
Abstract
Chimeric antigen receptor (CAR) T cell therapy has become an indispensable immunotherapy for the treatment of some hematologic cancers, but still faces numerous challenges in the form of antigen escape, variable patient responses, toxicities, limited CAR T cell persistence, and high cost, particularly against solid tumors. This Review discusses the potential role of the endogenous T cell receptor (TCR) as either a hindrance or partner to CAR T cell function. Specifically, we discuss the differences and similarities between CAR and TCR structure and function, findings supporting the value of TCR elimination in CAR T cells, and, in contrast, data in support of retaining and utilizing the endogenous TCR in CAR T cell therapy. We make the case that, while TCR-knockout systems may improve aspects such as the universality, cost, and CAR expression of CAR therapies, the endogenous TCR continues to play a significant role in maintaining CAR T cell persistence and can be used to augment CAR T cell therapeutic phenotypes. Overall, we highlight the uncertainties that persist within the field of CAR T cell therapy and outline emerging evidence and directions regarding the CAR T cell TCR that have the potential to transform patient outcomes.
Similar content being viewed by others
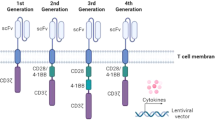
Introduction
Chimeric antigen receptor (CAR) T cell therapy has transformed the treatment of hematologic cancers, with remarkable efficacy against relapsed or refractory disease. Currently, seven US Food and Drug Administration (FDA)-approved CAR T cell therapies exist for the treatment of plasma cell and lymphoid malignancies [1]. As of 2024, about 35,000 patients had received commercial CAR T cell therapy in the US over a seven-year period, starting with the first FDA-approved CAR T therapy in 2017 [2]. The basic principle of CARs is to use recombinant antigen receptors to redirect the binding and cytotoxicity of T cells and other lymphocytes to directly target and eliminate cancer cells [3]. Additional trials are ongoing applying CAR T therapies to non-malignant diseases, including rheumatoid arthritis and systemic lupus erythematosus [4, 5]. Traditionally, a single-chain variable fragment (scFv) derived from the antigen-binding domains of monoclonal antibodies is joined via a hinge and transmembrane domain to intracellular signaling domains borrowed from downstream molecules of T cell receptor (TCR) signaling [6]. For clinical applications, T cells from whole peripheral blood mononuclear cells (PBMCs) or T cell-enriched PBMCs are engineered to express a CAR transgene via lentiviral or retroviral transduction. This method lacks transgene insertion-site specificity, a concern for mutagenesis and controlling CAR expression, and retains the expression of the endogenous TCR, necessitating autologous transfer [7]. Of note, insertional mutagenesis of the CAR has not thus far proven problematic [8]. Other options also exist for non-autologous CAR transfer, including NK and NKT cells [9, 10].
Despite the overall success of CAR T therapy, there remain numerous challenges, including variable patient responses, antigen escape, CAR T persistence, and cost [6, 11, 12]. To address at least some of these obstacles, the role of the endogenous TCR has been scrutinized, either as a way to lower cost by generating an “off-the-shelf” CAR through TCR knock out, or by promoting CAR persistence and tumor antigenicity by engaging the endogenous TCR [13,14,15,16]. By using gene editing techniques to either knock out the endogenous TCR or insert the CAR transgene into the TCR alpha chain (TRAC) locus, the hope is to replicate endogenous TCR activity and regulation of surface expression, in addition to preventing graft-versus-host disease (GvHD) [17, 18]. Meanwhile, studies capitalizing on retention of endogenous TCRs largely aim to use their proven functions of maintaining T cell proliferation and survival to improve CAR T cell persistence and engage costimulatory and differentiation pathways that remain elusive through CAR signaling [19, 20]. With the excitement in the field around TCR knockout CAR or guided CAR insertion into the TRAC locus, understandably so due to its potential cost and safety benefits, we propose that there is still much to learn about the interactions of the CAR and TCR and the potential benefits of engaging the endogenous TCR on CAR function. In this Review, we provide an analysis of our current understanding of CAR and TCR signaling and what the role of the TCR might be as CAR T therapies continue to evolve (Fig. 1).

Analysis of the interactions of activating receptors in a dual CAR or dual TCR system facilitates discussion of the role of the TCR in CAR T cell therapy. Targeting more than one antigen is an attractive premise driving interest in dual CAR, TCR, or CAR/TCR systems, in addition to potential benefits for migration and T cell activation. However, elimination of the endogenous TCR has its own benefits for allogeneic T cell transfer and exploiting more endogenous CAR regulation through TRAC integration.
TCR versus CAR signaling
TCR signaling
In the early 1980s, groups led by Ellis Reinherz, Philippa Marrack, and John Kappler first elucidated the structure and function of the TCR, demonstrating the clonal nature of the TCR, its association with CD3, and its function as an antigen receptor through peptide:MHC binding [21, 22]. We now know that the αβ TCR-CD3 complex is an octameric structure consisting of TCRαβ, CD3γε, CD3δε, and CD3ζζ subunits [23]. Upon peptide:MHC engagement, TCR signaling is initiated, recruiting Lck to the TCR complex upon CD4 or CD8 colocalization to peptide:MHC that phosphorylates immunoreceptor tyrosine-based activation motifs (ITAMs) within CD3 subunits. These phosphorylated ITAMs then recruit and bind Zap70, a kinase that initiates propagation of TCR signaling events through linker for activation of T cells (LAT), which serves as a starting point for numerous downstream cascades. Phosphorylation of LAT by Zap70 recruits other adaptor molecules (such as GRB2-related adaptor downstream of Shc (GADS), SH2-domain-containing leukocyte protein of 76 kDa (SLP76), adhesion and degranulation promoting adaptor protein (ADAP)) and subsequent effector signaling molecules, including phospholipase Cγ1 (PLCγ1), IL-2-inducible T cell kinase (ITK), and VAV1. These effector molecules ultimately lead to cytoskeletal reorganization and activation of transcription factors such as activator protein-1 (AP-1), nuclear factor-κB (NF-κB), and nuclear factor of activated T cells (NFAT), which regulate T cell proliferation, migration, and effector functions [24, 25].
