Alox12/15
Arachidonate 12/15-lipoxygenase (ALOX12/15) is a key enzyme involved in the metabolism of polyunsaturated fatty acids (PUFAs), primarily arachidonic acid, which is oxidized to produce bioactive lipids such as 12-hydroxyeicosatetraenoic acid (12-HETE) and 15-hydroxyeicosatetraenoic acid (15-HETE). These metabolites, along with others like prostaglandin E2 (PGE2) and eicosanoids, play a pivotal role in the immunosuppressive functions of MDSCs within the tumor microenvironment [39]. Research by Tang et al. has shown that IL-13 induces MDSCs to overexpress 15-lipoxygenase (ALOX15), leading to increased production of lipid mediators that enhance MDSC immunosuppressive activity [40]. Specifically, eicosanoids produced by ALOX15, such as 15-HETE, have been found to promote MDSC recruitment, growth, and activation, thereby contributing to the establishment of an immunosuppressive tumor microenvironment [10]. These eicosanoids dampen immune responses by inhibiting effector T cells and NK cells, and promoting the expansion of Tregs, which facilitates tumor progression. Further investigations have highlighted that deletion of the ALOX12/15 gene in tumor-associated PMN-MDSCs alters their metabolome and gene expression profiles [41]. The deletion enhances immune responses by upregulating genes involved in complement activation, neutrophil-mediated immunity, monocyte chemotaxis, and antigen presentation. Moreover, inhibition of ferroptosis, a form of iron defendant-regulated cell death mediated by ALOX12/15, has been shown to augment the anti-cancer effects of immune checkpoint inhibitors and to suppress tumor growth [42]. This suggests that ALOX12/15 not only supports MDSC-mediated immunosuppression but also plays a critical role in ferroptosis.
Given the central role of ALOX12/15 in MDSC biology, targeting this enzyme offers significant therapeutic potential. Blocking ALOX12/15 could disrupt the synthesis of lipid mediators that decrease immunity, such as PGE2 and 15-HETEs, reducing the immunosuppressive capacity of MDSCs and enhancing the immune system’s ability to attack the tumor. By impairing MDSC function, ALOX12/15 inhibitors could also promote a pro-inflammatory tumor microenvironment, facilitating the homing and activation of cytotoxic immune cells, including CD8+ T cells and NK cells. Furthermore, targeting ALOX12/15 could synergize with other immunotherapeutic approaches, such as cancer vaccines, immune checkpoint blockade, and adoptive cellular therapies. Further research on ALOX12/15 inhibitors is crucial to understanding their therapeutic potential and their role in MDSCs ferroptosis, which could enhance anti-tumor immunity and cancer immunotherapy efficacy.
PGE2
Prostaglandin E2 (PGE2) is a key mediator in the biology of MDSCs, playing a significant role in chronic inflammation and tumor progression. PGE2 facilitates MDSC expansion and enhances their suppressive impact on T cell-mediated immune responses [38]. It modulates the activity of enzymes associated with MDSC function, including iNOS, indoleamine 2,3-dioxygenase, and arginase 1, through binding to four distinct receptors: EP1 through EP4 [43]. Studies have demonstrated the pivotal role of the PGE2-COX2 signaling pathway in regulating MDSC immunobiology. Rodriguez et al. showed that PGE2-COX2 drives the expression of the immunosuppressive gene arginase 1 in MDSCs. Inhibiting COX-2 resulted in reduced arginase I expression and enhanced lymphocyte-mediated antitumor responses [43]. Additionally, COX2 activity in MDSCs is associated with the generation of mitochondrial ROS, and decreasing COX2 reduces ROS levels, compromising MDSCs’ inhibitory capacity [38, 39]. Moreover, PGE2 released by tumors upregulates CD73 expression in monocytic MDSCs, leading to increased suppression of T cell activity through elevated adenosine levels. Lowering adenosine levels with PEGylated adenosine deaminase has been shown to enhance CD8+ T cell activation and improve the response to immune checkpoint inhibitor therapy [44]. Furthermore, PGE2 secreted by MDSCs in ovarian cancer promotes the expression of PD-L1 and the emergence of cancer stem-like cells. It upregulates PD-L1 expression in ovarian cancer cells via the mTOR signaling pathway, contributing to tumor progression [45]. Targeting EP receptors, particularly EP2 and EP4, presents a promising approach for enhancing the effectiveness of adoptive immunotherapy. Clinical studies evaluating EP4 antagonists (such as E7046) and dual EP2/EP4 antagonists (such as TPST-1495) in advanced malignancies are underway. The COX-2-PGE2 signaling cascade plays a crucial role in modulating the tumor microenvironment (TME) by influencing immune cell polarization, activating cancer-associated fibroblasts, and promoting ECM remodeling, angiogenesis, and T cell exclusion. Its involvement in epithelial-to-mesenchymal transition (EMT) and crosstalk with pathways like Wnt/β-catenin and TGF-β highlights its importance in tumor progression. Understanding these mechanisms could help overcome resistance to COX-2 inhibitors and guide novel combination therapies.
COX
Cyclooxygenases (COX) are enzymes crucial to producing prostanoids, including prostaglandins, prostacyclins, and thromboxanes. There are two main isoforms: COX1, considered a “housekeeping” enzyme, and COX2, an inducible isoenzyme. COX2 is expressed at low levels in most tissues but can be highly expressed in response to various stimuli, including inflammatory mediators and tumor promoters [46]. Targeting the COX pathway in MDSCs has shown promise in enhancing immunotherapeutic efficacy. COX2 inhibitors reduce MDSC prevalence and impair their suppressive functions by decreasing ARG1 expression and inhibiting mitochondrial ROS production within MDSCs [47]. This highlights COX2 as a potential therapeutic target to mitigate MDSC-induced immunosuppression in the tumor microenvironment. Studies have explored the efficacy of targeting the end receptors of COX1/2 metabolites using selective EP4 receptor antagonists like MF-766 [48]. Furthermore, in mouse models, MF-766 enhances the efficacy of anti-PD-1 therapy by promoting the infiltration of CD8+ T cells, NK cells, and conventional dendritic cells into the tumor microenvironment, shifting macrophage phenotypes toward M1-like ones, and reducing the number of granulocytic MDSCs. Additionally, Li et al. found a correlation between COX-2 expression and key immunosuppressive genes in MDSCs in nasopharyngeal cancer (NPC). Inhibiting COX-2 attenuated the rise of COX-2 levels and epithelial-mesenchymal transition (EMT) scores in NPC cells driven by MDSCs, suggesting COX-2 as a critical checkpoint for MDSC–tumor cell interactions and tumor promotion [47]. In the same vein, Prima et al. reported a significant decrease in PD-L1 transcription after inhibiting PGE2 anabolism by COX2 inhibitors or enhancing PGE2 catabolism. This indicates the role of the COX2/mPGES1/PGE2 axis in controlling PD-L1 expression in myeloid cells within the tumor microenvironment [49]. Furthermore, Muthuswamy et al. demonstrated that COX2-specific inhibitor Celecoxib effectively counteracted the suppressive influence of myeloid cells on effector immune cells by inhibiting COX2, IDO, and IL-10 expression. In addition, Veltman et al. reported large numbers of infiltrating MDSCs were found to co-localize with COX-2 expression in areas of active tumor growth. Treatment with celecoxib was shown to effectively reduce prostaglandin E2 levels both in vitro and in vivo. In mesothelioma tumor-bearing mice, celecoxib administration prevented the local and systemic expansion of all MDSC subsets, leading to impaired MDSC function characterized by decreased levels of ROS and NO. Additionally, celecoxib treatment reversed T-cell tolerance, thereby enhancing the efficacy of immunotherapy [50]. Collectively, COX2 inhibitors show promise in diminishing the MDSC population and inhibiting their suppressive roles, positioning them as potential trajectories in cancer immunotherapy. However, further research is required to validate their safety and utility in combination with other immunotherapy.
ASAH2
Acylsphingosine amidohydrolase 2 (ASAH2) is a neutral ceramidase that plays a crucial role in sphingolipid metabolism in MDSCs, helping them resist ferroptosis. This hydrolytic enzyme is encoded by the ASAH2 gene which is critical in ceramide metabolism [51]. Multiple groups reported upregulation of ASAH2 expression in both colon cancer cells and intratumoral MDSCs. They found that ASAH2 functions as a survival factor for MDSCs via the p53 pathway [52]. The researchers developed a small molecule inhibitor to block ASAH2 function. Inhibition of ASAH2 led to increased p53 protein stability, upregulation of Hmox1 expression, suppression of ROS generation, and MDSC death via iron-dependent lipid peroxidation. Additionally, the inhibitor treatment reduced glutathione and cysteine uptake in MDSCs, indicating that it targets glutathione synthesis from cysteine [53]. These findings highlight the role of ASAH2 as an important checkpoint and ferroptosis regulator in MDSCs. Coant and colleagues demonstrated that inhibition of ASAH2 triggers the dephosphorylation of GSK3β, necessary for the phosphorylation and degradation of β-catenin. They found that ASAH2 inhibition in colorectal cancer cells leads to decreased GSK3β phosphorylation and activation of AKT, a critical cell growth target. This inhibition also causes AKT dephosphorylation and dysfunction. The study suggests that ASAH2 controls the basal activation of AKT, a major pathway employed by MDSCs to drive immunosuppressive phenotypes, making it a potential checkpoint for colon cancer treatment. In a xenograft model, ASAH2 inhibition resulted in elevated ceramide levels, reduced proliferation, and delayed tumor growth, confirming the functional requirement of neutral ceramidase in colon cancer control and progression [52]. These results highlight the significance of ASAH2 in controlling ferroptosis in MDSCs and imply its possibility as a target to overcome MDSCs-mediated immunosuppression.
SLFN4
Targeting Schlafen 4 (SLFN4) in MDSCs presents a promising strategy for overcoming immune suppression within the TME. SLFN4 is a member of the Schlafen family of proteins that plays a key regulatory role in the differentiation, recruitment, and activation of MDSCs [54]. Its expression is induced by type 1 interferons, particularly IFN-α, primarily produced by plasmacytoid dendritic cells (pDCs), and it has been shown to significantly enhance the immunosuppressive capacities of MDSCs [55]. In gastrointestinal malignancies, particularly during Helicobacter pylori infection, SLFN4 expression is upregulated and associated with the expansion of SLFN4+ MDSCs in the gastric TME. This process is mediated by the Sonic Hedgehog (SHH) signaling pathway, which promotes metaplastic transformation and enhances the immunosuppressive activity of MDSCs through mechanisms involving key suppressive factors such as Arg-1 and iNOS. SLFN4+ MDSCs also express microRNA miR130b, which mediates T-cell suppression, further contributing to the pro-tumorigenic environment [56]. The ability of SLFN4 to drive immune suppression by promoting MDSC polarization and activation suggests that it may play a similar role in other solid tumors. Indeed, SLFN4 knockdown experiments have shown that inhibition of this protein diminishes the immunosuppressive properties of MDSCs, reducing levels of Arg-1 and iNOS, and consequently restoring T cell function [57]. This suggests that targeting SLFN4 could impair MDSC function, alleviate immune suppression, and enhance the efficacy of other immunotherapeutic approaches. From a therapeutic standpoint, pharmacological inhibition of SLFN4 either through small molecules like sildenafil or genetic approaches has demonstrated efficacy in preclinical models. For instance, inhibiting SLFN4 in mouse models of H. pylori-induced gastric metaplasia not only attenuated MDSC-mediated immune suppression but also reduced the proliferation of cancer stem cells [58]. Targeting SLFN4 in MDSCs offers a dual benefit: it could reduce the immune suppression exerted by MDSCs, thereby lowering their inhibitory effects on T cells, while also modulating the TME to become less favorable for tumor growth. In this context, SLFN4 blockade could synergize with existing immunotherapies. For example, combining SLFN4 inhibition with immune checkpoint inhibitors (such as anti-PD-1 or anti-CTLA-4) could enhance T cell activity by not only relieving checkpoint-mediated inhibition but also reducing the immunosuppressive influence of MDSCs. Future research should explore the signaling pathways regulating SLFN4 expression and its effects on MDSC function, particularly STAT3 and NF-κB. Understanding SLFN4’s interactions could reveal how MDSCs maintain suppressive phenotypes and identify targetable metabolic vulnerabilities with SLFN4 blockade.
Targeting checkpoints involve in amino acid metabolism
CHOP
C/EBP homologous protein (CHOP), a crucial transcription factor, acts as a checkpoint for MDSCs in executing their immunosuppressive functions. Experimental evidence suggests that CHOP-deficient MDSCs can transition from immune suppressors to immune stimulators, acquiring some dendritic cell phenotypes and functionalities [59]. This functional polarization of MDSCs has been linked to factors such as endoplasmic reticulum (ER) stress, acidosis, and hypoxia, which significantly upregulate CHOP expression in intratumoral M-MDSCs and bone marrow-residing PMN-MDSCs [13]. The increase in CHOP expression is associated with peroxynitrite and ROS levels in the tumor microenvironment. Notably, CHOP is primarily expressed by myeloid cells infiltrating the tumor microenvironment, excluding macrophages [60]. Knocking out the Chop gene significantly inhibits tumor growth by altering MDSC activity, reducing their immunosuppressive functions, including diminished arginase-1 and ROS levels. Chop deficiency enhances IFN-γ-producing CD8+ T cell recruitment to tumors, reversing tumor growth when CD8+ T cells are reduced. MDSCs from Chop-deficient mice gain antigen-presenting capabilities, reflecting a functional shift. Additionally, CHOP deficiency reduces C/EBPβ, p-STAT3 activity, and IL-6 production, further impacting tumor progression [13]. Importantly, the restoration of tumor growth and MDSC suppressive activity in CHOP-deficient mice through IL-6 overexpression highlights CHOP’s regulatory role in MDSC behavior via IL-6 production. Notably, immunosuppressive activity is primarily observed in intratumoral and vascular MDSCs, whereas splenic MDSCs show minimal suppression of nonspecific T-cell proliferation. This distinction aligns with the specialized immunosuppressive role of MDSCs in the tumor microenvironment. Furthermore, lncRNAs play a critical role in modulating MDSC functions within the tumor, further emphasizing the complexity of their regulation [61]. A recently identified lncRNA, Lnc-CHOP, has been found to influence the immunosuppressive behavior of MDSCs. Lnc-CHOP promotes C/EBP activation and enhances the expression of immunosuppressive genes in MDSCs, whereas lnc-C/EBP suppresses C/EBP activation, leading to decreased immunosuppressive function and reduced MDSC differentiation [60]. Future research should explore CHOP’s role in MDSC-mediated immunosuppression during tumor progression, its interaction with cellular stress responses, immune checkpoint molecules, and tumor microenvironment. Understanding CHOP’s impact on T cell and NK cell functions is crucial. Identifying CHOP-related biomarkers could improve patient selection for CHOP-targeted therapies, making cancer immunotherapy more precise and effective.
