
Abstract
Acute kidney injury (AKI) caused by nephrotoxins, ischemia reperfusion (IR) or sepsis is associated with high morbidity and mortality. Unveiling new mechanisms underlying AKI can help develop new therapeutic strategy. Cullin 4B (CUL4B) is a scaffold protein in the CUL4B-RING E3 ubiquitin ligase (CRL4B) complex. Here, we demonstrate that CUL4B can protect kidneys from acute injury induced by cisplatin and IR. CUL4B is upregulated in mouse tubular epithelial cells (TECs) after cisplatin treatment or IR. Loss of CUL4B in kidneys exacerbates renal injury, inflammation, and apoptosis of TECs caused by cisplatin and IR. Transcriptome analysis reveals that Cul4b deficiency enhances injury-induced PAI-1 expression. CUL4B suppresses PAI-1 expression by promoting polyubiquitination and degradation of p53. Inhibition of either PAI-1 or p53 can prevent the aggravated renal injury and inflammation caused by loss of CUL4B. Our work has identified the kidney-protective role of CUL4B against acute injury.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI), defined by an abrupt decrease in kidney function occurring over 7 days or fewer, affects about 20% of hospitalized patients [1]. The common causes of AKI include exposure to nephrotoxins, sepsis, major surgery, renal hypoperfusion, etc. Patients with AKI usually exhibit increased serum creatinine, reduced urinary output, death of tubular epithelial cells (TECs) and inflammation [2]. Currently, therapies for AKI are very limited, making high mortality and poor prognosis of AKI patients. Identifying new regulatory molecules and therapeutic targets for AKI is in urgent need.
The Cullin-RING E3 ubiquitin ligase (CRL) complexes constitute the largest superfamily of E3 ligases [3]. Besides mediating polyubiquitination of protein substrates to direct their degradation, the CRL4B complexes, which use CUL4B as the scaffold protein, can also repress gene transcription by catalyzing monoubiquitination of H2AK119 [4, 5]. CUL4B has been shown to regulate a variety of developmental processes such as embryogenesis, spermatogenesis, osteogenesis, adipogenesis, neurogenesis via assembling the CRL4B complexes [6,7,8,9,10,11,12]. In addition, CUL4B plays important roles in diseases. Mutation in CUL4B gene is one of the top causes of human X-linked intellectual disability [13, 14]. In many types of cancer, CUL4B functions as an oncogene to promote cancer growth, metastasis and drug resistance [15]. Myeloid depletion of CUL4B exacerbates lipopolysaccharide (LPS)-induced peritonitis and septic shock [16, 17]. Recently, our group has reported that loss of CUL4B in macrophages ameliorates renal injury and fibrosis in diabetic mice by reducing macrophage infiltration [18]. However, whether CUL4B in kidney parenchymal cells participates in regulation of acute or chronic kidney injury is not clear.
Here, we show that CUL4B is upregulated in TECs in kidneys from both mice and patients with AKI. By generating mice with Cul4b deleted in kidneys, we demonstrate that CUL4B exerts a kidney-protective effect in response to cisplatin or IR through suppressing the p53-dependent PAI-1 expression.
Results
CUL4B is upregulated in kidneys with acute injury
CUL4B is expressed in both glomeruli and renal tubules (Supplementary Fig. S1). To investigate the role of CUL4B in AKI, we first examined the expression of CUL4B in the kidneys of mice after peritoneal injection of cisplatin at a dose of 20 mg/kg. The kidney injury was verified by the elevated levels of blood urea nitrogen (BUN) and creatinine in serum (Fig. 1A) and the increased expression of markers for tubular injury, KIM-1 and NGAL, as well as the tubular damage scoring (Supplementary Fig. S2A–C). We detected time-dependent upregulation of CUL4B in kidneys after cisplatin injection (Fig. 1B). Immunohistochemistry staining revealed that CUL4B was mainly elevated in the TECs (Fig. 1C). To figure out whether CUL4B upregulation is specific to cisplatin-induced injury, we adopted another commonly-used AKI model, ischemia reperfusion injury (IRI) (Fig. 1D, Supplementary Fig. S2D–F). CUL4B was gradually increased in kidneys from 24 to 72 h after IRI (Fig. 1E, F). Moreover, the level of CUL4B in the renal tubules from patients with acute tubular necrosis was much higher than that in the control samples (Fig. 1G). These data together suggest the involvement of CUL4B in AKI.

A The creatinine (Cr) and BUN levels in the serum collected from mice at 24, 48 or 72 h after peritoneal injection of 20 mg/kg cisplatin (Cis) or at 24 h after injection of the vehicle control (Con). N = 5. B Western blots showing the level of CUL4B protein in kidneys of the indicated mice. C Immunohistochemistry staining of CUL4B in kidneys of the indicated mice. The bar graph shows integrated density (intDen) relative to Con. 5 fields were imaged for each mouse and 3 mice were included in each group. Scale bar, 60 μm. D The creatinine (Cr) and BUN levels in the serum collected from mice at 24, 48 or 72 h after IRI or at 24 h after sham operation. N = 5. E Western blots showing the level of CUL4B protein in kidneys of the indicated mice. F Immunohistochemistry staining of CUL4B in kidneys of the indicated mice. The bar graph shows integrated density (intDen) relative to Sham. 5 fields were imaged for each mouse and 3 mice were included in each group. Scale bar, 60 μm. G Immunohistochemistry staining of CUL4B in kidneys from patients with acute renal failure (ARF) or acute nephritic syndrome (ANS) or the paracancerous tissues (Normal) obtained from individuals with kidney cancer and without other renal diseases who underwent nephrectomies. Scale bar, 60 μm. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Cul4b deficiency exacerbates AKI induced by cisplatin or ischemia reperfusion
To determine the function of CUL4B in AKI, we knocked out Cul4b in kidneys by crossing Pax8-Cre+/– mice with Cul4bflox/flox mice. Immunohistochemistry staining confirmed depletion of CUL4B in kidneys of Pax8-Cre+/– Cul4bflox/Y (Cul4bcKO) mice (Supplementary Fig. S3A). The histological morphology of Cul4bcKO kidneys was normal (Supplementary Fig. S3B). The body weight, kidney weight and serum creatinine and BUN of the eight-week Cul4bcKO mice were comparable to those of the age-matched controls (Cul4bflox/Y mice, designated as Cul4bCon) (Supplementary Fig. S3C). Seventy-two hours after cisplatin injection, Cul4bcKO mice exhibited significantly higher levels of serum creatinine and BUN than Cul4bCon mice (Fig. 2A). Loss of CUL4B enhanced cisplatin-induced tubular injury as demonstrated by the elevated expression of NGAL and KIM-1 (Fig. 2B, C) and the increased injury scores (Fig. 2D). Inflammation is an important step in AKI [19]. We found that the Cul4bcKO kidneys displayed upregulated expression of pro-inflammatory cytokines and chemokines (Fig. 2E) and elevated macrophage infiltration compared to the Cul4bcKO kidneys (Fig. 2F). The exacerbated renal injury and inflammation caused by Cul4b depletion was also observed in mice with IRI (Supplementary Fig. S4). These results indicate that CUL4B protects mice against AKI.