CAR signaling and distinctions from TCR signaling
Unlike TCRs, CAR molecules contain both the antigen-sensing and intracellular signaling domains within the same construct. All currently FDA-approved CAR therapies use a scFv, which recognizes an extracellular protein rather than a peptide: MHC complex, and are second-generation, meaning they contain one costimulatory molecule (either 4-1BB or CD28) in-line with CD3ζ. For normal TCR signaling, 4-1BB acts via recruitment of TNFR-associated factors (TRAFs), while CD28 activates the phosphatidylinositol 3-kinase (PI3K)–AKT pathway. These pathways are assumed to be preserved in CAR T cells, but their precise homology to native TCR pathways remains unclear, although we know that inclusion of 4-1BB versus CD28 alters CAR T fate and function [26, 27]. Additionally, CARs contain only one CD3ζ carrying three ITAMs, as opposed to the ten available ITAMs within the TCR-CD3 complex. The number of ITAMs and type of CD3 chain within a CAR have been shown to modulate T cell activation, a potentially important caveat for poorly expressed antigens and CAR persistence [28, 29]. Functionally, CAR sensitivity to antigen has been shown to be orders of magnitude lower than TCR, even in the absence of the CD8 coreceptor [30,31,32]. Signaling studies detected absent or weaker phosphorylation of CD3δ, CD3ε, and CD3ζ, in addition to LAT, likely from inefficient recruitment and activation of Zap70 by the CAR [30, 33]. Using LAT-deficient T cells, Dong et al. confirmed that CAR function can occur even independently of LAT, with CAR micro-clustering and recruitment of downstream GADS and SLP76 occurring in the absence of LAT [34]. CAR engagement also inefficiently utilizes accessory proteins such as CD2 and LFA-1, compared to that of the TCR [35]. Although the magnitude of activation is reduced, CAR signaling appears to induce more sustained T cell activation via calcium influx and IL2 production [36]. Furthermore, an analysis by Wong et al. on CAR and TCR complementarity-determining regions (CDRs) found significant structural differences between the two, notably increased TCR CDR flexibility and structural variation [37].
In terms of TCR and CAR interactions, it has been shown that neither CAR nor TCR recruits the other into its immune synapse or cross-signals via CD3ζ [38]. However, when both CAR and TCR bind their respective antigen, complementation can occur through potentiation of downstream activators. This “AND” gating implicates CAR activation in the modulation of TCR function in contexts where the CAR carries activating, inhibitory, or costimulatory signaling moieties [39]. Even in the absence of signaling, CAR molecules with CD28 transmembrane regions have been shown to form heterodimers with endogenous CD28 and become activated through CD80/CD86 interactions with separate CD28 homodimers [40]. Despite these studies, much remains unknown regarding CAR-TCR interactions and the interplay between CARs and endogenous T cell signaling molecules. Adding to this complexity, the structure of each CAR domain, for example, the length of the hinge region, and the avidity of the CAR itself, may modify interactions of the CAR with secondary CARs or endogenous TCRs through altered downstream signaling and protein-protein interactions [41]. With the jury still out on the nature of these interactions, it is therefore reasonable to pursue potentiation of T cell function via combinations of CARs, TCRs, and TCR knockouts.
Dual CARs and dual TCRs: more of a good thing?
T cells expressing two CARs were intended to address several issues plaguing CAR therapies for solid tumors: on-target/off-tumor effects, antigen escape, and survival within the immunosuppressive tumor microenvironment. For example, Zhang et al. developed dual-receptor CAR T cells with separate CARs containing either CD3ζ or 4-1BB for intracellular signaling. Upon either dual or single CAR antigen stimulation, only the dual antigen condition led to optimal CAR T tumor killing and proliferation, a benefit for tumor specificity and reduced off-target toxicity [42]. Interestingly, dual CARs have also shed light on the role of co-stimulation and spatial relationships between stimulatory molecules within CAR signaling. It was proposed that the stoichiometry of second-generation CD28 CARs forces an artificially high 1:1 CD28:CD3ζ interaction, pushing T cells towards a highly differentiated, less persistent fate [43]. Subsequently, dual CARs in which CD3ζ and/or costimulatory molecules CD28 and 4-1BB are activated in trans have been shown to enhance CAR T metabolic fitness by rapidly inducing effector-associated glycolysis while preserving oxidative functions, promote superior proliferation, and boost CAR T in vivo cytotoxicity and persistence [44,45,46]. Similar outcomes were observed when a second-generation CD28 CAR was co-expressed with 4-1BBL [47].
For a dual antigen context, various double CAR platforms are also being tested, including infusion of two mono-specific CARs, expression of two CARs on the same T cell (dual CARs), and tandem/bispecific CARs, with antigen specificity against two antigens within the same CAR molecule. Fernandez de Larrea et al. found that in the setting of one antigen, dual CARs could not induce the same degree of activation as mono-specific or tandem CARs, but with dual antigen expression, they were superior in vivo, likely due to increased avidity [48]. On the other hand, it has been shown that the increased genetic payload of dual CARs can lead to lower CAR protein expression, resulting in worse in vivo outcomes compared to lower payload tandem CARs [49].