PERK
Protein Kinase R-like Endoplasmic Reticulum Kinase (PERK) is a type I integral transmembrane protein crucial for the cellular unfolded protein response triggered by ER stress resulting from the accumulation of unfolded or misfolded proteins. Several studies have highlighted the capability of targeting the PERK axis to reprogram MDSCs from immunosuppressive to antitumoral phenotype [62]. Research by Mahadevan et al. demonstrated that when tumor cells undergo ER stress, they can induce stress in intratumoral myeloid cells, resulting in upregulation of various tumor-promoting genes like Arg-1 and iNOS, accelerating tumor growth [63]. Additionally, recent findings suggest that the integrated stress response can drive PD-L1 overexpression in lung cancer [64]. Interestingly, Mohammed et al. reported elevated PERK signaling in tumor-derived MDSCs, and inhibition of this signaling reprogrammed MDSCs into effector antitumoral myeloid cells [62]. These reprogrammed MDSCs engulfed tumor cells, co-activated anti-tumor CD8+ T-cell function, and served as professional antigen-presenting cells. The disturbance of NRF2-driven antioxidant capability and mitochondrial respiratory equilibrium in PERK-deficient tumor-MDSCs impaired their ability to suppress CD8+ T cells. Moreover, cytosolic mitochondrial DNA elevation induced by reduced NRF2 signaling in PERK-deficient MDSCs led to STING-dependent production of anti-tumor Type-I Interferon. The immunoinhibitory capability of PERK-ablated MDSCs was restored by blocking Type-I interferon receptor-I, conditionally via deleting STING or reactivating NRF2 signaling. Furthermore, intravenous administration of MDSC-specific PERK knockout resulted in significant tumor growth suppression [62]. Studies also evaluated the synergistic effect of PERK inhibitors with anti-PD-L1 in the B16F10 tumor model, showing promising outcomes. PERK has been linked to MDSCs' “well-being” and prevents STING activation through its antioxidant properties. Additionally, attenuated tumor growth and activated anti-tumor T cell immunity were observed when PERK was deleted or pharmacologically inhibited in mice harboring melanoma [65]. Recently, Liu et al. found that HSPC reprogramming into committed MDSC precursors in the spleen was mediated via PERK-ATF4-C/EBPβ signaling [66]. In addition, pharmacological and genetic suppression of this pathway prevented the differentiation of myeloid descendant cells into MDSCs, leading to significant tumor regression in mice [67].
Targeting mitochondrial metabolism and bioenergetics
AMPK alpha 1
AMP-activated protein kinase (AMPK) is a heterotrimeric serine/threonine kinase complex present in all animals, serving as a crucial metabolic sensor to maintain cellular energy balance during stress. This complex consists of catalytic subunits (AMPKα1 or AMPKα2) and regulatory subunits (AMPKβ and AMPKγ) [68]. Studies by Trillo Tinoco et al. revealed a significant association between AMPKα signaling and the immunosuppressive functions of MDSCs in tumors. They found heightened AMPKα activity in MDSCs from tumor-bearing mice and human ovarian cancer patients, driven by GM-CSF from cancer cells via STAT5-dependent transcription of the Ampkα1 gene. Inhibiting AMPKα slowed tumor growth, weakened MDSC suppression, activated antitumor CD8+ T-cell immunity, and enhanced the success of CAR T-cell therapy. Conversely, stimulating AMPKα signaling increased MDSC immunoregulatory functions [69]. Similarly, deleting AMPKα1 gene altered the differentiation pathway of M-MDSCs, leading them to adopt a cytotoxic role against tumors through nitric oxide synthase 2, highlighting the central role of AMPKα1 in MDSC-mediated immunosuppression. Various studies have demonstrated that AMPK activation inhibits the growth and negative functionality of MDSCs, suggesting it is a viable therapeutic target in immuno-oncology. Trikha et al. found that OSU-53 administration triggered AMPK phosphorylation, reducing nitric oxide production, MDSC migration, and IL-6 expression. Treatment with OSU53 decreased the immunosuppressive effects of murine MDSCs and increased T cell functionality. Moreover, AMPK is also essential for the differentiation of bone marrow cells into endothelial progenitor cells and mediates the differentiation of CD11b+/Gr-1+ MDSCs in tumor-bearing hosts. Inhibition of AMPK activity through Comp-C treatment reduced glucose uptake rates without impacting overall cell growth or viability [70]. Moreover, Adeshakin et al. demonstrated that metformin, an anti-diabetic drug, activates AMPK and enhances CHOP expression, thereby increasing stress responses in an anchorage-independent B16F10 melanoma model. This results in reduced antioxidant activity and the accumulation of misfolded proteins, sensitizing tumor cells to anoikis. This metabolic reprogramming could disrupt the supportive environment for MDSCs [71]. Consequently, targeting AMPKα1 in MDSCs, in combination with adaptive cellular therapies or immune checkpoint inhibitors (ICIs), could enhance the effectiveness of cancer treatments. However, care must be taken when targeting AMPKα1, as it plays a crucial role in maintaining energy balance across various cell types. Inhibiting AMPK in MDSCs may unintentionally disturb energy regulation in healthy tissues, potentially causing adverse side effects. Therefore, therapeutic strategies must carefully balance targeting AMPK in MDSCs while minimizing potential harm to non-cancerous cells.
PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) plays a pivotal role in regulating cell cycle processes, such as proliferation, quiescence, and energy metabolism. In the context of MDSCs, PI3K signaling is integral to several cellular functions, including cytokine production, homeostasis, glucose and lipid metabolism, proliferation, and recruitment [72]. These processes collectively drive the immunosuppressive capabilities of MDSCs. The PI3K/AKT/mTOR pathway is particularly important for the development and immunosuppressive behavior of MDSCs in both chronic inflammation and malignancies. The PI3K family includes PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ, with PI3Kδ and PI3Kγ being the primary isoforms implicated in promoting survival and immunosuppressive functions in myeloid cells [73]. This signaling pathway can shift cellular energy generation from glycolysis to oxidative phosphorylation (OXPHOS), which supports MDSC survival and their suppressive activity. PI3Kγ specific blockade to target these cells and improve tumor outcomes has been widely studied in recent years with propitious outcomes in the preclinical stage. Inhibiting the PI3K/AKT/mTOR circuit compromises the viability and function of MDSCs [74]. Dysregulation of PI3K/AKT signaling has been linked to the accumulation of MDSCs in multiple tissues, including the bone marrow, secondary lymphoid organs, sites of chronic inflammation, and tumors. Importantly, targeting specific isoforms of PI3K, such as δ and γ, using inhibitors like IPI-145, has been shown to partially reverse MDSC-mediated immunosuppression. This inhibition enhanced the clinical success of ICIs for head and neck malignancies [75]. However, high doses of IPI-145 can inadvertently inhibit antitumor T cell function, thereby negating the therapeutic benefits of anti-PD-L1 therapy. Moreover, PMN-MDSCs acquire their suppressive function through the upregulation of G-CSF, which is controlled by PI3K activation and regulated by interferon signaling. Interestingly, blocking PI3K or enhancing IFN-I signaling has been demonstrated to lower tumor growth by impairing the immunosuppressive function of MDSCs [34]. One challenge of PI3K inhibition is its potential to inadvertently impair T cell function. This highlights the need for further research into isoform-specific PI3K inhibitors to achieve better clinical outcomes with fewer off-target effects. Interestingly, artemisinin (ART), an antimalarial drug, has been shown to inhibit MDSC recruitment and function in breast cancer models, thereby enhancing the efficacy of anti-PD-L1 therapy both in vitro and in vivo. ART functions by blocking PI3K/AKT/mTOR signaling, which leads to reduced MDSC accumulation and increased T-cell infiltration into tumors [76]. Lastly, it is important to exercise caution when interpreting these findings, as tumors are known to promote MDSC expansion. Therefore, the observed reduction in MDSCs following treatment with a PI3Kδ inhibitor may be an indirect consequence of improved tumor control rather than a direct inhibitory effect on MDSCs. Furthermore, Tregs are also capable of driving MDSC expansion, and a decrease in MDSCs might reflect Treg inhibition rather than a direct action of the PI3Kδ inhibitor on MDSCs.
SIRT 1
Silent information regulator 1 (SIRT1) is a histone deacetylase that relies on NAD+ for regulating various biological functions, including cell proliferation, gene regulation, cellular viability, stress resilience, aging, programmed cell death, and metabolic processes [77]. It has been observed to deacetylate p53 and inhibit its transcriptional activity. Therefore, SIRT1 is considered a crucial epigenetic checkpoint in the reprogramming of MDSCs [78]. Numerous studies highlight its importance in controlling MDSC differentiation into the tumor-suppressive M1 subtype, facilitated by glycolytic reprogramming dependent on the mTOR/HIF-1α pathway [77]. Furthermore, SIRT1 plays a role in immunological modulation and metabolic reprogramming by deacetylating transcription factors like HIF1α. Targeting SIRT1 in MDSCs has been shown to regulate the production of TNFα and TGFβ1, aiding in the antigen-specific differentiation of TH1 and iTreg cells [77]. Additionally, SIRT1 acts as a checkpoint in MDSC differentiation into M1 or M2 lineages [79]. Interestingly, SIRT1 deficiency in MDSCs promotes a switch to the M1 lineage, reducing their immunosuppressive capacity and promoting a pro-inflammatory phenotype associated with attacking tumor cells. Furthermore, SIRT1 regulates T-cell-mediated immunity by preventing CD4+ T cells from differentiating into interleukin-9-secreting cells and downregulating transcription factors like NF-κB and AP-1, crucial for regulating T-cell responses [80]. These evidences highlights the critical role of SIRT1 in regulating MDSC activity and maturation, as well as its influence on T-cell immunity. Targeting SIRT1 presents a promising strategy for cancer immunotherapies aimed at overcoming MDSC-mediated immune suppression. Future research should focus on elucidating the precise mechanisms by which SIRT1 modulates MDSC function and identifying synergistic approaches that combine SIRT1 modulation with existing immunotherapies to enhance anti-tumor efficacy. Additionally, studies exploring SIRT1 inhibitors or activators in various cancer models could provide valuable insights into optimizing MDSC-targeted interventions.
Epigenetic and transcriptional regulators
C/EBPβ
C/EBPβ is one of the C/EBP family’s six transcription factors that has recently attracted significant attention for its role in MDSCs. While C/EBPα and C/EBPδ primarily regulate differentiation, growth, and apoptosis in myeloid cells, C/EBPβ is essential in Ly6C+ monocyte vitality and the broader regulation of MDSCs survival [81]. Studies have shown that C/EBPβ is essential for TanIIA-induced cell differentiation, indicated by the upregulation of CHOP, which controls the expression of key molecules in MDSCs. Furthermore, C/EBPβ collaborates with STAT3 during the production, expansion, and functional regulation of MDSCs, making it a critical transcription factor for MDSC gene expression and activation control. In the context of cancer-driven inflammation, the retinoic acid-related orphan receptor C (RORC) enhances the C/EBP-mediated expansion of MDSCs. Interestingly, this regulation is independent of IL-17A, a cytokine produced by RORC1/2, highlighting the unique pathways through which C/EBPβ drives MDSC proliferation [9]. Several studies have positioned C/EBPβ as a pivotal checkpoint in MDSC-mediated immunosuppression. For example, Qi and his colleagues revealed that C/EBPβ regulates MDSC function via the TIM-3 signaling pathway during Toxoplasma gondii infection. Additionally, C/EBPβ modulates the expression of immunosuppressive genes, including Arginase-1 and Nitric oxide synthase 2, which are essential for MDSC function [82]. In addition, energy metabolism and tumor glycolysis also play a role in C/EBPβ regulation, influencing the expression of G-CSF and GM-CSF via the C/EBPβ-LAP pathway. Inhibition of glycolysis using 2-deoxy-d-glucose has been shown to suppress MDSC proliferation and reduce their immunosuppressive effects in triple-negative breast cancer [11]. Notably, increased expression of C/EBPβ promotes the expansion of PMN-MDSCs while inhibiting the differentiation of M-MDSCs, which are more prevalent in tumors and exhibit higher immunosuppressive activity [83].