A The creatinine (Cr) and BUN levels in the serum collected from the Cul4bCon or Cul4bcKO mice at 72 h after injection of cisplatin (Cis) or the vehicle control (Con). N = 5. B Western blots of CUL4B and NGAL in the kidneys from the indicated mice. C The representative images of immunohistochemistry staining of KIM-1 and quantification of the percentage of KIM-1+ area in the indicated kidneys. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. D The representative images of Hematoxylin & Eosin (H & E) staining and the tubular damage scores of the kidneys from the indicated mice. 10 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. E The mRNA levels of the indicated inflammatory factors in the kidneys. N = 5. F The representative images of immunohistochemistry staining of the macrophage marker CD68 and quantification of the percentage of CD68+ area in kidneys. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. In all bar graphs, the grey bars represent Cul4bCon mice injected with vehicle control (Cul4bCon-Con); the white bars represent Cul4bcKO mice injected with vehicle control (Cul4bcKO-Con); the red bars represent Cul4bCon mice injected with cisplatin (Cul4bCon-Cis); the blue bars represent Cul4bcKO mice injected with cisplatin (Cul4bcKO-Cis). Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Loss of CUL4B renders TECs more susceptible to apoptosis
Next, we assessed the effect of Cul4b knockout on apoptosis in kidneys upon AKI. Without injury, both the Cul4bcKO and the Cul4bCon kidneys showed little apoptosis. After cisplatin treatment or IRI, the Cul4bcKO kidneys exhibited elevated TUNEL+ staining (Fig. 3A, B) and increased cleaved caspase-3 compared to the Cul4bCon kidneys (Fig. 3C, D). We further confirmed the role of CUL4B in apoptosis of TECs by knocking down CUL4B in the human proximal tubule epithelial cell line HK2 (Fig. 3E). RNA interference of CUL4B resulted in increased apoptosis in response to treatment with cisplatin or cobalt chloride (Fig. 3F, G). These data together suggest that CUL4B protects TECs from apoptosis.

A, B The representative images of TUNEL staining on kidneys and the quantification of the number of TUNEL+ cells in each field. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 85 μm. C, D Western blots showing the levels of cleaved caspase-3 in kidneys from the indicated groups. E Western blots showing the levels of CUL4B protein in HK2 cells expressing control shRNA (NS) or shRNA against CUL4B (shCUL4B). The representative images of Annexin V/7-AAD analysis of HK2 cells with or without CUL4B knockdown exposed to different doses of cisplatin (F) or cobalt chloride (G) and the quantification of the percentage of Annexin V+ cells. N = 3. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Cul4b deficiency enhances AKI through upregulating PAI-1 expression
To investigate the mechanism underlying CUL4B regulation of AKI, we performed RNA sequencing on kidneys from the Cul4bCon and Cul4bcKO mice with or without cisplatin treatment. Cisplatin treatment resulted in dramatic transcriptome changes in both Cul4bCon and Cul4bcKO mice (Supplementary Fig. S5A, B). Compared to the kidneys from the cisplatin-treated Cul4bCon mice, 561 genes were differentially expressed in the Cul4bcKO kidneys exposed to cisplatin (Supplementary Fig. S5B). Consistent with the elevated apoptosis and inflammation, genes upregulated in the cisplatin-treated Cul4bcKO group were enriched in GO terms related to apoptosis and inflammation (Supplementary Fig. S5C). Among the differentially expressed genes, Serpine1, which encodes plasminogen activator inhibitor-1 (PAI-1), drew our attention. PAI-1 is a member of the serine protease inhibitor family that can suppress urokinase-type plasminogen activator to inhibit fibrinolysis. PAI-1 is rarely expressed in healthy kidneys but has been reported upregulated in kidneys with acute or chronic injury [20]. RNA sequencing revealed strong upregulation of Serpine1 mRNA in kidneys from the cisplatin-treated Cul4bcKO mice compared to that in the cisplatin-treated Cul4bCon group, which was confirmed by RT-qPCR (Fig. 4A). Western blotting and immunohistochemistry staining demonstrated that PAI-1 protein was barely presented in the uninjured kidneys but induced in the TECs by cisplatin treatment. Loss of CUL4B dramatically elevated the injury-induced PAI-1 in kidneys (Fig. 4B, C). The enhanced PAI-1 expression by Cul4b deficiency was also observed in kidneys with IRI (Supplementary Fig. S6). To determine whether loss of CUL4B exacerbates AKI through upregulating PAI-1 expression, we evaluated the effect of the PAI-1 inhibitor tiplaxtinin (PAI-039) on the cisplatin-induced AKI in the Cul4bCon and the Cul4bcKO mice. Treatment with PAI-039 significantly ameliorated renal injury in the cisplatin-treated Cul4bcKO mice as demonstrated by the reduced BUN and creatinine levels in serum (Fig. 4D), the downregulated KIM1 protein (Fig. 4E) and decreased pathological injury scores (Fig. 4F). PAI-039 also reduced macrophage infiltration and expression of inflammatory factors in the cisplatin-treated Cul4bcKO group (Fig. 4G, H). We further evaluated the effect of PAI-1 inhibition on apoptosis. Inhibition of PAI-1 decreased the number of TUNEL+ cells in the kidneys from the cisplatin-treated Cul4bcKO mice (Fig. 4I). In addition, knocking down SERPINE1 markedly reduced the cisplatin-induced apoptosis in CUL4B knockdown cells (Fig. 4J). Taken together, these data suggest that CUL4B protects kidneys from cisplatin-induced renal injury, inflammation and apoptosis through suppressing PAI-1 expression.