In addition to dual CARs, dual TCR T cells and their role in antitumor immunity are currently an exciting area of research. The presence of T cells expressing two Vα chains, thus two separate, functional TCRs, was shown in the early 1990s. These dual TCR T cells were found to comprise up to a third of the mature T cell population post-TCR rearrangement [50]. Since then, these dual TCR T cells have been implicated in autoimmunity and anti-tumor immunity [51,52,53]. In the setting of an immunogenic tumor, dual TCR T cells disproportionately infiltrate the tumor and are more activated (CD44 + , OX40 + ) than their single TCR counterparts [54]. Single-cell characterization of these dual TCR T cells uncovered a more migratory, CXCL13 + , anti-tumor phenotype [55]. A proposed explanation for this favorable phenotype is the decrease in TCR avidity of each expressed TCR, preventing overactivation and subsequent exhaustion, whilst the total number of TCRs expressed on a T cell remains the same [56]. Dual TCRs are also more reactive against self-like neoantigens through escape of the secondary TCR from central and peripheral tolerance. This both contributes to pathogenesis in autoimmune disease and poses a potential benefit in expanding tumor-reactive T cells against self-like tumor antigens [52].
A case for leaving the CAR T cell TCR behind
To date, commercially available CAR T products use autologous T cells, which must be manufactured for each individual patient. The CAR T manufacturing process takes approximately 14 days, is costly, and outcomes can be variable due to differences in the fitness of patient lymphocytes [57]. Allogeneic CAR T cell transfer has been attempted, with some favorable outcomes. A clinical trial enrolling patients with persistent CD19 + B cell malignancies post allo-hematopoietic stem cell transfer (alloHSCT), and at least one donor lymphocyte infusion, demonstrated tumor regression in 3/10 patients and no observed GvHD with allogeneic anti-CD19 second-generation CARs. However, these patients were not lymphodepleted out of fear of GvHD and experienced precipitous declines in blood CAR levels to become undetectable by 30 days post-infusion [58]. A similar trial reported remission in 8/20 patients (six complete remissions and two partial remissions). These patients also did not undergo lymphodepletion prior to CAR infusion and experienced limited CAR persistence in the blood of less than four weeks [59]. A separate investigation in mice proposed compounded signaling through both the alloreactive TCR and anti-CD19 CAR as a mechanism for avoiding GvHD by promoting T cell exhaustion, diminished function, and clonal depletion [60]. Thus, given the seeming trade-off between GvHD and CAR persistence with allogeneic transfer, the benefits of TCR knockout in CAR T cells are still debatable.
TCR knockout has the potential to offer “off-the-shelf” CAR T cell therapy by minimizing the risk of GvHD, while using healthy donor T cells for the starting material. These healthy donor T cells are likely to be more robust than those of patients with malignancies, and this also decreases the risk of inadvertently transducing a malignant cell with activating receptors [61]. In addition, Yang et al. demonstrated a detrimental effect of endogenous TCR engagement on CAR T function. Engagement of the TCR in CD8, but not CD4, CAR T abrogated anti-tumor effects and reduced CD8 CAR T numbers with upregulated exhaustion and apoptotic markers [62]. Thus, several mechanisms exist to knock out or interrupt endogenous TCR expression. Zinc-finger nucleases, transcription activator-like effector nucleases (TALENs), and CRISPR-Cas9 have all been employed for TCR knockout [14, 18, 63]. These mechanisms are not 100% efficient, and even magnetic bead separation to purify TCRα/β- cells may not purify the final product enough to avoid GvHD; therefore, protocols such as anti-CD3 CAR NK elimination of TCR/CD3 + T cells prior to CAR transduction have been proposed [64].
In terms of efficacy, two studies using Sleeping Beauty transposon delivery of anti-CD19 CAR in the setting of TCR knockout confirmed CAR signaling and cytolytic activity similar to TCR wildtype CAR while avoiding GvHD in a murine allogeneic setting [14, 65]. In the context of CD7 + T cell malignancies where CAR T fratricide is a challenge, CRISPR-Cas9 deletion of both the TRAC and CD7 genes was used to generate "off-the-shelf” allogeneic CAR T resistant to fratricide [13]. To pursue further universality, additional genes can be targeted simultaneously with gene editing - for example, to knock out the TCR, β-2 microglobulin, and PD-1 from CAR T products [66]. In 2017, two infants were treated with TALEN-mediated TCR knockout anti-CD19 CAR for B cell acute lymphoblastic leukemia (B-ALL). Both achieved molecular remission within 28 days, with one participant experiencing GvHD of the skin and bone marrow, potentially due to infusion of remnant TCR + CAR T [18]. The same group then conducted a phase 1 trial of TCR/CD52 knockout CAR T for pediatric B-ALL patients, using healthy donor PMBCs and next-generation CRISPR-Cas9 encoded in a self-inactivating lentiviral vector to knock out simultaneously the TRAC and CD52 genes and insert an anti-CD19 CAR (refer to Table 1 for clinical trial references). CD52 knockout was included to provide a CAR T cell survival advantage in the presence of alemtuzumab-mediated lymphodepletion. CAR infusion in this case was intended to serve as a “bridge-to-transplant”, with the hope of achieving remission prior to allogeneic stem cell transfer. Of eight patients, one experienced grade IV neurotoxicity, post tocilizumab infusion for grade II cytokine release syndrome. Four patients achieved morphological complete remission upon bone marrow analysis, and for all patients, CARs became undetectable by day 28 [67]. The multinational CALM Phase 1 trial reported 3/25 patients with dose-limiting toxicities, with 6/25 experiencing grade 3 or higher neurotoxicity and 7/25 experiencing grade 3 or higher infection testing TALEN-mediated TRAC and CD52 knockout anti-CD19 CAR T cells for patients with B-ALL [68]. A separate clinical trial evaluating the safety of TRAC and SPPL3 knockout anti-CD19 CAR T cells for B-NHL reported grade 3 or higher cytokine release syndrome in 3/9 patients, with 2/9 experiencing grade 3 neurotoxicity. SPPL3 knockout was intended to alter the T cell glycan profile and reduce TCR alloreactivity. Interestingly, this trial observed expansion of CD3 + TCR + CAR T cells, which aligned with peripheral B cell depletion. These TCR-sufficient CAR T cells managed to expand over 100-fold and persist in the blood despite their low initial prevalence in the infusion product, constituting the majority of the total CAR T cells in the peripheral blood after day 10 [69]. This unexpected finding suggests a critical role for the TCR in CAR T cell persistence over time.
