
Abstract
Gastric cancer (GC) remains a leading cause of cancer-related mortality worldwide, with limited treatment options in advanced stages. Immunotherapy, particularly immune checkpoint inhibitors (ICIs) targeting PD1/PD-L1, has emerged as a promising therapeutic approach. However, a significant proportion of patients exhibit primary or acquired resistance, limiting the overall efficacy of immunotherapy. This review provides a comprehensive analysis of the mechanisms underlying immunotherapy resistance in GC, including the role of the tumor immune microenvironment, dynamic PD-L1 expression, compensatory activation of other immune checkpoints, and tumor genomic instability. Furthermore, the review explores GC-specific factors such as molecular subtypes, unique immune evasion mechanisms, and the impact of Helicobacter pylori infection. We also discuss emerging strategies to overcome resistance, including combination therapies, novel immunotherapeutic approaches, and personalized treatment strategies based on tumor genomics and the immune microenvironment. By highlighting these key areas, this review aims to inform future research directions and clinical practice, ultimately improving outcomes for GC patients undergoing immunotherapy.
Similar content being viewed by others
FACTS
-
Gastric cancer remains a highly lethal disease, with immunotherapy resistance being a major clinical challenge.
-
The tumor-immune microenvironment significantly impacts immune checkpoint inhibitor efficacy in gastric cancer.
-
Unique molecular and immune subtypes of gastric cancer influence resistance mechanisms and treatment outcomes.
-
Combination therapies are emerging as promising strategies to overcome immunotherapy resistance in gastric cancer.
Open questions
-
What are the precise mechanisms by which the tumor-immune microenvironment drives immunotherapy resistance in gastric cancer?
-
What are the key signaling pathways involved in ECM remodeling that hinder T-cell infiltration in gastric cancer?
-
Can personalized treatment strategies based on gastric cancer molecular subtypes effectively overcome immunotherapy resistance?
-
How can novel combination therapies be optimized to enhance the efficacy of immune checkpoint inhibitors in gastric cancer?
Introduction
GC remains one of the leading causes of cancer-related mortality worldwide, with advanced stages presenting particularly challenging therapeutic scenarios [1,2,3]. Despite advances in treatment modalities such as surgery, chemotherapy, and radiotherapy, the prognosis for advanced GC patients continues to be poor [1, 4]. Recently, immunotherapy, particularly ICIs targeting PD1/PD-L1, has emerged as a promising approach, offering new hope for improving patient outcomes [5, 6].
ICIs work by blocking the interaction between PD1 on immune cells and PD-L1 on tumor cells, thereby relieving the immune system’s suppression and allowing it to mount an effective antitumor response [7, 8]. In GC, PD1/PD-L1 inhibitors have demonstrated clinical efficacy, especially in patients with specific molecular subtypes [9, 10]. However, the benefits of immunotherapy are not universal. A significant proportion of patients experience either primary resistance, where the therapy fails to elicit a response, or acquired resistance, where the initial response diminishes over time [11, 12].
Understanding the mechanisms underlying immunotherapy resistance in GC is crucial for improving treatment outcomes. Resistance mechanisms are multifaceted, involving alterations in the tumor-immune microenvironment, dynamic changes in PD-L1 expression, compensatory activation of alternative immune checkpoints, and intrinsic tumor genomic instability [13, 14]. Additionally, GC-specific factors, such as distinct molecular subtypes and unique immune evasion mechanisms, including the role of Helicobacter pylori infection, further complicate the landscape of resistance [15].
This review aims to provide a comprehensive overview of the current understanding of immunotherapy resistance in GC. We will explore the intricate mechanisms contributing to resistance, discuss emerging strategies to overcome these barriers and highlight future directions for research and clinical practice. By elucidating these aspects, we hope to offer insights that will guide the development of more effective therapeutic strategies for GC patients undergoing immunotherapy.
Current status of immunotherapy in GC
The main ICIs currently used in the treatment of GC include PD1 inhibitors, PD-L1 inhibitors, and CTLA-4 inhibitors, each with specific indications. PD1 inhibitors such as pembrolizumab and nivolumab are commonly used in GC treatment [16,17,18]. Pembrolizumab has been approved for multiple cancer types [19, 20] and was first approved by the FDA in 2017 for patients with previously treated GC, especially those with PD-L1-positive tumors (CPS ≥ 1) [21, 22]. It has shown significant efficacy in patients with high microsatellite instability (MSI-H) or mismatch repair deficiency (dMMR) [23]. Nivolumab, another PD1 inhibitor, has been approved for GC in multiple countries. The CheckMate-649 study demonstrated that nivolumab combined with chemotherapy significantly extended overall survival (OS) in untreated advanced GC patients, particularly those with PD-L1 CPS ≥ 5, compared to chemotherapy alone. It is indicated for first-line treatment of advanced or metastatic GC in combination with chemotherapy and as monotherapy for recurrent or metastatic cases [9, 24].
The primary PD-L1 inhibitor studied in GC is atezolizumab, with extensive research focusing on its combination with other treatments. While not yet formally approved by the FDA or EMA for GC, it has shown promising outcomes in some clinical trials [25, 26]. CTLA-4 inhibitors, such as ipilimumab, are approved for melanoma and other cancers. In GC, studies on ipilimumab are still ongoing, often in combination with PD1 inhibitors like nivolumab. However, ipilimumab has not been approved for GC monotherapy, though combination treatments show potential [27, 28].
Multiple phase III trials have confirmed the efficacy and safety of ICIs in GC. The CheckMate-649 trial showed that nivolumab plus chemotherapy significantly improved OS and progression-free survival (PFS), with the most common adverse events being nausea, diarrhea, and peripheral neuropathy [29]. In the ATTRACTION-4 trial, nivolumab combined with oxaliplatin-based chemotherapy significantly improved PFS in Asian patients with HER2-negative, unresectable advanced or recurrent GC, though it did not improve OS. Common adverse events included neutropenia, thrombocytopenia, and decreased appetite [30]. The phase III KEYNOTE-062 trial showed that pembrolizumab monotherapy was non-inferior to chemotherapy in terms of OS for untreated advanced GC or gastroesophageal junction (GEJ) cancer patients with PD-L1 CPS ≥ 1, with fewer adverse events. However, neither pembrolizumab nor its combination with chemotherapy showed superiority in OS or PFS over chemotherapy alone [31]. The KEYNOTE-811 trial demonstrated that pembrolizumab combined with first-line trastuzumab and chemotherapy significantly improved PFS in patients with PD-L1 CPS ≥ 1 metastatic HER2-positive GC, with the most common treatment-related adverse events (TRAEs) being diarrhea, nausea, and anemia [32]. Finally, the KEYNOTE-859 trial indicated that pembrolizumab plus chemotherapy significantly improved OS with manageable toxicity, with the most common grade 3–5 adverse events being anemia and neutropenia [33].