The levels of Serpine1 mRNA (A) and PAI-1 protein (B) in kidneys from the indicated groups. N = 3 in (A). C Immunohistochemistry staining of PAI-1 in kidneys from the indicated groups. Scale bar, 60 μm. D The creatinine (Cr) and BUN levels in the serum collected from the Cul4bCon or Cul4bcKO mice at 72 h after injection of cisplatin (Cis) with or without pretreatment of PAI-039 (039), an inhibitor for PAI-1. N = 5. E Western blots showing the levels of CUL4B and KIM-1 in the indicated kidneys. F The representative images of H & E staining and the tubular damage scores of the kidneys from the indicated mice. 10 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. G The representative images of immunohistochemistry staining of the macrophage marker CD68 and quantification of the percentage of CD68+ area in kidneys. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. H The mRNA levels of the indicated inflammatory factors in the kidneys. N = 5. I The representative images of TUNEL staining on kidneys and the quantification of the number of TUNEL+ cells in each field. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 85 μm. In all bar graphs in (D)–(I), the bars from left to right represent Cul4bCon mice injected with cisplatin (Cul4bCon-Cis), Cul4bCon mice pretreated with PAI-039 before injection with cisplatin (Cul4bCon-Cis+039), Cul4bcKO mice injected with cisplatin (Cul4bcKO-Cis) and Cul4bcKO mice pretreated with PAI-039 before injection with cisplatin (Cul4bCon-Cis+039), respectively. J The representative images of Annexin V/7-AAD analysis of the cisplatin-treated HK2 cells expressing control shRNA (NS) or shRNA against CUL4B (shCUL4B) together with or without siRNA against SERPINE1 (siSERPINE1) and the quantification of the percentage of Annexin V+ cells. N = 3. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
CUL4B suppresses PAI-1 expression by promoting p53 degradation
The elevated Serpine1 mRNA in the cisplatin-injured Cul4bcKO kidneys indicates CUL4B regulates PAI-1 at the transcription level. p53 has been reported as a transcription activator for Serpine1 [21,22,23]. P53 is also a substrate for the CRL4B complex in cancer cells and fibroblasts [24,25,26]. To determine whether p53 mediates CUL4B suppression of PAI-1 in injured kidneys, we first assessed the level of p53 in kidneys after injury caused by cisplatin or IR. As reported in literature [27], p53 protein was upregulated in the injured kidneys. Importantly, the injured Cul4bcKO kidneys displayed dramatic elevation of p53 protein compared to the injured Cul4bCon kidneys (Fig. 5A, Supplementary Fig. S7A). Next, we treated the Cul4bCon and the Cul4bcKO mice with Pifithrin-α (PFT-α), which inhibits the transcription regulatory activity of p53, before cisplatin treatment or IR. Inhibition of p53 abolished the difference in the levels of Serpine1 mRNA and PAI-1 protein between the Cul4bCon and the Cul4bcKO kidneys (Fig. 5B, C, Supplementary Fig. S7B, C), suggesting that Cul4b knockout increased Serpine1 expression by upregulating p53. Moreover, inhibition of p53 prevented the exacerbated kidney injury (Fig. 5C–F, Supplementary Fig. S7C–F), inflammation (Fig. 5G, H, Supplementary Fig. S7G, H) and apoptosis (Fig. 5I & Supplementary Fig. S7I) in the Cul4bcKO mice after cisplatin treatment or IRI. To further confirm the role of p53 in CUL4B regulation of PAI-1, we interfered TP53 expression in HK2 cells with CUL4B knockdown. Knocking down TP53 suppressed both the increased SERPINE1 mRNA and the elevated apoptosis induced by reduced CUL4B (Supplementary Fig. S8). These results together indicate that CUL4B inhibits PAI-1 expression and ameliorates AKI by restraining p53 level.