Another approach to knocking out the endogenous TCR while expressing a CAR is to insert the CAR transgene into the TRAC locus. A study using homing endonucleases and an AAV6 vector to insert an anti-CD19 CAR into the TRAC locus achieved concurrent TCR/CD3 knockout, stable CAR expression within the TRAC locus, potent in vitro and in vivo activity against CD19+ tumors, and preserved T cell proliferation with CAR antigen [70]. This technique may provide additional benefit in further recapitulating native TCR behavior within the CAR. Another study combining CRISPR-Cas9 TCR knockout with AAV CAR gene delivery found that CAR insertion into the TRAC locus resulted in more uniform CAR expression, increased CAR T potency against murine leukemia models, and delayed CAR T exhaustion through reduction in CAR tonic signaling and efficient cycling of CAR surface expression versus retrovirally transduced CAR [17]. This protection from exhaustion has been observed more broadly with TRAC-targeted CAR integration, likely from decreased tonic signaling, which has also been correlated with preservation of a memory-like phenotype that can be enhanced under altered nutrient and cytokine conditions [71, 72]. In 2020, Fate Therapeutics initiated a phase 1 clinical trial testing an iPSC-derived TRAC-integrated anti-CD19 CAR T in patients with relapsed/refractory (r/r) BCL, CLL, and B-ALL. Among 12 patients receiving either a two-dose (90e6, 180e6 cells) or three-dose (30e6 cells each) regimen, no dose-limiting toxicities, neurotoxicity symptoms, GvHD, or grade ≥3 serious adverse events were observed [73, 74]. Other groups have also used CRISPR-Cas systems for TRAC integration of the CAR, in combination with β-2 microglobulin and/or PD-1 disruption for improved resilience, with Caribou Biosciences recently reporting comparable clinical performance of their allogeneic anti-CD19 CAR T product with standard autologous CAR T therapies for patients with r/r B-NHL in their ANTLER trial [75,76,77].
In summary, allogeneic, “off-the-shelf” CAR products have the exciting potential to reduce the cost of CAR T therapies, generate CAR T cells from healthy PBMC donors, and improve the availability of CAR therapies. Both allogeneic CAR T transfer and TCR knockout CAR demonstrate efficacy in patient trials, but with poor CAR persistence seen in the disappearance of peripheral CAR T cells within 30 days. Insertion of the CAR into the TRAC locus could provide benefit over traditional virally transduced CAR T cells, by appropriating endogenous TCR biology for the CAR to minimize tonic signaling, prevent exhaustion, and promote memory phenotypes - through more physiologic regulation of CAR expression - which are sought after in CAR T products [78]. These TRAC-targeted CAR also appear to be safe in phase 1 clinical trials, although more patient efficacy data is still needed. Despite initial safety and efficacy, loss of the endogenous TCR raises some concerns for CAR T persistence, especially in the absence of CAR antigen, and could result in the loss of a tool for reinvigorating transferred CAR T over time. There is also evidence that continued “tickling” of the TCR is important for T cell homeostasis and persistence, a phenomenon that would be lost with TCR knockout [79, 80]. While this “tickling” could be replaced by continued production of CD19 + B cells in the case of anti-CD19 CAR T, this would be harder to replicate in other contexts, particularly against solid tumors.
A case for keeping the CAR T cell TCR
One of the main predictors of CAR T cell efficacy in maintaining remission and relapse-free survival is CAR T persistence over time. Shiqi et al. correlated CAR T persistence of ≥3 or 6 months with significantly improved relapse-free survival in a cohort of 50 patients with CAR-responsive r/r ALL [81]. For a cohort of 30 patients with ALL treated with anti-CD19 CAR T at the University of Pennsylvania, it was concluded that limited CAR T persistence was unlikely to produce long-term remission without subsequent stem cell transplantation [82]. A potential drawback to TCR knockout in CAR T cells is restriction of persistence resulting from a loss of activating and proliferative signals typically provided by the endogenous TCR. In a murine system with tetracycline-based control of TCRα, stripping T cells of TCR expression led to loss of the CD8 compartment within 19 days [83]. As a therapy, antigen-specific cytotoxic T lymphocytes (CTLs) have been used in patients to combat EBV reactivation, with persistence ranging from 10 weeks to 9 years post-infusion in the context of viral clearance within two to four weeks [84,85,86,87]. The promise of using the endogenous TCR as a tool for augmenting CAR T survival and expansion appears especially promising given the 2023 study from the Masopust group, demonstrating sustained proliferation of T cells over 10 years through multiple generations of mice with repeated TCR stimulation through vaccination [88]. Additionally, Kondo et al., using an anti-CD19 OT-1 CAR T cell model, found that TCR activation with a strong antigen can potentiate CAR function against a separate antigen, augmenting both CAR T cell therapy and cytotoxicity against CD19-expressing targets [89].