We summarize the clinical trial results of ICIs in GC in Table 1 for a clearer overview.
Mechanisms of immunotherapy resistance in GC
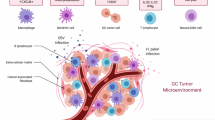
Multiple factors contribute to the complex process of immunotherapy resistance in GC. The tumor microenvironment (TME) weakens the antitumor-immune response through the interactions of immunosuppressive cells and signaling pathways, while the dynamic changes in PD-L1 expression exacerbate resistance to ICIs. The molecular classification of GC reveals differences in immunotherapy responses across subtypes, and unique immune evasion mechanisms further drive the development of resistance. (Fig. 1).

A Immunosuppressive tumor microenvironments (such as Tregs and MDSCs) promote immune escape and drug resistance in gastric cancer. B The heterogeneity of PD1/PD-L1 expression in tumor and TME affects the response of gastric cancer to immunotherapy. C Different molecular classifications (such as MSI-H, EBV positive, etc.) and subtypes of gastric cancer affect the efficacy of immunotherapy. D Factors specific to gastric cancer, such as H. pylori infection and gastric mucosal immune environment, affect the efficacy of immunotherapy.
Tumor-immune microenvironment
The tumor-immune microenvironment in GC is a complex and dynamic network of cells, signaling molecules, and extracellular matrix components that interact with tumor cells and play a critical role in modulating the immune response. The composition of the TME includes immune cells such as T lymphocytes, natural killer (NK) cells, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs), alongside fibroblasts and endothelial cells. While certain immune cells such as cytotoxic T cells and NK cells contribute to antitumor immunity, immunosuppressive components within the TME can promote immune evasion and resistance to therapies, including immunotherapy.
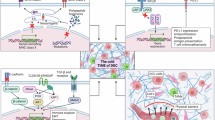
Cytotoxic T cells are key effectors in the antitumor-immune response, directly targeting and killing cancer cells. However, in the GC TME, their activity is often suppressed by immunosuppressive factors, leading to reduced effectiveness and contributing to immune evasion (Fig. 2A). Research has identified two distinct immune checkpoint expression patterns (ICEP1 and ICEP2) in GC. ICEP1 includes CD8 + T cells co-expressing PD1, CTLA-4, TIGIT, LAG-3, or CD38, while ICEP2 involves CD8 + T cells expressing NKG2A alone or co-expressing it with other checkpoints. The ICEP2 subgroup is associated with resistance to anti-PD1 therapy in GC, potentially mediated by the recruitment of LGMN+ macrophages via the CXCL16-CXCR6 signaling pathway [34]. Additionally, another study found that heat shock gene expression in intratumoral CD4/CD8 + T cells was significantly upregulated following immune checkpoint blockade (ICB) therapy, particularly in non-responsive tumors, suggesting that stress response T cells characterized by heat shock gene expression may be linked to immunotherapy resistance [35].

A Mechanisms of T cell involvement in immunotherapy resistance in gastric cancer. B Mechanisms of TAM involvement in immunotherapy resistance. C Mechanisms of MDSCs involvement in immunotherapy resistance. D Mechanisms of neutrophils involvement in immunotherapy resistance.
Cancer-associated fibroblasts (CAFs), as key components of the TME, exhibit significant heterogeneity and can be classified into distinct subtypes based on their phenotypic and functional characteristics. These subtypes play diverse roles in tumor progression and immune modulation, significantly influencing the efficacy of immunotherapy [36, 37]. The main subtypes include myofibroblast CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs), among others [38, 39]. MyCAFs are characterized by high α-SMA expression and are primarily involved in ECM remodeling [40, 41]. These cells enhance tissue stiffness, create physical barriers to immune cell infiltration, and contribute to tumor invasion [42, 43]. ICAFs exhibit low α-SMA expression and secrete pro-inflammatory cytokines such as IL-6 and CXCL1 [41, 44]. They promote an immunosuppressive TME by recruiting MDSCs and inhibiting T-cell activation [45, 46]. In GC, ICAFs may contribute to immune evasion and resistance to ICIs. Single-cell study has shown that iCAFs can interact with T cells by secreting IL-6 and CXCL12. iCAFs not only showed enhanced pro-invasion activity but also mobilized surrounding immune cells to build a microenvironment favorable to tumors. Therefore, inhibiting their activation inhibits the GC “seeds” while improving the GC soil [47]. ApCAFs express MHC II molecules and exhibit antigen-presenting potential [48, 49]. They might modulate T-cell responses [50]; however, their role remains controversial and requires further investigation. Although less studied in gastric cancer, their immunomodulatory role could influence response to immunotherapy. In addition, a study has shown that CAFs in GC promote immune evasion via the PDGF-C/D signaling pathway, specifically by recruiting suppressive myeloid cells through the increased expression of CXCL chemokines, leading to anti-PD1 therapy resistance. Blocking PDGFRα/β can reverse the immunosuppressive TME by remodeling the tumor stroma and, when combined with anti-PD1 therapy, synergistically suppresses the growth of fibrotic tumors [51]. A specific CAF subset in GC—CPT1C+ CAFs—promotes immunosuppression in the TME by secreting IL-6, enriching extracellular matrix molecules, and recruiting immunosuppressive cells, particularly M2 macrophages. High levels of CPT1C+ CAFs are associated with poor response to immunotherapy in GC patients [52]. Furthermore, multiple models utilizing fibroblast-associated gene markers have been developed to predict immunotherapy response, providing new avenues for overcoming immune resistance in GC [53,54,55]. Figure 3 illustrates some of the roles of CAFs in immunotherapy for GC.

CAFs contribute to an immunosuppressive tumor microenvironment through various mechanisms, including secretion of immunosuppressive factors, recruitment of immunosuppressive cells, and modulation of immune checkpoint signaling. Targeting key CAF-associated pathways, such as the PDGF-C/D and IL-6 signaling axes, may enhance the efficacy of immunotherapy and reverse resistance in gastric cancer.
