
Abstract
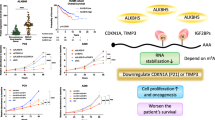
Cellular senescence is the major hallmark and therapeutic target of aging and age-related diseases. The role of ALKBH5, one of the main m6A demethylases, in cellular senescence emerges however remains contentious. Herein, we show the reversible ALKBH5 aggregation in cytoplasm promotes cellular senescence. Mechanically, ALKBH5 aggregation causes cytosolic retention, resulting in the m6A dysregulation and m6A hypermethylation of Cdk2, which promotes Cdk2 RNA instability to drive senescence. In addition, m6A imbalance aggravates ALKBH5 cytosolic aggregation in a feedback loop. We further demonstrate that ALKBH5 nuclear translocation required the formation of ALKBH5 droplet phase via binding Nucleoporin p62 (Nup62), while the aggregation of ALKBH5 traps with Nup62 in the cytoplasm. Reduced Nup62 prevents ALKBH5 nuclear entry leading to cellular senescence. Importantly, administration of m6A labeled RNA efficiently reverses ALKBH5 cytosolic aggregates and restores its nuclear entry to alleviate cellular senescence. Forced nuclear entry by NLS-ALKBH5 can prevent senescence in vitro and in vivo. Taken together, these findings unravel a novel paradigm for m6A epigenetic regulation in cellular senescence and offer promising therapeutic targets and strategies for the intervention of aging and age-associated diseases.

This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
All sequencing data generated for this paper have been deposited at NCBI’s Gene Expression Omnibus under accession number GSE264231.
References
Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–13.
Cai Y, Song W, Li J, Jing Y, Liang C, Zhang L, et al. The landscape of aging. Sci China Life Sci. 2022;65:2354–454.
Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28:1556–68.
McMahon M, Forester C, Buffenstein R. Aging through an epitranscriptomic lens. Nat Aging. 2021;1:335–46.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Wu Z, Shi Y, Lu M, Song M, Yu Z, Wang J, et al. METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res. 2020;48:11083–96.
Ye G, Li J, Yu W, Xie Z, Zheng G, Liu W, et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp Mol Med. 2023;55:1743–56.
Wu Z, Lu M, Liu D, Shi Y, Ren J, Wang S, et al. m6A epitranscriptomic regulation of tissue homeostasis during primate aging. Nat Aging. 2023;3:705–21.
Castro-Hernández R, Berulava T, Metelova M, Epple R, Centeno TP, Richter J, et al. Conserved reduction of m6A RNA modifications during aging and neurodegeneration is linked to changes in synaptic transcripts. Proc Natl Acad Sci USA. 2023;120:e2204933120.
Shafik AM, Zhang F, Guo Z, Dai Q, Pajdzik K, Li Y, et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 2021;22:17.
Wang J, Wang J, Gu Q, Ma Y, Yang Y, Zhu J, et al. The biological function of m6A demethylase ALKBH5 and its role in human disease. Cancer Cell Int. 2020;20:347.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 Is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 2021;49:5779–97.
Gao X, Liang X, Liu B, Hong Y, He H, Shen Y, et al. Downregulation of ALKBH5 rejuvenates aged human mesenchymal stem cells and enhances their therapeutic efficacy in myocardial infarction. FASEB J. 2023;37:e23294.
Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17:e12840.
Bie J, Li R, Li Y, Song C, Chen Z, Zhang T, et al. PKM2 aggregation drives metabolism reprograming during aging process. Nat Commun. 2024;15:5761.
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–77.
Chen H-X, Zhang Z, Ma D-Z, Chen L-Q, Luo G-Z. Mapping single-nucleotide m6A by m6A-REF-seq. Methods. 2021;203:392–8.
Zhang Z, Chen LQ, Zhao YL, Yang CG, Roundtree IA, Zhang Z, et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5:eaax0250.
Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98.
Qin X, Long Y, Bai X, Cao L, Yan H, Zhang K et al. The disordered C terminus of ALKBH5 promotes phase separation and paraspeckles assembly. J Biol Chem 2023;299:105071.
Lu J, Wu T, Zhang B, Liu S, Song W, Qiao J et al. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun Signal. 2021; 19. https://doi.org/10.1186/s12964-021-00741-y.
Fung HYJ, Fu SC, Chook YM. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. Elife. 2017;6:60.
Wang J, Pei G, Wang Y, Wu D, Liu X, Li G, et al. Phase separation of the nuclear pore complex facilitates selective nuclear transport to regulate plant defense against pathogen and pest invasion. Mol Plant. 2023;16:1016–30.
Louka A, Zacco E, Temussi PA, Tartaglia GG, Pastore A. RNA as the stone guest of protein aggregation. Nucleic Acids Res. 2020;48:11880–9.
Sun T, Zhang L, Feng J, Bao L, Wang J, Song Z, et al. Characterization of cellular senescence in doxorubicin-induced aging mice. Exp Gerontol. 2022;163:111800.
Xu C, Liu K, Tempel W, Demetriades M, Aik WS, Schofield CJ, et al. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289:17299–311.
D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–95.
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94:93–107.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61.
Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877.
Acknowledgements
We thank Dr. Ge Gao, Dr. Linsheng Wang, Dr. Deliang Zhu and Dr. Zhihong Chen for their assistance in this project. This work was supported by National Natural Science Foundation of China (81821003, 82273180 and 32300617), Guangdong Basic and Applied Basic Research Foundation (2024A1515011365, 2021B1515130004), Science and Technology Projects in Guangzhou (2025A03J4509), China Postdoctoral Science Foundation (2023T160133), Guangdong Provincial People’s Hospital, High-level Hospital Construction Project (KJ012021074, KJ012019517).
Author information
Authors and Affiliations
Contributions
GJ, ZZ, X-WB and LC conceived the study. LC, ZC, JM, QX, WL, HZ, YC and YZ generated reagents and conducted experiment design and execution, data collection and data analysis. ZC and LC performed bioinformatics analysis. GJ, ZZ, X-WB, and LC wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All procedures were performed in compliance with the relevant guidelines and regulations, as approved by the Ethics Review Committee of Guangdong Provincial People’s Hospital (KY2024-450-02).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, L., Chen, Z., Mo, J. et al. Reversible ALKBH5 cytosolic aggregation accelerates cellular senescence. Cell Death Differ 33, 171–187 (2026). https://doi.org/10.1038/s41418-025-01560-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41418-025-01560-1