A single-cell gene analysis of T cell clonotypes pre- and post-CD19 CAR T infusion in pediatric patients demonstrated moderate clonal expansion of specific pre-infusion clones, with expansion associated with increased granzyme B transcript abundance [90]. A study in adult patients reported contraction of TCRβ clonal diversity within anti-CD19 CAR T cells post-infusion, with clonal expansion primarily observed in infusion clusters higher in cytotoxicity and proliferation genes [91]. Thus, despite the presence of a CAR, the TCR on T cells remains a primary mediator of expansion and persistence, in addition to enhancing cytotoxicity-associated markers. To capitalize on a potential correlation of TCR clonotype with CAR T function and the ability of TCR stimulation repeatedly to induce T cell proliferation, several groups have attempted to employ the endogenous TCR to improve CAR T function. One approach by Kershaw et al. was to generate CAR T cells with alloreactive TCRs. Upon allogeneic cell immunization, these dual-specific CAR T cells expanded consistently by about 10% and exerted improved solid tumor control over non-immunized mice [92]. An alternative approach is to utilize a TCR against a viral or tissue antigen to drive targeted CAR T expansion. A common method of isolating anti-viral TCRs involves stimulation of donor PBMCs with antigenic peptides and either using the resulting enriched population or selecting cytokine-responsive T cells. In three separate studies transducing enriched anti-viral (EBV, CMV, or VZV) T cells with a CAR, CAR T cells reliably expanded with TCR stimulation, even after chronic CAR stimulation, but exhibited variable anti-tumor efficacy [15, 16, 93]. Against EBV-transformed lymphoblastoid cell lines, anti-CD19 CAR enriched for CMV TCR reactivity and boosted with pp65 peptide-loaded and irradiated T cells significantly restricted in vivo tumor growth compared to non-boosted CMV-reactive anti-CD19 CAR T [15]. On the other hand, VZV-reactive anti-GD2 CAR T appeared to lose CAR T function in vitro after one round of co-culture with neuroblastoma tumor cells. Stimulation of the TCR, though, with VZV pepmix-loaded DCs was able to restore CAR function upon subsequent rounds of tumor cell co-culture [16]. Similarly, Slaney et al. generated the CARaMEL mouse, with T cells encoding an anti-HER2 CAR and a TCR against gp100, a weak antigen expressed in normal human melanocytes. Following a regimen consisting of in vivo preconditioning with IL2 and live vaccina virus encoding gp100, CAReMEL T cells were highly effective against orthotopic breast cancer, subcutaneous sarcoma, and colon carcinoma in vivo. The CAR was required for anti-tumor effects, while vaccination resulted in CARaMEL expansion, activation, and tumor infiltration [94].
A 2007 phase 1 clinical trial applied allogeneic immunization of anti-folate receptor CAR T cells to the treatment of metastatic ovarian cancer. This allogeneic immunization consisted of the injection of the allogeneic PBMCs used to stimulate the CARs during ex vivo production. Patients who received allogeneic immunization reported mild side effects (grade 1 or 2), but unfortunately, experienced no reduction in tumor burden and limited CAR T tumor infiltration and persistence. The failure of allogeneic immunization to expand the transferred CAR T population was hypothesized to be due to suboptimal doses of allogeneic cells [95]. A later 2013 phase 1 trial instead used virus-specific T cells (VSTs) with reactivity to various CMV, EBV, and adenovirus peptides for anti-CD19 CAR generation. In patients with relapsed B-cell malignancies, allogeneic VST CAR T cells avoided GvHD and, in two patients with EBV reactivation, re-expanded in response to the virus. However, antitumor effects were modest, with objective responses in 2/6 patients. Of note, these patients did not receive pre-infusion lymphodepletion and the expansion process of the VSTs to obtain sufficient numbers was longer than typical CAR T timelines, being five to six weeks instead of two [96]. A separate Phase 1 trial administering anti-CD19 CAR T cells generated from allogeneic T cells reactive to EBV demonstrated safety and progression-free survival of 83% at 36 months, but limited CAR T cell persistence [97].
Finally, some evidence points to the presence of the endogenous TCR, in general, being important for CAR T persistence and the ability of the TCR to impart unique characteristics to CAR T cells upon priming. When comparing tumor control of Nalm6 tumors by anti-CD19 4-1BB second-generation CAR T cells, TCR beta chain (TRBC) knockout and wildtype showed comparable short-term efficacy, but TRBC wildtype CAR T persisted significantly longer in vivo, resulting in prolonged tumor control but with increased risk of GvHD [98]. This finding was also observed in patients with solid mesothelin+ (MSLN) tumors treated with double TRAC, PD-1 knockout anti-MSLN CD28 second-generation CAR T. Of the 15 patients treated, only two maintained stable disease (SD) over 8-12 weeks. This response rate was attributed to poor CAR persistence in the blood and tumor sites, but in the patients with SD, CD3 + CAR T accounted for the majority of circulating CAR T cells post-infusion (from <5% pre-infusion) [99]. This resurgence of TCR-competent CAR T cells from a TCR knockout product mirrors the findings of the TRAC/SPPL3 knockout CAR trial [69]. This suggests that the endogenous TCR, even without known specificity, may play a role in maintaining CAR T cell persistence. These endogenous TCRs of unknown specificity can also be engaged through stimuli, such as vaccination, to skew CAR T function and phenotype. Our group has shown that in vivo combination therapy against solid tumors using a third-generation anti-EGFRviii CAR and an oncolytic virus can generate anti-viral CAR T, clonally expanded from a naïve CAR T TCR pool. These anti-viral CAR T were more effector polarized with improved degranulation and effector cytokine production and were able to mediate tumor cures with an additional viral boost [100]. While the concept of boosting has also been investigated through the CAR itself, there is evidence that a durable cure is largely maintained through TCR engagement upon epitope spreading, especially in the setting of antigen loss [101,102,103].