TAMs often exhibit an immunosuppressive M2 phenotype in GC [56, 57]. They promote tumor growth and immune evasion, contributing to resistance to immunotherapy (Fig. 2B). Studies have revealed that GC cells secrete legumain (LGMN), which binds to integrin αvβ3 on macrophages, activating the PI3K/AKT/mTORC2 signaling pathway and driving the polarization of macrophages from an M1 to M2 phenotype, thereby promoting immune evasion and resistance to anti-PD1 therapy. Blocking LGMN or integrin αvβ3 can significantly inhibit this process [58]. Additionally, high levels of Siglec-10+ TAMs promote CD8 + T cell exhaustion, contributing to immune evasion and chemotherapy resistance. Blocking Siglec-10 reactivates antitumor immunity and shows synergistic effects when combined with anti-PD1 therapy [59].
MDSCs suppress T-cell activity and create an immunosuppressive environment, contributing to resistance to immunotherapy in GC [60] (Fig. 2C). Studies have shown that in the GC TME, infiltrating monocytic MDSCs (M-MDSCs) highly express immunosuppressive genes and are significantly enriched in GC tissues, with IER3 + M-MDSCs being closely associated with immunosuppression and treatment resistance [61].
Neutrophils in the TME can promote tumor growth and suppress antitumor immunity, contributing to immunotherapy resistance in GC (Fig. 2D). In anti-PD1 therapy for GC, overexpression of CXCL5 recruits tumor-associated neutrophils via the CXCL5/CXCR2 axis, which is a key factor in driving immunosuppression. Apatinib blocks this process, enhancing the effectiveness of anti-PD1 therapy [62].
In addition to the above, DCs and NK cells also play potential roles in immunotherapy resistance in GC [63,64,65]. In Fig. 1, we present a schematic of the immune microenvironment associated with immunotherapy resistance in GC for a clearer visualization.
The ECM has also been shown to be an important factor affecting the invasion and function of immune cells in tumors and the effect of immunotherapy [66, 67]. The ECM is a complex network of proteins, glycoproteins, and proteoglycans that provide structural and biochemical support to cells [68]. In GC, ECM undergoes significant remodeling, promotes tumor progression and immune escape, and plays a crucial role in regulating the infiltration of immune cells, especially T cells, thus affecting the efficacy of immunotherapy [47, 69]. Studies have shown that TRIM44 is highly expressed in gastric cancer and is associated with T-cell infiltration. TRIM44 inhibits gastric tumorigenicity by regulating T-cell-mediated antitumor immunity and LOXL2 protein levels. Mechanically, TRIM44 directly binds to LOXL2, affecting the stability of LOXL2, altering extracellular matrix remodeling, and affecting tumor immunity [70]. Key ECM components, such as collagen [71], laminin [72], and fibronectin [73], are often overexpressed in GC, leading to increased matrix stiffness and [74, 75]. This enhanced rigidity not only restricts the physical movement of T cells into the tumor core but also alters integrin-mediated signaling pathways, reducing the ability of T cells to adhere to and migrate through the ECM [76]. Additionally, the ECM can sequester immunosuppressive molecules like TGF-β, further suppressing T-cell activation and infiltration [77]. CAFs, which are abundant in GC, play a pivotal role in ECM remodeling. CAFs deposit excessive ECM components and promote crosslinking of collagen fibers via lysyl oxidase, exacerbating matrix stiffness [78]. ECM remodeling in cancer is driven by dynamic interactions between tumor cells, stromal cells, and soluble factors [79]. Matrix metalloproteinases are key enzymes that degrade ECM components, facilitating tumor invasion and altering immune cell accessibility [80]. However, paradoxically, excessive ECM degradation can release bioactive fragments called matrikines, which may promote tumor growth and immunosuppression [81]. In addition, the accumulation of hyaluronic acid and fibronectin in the extracellular matrix are also important factors affecting the tumor-immune microenvironment [82, 83]. In conclusion, in a variety of tumors, including gastric cancer, ECM is a key barrier to effective immunotherapy, primarily by limiting T-cell invasion and inducing an immunosuppressive environment. Understanding the mechanisms of ECM remodeling and its effects on immune cell dynamics is critical to developing new therapeutic strategies. By combining ECM-targeted therapy with existing immunotherapies, it is possible to enhance the immune response and improve outcomes in GC patients.
Dynamic changes of PD-L1 expression
The dynamic and heterogeneous expression of PD-L1 in GC plays a critical role in mediating resistance to immunotherapy. PD-L1 expression can vary spatially and temporally within the TME, influenced by intrinsic tumor factors and external signals such as inflammation and immune cell interactions. This heterogeneity complicates the prediction of therapeutic response to PD1/PD-L1 inhibitors, as fluctuating levels of PD-L1 can lead to inconsistent responses to treatment. Understanding the mechanisms driving these dynamic changes in PD-L1 expression is crucial for overcoming resistance and improving the efficacy of immunotherapy in GC.
A study analyzing 1014 GC specimens through immunohistochemistry evaluated the clinical significance of PD1 and its ligands (PD-L1 and PD-L2) in GC. The results showed that PD-L1 was expressed in 37.8% of tumor cell membranes and 74.9% of infiltrating immune cells. Higher PD-L1 expression was observed in patients without metastasis, Epstein-Barr virus (EBV)-positive patients, and those with elevated C-met and PCNA expression. Additionally, patients with high PD-L1 expression exhibited better survival rates. Increased PD-L1, PD-L2, and PD1 expression was found in patients with higher T-cell infiltration, which may be linked to adaptive immune resistance mechanisms [84].
Previous studies have shown that GC mesenchymal stem cells (MSCs) promote PD-L1 expression and lactate production via the IL-8/CXCR2 pathway, impairing the antitumor efficacy of PD1 immunotherapy. Blocking the IL-8/CXCR2 pathway or reducing PD-L1 expression and lactate production significantly restored the antitumor effects of PD1 antibodies [85]. Similar findings revealed that GC MSCs enhanced PD-L1 expression in GC cells, leading to resistance to CD8 + T-cell cytotoxicity. This resistance was mediated by IL-8 released from GC MSCs, which activated STAT3 and mTOR signaling pathways, promoting c-Myc induction and increasing PD-L1 expression in GC cells. Consistently, blocking IL-8 helped overcome immune evasion and improved the efficiency of immunotherapy [86].