The critical role of C/EBPβ in the immunoregulatory functions of MDSCs has been further demonstrated in several studies, which suggest that microRNA-181b and microRNA-21 expression promotes MDSC development in the spleen and bone marrow by enhancing C/EBPβ activity [84]. The vast majority agree that C/EBPβ is necessary for MDSC development and functioning. However, recent findings indicate that C/EBPα negatively regulates MDSC differentiation, making it a potential therapeutic target in myeloid cells. For instance, MTL-CEBPα, an RNA therapy targeting C/EBPα, has shown encouraging outcomes from a phase I clinical trial for hepatocellular carcinoma, where it suppressed CXCR4 expression on immune cells attracted to the tumor microenvironment [85]. These collective findings underscore the pivotal role transcription factors like the C/EBP family play in controlling MDSC function, suggesting that targeting these pathways could offer new therapeutic strategies. The precise stage at which C/EBPβ exerts its most significant impact on MDSCs remains to be elucidated.
c-Rel
c-Rel, a member of the NF-κB family, plays a pivotal role in regulating the function of both myeloid and lymphoid cells [86]. Despite its therapeutic potential, the mechanistic role of c-Rel in MDSCs is not fully understood. However, emerging research indicates that c-Rel may serve as a potential therapeutic checkpoint in MDSCs, selectively promoting pro-tumoral gene expression while repressing anti-tumoral genes via a c-Rel enhanceosome complex [87]. Studies have shown that c-Rel is crucial for the development and immunosuppressive function of MDSCs in cancer. For instance, research by Li et al. demonstrated that c-Rel deficiency reduces CD11b+ Gr-1+ MDSCs and their suppressive capacity, while its inhibition combined with PD-1 blockade enhances CD8+ T cell antitumor activity and reduces tumor growth. In support of this, c-Rel inhibitor c has been developed by the same group and shown to specifically block c-Rel activity, leading to improved anti-tumor immune responses. Moreover, deletion of c-Rel in MDSCs has been associated with reduced tumor progression in mouse models of melanoma and lymphoma. Tumor size and weight were significantly decreased, corresponding with a reduction in MDSC numbers and a loss of their immunosuppressive characteristics, while important anti-tumoral genes were upregulated [88]. Recent studies by the same group utilizing single-cell transcriptomics have identified a novel subset of highly immunosuppressive immature myeloid cells, referred to as Rel-dependent monocytes (rMos), which rely on c-Rel for their function. These rMos contribute to tumor-associated inflammation and suppress T cell activity and proliferation within the tumor microenvironment. Notably, rMos eventually differentiates into M2 macrophages, which are known to support tumor growth. These findings suggest that c-Rel is a critical promoter of MDSC-driven tumorigenesis and may represent a novel therapeutic target in cancer treatment.
STAT
The signal transducer and activator of the transcription STAT family comprise transcription factors that play essential roles in cellular signaling from the membrane to the nucleus, regulating gene expression, and modulating various biological processes. STAT proteins, particularly STAT3 and STAT5, play central roles in the expansion, activation, and immunosuppressive functions of MDSCs [89]. Aberrant activation of STAT3, triggered by cytokines drives the survival, proliferation, and differentiation of MDSCs while upregulating immunosuppressive molecules. Inhibition of STAT3 disrupts MDSC-mediated suppression by impairing their expansion and blocking transcription of immunosuppressive genes [90]. Similarly, STAT5 activation downstream of GM-CSF contributes to MDSC differentiation and functional stability. Research has shown that MDSCs can activate STAT signaling pathways to enhance their immunosuppressive functions. For instance, Peng et al. demonstrated that MDSCs promote the crosstalk between IL-6 and NO, activating the Notch and STAT3 signaling pathways in breast cancer cells. This interaction plays a key role in maintaining the stem-like properties of breast cancer cells [91]. Similarly, MDSCs release exosomal proteins like S100A9, which activate STAT3 signaling to sustain CSC phenotypes [92]. Moreover, Adeshakin et al. recently reported that STAT3 plays a critical role in helping cancer cells evade anoikis by promoting the expression of vacuolar ATPase (V-ATPase), which aids in their survival during detachment. By inhibiting STAT3, cancer cells can become more susceptible to anoikis, and simultaneously, a decrease in V-ATPase levels can lead to elevated reactive oxygen species and the accumulation of misfolded proteins within MDSCs [93]. This disruption may enhance the apoptosis of MDSCs, thereby lessening their ability to suppress T-cell activation. Ultimately, targeting the STAT3/V-ATPase pathway not only disrupts the survival mechanisms of detached tumor cells but also enhances anti-tumor immunity by promoting MDSC apoptosis, making it a promising strategy for preventing cancer metastasis and improving the therapeutic success of immunotherapy. The involvement of STAT family signaling extends to various cancer types, with distinct roles for different MDSC subpopulations. In lung cancer, M-MDSCs and MDSCs PMN-MDSCs activate different STAT pathways, with M-MDSCs upregulating STAT1 [94] and STAT3 signaling to induce CSC phenotypes and promote lung metastasis [95]. PMN-MDSCs, on the other hand, facilitate metastasis by restoring the epithelial-to-mesenchymal transition and inducing CSC properties [25]. Studies have shown that inhibiting STAT3, using inhibitors like Stattic, can reduce the accumulation of M-MDSCs and improve the immune response [96]. By inhibiting STAT3, it may be possible to prevent the differentiation of progenitors into MDSCs, thereby alleviating immune suppression within the TME. Furthermore, combining STAT3 blockers with conventional treatments, such as immune checkpoint inhibitors or chemotherapy, has shown synergistic effects, suggesting that dual targeting may improve therapeutic outcomes [97]. However, it is important to note that STAT3 inhibitors alone have not always been successful. For example, phase I clinical trials by Yoo et al. in hepatocellular carcinoma found limited benefit from STAT3 inhibitors as monotherapy. This highlights the complexity of STAT signaling and suggests that combination therapies or targeting multiple pathways simultaneously may be necessary to achieve optimal outcomes. However, due to the pleiotropic roles of STAT proteins in immune and non-immune cells, selective targeting of STAT pathways in MDSCs remains a challenge. To overcome some of the challenges of STAT-specific inhibition in MDSCs, several strategies can be employed. Targeted delivery systems, such as nanoparticles or MDSC-specific drug conjugates (e.g., CD11b or CD33 markers) can enhance selective delivery of STAT inhibitors to MDSCs within the TME. The use of prodrugs activated in the hypoxic or acidic TME, as well as siRNA or antisense oligonucleotides delivered via MDSC-specific carriers, can further improve precision. Developing next-generation selective STAT3 inhibitors targeting unique MDSC-specific pathways or combining upstream regulators (e.g., IL-6, JAK) with myeloid-restricted signals (e.g., S100A8/A9) can enhance specificity. Time-limited dosing strategies, biomarker-guided therapies, and dual pathway modulation approaches can ensure transient and context-specific inhibition of STAT3, minimizing off-target effects while restoring anti-tumor immunity.
TET2
Tet methylcytosine dioxygenase 2 (TET2) is an important epigenetic checkpoint enzyme in MDSC that regulates myelopoiesis and immune function through the hydroxylation of methylcytosine [98]. This epigenetic modification plays a critical role in modulating various immune cell functions, including the suppressive activity of MDSCs. Research has shown that targeting TET2 can disrupt the immunosuppressive activity of MDSCs and TAMs, thereby enhancing the efficacy of adaptive cellular therapies [99]. Increased TET2 expression within tumor-infiltrating myeloid cells has been implicated in maintaining the immunosuppressive environment of tumors. This upregulation, driven by IL-1R-MyD88 signaling, supports the suppressive functions of MDSCs and TAMs. However, knockdown or deletion of TET2 shifts the gene expression profiles of these myeloid cells toward a pro-inflammatory phenotype, impairing their suppressive capabilities and promoting anti-tumor immune responses [100]. Specifically, TET2 knockdown in myeloid cells has been demonstrated to slow the growth of tumors and improve the infiltration and function of CD8+ T cells in the TME [101]. Furthermore, in a mouse model, TET2 deletion resulted in the expansion of immunosuppressive granulocytic MDSCs through IL-6 signaling. Treatment with an anti-IL-6 antibody restored CD8+ T cell numbers and functions, countering the MDSC-mediated immune suppression and slowing tumor progression [102]. In addition to its role in MDSCs, TET2 has been linked to CAR-T cell efficacy. Research by Jain et al. demonstrated that TET2 deletion enhances the clonal expansion and tumor-penetrating ability of CAR-T cells, improving their antitumor activity in prostate cancer and leukemia models [103]. This suggests that targeting TET2 could be a dual weapon strategy to both improve CAR-T cell therapy outcomes and simultaneously reprogram MDSCs to weaken their suppressive influence within the TME. However, further research is needed to understand TET2’s role in myeloid cell regulation and develop effective inhibitors, which could improve cancer immunotherapies, especially in solid tumors.
Retinoblastoma gene 1
The retinoblastoma gene 1 (RB1) encodes the retinoblastoma protein (pRB), which plays a crucial role in regulating cell cycle progression, cell proliferation, and differentiation. Depletion of RB1 in MDSCs has been shown to promote the growth of MDSC populations and inhibit their differentiation into mature myeloid cells such as neutrophils, monocytes, dendritic cells, and macrophages [104]. Targeting RB1 may disrupt the control of the cell cycle in MDSCs, leading to reduced proliferation and accumulation within the TME, thereby weakening their capacity to inhibit anti-tumor immune responses by lymphoid and myeloid cells [31]. The hypophosphorylated form of the Rb protein binds to E2F transcription factors, acting as a potent transcriptional repressor. Additionally, Rb facilitates terminal maturation by enhancing the expression of tissue-specific genes [105]. Loss of Rb function has been found to moderately increase myeloid cell populations, although it does not significantly affect the cell cycle in hematopoietic stem cells (HSCs) under normal conditions. Additionally, Youn et al. reported that epigenetic silencing of the retinoblastoma gene (Rb) by histone deacetylase 2 (HDAC-2) critically influences the pathological differentiation of myeloid cells into MDSCs. Interestingly, inhibiting HDACs increases RB1 expression in MDSCs, halting their progression into immunosuppressive subsets and promoting their differentiation into cell types that enhance immune activity [106]. Collectively, these findings suggest that modulating RB1 and HDACs in MDSCs could be an effective strategy for controlling their proliferation and immunosuppressive activity within the tumor microenvironment, potentially improving the outcomes of cancer immunotherapy.
Targeting membrane receptor signaling
PD-1/PD-L
Programmed cell death protein 1 (PD-1), a member of the CD28 family is expressed by various immune cells, including macrophages, B cells, natural killer cells, activated T cells, and regulatory T cells, and serves as a crucial immune checkpoint regulating immune responses. [21]. The ligands for PD-1, namely PD-L1 and PD-L2 (members of the B7 family), are expressed on tumor cells and MDSCs [107], tumor-associated macrophages (TAMs), and M2 macrophages [22]. Upon binding to its ligands, PD-1 inhibits T cell function, reduces T cell proliferation, and promotes immune tolerance [108]. When PD-1 binds to PD-L1 or PD-L2, the intracellular domain of PD-1 becomes phosphorylated, leading to the recruitment of SHP-2 phosphatase enzymes. These enzymes dephosphorylate TCR signaling molecules, which in turn inhibit classical antigen-specific T cell receptor (TCR) function and downstream signaling via the PI3K-AKT and mTOR pathways. This results in reduced expression of critical cytokines, such as IL-2 and IFN-γ, which are necessary for T cell activation and adaptive immunity. Over time, this signaling cascade leads to impaired antitumor T cell function, fostering an environment conducive to tumor growth [109]. The PD-1/PD-L1 axis, a normal immune checkpoint pathway, is frequently upregulated in cancer, particularly within intratumoral and circulating MDSCs, due to hypoxia in the TME and the activity of transcription factors such as HIFα, as well as inflammatory cytokines like GM-CSF and IL-6 secreted by malignant and immune cells. These conditions enable tumor cells to evade immune surveillance. Extensive research is being conducted to improve the efficacy of immune checkpoint inhibitors, and clinical trials have highlighted the critical role of the PD-1 pathway in human tumors [108]. Following treatment with monoclonal antibodies (mAbs) targeting PD-1 or PD-L1, 17–28% of patients with advanced cancers exhibit partial or complete remissions [24].
Despite the availability of more than ten FDA-approved PD-1/PD-L1 inhibitors for cancer treatment, resistance, especially in solid tumors, remains a significant challenge. Recent studies have shown that anti-PD-L1 blocking agents are ineffective against PD-L1hi tumors. However, anti-PD-L1 therapy can control the growth of PD-L1-deficient tumors in wild-type mice, suggesting that targeting PD-L1 on host cells specifically MDSCs, rather than tumor cells, may be critical in enhancing anti-PD-1/PD-L1 therapy efficacy [110]. Some patients initially respond to ICIs but later develop resistance, further emphasizing the need for combination therapies that target multiple MDSC checkpoints to overcome MDSCs driving ICI resistance. Future strategies should focus on combination approaches that target MDSCs PD-L1 expression, especially at the transcriptional level. This would increase the effectiveness of PD-1/PD-L1 blockade, particularly in cases where tumors have developed resistance. By targeting both the tumor and immune components of the TME, such as MDSCs, TAMs, and other suppressive cells, the efficacy of immunotherapy can be improved, offering a broader clinical benefit.
CD300ld
CD300ld is a newly identified member of the CD300 family and has emerged as a significant contributor to immune suppression mediated by PMN-MDSCs in cancer. Recent research by Wang et al. highlights the elevated expression of CD300ld in PMN-MDSCs within the TME, compared to its expression in normal neutrophils. This upregulation in malignant conditions drives immune evasion through the activation of the STAT3-S100A8/A9 axis, a pathway known for promoting tumor progression and immune suppression. Targeting CD300ld in preclinical models has shown promising results. Deletion of CD300ld in mice with B16-F10 tumors reversed the immunosuppressive TME, leading to enhanced anti-tumor immune responses. Moreover, combining CD300ld knockout with anti-PD1 therapy produced a synergistic effect, resulting in robust tumor regression, far exceeding the efficacy of either treatment alone [111]. This evidence suggests that CD300ld plays a pivotal role in MDSC-mediated immune suppression, making it an attractive therapeutic target. Unlike broad-spectrum approaches such as S100A8/A9 inhibition, which can impact multiple cell types and lead to adverse effects like impaired dendritic cell differentiation, CD300ld offers a more specific approach. By selectively inhibiting CD300ld, PMN-MDSCs can be reprogrammed or depleted, potentially restoring immune surveillance while minimizing off-target effects. This strategy not only enhances the efficacy of immune checkpoint inhibitors in halting cancer progression like PD1 blockers but also holds potential in combination with adaptive cellular therapies. In a recent study, CD300ld was identified as a critical receptor involved in the recruitment and functionality of PMN-MDSCs through an in vivo CRISPR-Cas9 screen in tumor-bearing mice. CD300ld, a single-pass transmembrane protein, is part of the CD300 family and is highly expressed in neutrophils and PMN-MDSCs while showing low or no expression in other immune cells. Tumor-bearing conditions lead to the upregulation of CD300ld in PMN-MDSCs, making it a key marker for these cells. Interestingly, knockout of CD300ld in multiple tumor models significantly impaired tumor development by reducing PMN-MDSC presence and altering the TME from immunosuppressive to immune-active. Analysis of single-cell transcriptomes confirmed these changes, highlighting the pivotal role of CD300ld in fostering a tumor-promoting environment [112]. However, despite these promising findings, further research is required to elucidate the underlying mechanisms driving the overexpression of CD300ld in PMN-MDSCs and to identify its molecular ligands. Understanding these factors will be crucial for developing targeted therapies that can precisely modulate this pathway. Additionally, clinical studies are needed to validate the safety and efficacy of CD300ld inhibitors in human cancers and to assess their potential for integration into existing treatment protocols.