Overall, retaining the CAR T cell TCR appears to improve CAR T cell persistence both for solid and liquid tumors, and preserves a potential tool for CAR restimulation and re-expansion. Given the reported importance of CAR T persistence for patient outcomes, the seemingly abridged lifespan of TCR knockout CAR T may not position them for optimal therapy, despite the benefits in terms of cost, ease of manufacturing, time to treatment, and other benefits to CAR T cell phenotype with TRAC integration [71, 81, 98]. TCR-wildtype CAR T cells also carry their own burdens, including the requirement for autologous transfer, unknown significance of TCR specificity, and the difficulties of enriching/selecting for certain TCRs [96]. However, in a situation where persistence is the name of the game, TCR retention may be critical.
Conclusion
Much remains unknown regarding CAR signaling, CAR interactions with the TCR, and how multiple activating receptors, through either one or more CARs or TCRs, may affect T cell function. Our current understanding of how CARs may interact with TCRs, other CARs, and how CARs may or may not need a TCR for optimal therapy remains patchy. Persuasive arguments exist on both sides for keeping or losing the CAR T cell TCR, and further pre-clinical and clinical studies will determine the best course of action, which may be different for different clinical situations. However, given the current challenges facing TCR knockout CAR T cells, despite their potential manufacturing benefits, and the undeniable role of the endogenous TCR in maintaining CAR T persistence and amplifying effector phenotypes, we propose capitalizing on the TCR as a powerful tool for augmenting CAR T therapy.
Method
Ethics approval and consent to participate
This article is a review of previously published studies and does not contain any original research involving human participants or animals performed by the authors. Therefore, ethics approval and informed consent were not required.
References
Goyco Vera D, Waghela H, Nuh M, Pan J, Lulla P. Approved CAR-T therapies have reproducible efficacy and safety in clinical practice. Hum Vaccin Immunother. 2024;20:2378543.
Ghilardi G, Fraietta JA, Gerson JN, Van Deerlin VM, Morrissette JJD, Caponetti GC, et al. T cell lymphoma and secondary primary malignancy risk after commercial CAR T cell therapy. Nat Med. 2024;30:984–9.
Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98.
Li Y, Li S, Zhao X, Sheng J, Xue L, Schett G, et al. Fourth-generation chimeric antigen receptor T-cell therapy is tolerable and efficacious in treatment-resistant rheumatoid arthritis. Cell Res. 2025;35:220–23.
Mackensen A, Muller F, Mougiakakos D, Boltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022;28:2124–32.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69.
Ayala Ceja M, Khericha M, Harris CM, Puig-Saus C, Chen YY. CAR-T cell manufacturing: major process parameters and next-generation strategies. J Exp Med. 2024;221:e20230903.
Jadlowsky JK, Hexner EO, Marshall A, Grupp SA, Frey NV, Riley JL, et al. Long-term safety of lentiviral or gammaretroviral gene-modified T cell therapies. Nat Med. 2025;31:1134–44.
Li YR, Shen X, Zhu E, Li Z, Huang J, Le T, et al. Targeting orthotopic and metastatic pancreatic cancer with allogeneic stem cell-engineered mesothelin-redirected CAR-NKT cells. Proc Natl Acad Sci USA. 2025;122:e2517786122.
Wang W, Liu Y, He Z, Li L, Liu S, Jiang M, et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024;10:40.
Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023;20:359–71.
Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost- effectiveness of chimeric antigen receptor T-Cell therapy in multiply relapsed or refractory adult large B-Cell lymphoma. J Clin Oncol. 2019;37:2105–19.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–83.
Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell-based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705.
Wang X, Wong CW, Urak R, Mardiros A, Budde LE, Chang WC, et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin Cancer Res. 2015;21:2993–3002.
Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res. 2017;23:3499–509.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9:eaaj2013.
McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. 2019;97:664–74.
Lopez-Cantillo G, Uruena C, Camacho BA, Ramirez-Segura C. CAR-T cell performance: how to improve their persistence? Front Immunol. 2022;13:878209.
Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J Exp Med. 1983;157:1149–69.
Meuer SC, Fitzgerald KA, Hussey RE, Hodgdon JC, Schlossman SF, Reinherz EL. Clonotypic structures involved in antigen-specific human T cell function. Relationship to the T3 molecular complex. J Exp Med. 1983;157:705–19.
Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, et al. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature. 2019;573:546–52.
Courtney AH, Lo WL, Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci. 2018;43:108–23.
Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18:485–97.
Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. 2020;6:eaaz3223.
Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11:eaat6753.
James JR. Tuning ITAM multiplicity on T cell receptors can control potency and selectivity to ligand density. Sci Signal. 2018;11:eaat6753.
Wu W, Zhou Q, Masubuchi T, Shi X, Li H, Xu X, et al. Multiple signaling roles of CD3epsilon and its application in CAR-T cell therapy. Cell. 2020;182:855–71.e23.
Gudipati V, Rydzek J, Doel-Perez I, Goncalves VDR, Scharf L, Konigsberger S, et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat Immunol. 2020;21:848–56.
Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. 2018;200:1088–100.
Anikeeva N, Panteleev S, Mazzanti NW, Terai M, Sato T, Sykulev Y. Efficient killing of tumor cells by CAR-T cells requires a greater number of engaged CARs than TCRs. J Biol Chem. 2021;297:101033.
Salter AI, Rajan A, Kennedy JJ, Ivey RG, Shelby SA, Leung I, et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci Signal. 2021;14:eabe2606.
Dong R, Libby KA, Blaeschke F, Fuchs W, Marson A, Vale RD, et al. Rewired signaling network in T cells expressing the chimeric antigen receptor (CAR). EMBO J. 2020;39:e104730.
Burton J, Siller-Farfan JA, Pettmann J, Salzer B, Kutuzov M, van der Merwe PA, et al. Inefficient exploitation of accessory receptors reduces the sensitivity of chimeric antigen receptors. Proc Natl Acad Sci USA. 2023;120:e2216352120.
Wu L, Brzostek J, Sankaran S, Wei Q, Yap J, Tan TYY, et al. Targeting CAR to the peptide-MHC complex reveals distinct signaling compared to that of TCR in a Jurkat T cell model. Cancers. 2021;13:867.
Wong WK, Leem J, Deane CM. Comparative analysis of the CDR loops of antigen receptors. Front Immunol. 2019;10:2454.
Beppler C, Eichorst J, Marchuk K, Cai E, Castellanos CA, Sriram V, et al. Hyperstabilization of T cell microvilli contacts by chimeric antigen receptors. J Cell Biol. 2023;222:e202205118.
Barden M, Holzinger A, Velas L, Mezosi-Csaplar M, Szoor A, Vereb G, et al. CAR and TCR form individual signaling synapses and do not cross-activate; however, they can cooperate in T cell activation. Front Immunol. 2023;14:1110482.
Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol. 2021;12:639818.
Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: elements and their synergistic function. EBioMedicine. 2020;58:102931.
Zhang E, Yang P, Gu J, Wu H, Chi X, Liu C, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11:102.
Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25:82–8.
Hirabayashi K, Du H, Xu Y, Shou P, Zhou X, Fuca G, et al. Dual targeting CAR-T cells with optimal costimulation and metabolic fitness enhances antitumor activity and prevents escape in solid tumors. Nat Cancer. 2021;2:904–18.
Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B-cell non-Hodgkin’s lymphomas. Mol Ther. 2018;26:2727–37.
Chen C, Li K, Jiang H, Song F, Gao H, Pan X, et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol Immunother. 2017;66:475–89.
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–28.
Fernandez de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, et al. Defining an optimal dual-targeted Car T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020;1:146–54.
Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat Commun. 2020;11:2283.
Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor alpha chains: dual receptor T cells. Science. 1993;262:422–4.
Schuldt NJ, Auger JL, Spanier JA, Martinov T, Breed ER, Fife BT, et al. Cutting edge: dual TCRalpha expression poses an autoimmune hazard by limiting regulatory T cell generation. J Immunol. 2017;199:33–8.
Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol. 2010;11:628–34.
Ni PP, Solomon B, Hsieh CS, Allen PM, Morris GP. The ability to rearrange dual TCRs enhances positive selection, leading to increased Allo- and autoreactive T cell repertoires. J Immunol. 2014;193:1778–86.
Jang HJ, Caron C, Lee CK, Wang L, Jama B, Bui JD, et al. Dual receptor T cells mediate effective antitumor immune responses via increased recognition of tumor antigens. J Immunother Cancer. 2023;11:e006472.
Peng Q, Xu Y, Yao X. scRNA+ TCR-seq revealed dual TCR T cells antitumor response in the TME of NSCLC. J Immunother Cancer. 2024;12:e009376.
Muhowski EM, Rogers LM. Dual TCR-expressing T cells in cancer: how single-cell technologies enable new investigation. Immunohorizons. 2023;7:299–306.
Blache U, Popp G, Dunkel A, Koehl U, Fricke S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun. 2022;13:5225.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–39.
Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016;34:1112–21.
Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23:242–9.
Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24:1499–503.
Yang Y, Kohler ME, Chien CD, Sauter CT, Jacoby E, Yan C, et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. 2017;9:eaag1209.
Kamali E, Rahbarizadeh F, Hojati Z, Frodin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21:9.
Kath J, Du W, Martini S, Elsallab M, Franke C, Hartmann L, et al. CAR NK-92 cell-mediated depletion of residual TCR+ cells for ultrapure allogeneic TCR-deleted CAR T-cell products. Blood Adv. 2023;7:4124–34.
Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther. 2022;30:3155–75.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27:154–7.
Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14:eabq3010.
Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol. 2022;9:e833–e43.
Wu Z, Shi J, Lamao Q, Qiu Y, Yang J, Liu Y. et al. Glycan shielding enables TCR-sufficient allogeneic CAR-T therapy. Cell. 2025;188:6317–34.e21.
MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther. 2017;25:949–61.
Mueller KP, Piscopo NJ, Forsberg MH, Saraspe LA, Das A, Russell B, et al. Production and characterization of virus-free, CRISPR-CAR T cells capable of inducing solid tumor regression. J Immunother Cancer. 2022;10:e004446.
Cappabianca D, Pham D, Forsberg MH, Bugel M, Tommasi A, Lauer A, et al. Metabolic priming of GD2 TRAC-CAR T cells during manufacturing promotes memory phenotypes while enhancing persistence. Mol Ther Methods Clin Dev. 2024;32:101249.