A Western blots showing the level of p53 in the indicated kidneys. B The level of Serpine1 mRNA in kidneys from the cisplatin (Cis)-treated Cul4bCon or Cul4bcKO mice with or without pretreatment of PFT-α, an inhibitor for p53. N = 5. C Western blots showing the levels of PAI-1 and NGAL in the indicated kidneys. D The creatinine (Cr) and BUN levels in the serum collected from the indicated mice. N = 5. E The representative images of immunohistochemistry staining of KIM-1 in kidneys and the quantification of the percentage of KIM-1+ area. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. F The representative images of H & E staining and the tubular damage scores of the kidneys from the indicated mice. 10 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. G The representative images of immunohistochemistry staining of the macrophage marker CD68 and quantification of the percentage of CD68+ area in kidneys. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 60 μm. H The mRNA levels of the indicated inflammatory factors in the kidneys. N = 5. I The representative images of TUNEL staining on kidneys and the quantification of the number of TUNEL+ cells in each field. 5 fields were imaged for each mouse and 5 mice were included in each group. Scale bar, 85 μm. In all bar graphs, the bars from left to right represent Cul4bCon mice injected with cisplatin (Cul4bCon-Cis), Cul4bCon mice pretreated with PFT-α before injection with cisplatin (Cul4bCon-Cis+PFT-α), Cul4bcKO mice injected with cisplatin (Cul4bcKO-Cis) and Cul4bcKO mice pretreated with PFT-α before injection with cisplatin (Cul4bCon-Cis+PFT-α), respectively. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To figure out how CUL4B suppresses p53 in response to kidney injury, we first measured the mRNA level of Trp53 in the kidneys with or without injury. While cisplatin treatment induced upregulation of Trp53 mRNA, knockout of Cul4b showed no effect (Fig. 6A). In cisplatin-treated HK2 cells, we also observed elevation in p53 protein but not in mRNA after CUL4B knockdown (Fig. 6B, C). These data suggest that after kidney injury CUL4B regulates p53 at the post-transcriptional level. To determine whether the CRL4B complex mediates degradation of p53 in kidney cells, we first performed cycloheximide (CHX) chasing experiments to evaluate the effect of CUL4B knockdown on p53 degradation. We found that knocking down CUL4B in HK2 cells under cisplatin treatment resulted in stabilization of p53 (Fig. 6D). Next, through co-immunoprecipitation, we detected physical interaction between CUL4B, DDB1 and p53 in HK2 cells treated with cisplatin (Fig. 6E). Moreover, knocking down CUL4B reduced while overexpression of CUL4B increased polyubiquitination of p53 in the cisplatin-treated HK2 cells (Fig. 6F, G). To determine which lysine in p53 is ubiquitinated by CRL4B complex, we purified p53 from cells with CUL4B overexpressed and applied it to mass spectrometry analysis. Lysine 164 (K164) was identified as the major ubiquitination target residue of CRL4B (Supplementary Fig. S9). We then mutated K164 in p53 protein to arginine (R). Compared to wild type p53, the K164R mutant was more stable (Fig. 6H) and exhibited dramatically reduced polyubiquitination in CUL4B-overexpressing cells (Fig. 6I), suggesting that CRL4B regulates p53 stability mainly through ubiquitinating K164 in p53 protein. These data together support that CRL4B complex promotes p53 ubiquitination and degradation in injured kidney cells.

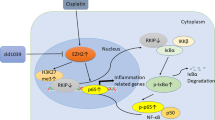
A The level of Trp53 mRNA in kidneys from the indicated mice. N = 5. B The level of TP53 mRNA in the indicated HK2 cells. N = 5. Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. ns: no significance. C Western blots showing the level of p53 protein in HK2 cells with CUL4B stably knocked down (shCUL4B) or control HK2 cells (NS) treated with 20 μg/ml cisplatin (20 μg/ml) or vehicle control (Con). D Western blots showing the level of p53 protein in the cisplatin-treated HK2 cells with or without CUL4B knocked down at different time points after treating with CHX and the quantification of the protein levels. E The co-immunoprecipitation (IP) assay showing p53 interacts with CUL4B and DDB1 in HK2 cells treated with cisplatin and MG132. Ubiquitination assay showing ubiquitination of p53 in HK2 cells stably knocking down CUL4B (F) or overexpressing CUL4B (G). Cells were transfected with HA-Ub plasmid and treated with MG132. The lysates were immunoprecipitated with anti-p53 antibody. H Western blots showing the level of wild type or K164R mutant p53 in HEK293T cells at different time points after treating with CHX and the quantification of the protein levels. Cells were transfected with 5×Myc: p53-WT or 5×Myc: p53-K164R. I Ubiquitination assay showing the levels of polyubiquitination on wild type or K164R mutant p53 in CUL4B-overexpressing HEK293T cells. Cells were transfected with the indicated plasmids and treated with MG132. The lysates were immunoprecipitated with anti-Myc antibody. J Schematic showing the molecular mechanism underlying CUL4B regulation of AKI.
Discussion
CUL4B has been identified as important regulators in cancer, X-linked intellectual disability and acute inflammatory diseases like peritonitis [13,14,15,16,17]. Yet, the role of CUL4B in kidney diseases is poorly investigated. Previously, our group has reported that loss of CUL4B in myeloid cells reduces infiltration of macrophages into diabetic kidneys, which in turn ameliorates kidney injury and fibrosis [18]. In this study, we, for the first time, demonstrate that CUL4B can protect kidneys from acute injury caused by cisplatin or IR. Upon exposure to cisplatin or IR, CUL4B is elevated in the kidney to promote degradation of p53, which slows down the induced elevation of p53 and the subsequent PAI-1 expression to restrain the injury (Fig. 6J).