TRAIL-Rs
TNF-related apoptosis-inducing ligand receptors (TRAIL-Rs), belonging to the TNF receptor superfamily, play a pivotal role in immune regulation by interacting with the TRAIL to mediate apoptosis in MDSCs [113]. There are four recognized TRAIL-Rs: TRAIL-R1, TRAIL-R2, TRAIL-R3, and TRAIL-R4, of which only TRAIL-R1 and TRAIL-R2 are capable of inducing apoptosis. In contrast, TRAIL-R3 and TRAIL-R4 act as decoy receptors, preventing apoptosis and maintaining cell viability [114]. Targeting TRAIL-Rs, particularly TRAIL-R1 and TRAIL-R2, in MDSCs represents a novel approach to overcome immune suppression and improve the success of therapies. Recent studies have shown that MDSCs in gastric cancer upregulate TRAIL-Rs, making them susceptible to apoptosis when TRAIL-R signaling is activated [115]. Clinical trials have further underscored the potential of TRAIL-R2 as a therapeutic target. For example, Dominguez et al. demonstrated that targeting TRAIL-R2 led to a significant reduction in PMN-MDSCs, which in turn amplified the effectiveness of ICIs [113]. The combination of TRAIL-R2 activation and ICIs resulted in greater tumor suppression compared to monotherapy approaches. Moreover, research by Rapoport et al. has confirmed that TRAIL-R2 signaling enhances ER stress, leading to apoptosis of MDSCs and a reduction in their immunosuppressive function [116]. The ability to selectively induce apoptosis in MDSCs through TRAIL-R1 and TRAIL-R2 activation provides a unique opportunity to dismantle the immunosuppressive TME. MDSCs play a central role in maintaining the TME by releasing pro-tumorigenic factors, such as NO and ROS which dampen T-cell responses and foster tumor growth [65]. Likewise, Chen et al. demonstrated that acetaminophen-induced liver injury is characterized by the recruitment of neutrophils that express TRAIL, which can trigger apoptosis in hepatocytes via the upregulation of the TRAIL receptor DR5. Blocking TRAIL signaling with a soluble DR5-Fc fusion protein has been shown to significantly reduce the recruitment and activity of MDSCs [117]. This intervention not only alleviates liver injury but also enhances anti-tumor immune responses. By disrupting TRAIL-mediated apoptosis in hepatocytes, the blockade may diminish the inflammatory environment that promotes MDSC expansion and activation. Further supporting this strategy, Andrés et al. demonstrated that inducing ER stress via TRAIL-Rs significantly decreased MDSC viability in tumor-bearing mice, contributing to the breakdown of the immunosuppressive environment. However, the therapeutic impact of targeting TRAIL-Rs in cancer has been moderate when used as a monotherapy.
In certain cancers, such as NSCLC, TRAIL receptor signaling has been associated with a pro-tumorigenic inflammatory profile. The release of IL-8 and CXCL8, induced by TRAIL-R signaling, promotes tumor growth and metastasis [114]. Thus, careful consideration must be given to the potential pro-tumorigenic effects of TRAIL-R signaling in specific contexts. Recent studies suggest that TRAIL-R activation may modulate STAT3 activity, either synergistically or antagonistically, depending on the cellular context [59]. One area ripe for investigation is the interaction between TRAIL-R signaling and the JAK/STAT3 pathway. Unraveling the specific molecular mechanisms by which TRAIL-R signaling interacts with STAT3 within MDSCs could lead to novel combinatorial discovery. Furthermore, the interaction between TRAIL-Rs and UPR pathways within the ER presents a promising mechanism for enhancing MDSC apoptosis. TRAIL-R-induced ER stress has been shown to promote apoptosis, suggesting that targeting UPR components, such as PERK, IRE1, and ATF6, could identify synergistic therapeutic strategies. Combining TRAIL-R agonists with UPR modulators or ER stress-enhancing compounds might increase MDSC vulnerability in the tumor microenvironment. Additionally, TRAIL-R signaling may disrupt key metabolic pathways like glycolysis and fatty acid oxidation, impairing MDSC function. Exploring how metabolic checkpoints, such as AMPK or mTOR, influence TRAIL-R-induced apoptosis could pave the way for innovative metabolic-immunotherapeutic approaches.
β2-AR
βeta-2 adrenergic receptor (β2-AR) is a G protein-coupled receptor activated by stress hormones like epinephrine and norepinephrine, regulates numerous cellular processes, and plays a crucial role in immune modulation within the TME. Recent studies have demonstrated that β2-AR signaling can significantly impact the functionality of immune cells, particularly MDSCs, which are key players in creating an immunosuppressive TME [118]. By modulating β2-AR signaling in MDSCs, there is potential to overcome the immune suppression that hinders the effectiveness of other cancer immunotherapies [119]. Chronic stress-induced β2-AR signaling has been shown to promote the differentiation and immunosuppressive function of MDSCs through metabolic reprogramming. These stress signals enhance MDSC proliferation and activation within the TME, further facilitating tumor growth and survival. β2-AR activation not only drives MDSC expansion but also increases their mobilization from the bone marrow into the vasculature, exacerbating their accumulation in tumors [120]. This suggests that blocking β2-AR signaling could inhibit MDSC trafficking and reduce their immunosuppressive influence.
Inhibiting β2-AR signaling using non-selective beta-blockers like propranolol has become a viable therapeutic approach. Studies in mouse cancer models have shown that combining propranolol with chemotherapy or immunotherapy significantly reduces tumor growth and improves survival rates [121]. This is accompanied by a reduction in both granulocytic and monocytic MDSCs, highlighting the potential of β2-AR inhibition to impair MDSC function and alleviate immunosuppression in the TME. For instance, propranolol has been shown to stunt tumor growth when used alongside a tumor vaccine in an in vivo breast cancer model [122], suggesting synergy between β2-AR inhibition and immunotherapies. Blocking β2-adrenergic receptor (β2-AR) signaling in adaptive cellular therapy could enhance immunotherapy outcomes. Interestingly, administering β2-AR blockers coupled with CAR-T would also prevent PKA-dependent inhibition of ZAP70 phosphorylation, thereby boosting T cell activation, co-stimulation, and cytotoxicity [123]. Exploring how β2-AR modulates the expression and function of immune checkpoints, including CTLA-4 and TIM-3, across both T cells and MDSCs could provide critical insights. Dual inhibition of β2-AR signaling and immune checkpoints may synergistically enhance immunotherapy efficacy by concurrently alleviating MDSC-mediated immunosuppression and reinvigorating T-cell responses.
LILRB Family
The leukocyte immunoglobulin-like receptor (LILRB) family consists of cell surface receptors found on lymphoid and myeloid cells, playing essential roles in regulating innate immunity and fetomaternal tolerance. This family is divided into inhibitory receptors (LILRB1-LILRB5) and activating receptors (LILRA1-LILRA6) [124]. Among them, LILRB2 stands out as a key immune regulatory protein, with its overexpression being notably observed in septic shock patients following LPS stimulation [125]. LILRB2 interacts with a variety of ligands, including SEMA4A, CD1, ANGPTL2/5, and classical and non-classical MHC I, all of which have been implicated in promoting immune inhibition within the TME [126]. Targeting LILRB presents a promising strategy for overcoming MDSC-mediated immune suppression in the TME. Studies have shown that blocking LILRB2 with therapeutic antibodies disrupts its interaction with inhibitory ligands, shifting macrophages toward a pro-inflammatory, M1 phenotype and reducing the expansion of MDSCs and Tregs recruitment [126, 127]. By inhibiting LILRB2, the suppressive functions of MDSCs are mitigated, leading to enhanced T-cell activity and immune responses. This approach has been associated with a reduction in the recruitment of MDSCs and Tregs to the TME, as well as a shift in the MDSC phenotype toward an anti-tumoral state. Inhibition of LILRB2 also plays a critical role in reprogramming the tumor-associated macrophage population toward a more active, inflammatory phenotype, which can further disrupt the immunosuppressive environment [126]. This shift has been shown to restore the ability of MDSCs, particularly M-MDSCs, to support immune activation, enhancing their ability to kill intracellular pathogens and facilitating better anti-tumor immune responses. Despite its potential, the role of LILRB2 blockade in combination with advanced immunotherapies remains underexplored. Mechanistic studies investigating how LILRB2 inhibition may enhance the effectiveness of these therapies are needed. Specifically, examining how LILRB2 signaling influences subcellular communication between myeloid and lymphoid cells could shed light on potential synergies between LILRB2 blockade and CAR-based therapies.
VISTA
V-domain Ig suppressor of T-cell activation (VISTA) is a member of the B7 family of immune checkpoint proteins expressed by MDSCs, TAMs, and regulatory T cells [128]. It plays a crucial role in suppressing the functions of CD4+ and CD8+ T cells, as well as in regulating myeloid cell populations. A study by Xu et al. indicated that VISTA blockade reprograms MDSCs, reducing their T cell-suppressive activity and enabling a more proinflammatory and immunostimulatory tumor microenvironment. This shift not only disrupts the suppressive influence of MDSCs but also enhances T-cell infiltration and activation, which can significantly amplify the efficacy of T-cell-based immunotherapies. Therefore, VISTA inhibition could serve as a double-edge strategy to overcome MDSC-driven immune resistance, fostering robust antitumor immunity in otherwise resistant cancers [129]. Anti-VISTA therapy alone has shown efficacy in slowing tumor growth in colorectal tumor models and in conjunction with anti-PD-1/CTLA-4 therapy [130]. This combination therapy improved myeloid cell antigen presentation, decreased immune suppression by MDSCs, promoted T-cell activation, and decreased its quiescence. Furthermore, research has revealed intricate mechanisms by which CNS-native myeloid cells and bone marrow-derived myeloid cells regulate brain immunity. Inhibiting signaling pathways involving VISTA and PD-L1 in CNS-native myeloid cells reduced brain metastases, suggesting a potential strategy to overcome immunosuppressive tumor microenvironment in solid tumors [131]. In summary, targeting VISTA in immunosuppressive MDSCs holds promise for improving the therapeutic efficacy of ICIs and adaptive cellular therapies. Further research into the mechanisms of VISTA inhibition in MDSCs and the development of potential combination therapies is warranted. Future studies should investigate the impact of VISTA inhibition on the metabolic reprogramming of MDSCs and its effects on proinflammatory cytokine production could provide new insights into modulating MDSC function. Identifying biomarkers that predict responsiveness to VISTA-targeted therapies would further optimize treatment strategies, potentially enhancing efficacy.
Prokineticin 2 (BV8) receptor
Prokineticin 2, initially identified from the venom of the European viper, exhibits potent angiogenic properties, signifying its ability to promote neovascularization. BV8 is a small, secreted protein that functions as a ligand for the prokineticin receptors (PKR1 and PKR2). While both receptors can bind BV8, PKR2 is often considered more influential in promoting immunosuppressive pathways [109]. BV8 is particularly notable for its involvement in the tumor microenvironment, where it contributes to the recruitment and activation of MDSCs [132]. Importantly, BV8’s involvement in MDSC-driven angiogenesis and immune suppression has highlighted it as a significant checkpoint in the immunosuppressive landscape of various cancers, including thoracic and colorectal malignancies [107]. Nguyen et al. demonstrated that S-1, efficiently eliminates MDSCs by regulating S1008 and BV8 from thoracic tumors thereby suppressing tumor progression. Notably, treatment with S-1 increased tumor-infiltrating effector T cells and dendritic cells, thereby fostering an anti-tumor immune response. Additionally, the combination of this drug and PD-1 blocking antibodies significantly inhibited MDSC recruitment, enhancing therapeutic efficacy [107]. However, these treatments did not directly affect MDSC survival or maturation in vitro, suggesting that their primary mode of action lies in disrupting MDSC recruitment to the TME. Importantly, 5-FU and S-1 were found to reduce the expression of tumor-derived BV8 and S100A8, another key player in MDSC function. BV8 inhibition, along with S100A8 blockade, led to diminished tumor growth and decreased MDSC infiltration in vivo, providing strong evidence that BV8 is a critical driver of MDSC-mediated tumor progression. Given the multifaceted roles of BV8 in angiogenesis, immunosuppression, and MDSC recruitment, it represents a promising therapeutic target. Strategies aimed at inhibiting BV8 action on MDSC could enhance the efficacy of immunotherapies. Notably, blocking BV8 has been shown to sensitize previously resistant tumors to anti-PD1 therapy, converting them into PD1-responsive phenotypes [133]. This shift is attributed to the disruption of BV8-mediated MDSC recruitment and immunosuppressive activity within the tumor microenvironment. By inhibiting the BV8 receptor on MDSC, the immunosuppressive barrier is reduced, allowing immune checkpoint inhibitors like anti-PD1 to more effectively promote T cell-mediated anti-tumor responses. This finding highlights BV8 receptor blockade as a promising strategy to overcome immune resistance in tumors and improve the efficacy of immunotherapies. However, the complexity of BV8 roles in both physiological and pathological processes warrants careful consideration in drug design. Optimizing BV8-targeting agents, especially in combination with other checkpoint inhibitors or cellular therapies, could provide a multifaceted approach to disrupt tumor-promoting mechanisms and improve clinical outcomes in cancer patients.