Chang C, Van Der Stegen S, Mili M, Clarke R, Lai Y-S, Witty A, et al. FT819: translation of off-the-shelf TCR-less trac-1XX CAR-T cells in support of first-of-kind phase I clinical trial. Blood. 2019;134:4434.
Mehta A, Farooq U, Chen A, McGuirk JP, Ly T, Wong L, et al. Interim phase I clinical data of FT819-101, a study of the first-ever, off-the-shelf, iPSC-derived TCR-less CD19 CAR T-cell therapy for patients with relapsed/refractory B-cell malignancies. Blood. 2022;140:4577–8.
Ghobadi A, McGuirk JP, Shaughnessy P, Tam CS, Allen M, Pan C, et al. CTX112, a next-generation allogeneic CRISPR-Cas9 engineered CD19 CAR T cell with novel potency edits: data from phase 1 dose escalation study in patients with relapsed or refractory B-cell malignancies. Blood. 2024;144:4829.
Daver NG, Maiti A, Sallman DA, Roboz GJ, Solh MM, Milano F, et al. A first-in-human phase 1, multicenter, open-label study of CB-012, a next-generation CRISPR-edited allogeneic anti-CLL-1 CAR-T cell therapy for adults with relapsed/refractory acute myeloid leukemia (AMpLify). J Clin Oncol. 2024;42:TPS6586.
Berdeja JG, Martin TG, Rossi A, Essell JH, Siegel DSD, Mailankody S, et al. A first-in-human phase 1, multicenter, open-label study of CB-011, a next-generation CRISPR-genome edited allogeneic anti-BCMA immune-cloaked CAR-T cell therapy, in patients with relapsed/refractory multiple myeloma (CAMMOUFLAGE trial). J Clin Oncol. 2023;41:TPS8063-TPS.
Tantalo DG, Oliver AJ, von Scheidt B, Harrison AJ, Mueller SN, Kershaw MH, et al. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J Immunother Cancer. 2021;9:e002555.
Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol. 2004;172:40–4.
Hao Y, Legrand N, Freitas AA. The clone size of peripheral CD8 T cells is regulated by TCR promiscuity. J Exp Med. 2006;203:1643–9.
Shiqi L, Jiasi Z, Lvzhe C, Huailong X, Liping H, Lin L, et al. Durable remission related to CAR-T persistence in R/R B-ALL and long-term persistence potential of prime CAR-T. Mol Ther Oncolytics. 2023;29:107–17.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Labrecque N, Whitfield LS, Obst R, Waltzinger C, Benoist C, Mathis D. How much TCR does a T cell need? Immunity. 2001;15:71–82.
Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13.
Roskrow MA, Suzuki N, Gan Y, Sixbey JW, Ng CY, Kimbrough S, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin’s disease. Blood. 1998;91:2925–34.
Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–5.
Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–35.
Soerens AG, Kunzli M, Quarnstrom CF, Scott MC, Swanson L, Locquiao JJ, et al. Functional T cells are capable of supernumerary cell division and longevity. Nature. 2023;614:762–6.
Kondo T, Bourassa FXP, Achar S, DuSold J, Cespedes PF, Ando M, et al. Engineering TCR-controlled fuzzy logic into CAR T cells enhances therapeutic specificity. Cell. 2025;188:2372–89.e35.
Wilson TL, Kim H, Chou CH, Langfitt D, Mettelman RC, Minervina AA, et al. Common trajectories of highly effective CD19-specific CAR T cells identified by endogenous T-cell receptor lineages. Cancer Discov. 2022;12:2098–119.
Sheih A, Voillet V, Hanafi LA, DeBerg HA, Yajima M, Hawkins R, et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat Commun. 2020;11:219.
Kershaw MH, Westwood JA, Hwu P. Dual-specific T cells combine proliferation and antitumor activity. Nat Biotechnol. 2002;20:1221–7.
Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–16.
Slaney CY, von Scheidt B, Davenport AJ, Beavis PA, Westwood JA, Mardiana S, et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res. 2017;23:2478–90.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15.
Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–73.
Shahid S, Prockop SE, Flynn GC, Mauguen A, White CO, Bieler J, et al. Allogeneic off-the-shelf CAR T-cell therapy for relapsed or refractory B-cell malignancies. Blood Adv. 2025;9:1644–57.
Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136:1407–18.
Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18:2188–98.
Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med. 2022;14:eabn2231.
Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–8.
Ma L, Hostetler A, Morgan DM, Maiorino L, Sulkaj I, Whittaker CA, et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell. 2023;186:3148–65.e20.
Rakhshandehroo T, Mantri SR, Moravej H, Louis BBV, Salehi Farid A, Munaretto L. A CAR enhancer increases the activity and persistence of CAR T cells. Nat Biotechnol. 2024;43:948–59.
Acknowledgements
We would like to thank Toni L. Woltman for valuable secretarial assistance.
Funding
Funding was provided by the Joan Reece Chair of Immuno-oncology, King’s College, London; the National Institutes of Health R01 AI170535 (RV); R01 CA269384 (RV); P50 CA210964-05 (RV), CA210964-06A1 P3 (RV), The Richard M. Schulze Family Foundation (RV), the Mayo Foundation (RV), the Shannon O’Hara Foundation (RV), and Hyundai Hope on Wheels (RV). OL was supported by the Mayo Clinic MSTP T32 GM145408.
Author information
Authors and Affiliations
Contributions
OL and RV wrote and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liseth, O., Vile, R. The TCR in CAR T cell therapy: use it or lose it?. Cancer Gene Ther (2026). https://doi.org/10.1038/s41417-026-01018-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41417-026-01018-7