Certain microRNAs, such as miR-105-5p, can inhibit post-transcriptional PD-L1 expression by binding to a key cis-acting element in the PD-L1 3’ untranslated region. This reduces PD-L1 protein and surface expression, promoting CD8 + T-cell activation. The expression of miR-105-5p is regulated by DNA methylation of its host gene GABRA3 promoter [87]. Additionally, all-trans retinoic acid (ATRA) enhances PD-L1 expression by increasing its protein stability and synthesis. ATRA-induced PD-L1 upregulation strongly confers resistance to activated T-cell cytotoxicity in GC cells and antagonizes the effects of PD-L1 antibodies [88].
Molecular classification and immunotherapy resistance
In recent years, the molecular classification of GC has provided valuable insights into the mechanisms of immunotherapy resistance, helping to identify distinct tumor subtypes with varying responses to ICIs. These classifications are based on genetic, epigenetic, and immune profiling, offering a more precise understanding of tumor heterogeneity and its role in immune evasion. Different molecular subtypes exhibit varying levels of sensitivity to immunotherapy [89,90,91]. For example, based on the characteristics of immune cell infiltration and their functional states, it can be classified into inflamed, immune-excluded, and immune-desert subtypes [92,93,94]. The inflamed subtype is characterized by high levels of T-cell and NK cell infiltration, active interferon signaling, and frequent PD-L1 overexpression, which generally makes it sensitive to ICIs [95]. The immune-excluded subtype, although containing immune cells, is marked by their confinement to the tumor periphery, potentially due to barriers imposed by CAFs and the ECM, leading to resistance to ICIs [94, 96,97,98]. The immune-desert subtype shows minimal immune cell infiltration, typically associated with low tumor immunogenicity and immune evasion mechanisms, and represents a prototypical feature of resistance to ICIs [99, 100]. These subtypes also significantly affect the response of gastric cancer to immunotherapy. Understanding these molecular classifications is crucial for developing more effective strategies to overcome resistance and tailor immunotherapy approaches to individual patients with GC.
A study explored the efficacy of neoadjuvant immunotherapy with nivolumab and ipilimumab in patients with resectable dMMR/MSI-H gastric or GEJ adenocarcinoma. The findings demonstrated the potential efficacy and safety of this immunotherapy regimen in this subset of gastric/GEJ adenocarcinoma [101]. Similarly, studies have shown that tumors with MSI-H tend to be resistant to chemotherapy but may exhibit durable responses to immunotherapy. In some cases, EBV-positive patients achieved complete long-term responses to immunotherapy [102]. However, dMMR/MSI-H gastrointestinal cancers with peritoneal metastasis and ascites respond poorly to ICIs [103]. A high number of mutations in the PI3K-AKT-mTOR pathway (NMP) genes may predict primary resistance to ICIs in dMMR/MSI-H gastric adenocarcinoma, and the use of PI3K-AKT-mTOR inhibitors as adjuncts to immunotherapy is recommended for patients with high NMP mutations [104]. Nonetheless, real-world cases suggest that MMR status and microsatellite stability may not fully predict GC resistance to anti-PD1 therapy [105].
For HER2-positive patients, studies indicate a good objective response rate to ICI treatment. The efficacy of ICIs in patients with liver metastases from GC is associated with peritoneal metastasis status, and HER2-positive patients may derive greater clinical benefit [106]. In metastatic/unresectable HER2-negative GC patients, those with a higher relative abundance of Lactobacillus exhibited better responses to immunotherapy and longer PFS, suggesting that Lactobacillus may serve as a novel adjuvant to enhance the efficacy of immunotherapy in GC [107].
Beyond the traditional classifications, some studies have reclassified GC patients to provide guidance for immunotherapy and precision medicine [108,109,110,111,112,113,114,115,116], such as the immune-inflamed, immune-excluded, and immune-desert phenotypes mentioned above. In immune-desert GC, epithelial-mesenchymal transition (EMT) signaling is highly enriched, rendering these tumors insensitive to CTLA-4 blockade [117]. Another study utilized a sample-specific edge perturbation matrix based on global immune gene network backgrounds to identify four molecular network subtypes of GC (MNG). Among these, MNG-1 exhibited the best prognosis with robust cell cycle activity, while MNG-2 was enriched for the immune-hot phenotype, showing potential responsiveness to immunotherapy. MNG-3 and MNG-4 were associated with EMT and had poorer prognoses. Notably, MNG-4 displayed chromosomal instability and an immune-desert microenvironment, showing a propensity for metastasis and resistance to immunotherapy [118].
Additionally, several studies have developed predictive models to classify GC patients based on their immunotherapy outcomes, helping to predict prognosis and treatment response [119,120,121]. For instance, the epigenetic modification disorder score, characterized by high FTO expression and low HDAC1 expression, showed features of immune suppression [122]; the immunogenic cell death-related gene risk score (ICDRS), where patients with low ICDRS had better prognoses and were more sensitive to immunotherapy [123]; the DNA damage repair (DDR) signature score, where patients with low DDR signature scores may not benefit from adjuvant chemotherapy or anti-PD1 monoclonal antibody treatment [124]; the ICI score system, where a low ICI score was associated with increased tumor mutational burden (TMB) and served as a potential prognostic and predictive biomarker for chemotherapy and immunotherapy [125]; and the stromal score, where patients with low stromal scores had higher TMB and MSI, making them more sensitive to PD1/PD-L1 ICIs. Conversely, high stromal score subtypes exhibited activation of transforming growth factors and EMT, potentially leading to T-cell suppression and resistance to immunotherapy [126].
The role of gut microbiota in immunotherapy resistance in GC
The gut microbiota has emerged as a crucial factor in modulating immune responses and significantly influencing the efficacy of immunotherapy [127, 128]. In the context of GC, dysbiosis (microbial imbalance) can impact the tumor-immune microenvironment, contributing to resistance to ICIs [129, 130]. Specific microbial signatures have been shown to influence the effectiveness of ICIs by modulating T-cell function, antigen presentation, and immune checkpoint expression [131]. Gut microbes such as Bacteroides fragilis and Faecalibacterium prausnitzii have been associated with a robust T-cell response. These microbes promote the differentiation of CD4+ and CD8 + T cells into effector T cells, enhancing antitumor immunity [132, 133]. On the other hand, dysbiosis, characterized by an overgrowth of bacteria like Enterococcus faecalis or Fusobacterium nucleatum, may impair T-cell activation and reduce T-cell infiltration into the tumor, contributing to immunotherapy resistance [134, 135].