CD74
CD74 is also known as the major histocompatibility complex (MHC) II-associated invariant chain (Ii), is a transmembrane protein primarily expressed in antigen-presenting cells (APCs) such as macrophages and dendritic cells. It is the main receptor for macrophage migration inhibitory factor (MIF) and plays a significant role in MDSCs-mediated immune suppression. MIF was first discovered to be a T lymphocyte-induced inhibitor of macrophage motility but is now overexpressed in the majority of solid and hematogenous malignant tumors and expressed by almost all immune cell types studied [134]. MIF binding to CD74 on MDSCs has been shown to activate various cell signaling pathways, including the PI3K/AKT and MAPK pathways, which are associated with immunosuppressive functions [135]. The interaction between MIF and CD74 triggers the activation of suppressive pathways in MDSCs, ultimately leading to the inhibition of T cell effector functions. Evidence indicates that MIF-CD74 signaling plays a critical role in the accumulation and immunosuppressive functions of MDSCs within the tumor microenvironment. Targeting this interaction has emerged as a potential strategy to mitigate MDSC-mediated immune suppression and enhance antitumor immune responses. Notably, an IG-CDR-based peptide designed to disrupt MIF-CD74 signaling has been shown to reduce the accumulation of M-MDSCs in metastatic melanoma lesions [136]. This supports the findings of the GBM study that MIF-CD74 serves a specialized role in promoting the intratumoral accumulation of M-MDSC and, most likely, immune suppression [137]. Recent findings reveal that circulating MDSCs isolated from late-stage melanoma patients depend heavily on MIF for their immunosuppressive activity, particularly in suppressing antigen-independent T-cell activation. Additionally, MIF plays a crucial role in promoting ROS production within these cells. Pharmacological inhibition of MIF not only diminishes these suppressive functions but also drives a phenotypic shift in MDSCs, transforming them into immunostimulatory, dendritic cell (DC)-like cells. This functional reversion is partly attributed to a decrease in PGE2 levels, a mediator of MDSC-induced immune suppression. These findings suggest that monocyte-derived MIF is a key driver of the induction and suppressive function of monocytic MDSCs [138]. Targeting the MIF pathway could therefore represent a promising therapeutic strategy, converting immunosuppressive MDSCs into pro-inflammatory, antitumor DC-like cells, and thereby reinvigorating antitumor immune responses in late-stage melanoma patients. Another study found that shRNA suppression of MIF in murine breast cancer cell lines inhibited primary tumor expansion and decreased lung micro-metastases in immunocompetent Balb/c mice, but not in immunodeficient SCID mice. This anti-tumor phenotype was linked to MIF-deficient 4T1 breast cancer tumors [139]. These studies highlight the critical role of MIF in promoting tumor-associated MDSC phenotypes. However, the mechanisms and specific sources of MIF regulating MDSC activities such as differentiation, homing, expansion, motility, and immune suppression are not fully understood. Future research should focus on exploring the signaling pathways downstream of CD74 and its co-receptors in MDSCs, identifying the cellular and tumor-derived sources of MIF, and understanding how MIF signaling maintains MDSC function in various tumor microenvironments.
Targeting TLR/NF-kB and MAPK signaling
TIPE2
TIPE2 (tumor necrosis factor-α induced protein 8 (TNFAIP8)-like 2) has emerged as a complex regulator of immune responses in tumor progression and immune suppression.TIPE2 belongs to the TNFAIP8 protein family, which consists of four classes: TIPE, TIPE1, TIPE2, and TIPE3 [140]. While TIPE and TIPE1 are broadly expressed in most mammalian cells, TIPE2 is primarily found in hematopoietic stem cells, and TIPE3 is limited to glandular epithelial cells. Despite structural similarities among TNFAIP8 family members, their biological activities differ significantly. In the context of cancer, TIPE2 has emerged as a key regulator of both pro-tumoral and anti-tumoral immune homeostasis, positioning it as an important checkpoint in MDSCs. Tumor-derived cytokines and exosomes play a major role in recruiting and activating MDSCs. TIPE2 expression, which is regulated in a ROS-dependent manner, influences the balance between pro-tumoral and anti-tumoral activities by controlling the expression of genes like CEBPβ (pro-tumoral) and IFN-γ (anti-tumoral), driving the polarization of MDSCs [141].
Research highlights the potential therapeutic significance of targeting TIPE2 in cancer treatment. Yan et al. demonstrated that global TIPE2 knockout mice with established tumors exhibited reduced tumor progression and lung metastasis. Moreover, MDSC-specific TIPE2 knockout significantly improved anti-tumor immune responses, further underscoring its potential as a therapeutic target [141]. In addition to MDSCs, TIPE2 plays a critical role in natural killer (NK) cells. A study by Bi et al. demonstrated that elevated TIPE2 expression in NK cells correlates with NK cell exhaustion, contributing to poor prognoses in both human and mouse cancer models. As expected, TIPE2 deletion enhanced NK cell maturation and restored their anti-tumor activity [142]. While these findings underscore TIPE2’s role in NK cell function, they also suggest that TIPE2 plays a broader role in shaping the immunosuppressive tumor microenvironment. Given its regulatory effect on both NK cells and MDSCs, TIPE2 represents a critical therapeutic target in the context of MDSC-mediated immunosuppression. Targeting TIPE2 may enhance the efficacy of combination immunotherapies by blocking MDSC immunosuppressive pathways, ultimately promoting stronger anti-tumor immune responses. Interestingly, TIPE2’s role appears context-dependent. Contrary to its pro-tumoral function in some immune cells, Zhu et al. found that TIPE2 inhibits the Wnt/β-catenin signaling pathway, thereby reducing growth and tumorigenesis in esophageal cancer [143]. Furthermore, overexpression of TIPE2 in breast cancer cell lines suppressed proliferation and metastasis, likely by enhancing the cytotoxic activity of NK cells and CD8+ T cells and inhibiting the immunosuppressive function of MDSCs [144]. The potential of TIPE2 as a therapeutic target extends to other cancer types as well. Overexpression of TIPE2 has been shown to inhibit ovarian cancer growth by promoting apoptosis through the blockade of the PI3K/Akt signaling pathway [145]. Further investigation is needed to clarify the cellular signaling pathways linking ROS-mediated MDSC polarization to TIPE2 activity. While TIPE2 can act as a tumor suppressor in some contexts, its activity in MDSCs and NK cells appears to support immune evasion and tumor progression.
S100A8/A9 protein
S100A8 and S100A9, collectively referred to as calprotectin, are tiny molecular weight calcium-binding proteins that belong to the S100 protein family expressed predominantly by myeloid cells during inflammation and tumor progression. Their function in the immunosuppressive duties of MDSCs has garnered attention as a potential therapeutic target in cancer immunotherapy. S100A9 is typically expressed in PMN-MDSCs and forms heterodimers with S100A8, enhancing its immunosuppressive capabilities [146]. The interaction of S100A9 with cell surface receptors leads to the activation of various intracellular signaling pathways that promote immunosuppression. In PMN-MDSCs, activating transcription factor 3 (ATF3) mediates the expression of S100A9, driving their immunosuppressive functions. Silencing S100A9 has demonstrated a decrease in the proliferation and differentiation of PMN-MDSCs, suggesting its crucial role in maintaining their immunosuppressive phenotype [92]. Macrophages derived from M-MDSCs exhibit similar immunosuppressive characteristics, with sustained production of S100A9 essential for their ability to inhibit immune responses and promote M2 polarization [147]. Notably, S100A9 expression is absent in normal monocyte-derived or tissue-resident macrophages, which highlights its potential as a target. High levels of S100A9-positive macrophages in tumors have been correlated with poor patient outcomes in conditions like head and neck cancer and metastatic melanoma, indicating its association with reduced survival rates and diminished response to anti-PD-1 immunotherapy [148]. S100A9 has been implicated in myelodysplastic syndrome through its role in promoting the too-early death of progenitor cells, and hematopoietic stem along with increasing the expression of immune checkpoints such as PD-L1 on MDSCs. Moreover, the ERK1/2 signaling pathway is activated by S100A9, promoting tumor immune escape through upregulation of PD-L1 expression, which further enhances the immunosuppressive environment [149]. Conclusively, targeting S100A8/A9 could serve as a checkpoint in MDSC biology, disrupting their immunosuppressive functions and enhancing anti-tumor immunity.
TNF-α
Targeting tumor necrosis factor-alpha (TNF-α) presents a compelling therapeutic approach to overcoming MDSC-mediated immune suppression within the TME. TNF-α, a proinflammatory cytokine, is primarily produced by T lymphocytes, NK cells, and activated macrophages during inflammation and tumorigenesis. Experimental evidence suggests that TNF-α, not TNF-β, is the main driver of MDSC expansion, chemotaxis, and suppressive capacity within the TME [150]. This makes TNF-α, a key regulator of MDSC activity and a promising target for reversing MDSC-mediated immune suppression. TNF-α exerts its biological effects through two receptors: TNFR1 and TNFR2. Both receptors are expressed on MDSCs, though the literature suggests that TNFR2 plays a predominant role in regulating MDSC function, particularly in preventing excessive inflammation [151]. The higher expression of TNFR2 on MDSCs highlights its importance in promoting the immunosuppressive capacity of these cells, leading to their accumulation within the TME and their role in tumor progression. However, some studies suggest that TNFR1 may also contribute to MDSC expansion, leaving room for further exploration into receptor-specific signaling pathways in these cells [152]. It has been reported that systemic neutralization of TNF-α using monoclonal antibodies has been shown to significantly reduce MDSC accumulation and suppressive activity, which correlates with reduced tumor growth and volume in preclinical studies [153]. Despite the potential of TNF-α ablation therapies, caution is required in their application because TNF-α is critical for the anti-tumor activity of cytotoxic NK and T cells, and its complete neutralization may inadvertently impair these immune cells’ functions. Further mechanistic studies should investigate the precise signaling cascades downstream of TNFR2 in MDSCs and their contribution to immune suppression. This includes exploring the transcription pathways that TNF-α activates in MDSCs to maintain their suppressive phenotype. Additionally, studying how TNF-α antagonists reshape the metabolic programming of MDSCs could reveal new therapeutic targets.
CD45 Phosphatase
CD45 tyrosine phosphatase, also known as leukocyte common antigen (LCA), is a key regulator of immune cell activation and development, particularly in myeloid cells [154]. As a member of the protein tyrosine phosphatase (PTP) family, CD45 plays a crucial role in controlling signal transduction across cell membranes, significantly influencing various signaling pathways involved in the function of immune cells, including the development and expansion of MDSCs. One of the primary signaling pathways regulated by CD45 phosphatase is the STAT3 pathway [94], which has been implicated in promoting the immunosuppressive activity of MDSCs. Studies have shown that hypoxic conditions within the TME reduce STAT3 activity in myeloid cells, a process linked to increased CD45 phosphatase activity. This was mediated by the disruption of sialic acid-dependent dimerization of the CD45 protein, a key regulatory mechanism. In this context, CD45 phosphatase dephosphorylates and inactivates STAT3, thereby attenuating the expansion and suppressive function of MDSCs. Conversely, research by Huang et al. revealed that the deletion of SENP1, a small ubiquitin-like modifier protease, in myeloid cells enhanced MDSC proliferation and tumor progression through STAT3-dependent pathways. They identified that SUMOylation of CD45 reduced its phosphatase activity, consequently enhancing STAT3 activation and contributing to the immunosuppressive nature of MDSCs [155]. Pharmacological inhibition of CD45 phosphatase presents a promising therapeutic strategy. In preclinical models, the selective inhibition of CD45 phosphatase with compound 211 significantly delayed tumor growth and reduced metastasis without impairing the function or number of resting T cells [156]. CD45 phosphatase regulation may vary across different MDSC subsets. Therefore, future research should focus on detailed profiling of MDSC subsets in tumor-bearing hosts to better understand how selective inhibition of CD45 phosphatase impacts their function and interactions with other immunotherapies. By identifying which MDSC subsets are most responsive to CD45 inhibition, therapeutic strategies could be refined to achieve more targeted and effective outcomes.
PLCγ 2
Phosphatidylinositol-specific phospholipase C gamma 2 (PLCγ2) has emerged as a significant regulatory enzyme in cellular signaling, primarily through its catalytic role in breaking down membrane-bound phospholipids to produce diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) [157]. These products act as second messengers, initiating cascades that regulate immune functions, including calcium release and protein kinase C activation, processes critical for the immunosuppressive functioning of MDSCs. Capietto et al. demonstrated that MDSCs downregulate PLCγ2 during early tumor growth, a process that enhances their maturation and immunosuppressive function. Specifically, MDSCs with reduced PLCγ2 expression exhibit increased production of ROS and NO, which directly suppress CD8+ T cell activity and promote tumor growth. When MDSCs lacking PLCγ2 were transferred into wild-type mice, the result was increased tumor progression, underscoring the enzyme’s critical role in MDSC regulation. This finding highlights PLCγ2 as a checkpoint molecule within MDSCs that can be targeted to alleviate immune suppression [158]. Therapeutically restoring PLCγ2 activity in MDSCs could serve as an important strategy to overcome the immunosuppressive TME, which is one of the primary obstacles to effective cancer immunotherapy. By reactivating PLCγ2, MDSC function can be skewed away from immunosuppression, reducing their inhibitory effects on effector CD8+ T cells. This strategy aligns with the broader goal of enhancing T cell-mediated anti-tumor responses, particularly in tandem with immune checkpoint inhibitors, which have shown remarkable but inconsistent efficacy in solid tumors. This strategy is particularly promising when used in combination with immune checkpoint inhibitors, such as anti-PD-1/PD-L1 antibodies, which have shown success in some cancers but have inconsistent effectiveness in solid tumors because of the TME’s strong immunosuppressive properties [159]. By restoring PLCγ2 activity, researchers can inhibit MDSC-mediated suppression, thereby enhancing the efficacy of both checkpoint inhibitors and CAR-T cell therapies.