In this study, we found that Cul4b deficiency upregulated cisplatin or IR-induced PAI-1 expression in TECs. More importantly, the elevated PAI-1 expression accounts for the aggravated tubular injury and renal inflammation caused by loss of CUL4B. Previous reports have linked PAI-1 to AKI. Upregulation of urinary PAI-1 (uPAI-1) was detected in animal models or patients with AKI induced by sepsis, nephrotoxin or IR [28, 29]. In healthy kidneys, PAI-1 is rarely expressed, but elevated PAI-1 expression is common in injured kidneys [30]. Single-cell transcriptome analysis of kidneys from mice injected with LPS revealed that PAI-1 was mainly upregulated in endothelial cells [31]. Consistently, in vitro treatment of LPS only resulted PAI-1 upregulation in endothelial cells but not in TECs, suggesting that endothelial cells are responsible for PAI-1 production in LPS-injured kidneys [29]. However, in kidneys from murine with STZ-induced diabetes or unilateral ureteral obstruction, PAI-1 protein was upregulated in the tubules [32, 33]. Through in situ renal perfusion, Paniagua-Sancho et al. showed that the uPAI-1 in rat models with cisplatin, gentamicin or IR-induced AKI was produced by the damaged tubules and secreted into the urine [28]. In our work, by immunohistochemistry, we also found that in both the cisplatin and IR-injured kidneys PAI-1 was elevated in the TECs. The abovementioned work suggests that although PAI-1 is commonly upregulated in kidneys with acute injury, the cells responsible for producing PAI-1 vary in different AKI models. Exposure to nephrotoxins, IR or high glucose results in upregulation of PAI-1 in TECs, while LPS triggers PAI-1 expression in endothelial cells. In this study, we demonstrated that treatment with PAI-1 inhibitor ameliorated tubular injury and inflammation in both Cul4bCon and Cul4bcKO kidneys. Similar pro-injury and pro-inflammation role of PAI-1 has also been reported in models for other kidney diseases. For example, treatment with PAI-1 inhibitor or knockout of Serpine1 prevented tubular injury, apoptosis and inflammation in kidneys after LPS treatment or IR [29, 34, 35].
PAI-1 has been reported to suppress spontaneous or induced apoptosis in a variety of cell types including pulmonary fibroblasts [36, 37], endothelial cells [38], neutrophils [39], neurons [40] and vascular smooth muscle cells [41]. However, in some other cell types, PAI-1 exerts a pro-apoptotic effect. For example, Jiang et al. showed that PAI-1 was elevated with age in both pulmonary fibroblasts and in alveolar type 2 (ATII) cells. It protected fibroblasts from bleomycin or hydrogen peroxide-induced apoptosis but promoted apoptosis through upregulation of p53 in ATII cells [37]. By applying PAI-1 inhibitor to mice with cisplatin treatment or IRI, we showed that inhibition of PAI-1 suppressed the enhanced tubular apoptosis caused by loss of CUL4B in response to cisplatin treatment or IR. In vitro, knocking down Serpine1 also reduced cisplatin-induced apoptosis in HK2 cells with or without CUL4B knockdown, indicating that PAI-1 has autonomous effect on apoptosis in TECs. Consistent with our finding, Gifford et al. reported that overexpression of PAI-1 in HK2 cells induced apoptosis. They found that the PAI-1-induced apoptosis was mediated by upregulation of p53 [42]. Interestingly, p53 is a known transcription factor that can activate transcription of Serpine1 gene. In this study, we found that inhibition of p53 abolished upregulation in Serpine1 mRNA and PAI-1 protein in Cul4b knockout kidneys, suggesting that p53 mediates CUL4B regulation of PAI-1. Therefore, p53 and PAI-1 may form a positive feedback loop to aggravate apoptosis in TECs. Disturbing of this loop, as what the injury-induced CUL4B does, can reduce tubular injury.
The CRL4B complex can promote protein degradation by polyubiquitinating protein substrates or repress gene transcription by catalyzing monoubiquitination of H2AK119 [15]. We found that in TECs, the CRL4B complex catalyzed polyubiquitination of p53 and promoted its degradation. And we identified that CRL4B catalyzes polyubiquitination of the K164 residue in p53. CRL4B-mediated p53 degradation also occurs in cancer cells, underlying the pro-tumor effect of CUL4B [24, 26]. CUL4B can protect fibroblasts from radiation-induced senescence by promoting degradation of p53 [25]. These studies, together with our work, indicate that p53 may be a common substrate in different cell types.
In summary, our work, for the first time, reveal that CUL4B protects against AKI associated with cisplatin or IR. The nephroprotective effect of CUL4B is achieved by suppression of p53/PAI-1 signaling, which leads to reduced apoptosis and inflammation. Modulating this pathway may have therapeutic implications to reduce mortality and improve the prognosis after AKI.
Materials and methods
Human renal biopsy samples
Renal biopsies were obtained from Department of Pathology, Qilu Hospital of Shandong University. Patients with acute renal failure or acute nephritis syndrome were diagnosed by Department of Nephrology, Qilu Hospital of Shandong University. The control samples were the paracancerous tissues from the patients with kidney cancers who underwent tumor nephrectomies. All the investigation was conducted in accordance with the Declaration of Helsinki and were approved by the Research Ethics Committee of Shandong University. The informed consent was obtained.
Mouse
All animal experiments were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee, School of Basic Medical Sciences, Shandong University. C57BL/6J mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Floxed Cul4b mice [6] were crossed to Pax8-Cre mice (The Jackson Laboratory, Strain# 028196) to generate Pax8-Cre+/-; Cul4bflox/Y (Cul4bcKO) and Cul4bflox/Y (Cul4bCon) mice. The mice were housed in a specific-pathogen-free facility in plastic cages at 22 °C and 40–50% humidity, with a 12/12 h light-dark cycle. Mice with the same genotype were allocated randomly into different treatment groups.
AKI mouse model
For cisplatin-induced AKI model, 8 to 10-week-old male mice were injected peritoneally with cisplatin (dissolved in 10% DMSO) at a dose of 20 mg per kg of mouse weight. Mice injected with the same volume of 10% DMSO were served as vehicle control. IRI was performed as previously reported [43]. In brief, after anesthetized with pentobarbital sodium, the mice were placed on a heat pad of 37 °C and a midline abdominal incision was made. The bilateral renal pedicles were clipped for 35 min with vascular clamps (FINE SCIENCE TOOLS, Cat#18055-03). At the end of the ischemia, the clamps were removed to allow reperfusion. Then the abdominal incision was closed, and the mice were placed in a 37 °C incubator to allow recovery. At the desired time after injury, the blood samples were collected from anesthetized mice and serum creatinine and BUN levels were measured by KingMed Diagnostics (Jinan, China). Then the mice were sacrificed, and kidneys were collected, weighted and stored in –80 °C or fixed in 4% paraformaldehyde (PFA) (Servicebio, Cat#G1101).