In GC, helicobacter pylori (HP) infection upregulates the expression of CD80 and CD86 in gastric epithelial cells and activates T-cell response [136]. In addition, previous studies have shown that HP inhibits the proliferation of CD4 + T cells and reduces the synthesis of IL-2 and IFN-g by upregulating the expression of PD-L1 on gastric epithelial cells [137, 138]. It has also been shown that HP infection can induce IgA production by B cells by activating Group 2 innate lymphocytes [139]. In addition, Methylbacterium in gastric cancer tissue inhibited CD8+ tissue-resident memory T cells in TME while limiting TGF-b expression [140]. A retrospective study showed that Stenotrophomonas and Selenomonas were positively associated with BDCA2+ plasmacytoid DC (pDC) and Foxp3+ Treg. Comamonas is negatively correlated with BDCA2+ pDC, which is involved in the immune escape of GC cells [141]. In addition to the gut microbiota itself, its metabolites also play an important role in cancer immunity. Studies have shown that intestinal microbial metabolites can regulate immune cell phenotype and function by regulating the secretion of immunosuppressive cytokines [142]. These metabolites can enhance immune cell function by binding to immune cells [143,144,145]. For example, studies have shown that short chain fatty acids (SCFAs) can maintain intestinal homeostasis by promoting IL-10 production in Th1 cells [144, 146, 147]. Another study has shown that SCFAs inhibits histone deacetylase by binding to GPR41, and promotes the production of IL22 by CD4 + T cells, thereby inhibiting inflammation [148].
In addition to regulating tumor immunity, intestinal flora can affect the efficacy of tumor immunotherapy. Choi et al. found that ICB therapy induces an enhanced antitumor-immune response by metastasizing to secondary lymphatic organs of tumors and intestinal bacteria such as bifidobacterium, streptococcus, and Lactobacillus [149]. Several studies have confirmed that the microbiome and its metabolites may have a broad impact on anti-gastric cancer immunotherapy mediated by cytokine secretion and enhanced T-cell infiltration [150, 151]. GC can be divided into four types: EBV positive, MSI, genomic stability, and chromosomal instability [152]. GC large-scale microbiota profiles from two demographically distinct cohorts showed that Selenoides, Bacteroides, and porphyromonas were the top three microorganisms in MSI high GC patients [153]. In addition, in addition to high levels of MSI and EBV-positive status, HP infection is not only an indicator of high PD-L1 expression but also an indicator of poor prognosis after immunotherapy [138, 154]. This may be a predictor of immunotherapy efficacy in GC patients.
Strategies to overcome immunotherapy resistance
Targeting potential therapeutic resistance pathways
Overcoming immunotherapy resistance in GC requires a precise approach that targets specific molecular pathways contributing to immune evasion. Several potential targets have been identified, including immune checkpoint molecules, immunosuppressive cells, and signaling pathways involved in the TME. By focusing on these key pathways, it is possible to modulate the immune response and improve the efficacy of existing treatments. Identifying and inhibiting these resistance mechanisms holds promise for improving patient outcomes and overcoming the limitations of current immunotherapies.
Several classical tumor targets also play crucial roles in immunotherapy resistance in GC. VISTA, predominantly expressed on TAMs, is linked to poor clinical prognosis and reduced response to immunotherapy. In GC, VISTA+ TAMs exhibit a mixed phenotype that impairs CD8 + T-cell function. Blocking VISTA can reprogram TAMs into a pro-inflammatory state, thereby reactivating CD8 + T cells, promoting tumor cell apoptosis, and enhancing the efficacy of PD1 inhibitors [155]. The loss of Smad4 in GC confers an immune evasion advantage. Unlike their Smad4-expressing counterparts, Smad4-deficient gastric organoids form tumors in immunocompetent mice. These GC cells secrete CXCL1, inhibiting DC differentiation and promoting granulocytic MDSC (G-MDSC) accumulation, while also enhancing CD133+ cancer stem cell-like populations. Moreover, Smad4 deficiency upregulates PD-L1 expression and downregulates 4-1BBL, leading to immune evasion. Dual checkpoint blockade with anti-PD-L1 and anti-CTLA-4 antibodies or treatment with agonistic anti-4-1BB antibodies effectively targets Smad4-deficient xenografts [156]. Elevated VCAN expression is associated with poor prognosis in GC and resistance to immunotherapy. Patients with low VCAN expression benefit more from adjuvant chemotherapy and radiotherapy. High VCAN expression correlates with increased CAF infiltration and enrichment of stroma-related pathways, suggesting VCAN is a promising biomarker for predicting treatment response [157].
In GC, CAFs secrete SERPINE2, promoting an immunosuppressive microenvironment and contributing to immune evasion and treatment resistance. Targeting CAF-derived SERPINE2 could be a potential strategy to overcome immunotherapy resistance [158]. Additionally, MFAP2+ CAFs, through the release of macrophage migration inhibitory factors, influence T cells, B cells, and macrophages to create an immunosuppressive environment, further promoting treatment resistance. These findings highlight the potential of MFAP2+ CAFs as therapeutic targets in GC [159]. Overexpression of COX7A1 in GC regulates fibroblast abundance and communication with immune cells, inducing immune evasion. Monitoring COX7A1 expression may help predict prognosis, chemotherapy resistance, and immunotherapy outcomes [160]. Furthermore, DAZ-interacting zinc finger protein 1 (DZIP1) is upregulated in both CAFs and malignant epithelial cells in GC and is strongly associated with the mesenchymal phenotype. DZIP1 promotes CAF proliferation and enhances EMT in GC cells, driving angiogenesis and invasion. It is also linked to immunosuppressive TME, leading to poor responses to immunotherapy, making DZIP1 a potential target for overcoming resistance [161] (Fig. 3).
Notably, ATRX mutations are more frequent in female GC patients than in males. Female patients with ATRX mutations exhibit higher MSI, TMB, and PD-L1 expression, as well as increased anti-cancer immune indicators such as IFN-γ signaling, cytolytic activity, and antigen-presentation machinery scores. ATRX mutations may enhance immunogenicity by affecting DDR pathways, suggesting that ATRX could serve as a potential predictive biomarker for ICI therapy in female GC patients [162].