Targeting hypoxia and oxidative stress
HIF-1α
Hypoxia-inducible factor 1 (HIF-1) signaling plays a crucial role in cellular responses to low oxygen levels, a common feature of the tumor microenvironment [22]. Hypoxic conditions within the TME not only promote tumor survival and progression but also recruit various immunosuppressive cells, including MDSCs, tumor-associated macrophages, and regulatory T cells [160]. Under these conditions, both tumor cells and MDSCs upregulate HIF-1α, which drives immunosuppressive activities. Noman et al. demonstrated that HIF-1α, rather than HIF-2α, plays a key role in the upregulation of PD-L1 on MDSCs and other myeloid cells under hypoxia [20]. This promotes tumor immune evasion by allowing MDSCs to suppress effector T-cell activity through PD-L1-mediated pathways. Additionally, hypoxia induces the expression of other immunosuppressive genes, such as iNOS, and Arg-1 which further enhance the suppressive function of MDSCs [161]. Targeting HIF-1α represents a promising strategy to inhibit these immunosuppressive mechanisms. For instance, Salman et al. reported that a low molecular weight compound, 32-134D, effectively inhibits both HIF-1α and HIF-2α in an in vitro hepatocellular carcinoma model. Treatment with 32-134D reduced the hypoxia-induced expression of genes associated with tumor metabolism and immune evasion, such as PD-L1, while enhancing the activity of CD8+ IFN-γ+ T cells and NK cells, key players in antitumor immunity [22]. Mutations in the isocitrate dehydrogenase 1 (IDH1) gene, frequently observed in gliomas, have been linked to HIF-1α stabilization. Mutant IDH1 leads to the accumulation of the metabolite 2-hydroxyglutarate (2-HG), which stabilizes HIF-1α, further promoting immune suppression in the TME. Moreover, HIF-1α plays a central role in regulating immune evasion, glycolysis, metastasis, and tumor progression, particularly in triple-negative breast cancer (TNBC), highlighting its broad impact on tumor biology [162]. Collectively, targeting HIF-1α disrupts MDSC-mediated immune suppression by reducing PD-L1 expression and restoring T cell and NK cell activity.
Dkk1
Dickkopf-related protein 1 (DKK1) serves as a secretory inhibitor of the Wnt signaling pathway, playing a complex role in tumorigenesis [163]. While initially regarded as a tumor suppressor for its ability to inhibit canonical Wnt/β-catenin signaling by binding to low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6), recent research has revealed that DKK1 can also promote tumor growth by influencing MDSCs and the immune microenvironment [164]. Elevated DKK1 levels in the bloodstream and bone microenvironment have been linked to enhanced cancer development through the modulation of β-catenin levels in MDSCs, which can inhibit their immunosuppressive functions and support tumor growth [163]. Recent studies underscore the therapeutic potential of DKK1 antibodies, which have shown efficacy in inhibiting both melanoma and metastatic breast cancer growth, particularly in the presence of NK cells. Unlike NK cells, B cells and T cells appear crucial for the effectiveness of DKK1 antibodies, which promote immune cell recruitment and the release of NK-activating cytokines. This immune activation results in a reduction of MDSC populations and an upregulation of PD-L1 in MDSCs. Importantly, combining DKK1 antibodies with anti-PD-L1 treatment has yielded promising results in vivo, leading to significant 4T1 tumor volume reductions [165]. This synergy suggests that targeting DKK1 can enhance anti-tumor immune responses and improve treatment outcomes. Furthermore, high DKK1 expression correlates with an immunosuppressive microenvironment, often associated with lower survival rates in cancer patients [17]. Silencing DKK1 has demonstrated the potential to inhibit tumor development in a CD8+ T cell-dependent approach, indicating its role in immune modulation. Overexpression of DKK1 in conditions like HNSCC and HCC correlates with increased tumor growth and elevated PD-L1 expression, indicative of a cold tumor phenotype characterized by MDSC abundance and diminished CD8+ T cell infiltration [166].
Combination of MDSCs checkpoint blockade with other immunotherapies
Combination immunotherapy, which leverages the immune system to combat cancer cells, has emerged as a powerful strategy for cancer treatment. By integrating therapies that target different components of the tumor microenvironment, researchers aim to achieve more robust and durable responses [29]. One of the most promising approaches involves combining immune checkpoint inhibitors with adoptive cell therapies such as CAR-T, CAR-NK, or CAR-Macrophages. However, while ICIs can block inhibitory signals that dampen T-cell responses, they alone may not be sufficient to induce sustained T-cell infiltration and effector activity within solid tumors. Thus, overcoming the immunosuppressive barriers within the TME remains critical for maximizing therapeutic outcomes. One of the key limitations of current MDSC-targeting strategies is the fact that MDSCs express multiple immunosuppressive molecules that collectively contribute to tumor immune evasion (Fig. 3). While several checkpoint inhibitors have been tested, the combination of multiple checkpoint inhibitors that target distinct pathways on MDSCs holds promise in further. Targeting various MDSC-mediated checkpoints highlighted previously has shown potential in overcoming MDSC mediate potentiating the anti-tumor immune response. Future therapies may focus on the simultaneous blockade of these checkpoints, thereby disrupting the multifaceted mechanisms by which MDSCs impair immune cell function and enhance the immune response within the TME (Fig. 4).

This figure presents an overview of MDSCs and their immunosuppressive checkpoints, organized by subcellular location (cell surface, cytoplasm, and nucleus). It illustrates how these checkpoints contribute to the formation of an immunosuppressive TME, ultimately impairing immunotherapy efficacy.

This figure illustrates how MDSC checkpoint blockade therapy can transform TME from immunosuppressive to immunostimulatory, which restores the endogenous immune system and resensitizes cancer immunotherapy.
Combining MDSC checkpoint blockade with ICIs could therefore break the immune resistance conferred by MDSCs and facilitate deeper penetration of T cells, CAR-T cells, CAR-NK cells, or CAR- Macrophages into the tumor site. Immune checkpoint inhibitors, such as anti-PD-1 and anti-CTLA-4, are designed to reactivate exhausted T cells and restore their cytotoxic function against tumor cells [9, 130, 167]. However, in tumors characterized by a high prevalence of MDSCs, the efficacy of ICIs can be significantly hindered [168]. MDSCs secrete immunosuppressive cytokines[19, 83], such as PGE2 and TGF-β [38], which contribute to immune suppression within the TME [98]. This persistent immune suppression not only dampens T-cell activity but also diminishes the overall effectiveness of ICIs. Therefore, combining MDSC blockade with ICIs presents a compelling strategy to overcome these limitations and enhance anti-tumor responses.
Targeting MDSC checkpoints like PD-L1, CSF1R, and Arginase-1 may boost ICI activity but could also trigger compensatory mechanisms that affect immune suppression. Careful consideration is needed to avoid immune-related toxicities, as MDSCs are also involved in tissue repair and inflammation regulation. Optimizing the timing, dosing, and combination of these therapies is critical, alongside identifying predictive biomarkers for personalized treatment. Additionally, the combination of oxaliplatin or cyclophosphamide with anti-PD-L1 therapy has shown a synergistic effect in enhancing tumor control [169]. However, despite sustained anti-PD-1/PD-L1 treatment, persistent T-cell dysfunction was observed, indicating that simply blocking these immune checkpoints may not be sufficient for achieving durable tumor regression. This underscores the necessity of targeting additional immune inhibitory pathways, particularly those mediated by MDSCs. This combination of chemotherapeutic agents, which modulate the TME, with CAR-T therapy and ICIs presents a compelling strategy to enhance treatment efficacy. Incorporating MDSC checkpoint blockade into this type of regime would further enhance the immune response by reducing the immunosuppressive activity of MDSCs, allowing for greater CAR-T cell infiltration, improved T cell activation, and more effective anti-tumor response.
Furthermore, combining MDSC checkpoint blockade with CAR-based therapies holds great potential for enhancing anti-tumor responses. In preclinical studies, targeting MDSC-enriched tumors with a TRAIL-R2 blocker alongside CAR-T cells engineered to target tumor-associated mucin 1 (MUC1) resulted in increased tumor cell death and diminished immunosuppressive activity within the tumor microenvironment [170]. This highlights the synergistic effect of incorporating MDSC-targeting strategies with CAR therapies, particularly in solid tumors where MDSCs are highly prevalent.
MDSC checkpoint blockade combined with CAR-NK holds significant potential for improving therapeutic outcomes in solid tumors. CAR-NK, with their innate ability to bypass MHC restrictions, offer an advantage in reducing the risk of immune evasion and cytokine release syndrome compared to CAR-T cells [171]. The addition of MDSC checkpoint inhibitors could further enhance this therapeutic strategy by reducing immune suppression within the tumor microenvironment. MDSC blockade would likely diminish the secretion of anti-inflammatory cytokines such as prostaglandin E2 and nitric oxide, which are known to inhibit NK cell cytotoxicity. This reduction in MDSC-driven immunosuppression could promote a more pro-inflammatory environment, allowing CAR-NK cells to more effectively target and kill tumor cells.
In the context of CAR-Ms, these engineered cells not only attack tumor cells directly through phagocytosis but also reshape the immunosuppressive tumor microenvironment by enhancing antigen presentation and recruiting additional immune effectors. Klichinsky et al. Demonstrated that a single dose of CAR-Ms significantly reduced tumor burden and improved overall survival in solid tumor models [172]. Combining CAR-M therapies with MDSC checkpoint inhibitors could further enhance this therapeutic effect by reducing immune suppression within the TME. By blocking MDSC-mediated immune suppression, the secretion of anti-inflammatory cytokines, such as IL-10 and TGF-β, which are known to support an M2-like macrophage phenotype, would be diminished. This reduction in anti-inflammatory signaling could prevent the repolarization of CAR macrophages from a pro-inflammatory (M1) state back to an immunosuppressive (M2) state, thus maintaining their tumoricidal activity and reinforcing their ability to promote a sustained anti-tumor immune response.
Ultimately, strategic combinations of MDSC checkpoint blockades with ICIs, and CAR-based therapies, have the potential to overcome the various immunosuppressive mechanisms present in tumors. Such combinatorial approaches could improve the penetration of effector cells into the tumor, counteract immune evasion, and increase the durability of anti-cancer responses. As understanding of tumor heterogeneity deepens, selecting and prioritizing combination strategies tailored to specific TME profiles such as tumors with high MDSC infiltration or varying levels of PD-L1 expression will be critical for advancing cancer immunotherapy.
Challenges and future directions
Combining MDSC checkpoint blockade with immune checkpoint inhibitors and adaptive cellular therapies offers a promising strategy to overcome immune suppression in the tumor microenvironment, especially in solid tumors. However, several challenges must be addressed to fully realize this potential. One major obstacle is the heterogeneity of MDSCs, which complicates targeting efforts as monocytic and granulocytic subsets have distinct immunosuppressive mechanisms. Inhibiting MDSCs also poses the risk of off-target effects and immune dysregulation, raising concerns about potential toxicity and autoimmune reactions. Furthermore, tumors may develop resistance through compensatory mechanisms, making it essential to block multiple immunosuppressive pathways simultaneously to achieve long-lasting effects. Optimizing the dosing, timing, and sequence of therapies is another hurdle, as improper administration could result in antagonistic interactions rather than synergistic benefits. Additionally, the effectiveness of MDSC-targeting strategies may vary across different tumor types and TMEs, necessitating personalized approaches and biomarker development to guide treatment. Integrating MDSC blockade with ICIs and cellular therapies, such as CAR-T and CAR-M, presents technical challenges in ensuring these therapies complement each other and maximize therapeutic efficacy. There is also a risk of exacerbating immune-related adverse events (irAEs) [10], requiring careful monitoring and mitigation strategies in clinical trials. One potential approach to mitigate some of these challenges is the development of more selective targeting mechanisms, such as the use of bispecific antibodies or CAR-Ms engineered to recognize tumor-specific antigens, thereby minimizing off-target effects. Additionally, utilizing controlled delivery systems, like nanoparticles or exosome-based platforms, can help ensure that therapeutic agents are specifically delivered to the tumor microenvironment, reducing systemic exposure and toxicity. Incorporating checkpoint inhibitors that are specifically activated in the presence of inflammatory signals or within the tumor niche could further enhance specificity. Moreover, combining MDSC-targeted therapies with agents that modulate immune responses, like cytokines or immune modulators, may enhance efficacy while balancing safety. Finally, close monitoring of immune responses during treatment can help identify and manage potential adverse effects early, improving the overall therapeutic index. Future research should focus on identifying biomarkers to predict patient response, fine-tuning combination therapy regimens, and conducting large-scale clinical trials to validate the safety and efficacy of these approaches. Addressing these challenges will be critical for harnessing the full potential of MDSC-targeting therapies to overcome immune suppression and enhance immune cell infiltration and activation in solid tumors, offering a potential breakthrough in overcoming current immunotherapy limitations.
Conclusion
MDSCs are pivotal mediators of immune evasion in cancer, playing a dual role in both immunosuppressive and non-immunosuppressive tumor-promoting activities, such as supporting cancer stem cell survival, inducing angiogenesis, driving the epithelial–mesenchymal transition, and contributing to the formation of pre-metastatic niches. These diverse and dynamic functions make MDSCs a formidable barrier to effective cancer immunotherapy. MDSC populations are closely associated with poor prognosis, correlating with tumor progression, metastasis, and resistance to treatment. However, the lack of well-defined and universal biomarkers for MDSCs, combined with their inherent plasticity and heterogeneity, presents a significant challenge in developing targeted therapies. To address these issues, a deeper understanding of the molecular and functional heterogeneity of MDSCs across different tumor types is critical.
Emerging high-throughput single-cell multi-omics technologies provide unprecedented insights into MDSC diversity, allowing for the identification of novel markers and therapeutic targets. These advances offer a roadmap for the development of more precise MDSC-targeted therapies. As such, prioritizing research into MDSC phenotypic and functional characterization, particularly within distinct tumor microenvironments, will be essential for identifying the most relevant MDSC subsets for targeting. Additionally, a concerted focus on the immune and non-immune contributions of MDSCs in solid tumors is required, as these cells facilitate both immune suppression and tumor progression through diverse mechanisms.