Tubular damage scoring
After 24 h fixation in 4% PFA at room temperature, the kidney tissues were embedded in paraffin. The embedded tissues were cut into 4 μm thick sections, adhered onto glass slides, deparaffinized, and rehydrated by decreasing ethanol concentrations. Tissue-embedded slides were stained with hematoxylin and eosin (H & E) and imaged on a microscope (KEYENEC, Cat#BZ-X810). The tubular scoring was performed double blinded according to the presence and the extent of TEC flattening, brush border loss, cell membrane bleb formation, interstitial edema, cytoplasmic vacuolization, necrosis and tubular lumen obstruction [44].
Immunohistochemistry and immunofluorescent staining
For immunostaining, tissue-embedded slides were washed with PBS, boiled in citrate solution, and blocked with 10% goat serum before overnight incubation at 4 °C with primary antibodies. For immunohistochemistry, after incubation with primary antibodies, the sections were applied with Immunohistochemistry kit (ZSGB-Bio, Cat# PV9001 and PV9000). For immunofluorescent staining, after incubation with primary antibodies, the slides were stained with secondary antibodies (Invitrogen, Cat# A11029 and A11011) for 60 min at room temperature in the dark. Nucleus was labeled with DAPI (Abcam, Cat# ab104139). The TUNEL staining was performed using TUNEL kit (KeyGEN BioTECH, Cat# KGA1405-50) according to the manufacturer’s instruction. Images were collected with a microscope (KEYENEC, Cat#BZ-X810). The analyses of the images were performed using ImageJ. All the antibodies used are listed in Supplementary Table S1.
Cell culture
HK2 cells were purchased from FuHeng Biology (Cat# FH0228). Cells were maintained in a humidified atmosphere at 37 °C with 5% CO2 and routinely tested for Mycoplasma. To generate HK2 cells stably expressing control shRNA or shRNA targeting CUL4B, the shRNA sequence [45] was inserted into pGIPZ vector. The constructs were packed into lentivirus and applied to HK2 cells. Forty-eight hours after infection, the cells were subjected to selection with Blasticidin S hydrochloride (Solarbio, Cat# B9300). To generate HK2 cells stably overexpressing CUL4B, lentivirus carrying CUL4B were applied to HK2 cells. The infected cells were selected with Puromycin (InvivoGen, Cat# QLL-44-04). When evaluating effects of different treatments, cells were randomly allocated into different groups.
Transfection
siRNA transfection was performed using Lipofectamine™ RNAiMAX (Invitrogen, Cat# 137781050). Cells were collected at 24 or 48 h after transfection. Plasmid transfection was performed using Lipofectamine™ 3000 (Invitrogen, Cat# L3000015). Cells were collected at 24 h after transfection.
Annexin V/7-AAD analysis of apoptosis
Cells were collected and suspended in PBS. The Annexin V-PE/7-AAD staining was performed using Apoptosis Detection Kit (Vazyme, Cat# A213-02) according to the manufacturer’s manual. The stained cell suspension was then applied to flow cytometry analysis using CytoFLEX (Beckman Coulter). The data were analyzed using FlowJo 10.8.1 software.
CHX chasing experiment
To evaluate protein stability, 50 μg/ml CHX (GLPBIO, Cat# GC17198) were applied to cells to inhibit protein synthesis. Cells were collected and lysed in RIPA buffer (Sigma, Cat# R0278) supplemented with protease inhibitor (Solarbio, Cat# P0100). The protein samples were applied to Western blotting.
Ubiquitination assay
HK2 or HEK293T Cells were transfected with plasmids and treated with 20 μM MG132 for 5 h at 48 h post transfection. Cells were then lysed and precipitated with anti-Myc antibody or anti-p53 antibody. The precipitates were applied to Western blotting.
Identification of ubiquitination sites
HEK293T cells were transfected with plasmids expressing 5×Myc-p53, HA-Ub and GFP-CUL4B. Forty-eight hours after transfection, cells were treated with 20 μM MG132 for 5 h. Then cells were collected and lysed. p53 was precipitated with anti-Myc antibody and applied to 10% SDS-PAGE. After Coomassie Blue staining, the protein bands were cut and sent to mass spectrometry analysis. Mass spectrometry analysis was performed by Qinglianbio Biotech (Beijing China). Site-Directed Mutagenesis kit (NEB, Cat# E0552S) was used to generate K164R mutant of p53.
Co-immunoprecipitation
The cells were lysed in lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH7.4), 1% Triton X-100. After centrifugation, the supernatant was collected and incubated with CUL4B antibody, p53 antibody or control IgG (listed in Supplementary Table S1) at 4 °C overnight. Protein A/G Magnetic beads (Vazyme, Cat# PB101) were added to the protein/antibody mixture and incubated at room temperature for 2 h. The beads were then washed with PBST buffer (PBS containing 0.5% Tween-20) for 5 times and boiled in loading buffer for 10 min. The supernatant was subjected to Western blotting.
Western blotting
Protein samples from kidney tissues were collected by homogenizing the frozen tissues and lysing in RIPA buffer. Protein samples from cells were prepared as mentioned in the above sections. Protein concentrations were quantified using BCA kit (Vazyme, Cat# E112-02). Equal mass of protein samples and protein markers (Beyotime, Cat# P0069; Vazyme, Cat# MP102) was applied to 10% gels, transferred to PVDF membrane (Millipore, Cat# ISEQ00010), and probed with primary antibodies and secondary antibodies. The bands were detected using Chemiluminescent HRP Substrate (Millipore, Cat# WBKLS0100) and imaged using Tanon 5200 Multi.