A comprehensive summary of additional potential targets to enhance immunotherapy sensitivity in GC is provided in Table 2 for easier reference.
Combination therapies
While ICIs have shown significant therapeutic potential, many patients experience limited or short-lived responses due to the complexity of tumor-immune interactions and the presence of immunosuppressive mechanisms within the TME. To address these challenges, combination approaches that incorporate ICIs with other therapeutic modalities, such as chemotherapy, targeted therapies, radiation, or additional immune-modulating agents, have been explored. These combinations aim to enhance antitumor immunity, overcome intrinsic and acquired resistance, and ultimately improve clinical outcomes. By targeting multiple pathways simultaneously, combination therapies have the potential to convert immunologically “cold” tumors into “hot” ones, thereby increasing the likelihood of a sustained response to immunotherapy [163].
Chemotherapy has been shown to have an immunomodulatory effect, enhancing tumor immunogenicity by promoting immunogenic cell death and increasing the release of tumor antigens [164,165,166]. Studies have demonstrated that combining PD1/PD-L1 inhibitors with chemotherapeutic agents like fluoropyrimidine, oxaliplatin, and irinotecan improves immune recognition by upregulating MHC-I expression and reducing tumor-associated immunosuppressive cells, such as MDSCs and Tregs. For instance, in the KEYNOTE-062 trial, pembrolizumab combined with chemotherapy showed promising results in improving the overall response rate and PFS in advanced GC patients [167,168,169,170,171]. The clinical trials of ICIs combined with chemotherapy in GC are summarized in Table 1.
In the previous section, we discussed the molecular targets associated with ICI resistance in GC. Targeted therapies against these pathways can modulate the TME and enhance immune responses. For instance, anti-HER2 drugs combined with immunotherapy significantly increase the infiltration of NK cells, CD8 + T cells, and B lymphocytes in GC. In responsive patients, the interactions between these cells are strengthened, particularly through the CCL3/CCL4-CCR5 signaling pathway, where NK cells recruit CD8 + T cells. Meanwhile, B lymphocytes interact with M2 macrophages and Tregs via multiple signaling pathways, inhibiting immune resistance [64]. Similarly, anti-VEGF drugs like ramucirumab normalize aberrant tumor vasculature, improving T-cell infiltration and reducing the immunosuppressive environment. The combination of ramucirumab with pembrolizumab has been explored in several studies [3]. Relevant clinical trials of ICIs combined with targeted therapies are summarized in Table 1.
Anti-angiogenic therapies that target VEGF pathways have been shown to enhance the efficacy of ICIs by reducing the immunosuppressive effects of TME. VEGF not only promotes tumor angiogenesis but also impairs immune cell trafficking and promotes Treg and MDSC infiltration. By combining VEGF inhibitors with ICIs, the normalization of blood vessels can enhance the immune system’s ability to access the tumor [172,173,174]. Trials such as the REGONIVO study, which combined regorafenib (a multi-kinase inhibitor) with nivolumab (a PD1 inhibitor), demonstrated promising activity in heavily pretreated GC patients, suggesting that anti-angiogenic agents may restore immune surveillance and improve ICI efficacy [175].
Radiotherapy can induce a systemic immune response known as the “abscopal effect,” where localized radiation leads to the destruction of distant, non-irradiated tumor sites through immune-mediated mechanisms [176, 177]. In GC, radiotherapy has been shown to increase the release of tumor-associated antigens and enhance antigen presentation, thus sensitizing tumors to ICIs. The combination of radiation and ICIs is currently being explored in clinical settings, with early results showing enhanced antitumor effects through increased T-cell activation and inhibition of immune-suppressive pathways [178,179,180].
Epigenetic alterations are key drivers of immune resistance in cancer. Agents targeting epigenetic modifications, such as DNA methyltransferase inhibitors and histone deacetylase inhibitors, can reprogram the TME to become more immunogenic [181,182,183]. For example, HDAC inhibitors can enhance the expression of immune-related genes, promote the activity of NK cells, and increase tumor antigen presentation [181]. Combining HDAC inhibitors like vorinostat or romidepsin with PD1 inhibitors has shown preclinical promise, offering a rationale for clinical trials to test their efficacy in reversing resistance to ICIs [184, 185].
Dual ICI therapy is another promising combination approach [186]. The combination of anti-PD1 and anti-CTLA-4 therapies can increase the infiltration of tumor-specific CD8 + T cells, although many of these T cells exhibit an exhausted phenotype. Studies have shown that in resistant tumors, abnormal activation of the JAK-STAT pathway is observed, along with infiltration of macrophages, neutrophils, and Tregs. Introducing JAK inhibitors has been shown to restore CD8 + T-cell function and reshape the immunosuppressive TME, further enhancing the efficacy of dual ICI therapy [187]. Additionally, a clinical case reported complete remission in a HER2-positive advanced GEJ cancer patient through dual PD1/CTLA-4 bispecific immunotherapy combined with chemotherapy, providing a novel and effective treatment option for HER2-positive patients. This approach should be considered as an alternative when trastuzumab is not feasible [188].
Emerging immunotherapy
Emerging therapeutic strategies aim to develop novel immunotherapy approaches or integrate innovative techniques within existing frameworks to enhance the response of GC patients to immunotherapy. In recent years, several promising cutting-edge strategies have rapidly evolved, particularly in the regulation of antitumor-immune responses and personalized treatment.
Cell-based therapies, such as chimeric antigen receptor T-cell (CAR-T) therapy, have achieved significant success in hematologic malignancies and are now being extended to solid tumors. For GC, CAR-T cells targeting specific antigens like CLDN18.2 and MET are under development and testing [189,190,191,192,193]. Additionally, new immune effector cell therapies, such as CAR-NK cell therapy, have demonstrated favorable safety profiles and preclinical efficacy [194, 195]. Compared to CAR-T cells, CAR-NK cells offer advantages such as lower toxicity and the lack of need for matched donor sources. Specifically, in the TME of GC, CAR-NK cells enhance immune cell infiltration and antitumor activity.
The gut microbiota plays a crucial role in host immune responses and is closely linked to the efficacy of ICIs [149, 196, 197]. Studies have shown that the abundance of certain gut microbes positively correlates with the response of GC patients to ICIs. For instance, the increased presence of Lactobacillus and Bifidobacterium is associated with improved efficacy of PD1/PD-L1 inhibitors [107, 198,199,200]. By modulating the gut microbiota through methods such as probiotics, fecal transplantation, or selective amplification of specific microbial populations, the TME can be improved, thereby enhancing the sensitivity of patients to immunotherapy [129, 201, 202]. These microbiota-based therapies have entered clinical trials and may become an important component of GC immunotherapy in the future [203, 204].