MDSC checkpoint blockade presents a promising strategy to overcome both immune suppression and the tumor-promoting effects of MDSCs. However, achieving clinical success will require overcoming challenges related to the selective targeting of these cells without affecting normal immune function. Combining MDSC checkpoint blockade with immune checkpoint inhibitors (such as PD-1/PD-L1 blockade), adaptive cellular therapies (e.g., CAR-T and CAR-NK cells) and multiple MDSC checkpoints targeting strategies could create synergistic therapeutic strategies to enhance anti-tumor immunity while mitigating the negative impact of MDSCs. A critical future research direction should involve optimizing combination therapy regimens, defining the optimal timing and dosing strategies, and developing specific targeting modalities that minimize off-target effects. In addition to therapeutic targeting, the development of predictive biomarkers to assess the presence and activity of MDSCs in patients will be crucial for patient stratification and monitoring treatment efficacy. Develop physiologically relevant tumor models that better mimic the human tumor microenvironment. In addition, early-phase clinical trials focused on MDSC-targeted therapies, especially those incorporating advanced delivery systems like nanoparticles or exosomes should be prioritized, moreover, integration of systems biology and artificial intelligence can provide predictive models for therapy optimization, further advancing the field of MDSC-targeted immunotherapy. By addressing these gaps, we are poised to significantly enhance the efficacy of current immunotherapies and provide durable responses in cancer patients. With continued progress in MDSC biology, therapeutic innovation, and personalized medicine, we can look forward to more effective and sustainable treatment options for patients, particularly in the context of solid tumors where MDSCs are major contributors to therapy resistance and disease progression.
References
McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Shi G, Li D, Zhang D, Xu Y, Pan Y, Lu L, et al. IRF-8/miR-451a regulates M-MDSC differentiation via the AMPK/mTOR signal pathway during lupus development. Cell Death Discov. 2021;7:179.
Abdin SM, Paasch D, Morgan M, Lachmann N. CARs and beyond: tailoring macrophage-based cell therapeutics to combat solid malignancies. J Immunother Cancer. 2021;9:e002741.
Li Y-R, Wilson M, Yang L. Target tumor microenvironment by innate T cells. Front Immunol. 2022;13:999549.
Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, et al. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: Current Progress and New Prospects. Clin Med Insights Oncol. 2021;15:11795549211035540.
Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021;359:104254.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8.
Akkari L, Amit I, Bronte V, Fridlender ZG, Gabrilovich DI, Ginhoux F, et al. Defining myeloid-derived suppressor cells. Nat Rev Immunol. 2024;24:850–7.
Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020;17:1–12.
Sui H, Dongye S, Liu X, Xu X, Wang L, Jin CQ, et al. Immunotherapy of targeting MDSCs in tumor microenvironment. Front Immunol. 2022;13:990463.
Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Liu G, Jin Z, Lu X. Differential targeting of Gr-MDSCs, T cells and prostate cancer cells by dactolisib and dasatinib. Int J Mol Sci. 2020;21:2337.
Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. 2014;41:389–401.
Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. 2017;8:93.
Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol. 2018;9:2499.
Chen N, Xu Y, Mou J, Rao Q, Xing H, Tian Z, et al. Targeting of IL-10R on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood Cancer J. 2021;11:144.
Sarkar OS, Donninger H, Al Rayyan N, Chew LC, Stamp B, Zhang X, et al. Monocytic MDSCs exhibit superior immune suppression via adenosine and depletion of adenosine improves efficacy of immunotherapy. Sci Adv. 2023;9:eadg3736.
Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharm Sci. 2019;40:4–7.
Hou A, Hou K, Huang Q, Lei Y, Chen W. Targeting myeloid-derived suppressor cell, a promising strategy to overcome resistance to immune checkpoint inhibitors. Front Immunol. 2020;11:783.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci. 2019;76:1901–18.
Salman S, Meyers DJ, Wicks EE, Lee SN, Datan E, Thomas AM, et al. HIF inhibitor 32-134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J Clin Invest. 2022;132:e156774.
Liu M, Wei F, Wang J, Yu W, Shen M, Liu T, et al. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1-PD-L1+ Bregs through PD-L1/PI3K/AKT/NF-κB axis in breast cancer. Cell Death Dis. 2021;12:465.
Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200:422–31.
Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28.
Lu T, Ramakrishnan R, Altiok S, Youn J-I, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121:4015–29.
Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12:1717.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54.
Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11:702–11.
Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22:2908–18.
Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta. 2014;1846:55–65.
Black PN, DiRusso CC. Yeast acyl-CoA synthetases at the crossroads of fatty acid metabolism and regulation. Biochim Biophys Acta. 2007;1771:286–98.
Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur J Immunol. 2013;43:2943–55.
Sun Y, Han X, Shang C, Wang Y, Xu B, Jiang S, et al. The downregulation of type I IFN signaling in G-MDSCs under tumor conditions promotes their development towards an immunosuppressive phenotype. Cell Death Dis. 2022;13:36.
Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73–8.
Schuster C, Akslen LA, Straume O. β2-adrenergic receptor expression in patients receiving bevacizumab therapy for metastatic melanoma. Cancer Med. 2023;12:17891–900.
Mohammadpour H, MacDonald CR, McCarthy PL, Abrams SI, Repasky EA. β2-adrenergic receptor signaling regulates metabolic pathways critical to myeloid-derived suppressor cell function within the TME. Cell Rep. 2021;37:109883.
Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K, Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest. 2012;41:635–57.
Jin K, Qian C, Lin J, Liu B. Cyclooxygenase-2-prostaglandin E2 pathway: a key player in tumor-associated immune cells. Front Oncol. 2023;13:1099811.
Tang X, Spitzbarth N, Kuhn H, Chaitidis P, Campbell WB. Interleukin-13 upregulates vasodilatory 15-lipoxygenase eicosanoids in rabbit aorta. Arterioscler Thromb Vasc Biol. 2003;23:1768–74.
Du S, Zeng F, Deng G. Tumor neutrophils ferroptosis: a targetable immunosuppressive mechanism for cancer immunotherapy. Signal Transduct Target Ther. 2023;8:77.
Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338–46.
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–9.
King RJ, Shukla SK, He C, Vernucci E, Thakur R, Attri KS, et al. CD73 induces GM-CSF/MDSC-mediated suppression of T cells to accelerate pancreatic cancer pathogenesis. Oncogene. 2022;41:971–82.
Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. 2020;69:2477–99.
Wong JL, Obermajer N, Odunsi K, Edwards RP, Kalinski P. Synergistic COX2 Induction by IFNγ and TNFα self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol Res. 2016;4:303–11.
Li Z-L, Ye S-B, OuYang L-Y, Zhang H, Chen Y-S, He J, et al. COX-2 promotes metastasis in nasopharyngeal carcinoma by mediating interactions between cancer cells and myeloid-derived suppressor cells. Oncoimmunology. 2015;4:e1044712.
Wang Y, Cui L, Georgiev P, Singh L, Zheng Y, Yu Y, et al. Combination of EP4 antagonist MF-766 and anti-PD-1 promotes anti-tumor efficacy by modulating both lymphocytes and myeloid cells. Oncoimmunology. 2021;10:1896643.
Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. 2017;114:1117–22.
Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JGJV, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer. 2010;10:464.
Maltesen HR, Troelsen JT, Olsen J. Identification of a functional hepatocyte nuclear factor 4 binding site in the neutral ceramidase promoter. J Cell Biochem. 2010;111:1330–6.
García-Barros M, Coant N, Kawamori T, Wada M, Snider AJ, Truman J-P, et al. Role of neutral ceramidase in colon cancer. FASEB J. 2016;30:4159–71.
Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, et al. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol. 2021;206:1395–404.
Ding L, Hayes MM, Photenhauer A, Eaton KA, Li Q, Ocadiz-Ruiz R, et al. Schlafen 4-expressing myeloid-derived suppressor cells are induced during murine gastric metaplasia. J Clin Invest. 2016;126:2867–80.
Xiang X, Wu Y, Li H, Li C, Yan L, Li Q. Plasmacytoid dendritic cell-derived type I interferon is involved in helicobacter pylori infection-induced differentiation of schlafen 4-expressing myeloid-derived suppressor cells. Infect Immun. 2021;89:e0040721.
Ding L, Li Q, Chakrabarti J, Munoz A, Faure-Kumar E, Ocadiz-Ruiz R, et al. MiR130b from Schlafen4+ MDSCs stimulates epithelial proliferation and correlates with preneoplastic changes prior to gastric cancer. Gut. 2020;69:1750–61.
Geserick P, Kaiser F, Klemm U, Kaufmann SHE, Zerrahn J. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol. 2004;16:1535–48.
Ding L, Sheriff S, Sontz RA, Merchant JL. Schlafen4+-MDSC in Helicobacter-induced gastric metaplasia reveals role for GTPases. Front Immunol. 2023;14:1139391.
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25.
Gao Y, Wang T, Li Y, Zhang Y, Yang R. Lnc-chop promotes immunosuppressive function of myeloid-derived suppressor cells in tumor and inflammatory environments. J Immunol. 2018;200:2603–14.
Liu X, Zhao S, Sui H, Liu H, Yao M, Su Y, et al. MicroRNAs/LncRNAs modulate MDSCs in tumor microenvironment. Front Oncol. 2022;12:772351.
Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. 2020;52:668–82.e7.
Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. 2011;108:6561–6.
Suresh S, Chen B, Zhu J, Golden RJ, Lu C, Evers BM, et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat Cancer. 2020;1:533–45.
Mandula JK, Chang S, Mohamed E, Jimenez R, Sierra-Mondragon RA, Chang DC, et al. Ablation of the endoplasmic reticulum stress kinase PERK induces paraptosis and type I interferon to promote anti-tumor T cell responses. Cancer Cell. 2022;40:1145–60.e9.
Liu M, Wu C, Luo S, Hua Q, Chen H-T, Weng Y, et al. PERK reprograms hematopoietic progenitor cells to direct tumor-promoting myelopoiesis in the spleen. J Exp Med. 2022;219:e20211498.
Kny M, Fielitz J. Hidden Agenda—the involvement of endoplasmic reticulum stress and unfolded protein response in inflammation-induced muscle wasting. Front Immunol. 2022;13:878755.
Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol. 2023;24:255–72.
Trillo-Tinoco J, Sierra RA, Mohamed E, Cao Y, de Mingo-Pulido Á, Gilvary DL, et al. AMPK Alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res. 2019;79:5034–47.
Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012;13:18.
Adeshakin FO, Adeshakin AO, Liu Z, Cheng J, Zhang P, Yan D, et al. Targeting oxidative phosphorylation-proteasome activity in extracellular detached cells promotes anoikis and inhibits metastasis. Life (Basel). 2021;12:42.
Xie Y, Shi X, Sheng K, Han G, Li W, Zhao Q, et al. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol Med Rep. 2019;19:783–91.
Scott J, Rees L, Gallimore A, Lauder SN. PI3K isoform immunotherapy for solid tumours. Curr Top Microbiol Immunol. 2022;436:369–92.
Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. 2019;10:172.
Chandrasekaran S, Funk CR, Kleber T, Paulos CM, Shanmugam M, Waller EK. Strategies to overcome failures in T-cell immunotherapies by targeting PI3K-δ and -γ. Front Immunol. 2021;12:718621.
Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol. 2019;10:1399.
Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 2014;74:727–37.
Dong L, Bi Y, Jia A, Yu Q, Wang Y, Wang Y, et al. Crucial role of histone deacetylase SIRT1 in myeloid-derived suppressor cell-mediated reprogramming of CD4+ T-cell differentiation. Cell Mol Immunol. 2020;17:785–7.
Kim M, Kwon YE, Song JO, Bae SJ, Seol JH. CHFR negatively regulates SIRT1 activity upon oxidative stress. Sci Rep. 2016;6:37578.
Gao Y, Sun W, Shang W, Li Y, Zhang D, Wang T, et al. Lnc-C/EBPβ negatively regulates the suppressive function of myeloid-derived suppressor cells. Cancer Immunol Res. 2018;6:1352–63.
Mildner A, Schönheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, et al. Genomic characterization of murine monocytes reveals C/EBPβ transcription factor dependence of Ly6C-cells. Immunity. 2017;46:849–62.e7.
Qi H, Li Y, Liu X, Jiang Y, Li Z, Xu X, et al. Tim-3 regulates the immunosuppressive function of decidual MDSCs via the Fyn-STAT3-C/EBPβ pathway during Toxoplasma gondii infection. PLoS Pathog. 2023;19:e1011329.
Gao X, Sui H, Zhao S, Gao X, Su Y, Qu P. Immunotherapy targeting myeloid-derived suppressor cells (MDSCs) in tumor microenvironment. Front Immunol. 2020;11:585214.
Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell. 2015;28:253–69.
Sarker D, Plummer R, Meyer T, Sodergren MH, Basu B, Chee CE, et al. MTL-CEBPA, a small activating RNA therapeutic upregulating C/EBP-α, in patients with advanced liver cancer: a First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin Cancer Res. 2020;26:3936–46.
Li T, Bou-Dargham MJ, Fultang N, Li X, Pear WS, Sun H, et al. c-Rel-dependent monocytes are potent immune suppressor cells in cancer. J Leukoc Biol. 2022;112:845–59.
Fultang N, Li X, Li T, Chen YH. Myeloid-derived suppressor cell differentiation in cancer: transcriptional regulators and enhanceosome-mediated mechanisms. Front Immunol. 2020;11:619253.
Merika M, Thanos D. Enhanceosomes. Curr Opin Genet Dev. 2001;11:205–8.
Cao Y, Wang J, Jiang S, Lyu M, Zhao F, Liu J, et al. JAK1/2 inhibitor ruxolitinib promotes the expansion and suppressive action of polymorphonuclear myeloid-derived suppressor cells via the JAK/STAT and ROS-MAPK/NF-κB signalling pathways in acute graft-versus-host disease. Clin Transl Immunol. 2023;12:e1441.
Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014;193:2574–86.
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016;76:3156–65.
Wang Y, Yin K, Tian J, Xia X, Ma J, Tang X, et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv Sci (Weinh). 2019;6:1901278.
Adeshakin FO, Adeshakin AO, Liu Z, Lu X, Cheng J, Zhang P, et al. Upregulation of V-ATPase by STAT3 activation promotes anoikis resistance and tumor metastasis. J Cancer. 2021;12:4819–29.
Bitsch R, Kurzay A, Özbay Kurt F, De La Torre C, Lasser S, Lepper A, et al. STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J Immunother Cancer. 2022;10:e004384.
Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8:14979.
Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016;44:303–15.
Hopkins JL, Zou L. Induction of BRCAness in triple-negative breast cancer by a CDK12/13 Inhibitor improves chemotherapy. Cancer Cell. 2019;36:461–3.