RNA isolation and RT-qPCR
Total RNA was extracted with Trizol reagents (Invitrogen, Cat# 15596018). Reverse transcription was performed with HiScript RT SuperMix for qPCR kit (Vazyme, Cat# R32). RT-qPCR was run using 2×ChamQ SYBR Mix (Vazyme, Cat# Q411) on Analytikjena qTOWER. Primer sequences are listed in Supplementary Table S2.
RNA sequencing
RNA sequencing was performed using kidney tissues from Cul4bCon and Cul4bcKO mice collected at 72 h after treatment with cisplatin or vehicle controls. 3 mice were included in each group. The sequencing was performed by Beijing Biomarker Technologies Co., LTD (Beijing, China) and analyzed using the BMKCloud Bioinformatics analysis platform.
Statistical analysis
All data are presented as mean ± SD. Statistical analysis was done using GraphPad Prism 8. Statistical significance was determined using unpaired two-tailed t-test for two-sample comparison or one-way ANOVA with Tukey’s multiple comparisons test for comparison between three or more groups. The assumption of equal variance was validated by F-test. A P value <0.05 was considered significant. The sample sizes were chosen empirically based on the observed effects and previous reports. The sample size for each experiment is listed in the figure legends. When scoring tubular injury and analyzing data of RT-qPCR and immunohistochemistry, the investigators were blinded to the group allocation.
Data availability
The raw data of RNAseq were deposited at NCBI Bioproject with accession number PRJNA1108044. The original images of Western blots were provided as a supplementary file. All the other raw data supporting the findings of this study are available from the corresponding authors upon request.
References
Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7:52.
Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019;394:1949–64.
Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12:220.
Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34:562–70.
Hu H, Yang Y, Ji Q, Zhao W, Jiang B, Liu R, et al. CRL4B catalyzes H2AK119 monoubiquitination and coordinates with PRC2 to promote tumorigenesis. Cancer Cell. 2012;22:781–95.
Jiang B, Zhao W, Yuan J, Qian Y, Sun W, Zou Y, et al. Lack of Cul4b, an E3 ubiquitin ligase component, leads to embryonic lethality and abnormal placental development. PLoS ONE. 2012;7:e37070.
Yin Y, Liu L, Yang C, Lin C, Veith GM, Wang C, et al. Cell autonomous and nonautonomous function of CUL4B in mouse spermatogenesis. J Biol Chem. 2016;291:6923–35.
Lin CY, Chen CY, Yu CH, Yu IS, Lin SR, Wu JT, et al. Human X-linked intellectual disability factor CUL4B is required for post-meiotic sperm development and male fertility. Sci Rep. 2016;6:20227.
Li P, Song Y, Zan W, Qin L, Han S, Jiang B, et al. Lack of CUL4B in adipocytes promotes ppargamma-mediated adipose tissue expansion and insulin sensitivity. Diabetes. 2017;66:300–13.
Mi J, Wang S, Liu P, Liu C, Zhuang D, Leng X, et al. CUL4B upregulates RUNX2 to promote the osteogenic differentiation of human periodontal ligament stem cells by epigenetically repressing the expression of miR-320c and miR-372/373-3p. Front Cell Dev Biol. 2022;10:921663.
Yu R, Han H, Chu S, Ding Y, Jin S, Wang Y, et al. CUL4B orchestrates mesenchymal stem cell commitment by epigenetically repressing KLF4 and C/EBPdelta. Bone Res. 2023;11:29.
Ma Y, Liu X, Zhou M, Sun W, Jiang B, Liu Q, et al. CUL4B mutations impair human cortical neurogenesis through PP2A-dependent inhibition of AKT and ERK. Cell Death Dis. 2024;15:121.
Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80:561–6.
Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet. 2007;80:345–52.
Li Y, Wang X. The role of cullin4B in human cancers. Exp Hematol Oncol. 2017;6:17.
Hung MH, Jian YR, Tsao CC, Lin SW, Chuang YH. Enhanced LPS-induced peritonitis in mice deficiency of cullin 4B in macrophages. Genes Immun. 2014;15:404–12.
Song Y, Li P, Qin L, Xu Z, Jiang B, Ma C, et al. CUL4B negatively regulates Toll-like receptor-triggered proinflammatory responses by repressing Pten transcription. Cell Mol Immunol. 2021;18:339–49.
Jin S, Song Y, Zhou L, Jiang W, Qin L, Wang Y, et al. Depletion of CUL4B in macrophages ameliorates diabetic kidney disease via miR-194-5p/ITGA9 axis. Cell Rep. 2023;42:112550.
Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2016;27:371–9.
Eddy AA. Plasminogen activator inhibitor-1 and the kidney. Am J Physiol Renal Physiol. 2002;283:F209–20.
Kunz C, Pebler S, Otte J, von der Ahe D. Differential regulation of plasminogen activator and inhibitor gene transcription by the tumor suppressor p53. Nucleic Acids Res. 1995;23:3710–7.
Kawarada Y, Inoue Y, Kawasaki F, Fukuura K, Sato K, Tanaka T, et al. TGF-beta induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci Rep. 2016;6:35483.
Higgins SP, Tang Y, Higgins CE, Mian B, Zhang W, Czekay RP, et al. TGF-beta1/p53 signaling in renal fibrogenesis. Cell Signal. 2018;43:1–10.
Thirunavukarasou A, Singh P, Govindarajalu G, Bandi V, Baluchamy S. E3 ubiquitin ligase Cullin4B mediated polyubiquitination of p53 for its degradation. Mol Cell Biochem. 2014;390:93–100.
Wei Z, Guo H, Liu Z, Zhang X, Liu Q, Qian Y, et al. CUL4B impedes stress-induced cellular senescence by dampening a p53-reactive oxygen species positive feedback loop. Free Radic Biol Med. 2015;79:1–13.
Zhong M, Zhou L, Zou J, He Y, Fang Z, Xiang X. Cullin-4B promotes cell proliferation and invasion through inactivation of p53 signaling pathway in colorectal cancer. Pathol Res Pract. 2021;224:153520.