RNA vaccine technology, such as mRNA vaccines, has garnered attention due to the successful development of COVID-19 vaccines [205, 206]. Beyond infectious disease prevention, mRNA vaccines hold great potential in cancer immunotherapy [207, 208]. Neoantigen vaccines for GC can be personalized based on specific mutations or antigens in the patient’s tumor, designed to trigger a stronger antitumor-immune response [209, 210]. This personalized vaccine strategy enhances T-cell recognition of GC-specific antigens and promotes the generation of memory T cells, thereby improving long-term immune surveillance. Moreover, mRNA vaccines can be combined with ICIs to maximize their therapeutic benefits [211, 212].
The regulation of metabolic pathways within the TME also affects the efficacy of immunotherapy. Studies have found that metabolic competition in the immunosuppressive TME, such as lactate accumulation and glucose depletion, inhibits the function of effector T cells [213,214,215]. By using metabolic modulators, such as inhibitors of lactate dehydrogenase or glucose transporter protein, the metabolic state of the TME can be reprogrammed, restoring T-cell antitumor activity [216, 217]. Furthermore, regulating mitochondrial function in tumor cells is also considered a potential approach to enhancing the efficacy of immunotherapy [218, 219].
Additionally, advances in CRISPR-Cas9 technology offer new possibilities for GC treatment, especially in enhancing immunotherapy sensitivity and overcoming resistance. Through gene editing, inhibitory receptors like PD1 can be knocked out in T cells, or resistance-related genes can be knocked out in tumor cells, thereby improving responsiveness to immunotherapy [220, 221]. These emerging therapeutic strategies provide new opportunities to overcome resistance to GC immunotherapy. As technology continues to advance, frontier areas such as cell therapy, gene editing, gut microbiota modulation, RNA vaccines, and metabolic regulation will further expand the applications of GC immunotherapy and provide more evidence for individualized treatment approaches.
Future perspectives and challenges
Despite notable progress with ICIs in the treatment of GC, several limitations persist. First, the heterogeneity of GC leads to significant variability in patient responses to immunotherapy, and current biomarkers are insufficient for effectively predicting therapeutic outcomes. Most studies focus on PD-L1 expression levels, but this singular marker does not adequately explain the complex mechanisms of resistance. Moreover, immune-suppressive factors within the TME, such as TAMs, Treg, and metabolite accumulation, have not been thoroughly investigated, hindering our understanding of their role in ICI resistance. These gaps in knowledge obstruct the development of more precise immunotherapeutic strategies.
Future research should prioritize uncovering new resistance mechanisms and biomarkers. For instance, besides PD-L1, other immune evasion pathways—such as tumor neoantigen burden, metabolic pathway abnormalities, and gut microbiome influences—should be the focus of investigation. Integrating multi-omics data, including genomics, transcriptomics, metabolomics, and single-cell sequencing technologies, could help identify additional key molecules associated with immunotherapy sensitivity and resistance.
Optimizing combination therapies is also a crucial future direction. While dual ICI therapies targeting PD1/PD-L1 and CTLA-4 have shown potential, issues related to toxicity and tolerability remain unresolved. Future studies should explore combinations with metabolic inhibitors, gut microbiome modulators, or agents targeting the TME to enhance the efficacy of immunotherapy.
Translating laboratory research findings into clinical applications presents several challenges. Clinical trial designs and patient recruitment must account for GC’s heterogeneity and treatment response variations among different subtypes. Although preclinical models have demonstrated the potential of combination therapies, their effectiveness, and safety in actual patients require validation through large-scale clinical trials. Developing universal protocols that can be applied to the majority of patients remains challenging due to the individualized nature of immunotherapy.
Furthermore, toxicity management is a critical challenge in the clinical translation of immunotherapy. Particularly with dual ICB or combined with other therapies, the risk of adverse effects complicates clinical application. Future clinical research needs to focus on balancing efficacy with safety, and exploring safer and more effective treatment combinations and dosing regimens.
In summary, future research must address resistance mechanisms in GC immunotherapy, refine personalized combination treatment strategies, and overcome translational obstacles from laboratory to clinical practice. Despite the challenges, ongoing innovation and multidisciplinary collaboration hold promise for advancing the effectiveness of immunotherapy for GC patients.
Conclusion
In conclusion, overcoming immunotherapy resistance in GC requires a multifaceted approach. While ICIs have made strides in treatment, significant challenges remain, including tumor heterogeneity and insufficient biomarkers for predicting response. Future research should focus on identifying novel resistance mechanisms, exploring new biomarkers, and optimizing combination therapies to enhance efficacy. Translating these findings into clinical practice presents additional hurdles, such as managing toxicity and designing effective clinical trials. Nonetheless, continued innovation and collaborative efforts are essential to advancing therapeutic strategies and improving outcomes for GC patients.
References
Zeng Y, Jin RU. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin Cancer Biol. 2022;86:566–82.
Lopez MJ, Carbajal J, Alfaro AL, Saravia LG, Zanabria D, Araujo JM, et al. Characteristics of gastric cancer around the world. Crit Rev Oncol Hematol. 2023;181:103841.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–48.
Kim HD, Ryu MH, Kang YK. Adjuvant treatment for locally advanced gastric cancer: an Asian perspective. Gastric Cancer. 2024;27:439–50.
Jin X, Liu Z, Yang D, Yin K, Chang X. Recent progress and future perspectives of immunotherapy in advanced gastric cancer. Front Immunol. 2022;13:948647.
Guan WL, He Y, Xu RH. Gastric cancer treatment: recent progress and future perspectives. J Hematol Oncol. 2023;16:57.
Zhu Z, Jin Y, Zhou J, Chen F, Chen M, Gao Z, et al. PD1/PD-L1 blockade in clear cell renal cell carcinoma: mechanistic insights, clinical efficacy, and future perspectives. Mol Cancer. 2024;23:146.
Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ. 2018;362:k3529.
Takei S, Kawazoe A, Shitara K. The new era of immunotherapy in gastric cancer. Cancers. 2022;14:1054.
Akin Telli T, Bregni G, Camera S, Deleporte A, Hendlisz A, Sclafani F. PD-1 and PD-L1 inhibitors in oesophago-gastric cancers. Cancer Lett. 2020;469:142–50.