Guan Y, Tiwari AD, Phillips JG, Hasipek M, Grabowski DR, Pagliuca S, et al. A therapeutic strategy for preferential targeting of TET2 mutant and TET-dioxygenase deficient cells in myeloid neoplasms. Blood Cancer Discov. 2021;2:146–61.
Jain N, Zhao Z, Feucht J, Koche R, Iyer A, Dobrin A, et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature. 2023;615:315–22.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24.
Li S, Feng J, Wu F, Cai J, Zhang X, Wang H, et al. TET2 promotes anti-tumor immunity by governing G-MDSCs and CD8+ T-cell numbers. EMBO Rep. 2020;21:e49425.
Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Béguelin W, Fontán L, et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 2018;8:1632–53.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12.
Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, et al. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008;3:416–28.
Li F, Kitajima S, Kohno S, Yoshida A, Tange S, Sasaki S, et al. Retinoblastoma inactivation induces a protumoral microenvironment via enhanced CCL2 secretion. Cancer Res. 2019;79:3903–15.
Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. 2016;44:1337–49.
Nguyen NT, Mitsuhashi A, Ogino H, Kozai H, Yoneda H, Afroj T, et al. S-1 eliminates MDSCs and enhances the efficacy of PD-1 blockade via regulation of tumor-derived Bv8 and S100A8 in thoracic tumor. Cancer Sci. 2023;114:384–98.
Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6:8.
Benguigui M, Vorontsova A, Timaner M, Levin S, Haj-Shomaly J, Deo A, et al. Bv8 blockade sensitizes anti-PD1 therapy resistant tumors. Front Immunol. 2022;13:903591.
Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kδ/γ. Cancer Res. 2017;77:2607–19.
Wang C, Zheng X, Zhang J, Jiang X, Wang J, Li Y, et al. CD300ld on neutrophils is required for tumour-driven immune suppression. Nature. 2023;621:830–9.
Li Y, Wang C, Lu Z, Luo M. Targeting PMN-MDSCs via CD300ld receptor for cancer immunotherapy. Clin Transl Med. 2024;14:e1534.
Condamine T, Kumar V, Ramachandran IR, Youn J-I, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Investig. 2014;124:2626–39.
Favaro F, Luciano-Mateo F, Moreno-Caceres J, Hernández-Madrigal M, Both D, Montironi C, et al. TRAIL receptors promote constitutive and inducible IL-8 secretion in non-small cell lung carcinoma. Cell Death Dis. 2022;13:1046.
Tang Y, Zhou C, Li Q, Cheng X, Huang T, Li F, et al. Targeting depletion of myeloid-derived suppressor cells potentiates PD-L1 blockade efficacy in gastric and colon cancers. Oncoimmunology. 2022;11:2131084.
Rapoport BL, Steel HC, Theron AJ, Smit T, Anderson R. Role of the neutrophil in the pathogenesis of advanced cancer and impaired responsiveness to therapy. Molecules. 2020;25:1618.
Chen Q, Yan D, Zhang Q, Zhang G, Xia M, Li J, et al. Treatment of acetaminophen-induced liver failure by blocking the death checkpoint protein TRAIL. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165583.
Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL, et al. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J Clin Invest. 2019;129:5537–52.
Pani B, Ahn S, Rambarat PK, Vege S, Kahsai AW, Liu A, et al. Unique positive cooperativity between the β-arrestin-biased β-blocker carvedilol and a small molecule positive allosteric modulator of the β2-adrenergic receptor. Mol Pharm. 2021;100:513–25.
Ammons DT, MacDonald CR, Chow L, Repasky EA, Dow S. Chronic adrenergic stress and generation of myeloid-derived suppressor cells: Implications for cancer immunotherapy in dogs. Vet Comp Oncol. 2023;21:159–65.
Ashrafi S, Shapouri R, Mahdavi M. Immunological consequences of immunization with tumor lysate vaccine and propranolol as an adjuvant: a study on cytokine profiles in breast tumor microenvironment. Immunol Lett. 2017;181:63–70.
Iñigo-Marco I, Alonso MM. Destress and do not suppress: targeting adrenergic signaling in tumor immunosuppression. J Clin Invest. 2019;129:5086–8.
Farooq MA, Ajmal I, Hui X, Chen Y, Ren Y, Jiang W. β2-adrenergic receptor mediated inhibition of T cell function and its implications for CAR-T cell therapy. Int J Mol Sci. 2023;24:12837.
Zhang Z, Hatano H, Shaw J, Olde Nordkamp M, Jiang G, Li D, et al. The leukocyte immunoglobulin-like receptor family member LILRB5 binds to HLA-Class I heavy chains. PLoS One. 2015;10:e0129063.
Venet F, Schilling J, Cazalis M-A, Demaret J, Poujol F, Girardot T, et al. Modulation of LILRB2 protein and mRNA expressions in septic shock patients and after ex vivo lipopolysaccharide stimulation. Hum Immunol. 2017;78:441–50.
Chen H-M, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Investig. 2018;128:5647–62.
Zeller T, Lutz S, Münnich IA, Windisch R, Hilger P, Herold T, et al. Dual checkpoint blockade of CD47 and LILRB1 enhances CD20 antibody-dependent phagocytosis of lymphoma cells by macrophages. Front Immunol. 2022;13:929339.
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74:1933–44.
Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell-mediated inflammation and immunosuppression. Cancer Immunol Res. 2019;7:1497–510.
Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA. 2015;112:6682–7.
Zhang Y, Hu J, Ji K, Jiang S, Dong Y, Sun L, et al. CD39 inhibition and VISTA blockade may overcome radiotherapy resistance by targeting exhausted CD8+ T cells and immunosuppressive myeloid cells. Cell Rep. Med. 2023;4:101151.
Shojaei F, Wu X, Zhong C, Yu L, Liang X-H, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–31.
Li X, Chang E, Cui J, Zhao H, Hu C, O’Dea KP, et al. Bv8 mediates myeloid cell migration and enhances malignancy of colorectal cancer. Front Immunol. 2023;14:1158045.
Noe JT, Mitchell RA. MIF-dependent control of tumor immunity. Front Immunol. 2020;11:609948.
Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–76.
Figueiredo CR, Azevedo RA, Mousdell S, Resende-Lara PT, Ireland L, Santos A, et al. Blockade of MIF-CD74 signalling on macrophages and dendritic cells restores the antitumour immune response against metastatic melanoma. Front Immunol. 2018;9:1132.
Alban TJ, Bayik D, Otvos B, Rabljenovic A, Leng L, Jia-Shiun L, et al. Glioblastoma myeloid-derived suppressor cell subsets express differential macrophage migration inhibitory factor receptor profiles that can be targeted to reduce immune suppression. Front Immunol. 2020;11:1191.
Yaddanapudi K, Rendon BE, Lamont G, Kim EJ, Al Rayyan N, Richie J, et al. MIF Is necessary for late-stage melanoma patient mdsc immune suppression and differentiation. Cancer Immunol Res. 2016;4:101–12.
Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol. 2012;189:5533–40.
Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–26.
Yan D, Wang J, Sun H, Zamani A, Zhang H, Chen W, et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J Exp Med. 2020;217:e20182005.
Bi J, Huang C, Jin X, Zheng C, Huang Y, Zheng X, et al. TIPE2 deletion improves the therapeutic potential of adoptively transferred NK cells. J Immunother Cancer. 2023;11:e006002.
Zhu L, Zhang X, Fu X, Li Z, Sun Z, Wu J, et al. TIPE2 suppresses progression and tumorigenesis of esophageal carcinoma via inhibition of the Wnt/β-catenin pathway. J Transl Med. 2018;16:7.
Zhang Z, Liu L, Cao S, Zhu Y, Mei Q. Gene delivery of TIPE2 inhibits breast cancer development and metastasis via CD8+ T and NK cell-mediated antitumor responses. Mol Immunol. 2017;85:230–7.
Xu S, Gao X, Qiu J, Hong F, Gao F, Wang X, et al. TIPE2 acts as a tumor suppressor and correlates with tumor microenvironment immunity in epithelial ovarian cancer. Aging (Albany NY). 2023;15:1052–73.
Wang L, Chang EWY, Wong SC, Ong S-M, Chong DQY, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol. 2013;190:794–804.
Dai J, Kumbhare A, Youssef D, McCall CE, El Gazzar M. Intracellular S100A9 promotes myeloid-derived suppressor cells during late sepsis. Front Immunol. 2017;8:1565.
Cheng P, Eksioglu EA, Chen X, Kandell W, Le Trinh T, Cen L, et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia. 2019;33:2034–46.
Zhong C, Niu Y, Liu W, Yuan Y, Li K, Shi Y, et al. S100A9 derived from chemoembolization-induced hypoxia governs mitochondrial function in hepatocellular carcinoma progression. Adv Sci (Weinh). 2022;9:e2202206.
Yu K, Yu C, Jiao L, Miao K, Ni L, Rao X, et al. The Function and therapeutic implications of TNF signaling in MDSCs. Biomolecules. 2022;12:1627.
Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol. 2014;192:1320–31.
Ahmad S, Azid NA, Boer JC, Lim J, Chen X, Plebanski M, et al. The key role of TNF–TNFR2 interactions in the modulation of allergic inflammation: a review. Front Immunol. 2018;9:2572.
Atretkhany K-SN, Nosenko MA, Gogoleva VS, Zvartsev RV, Qin Z, Nedospasov SA, et al. TNF neutralization results in the delay of transplantable tumor growth and reduced MDSC accumulation. Front Immunol. 2016;7:147.
Al Barashdi MA, Ali A, McMullin MF, Mills K. Protein tyrosine phosphatase receptor type C (PTPRC or CD45). J Clin Pathol. 2021;74:548–52.
Huang X, Zuo Y, Wang X, Wu X, Tan H, Fan Q, et al. SUMO-specific protease 1 Is critical for myeloid-derived suppressor cell development and function. Cancer Res. 2019;79:3891–902.
Perron M, Saragovi HU. Inhibition of CD45 phosphatase activity induces cell cycle arrest and apoptosis of CD45+ lymphoid tumors ex vivo and in vivo. Mol Pharm. 2018;93:575–80.
Yagisawa H, Hirata M, Kanematsu T, Watanabe Y, Ozaki S, Sakuma K, et al. Expression and characterization of an inositol 1,4,5-trisphosphate binding domain of phosphatidylinositol-specific phospholipase C-delta 1. J Biol Chem. 1994;269:20179–88.
Capietto A-H, Kim S, Sanford DE, Linehan DC, Hikida M, Kumosaki T, et al. Down-regulation of PLCγ2-β-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. 2013;210:2257–71.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61.
Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–2793.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53.
Korbecki J, Simińska D, Gąssowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, et al. Chronic and cycling hypoxia: drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: a review of the molecular mechanisms. Int J Mol Sci. 2021;22:10701.
Niu J, Li X-M, Wang X, Liang C, Zhang Y-D, Li H-Y, et al. DKK1 inhibits breast cancer cell migration and invasion through suppression of β-catenin/MMP7 signaling pathway. Cancer Cell Int. 2019;19:168.
Sui Q, Liu D, Jiang W, Tang J, Kong L, Han K, et al. Dickkopf 1 impairs the tumor response to PD-1 blockade by inactivating CD8+ T cells in deficient mismatch repair colorectal cancer. J Immunother Cancer. 2021;9:e001498.
Li H, Wu C, Sun Z, Yu X, Liu W, Liu D, et al. DKK1 impairs the tumor response to αPD-1 immunotherapy by inactivating CD8+ T cells and recruiting MDSC cells in HNSC. Adv Ther. 2023;6:2300313.
D’Amico L, Mahajan S, Capietto A-H, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. 2016;213:827–40.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236–47.
Peranzoni E, Ingangi V, Masetto E, Pinton L, Marigo I. Myeloid cells as clinical biomarkers for immune checkpoint blockade. Front Immunol. 2020;11:1590.
Nalawade SA, Shafer P, Bajgain P, McKenna MK, Ali A, Kelly L, et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J Immunother Cancer. 2021;9:e003237.
Dagher OK, Posey AD. Forks in the road for CAR T and CAR NK cell cancer therapies. Nat Immunol. 2023;24:1994–2007.
Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14:73.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947–53.
Funding
This work was supported by National Key R&D Program of China (2019YFA0906100 and 2021YFC3300101), National Natural Science Foundation of China (82071772), Guangdong Basic and Applied Basic Research Foundation (2022A1515010070), Shenzhen Basic Science Research Project (JCYJ20220818102018038, JCYJ20220531095612029, JSGGZD20220822100005011), Shenzhen Medical Research Fund (A2301047), and Futian Healthcare Research Project (FTWS2023066).
Author information
Authors and Affiliations
Contributions
Abdulrahman Ibrahim completed the literature search, diagrams and figures preparation, and manuscript writing. Dehong Yan conceptualized the review, wrote and revised the manuscript. Shu Xu, Zhiming Xu, and Xiaochun Wan provided advice and revised the manuscript. Nada Mohamady Farouk Abdalsalam, Zihao Liang, Hafiza Kashaf Tariq, Rong Li, Lukman O. Afolabi, Lawan Rabiu, and Xuechen Chen provided some help in the figures and manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ibrahim, A., Mohamady Farouk Abdalsalam, N., Liang, Z. et al. MDSC checkpoint blockade therapy: a new breakthrough point overcoming immunosuppression in cancer immunotherapy. Cancer Gene Ther 32, 371–392 (2025). https://doi.org/10.1038/s41417-025-00886-9
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41417-025-00886-9
This article is cited by
-
Neurotoxicity in central nervous system tumors treated with CAR T cell therapy: a review
Journal of Neuro-Oncology (2026)
-
Immune checkpoint blockade in cancer: current insights and future horizons
Discover Oncology (2026)
-
CD86 costimulation enhances the antitumor activity of NKG2D CAR-Macrophages and synergizes with Anti-PD-L1 therapy to suppress prostate cancer progression
Cell Communication and Signaling (2025)
-
Advancing our understanding of the influence of myeloid-derived suppressor cells in chronic myeloid leukemia
Journal of Cancer Research and Clinical Oncology (2025)
-
PI3 K/AKT/mTOR pathway and its role in breast cancer stem cells
Naunyn-Schmiedeberg's Archives of Pharmacology (2025)