Tang C, Ma Z, Zhu J, Liu Z, Liu Y, Liu Y, et al. P53 in kidney injury and repair: mechanism and therapeutic potentials. Pharmacol Ther. 2019;195:5–12.
Paniagua-Sancho M, Quiros Y, Casanova AG, Blanco-Gozalo V, Agueros-Blanco C, Benito-Hernandez A, et al. Urinary plasminogen activator inhibitor-1: a biomarker of acute tubular injury. Am J Nephrol. 2021;52:714–24.
Tanaka K, Harada H, Kamuro H, Sakai H, Yamamoto A, Tomimatsu M, et al. Arid5a/IL-6/PAI-1 signaling is involved in the pathogenesis of lipopolysaccharide-induced kidney injury. Biol Pharm Bull. 2023;46:1753–60.
Huang Y, Noble NA. PAI-1 as a target in kidney disease. Curr Drug Targets. 2007;8:1007–15.
Janosevic D, Myslinski J, McCarthy TW, Zollman A, Syed F, Xuei X, et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife. 2021;10:e62270.
Jeong BY, Uddin MJ, Park JH, Lee JH, Lee HB, Miyata T, et al. Novel plasminogen activator inhibitor-1 inhibitors prevent diabetic kidney injury in a mouse model. PLoS ONE. 2016;11:e0157012.
Duymelinck C, Dauwe SE, De Greef KE, Ysebaert DK, Verpooten GA, De Broe ME. TIMP-1 gene expression and PAI-1 antigen after unilateral ureteral obstruction in the adult male rat. Kidney Int. 2000;58:1186–201.
Kim JI, Jung KJ, Jang HS, Park KM. Gender-specific role of HDAC11 in kidney ischemia- and reperfusion-induced PAI-1 expression and injury. Am J Physiol Renal Physiol. 2013;305:F61–70.
Gupta KK, Donahue DL, Sandoval-Cooper MJ, Castellino FJ, Ploplis VA. Abrogation of plasminogen activator inhibitor-1-vitronectin interaction ameliorates acute kidney injury in murine endotoxemia. PLoS ONE. 2015;10:e0120728.
Zhang YP, Wang WL, Liu J, Li WB, Bai LL, Yuan YD, et al. Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca(2+) signaling. Thromb Res. 2013;131:64–71.
Jiang C, Liu G, Cai L, Deshane J, Antony V, Thannickal VJ, et al. Divergent regulation of alveolar type 2 cell and fibroblast apoptosis by plasminogen activator inhibitor 1 in lung fibrosis. Am J Pathol. 2021;191:1227–39.
Bajou K, Peng H, Laug WE, Maillard C, Noel A, Foidart JM, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14:324–34.
Zmijewski JW, Bae HB, Deshane JS, Peterson CB, Chaplin DD, Abraham E. Inhibition of neutrophil apoptosis by PAI-1. Am J Physiol Lung Cell Mol Physiol. 2011;301:L247–54.
Soeda S, Oda M, Ochiai T, Shimeno H. Deficient release of plasminogen activator inhibitor-1 from astrocytes triggers apoptosis in neuronal cells. Brain Res Mol Brain Res. 2001;91:96–103.
Chen Y, Kelm RJ Jr., Budd RC, Sobel BE, Schneider DJ. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem. 2004;92:178–88.
Gifford CC, Lian F, Tang J, Costello A, Goldschmeding R, Samarakoon R, et al. PAI-1 induction during kidney injury promotes fibrotic epithelial dysfunction via deregulation of klotho, p53, and TGF-beta1-receptor signaling. FASEB J. 2021;35:e21725.
Zhou D, Tan RJ, Lin L, Zhou L, Liu Y. Activation of hepatocyte growth factor receptor, c-met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int. 2013;84:509–20.
Paller MS, Hoidal JR, Ferris TF. Oxygen free radicals in ischemic acute renal failure in the rat. J Clin Invest. 1984;74:1156–64.
Wang Y, Pan X, Li Y, Wang R, Yang Y, Jiang B, et al. CUL4B renders breast cancer cells tamoxifen-resistant via miR-32-5p/ER-alpha36 axis. J Pathol. 2021;254:185–98.
Acknowledgements
We thank Translational Medicine Core Facility of Shandong University and the School of Basic Medical Sciences Core Facility of Shandong University for consultation and instrument availability that supported this work.
Funding
This work was supported by National Natural Science Foundation of China to G.L. (No. 81770660) and G.S. (No. 32270869).
Author information
Authors and Affiliations
Contributions
Kaixuan Liu conducted the experiments and wrote the draft of the manuscript. Xiaoyu Hao and Yangfan Gao helped with the animal experiments and data analysis. Zhiyuan Cao and Min Hou helped with molecular cloning and experiments with lentivirus. Lining Qin helped with the animal experiments. Yu Song and Molin Wang were responsible for data curation and helped edit and revise manuscript. Baichun Jiang and Qiao Liu provided technical support for the animal experiments. Yongxin Zou and Yaoqin Gong helped revise and edit manuscript. Guangyi Liu and Gongping Sun supervised the project, designed the experiments and revised manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Francesca Bernassola
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, K., Hao, X., Gao, Y. et al. CUL4B protects kidneys from acute injury by restraining p53/PAI-1 signaling. Cell Death Dis 15, 915 (2024). https://doi.org/10.1038/s41419-024-07299-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-024-07299-w
This article is cited by
-
Acute kidney injury: pathogenesis and therapeutic interventions
Molecular Biomedicine (2025)
-
CUL4B promotes hepatocellular carcinoma progression and oxaliplatin resistance by facilitating FUS degradation
Cell Death & Disease (2025)
-
A systematic review of protein post-translational modifications in sepsis
Molecular Biology Reports (2025)