Mestrallet G, Brown M, Bozkus CC, Bhardwaj N. Immune escape and resistance to immunotherapy in mismatch repair deficient tumors. Front Immunol. 2023;14:1210164.
Yu Y. Multi-target combinatory strategy to overcome tumor immune escape. Front Med. 2022;16:208–15.
Liu Y, Li C, Lu Y, Liu C, Yang W. Tumor microenvironment-mediated immune tolerance in development and treatment of gastric cancer. Front Immunol. 2022;13:1016817.
Liu K, Yuan S, Wang C, Zhu H. Resistance to immune checkpoint inhibitors in gastric cancer. Front Pharmacol. 2023;14:1285343.
Zhu L, Huang Y, Li H, Shao S. Helicobacter pylori promotes gastric cancer progression through the tumor microenvironment. Appl Microbiol Biotechnol. 2022;106:4375–85.
Xu X, Chen J, Li W, Feng C, Liu Q, Gao W, et al. Immunology and immunotherapy in gastric cancer. Clin Exp Med. 2023;23:3189–204.
Zeng D, Wu J, Luo H, Li Y, Xiao J, Peng J, et al. Tumor microenvironment evaluation promotes precise checkpoint immunotherapy of advanced gastric cancer. J Immunother Cancer. 2021;9:e002467.
Li K, Zhang A, Li X, Zhang H, Zhao L. Advances in clinical immunotherapy for gastric cancer. Biochim Biophys Acta Rev Cancer. 2021;1876:188615.
Kwapisz D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol Immunother. 2021;70:607–17.
Yamamoto S, Kato K. Pembrolizumab for the treatment of esophageal cancer. Expert Opin Biol Ther. 2020;20:1143–50.
Alsina M, Arrazubi V, Diez M, Tabernero J. Current developments in gastric cancer: from molecular profiling to treatment strategy. Nat Rev Gastroenterol Hepatol. 2023;20:155–70.
Mishima S, Kawazoe A, Shitara K. Safety of pembrolizumab in recurrent or advanced gastric cancer expressing PD-L1 refractory to platinum and fluoropyrimidine. Expert Opin Drug Saf. 2020;19:1063–8.
Gervaso L, Ciardiello D, Oliveira RA, Borghesani M, Guidi L, Benini L, et al. Immunotherapy in the neoadjuvant treatment of gastrointestinal tumors: is the time ripe? J Immunother Cancer. 2024;12:e008027.
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:2461–71.
Buder-Bakhaya K, Hassel JC. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-a review from the melanoma perspective and beyond. Front Immunol. 2018;9:1474.
Verschoor YL, van de Haar J, van den Berg JG, van Sandick JW, Kodach LL, van Dieren JM, et al. Neoadjuvant atezolizumab plus chemotherapy in gastric and gastroesophageal junction adenocarcinoma: the phase 2 PANDA trial. Nat Med. 2024;30:519–30.
Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032 Study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol. 2018;36:2836–44.
Shitara K, Ajani JA, Moehler M, Garrido M, Gallardo C, Shen L, et al. Nivolumab plus chemotherapy or ipilimumab in gastro-oesophageal cancer. Nature. 2022;603:942–8.
Janjigian YY, Shitara K, Moehler M, Garrido M, Salman P, Shen L, et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet. 2021;398:27–40.
Kang YK, Chen LT, Ryu MH, Oh DY, Oh SC, Chung HC, et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): a randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2022;23:234–47.
Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1571–80.
Janjigian YY, Kawazoe A, Bai Y, Xu J, Lonardi S, Metges JP, et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet. 2023;402:2197–208.
Rha SY, Oh DY, Yanez P, Bai Y, Ryu MH, Lee J, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for HER2-negative advanced gastric cancer (KEYNOTE-859): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24:1181–95.
Li G, Liu X, Gu C, Ma G, Li S, Ma Z, et al. Mutual exclusivity and co-occurrence patterns of immune checkpoints indicate NKG2A relates to anti-PD-1 resistance in gastric cancer. J Transl Med. 2024;22:718.
Chu Y, Dai E, Li Y, Han G, Pei G, Ingram DR, et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat Med. 2023;29:1550–62.
Chen M, Chen F, Gao Z, Li X, Hu L, Yang S, et al. CAFs and T cells interplay: the emergence of a new arena in cancer combat. Biomed Pharmacother. 2024;177:117045.
Rimal R, Desai P, Daware R, Hosseinnejad A, Prakash J, Lammers T, et al. Cancer-associated fibroblasts: origin, function, imaging, and therapeutic targeting. Adv Drug Deliv Rev. 2022;189:114504.
Cords L, Tietscher S, Anzeneder T, Langwieder C, Rees M, de Souza N, et al. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat Commun. 2023;14:4294.
Chen B, Chan WN, Xie F, Mui CW, Liu X, Cheung AHK, et al. The molecular classification of cancer-associated fibroblasts on a pan-cancer single-cell transcriptional atlas. Clin Transl Med. 2023;13:e1516.
Kennel KB, Bozlar M, De Valk AF, Greten FR. Cancer-associated fibroblasts in inflammation and antitumor immunity. Clin Cancer Res. 2023;29:1009–16.
Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–96.
Yoshida GJ. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res. 2020;39:112.
Tschumperlin DJ, Lagares D. Mechano-therapeutics: targeting mechanical signaling in fibrosis and tumor stroma. Pharmacol Ther. 2020;212:107575.
Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013;437:397–402.
Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715.
Wang W, Li T, Xie Z, Zhao J, Zhang Y, Ruan Y, et al. Integrating single-cell and bulk RNA sequencing data unveils antigen presentation and process-related CAFS and establishes a predictive signature in prostate cancer. J Transl Med. 2024;22:57.
Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics. 2022;12:620–38.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–23.
Dempsey LA. Antigen-presenting CAFs. Nat Immunol. 2022;23:645.
Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 2022;40:656–73.e7.
Akiyama T, Yasuda T, Uchihara T, Yasuda-Yoshihara N, Tan BJY, Yonemura A, et al. Stromal reprogramming through dual PDGFRalpha/beta blockade boosts the efficacy of anti-PD-1 immunotherapy in fibrotic tumors. Cancer Res. 2023;83:753–70.
Wei R, Song J, Pan H, Liu X, Gao J. CPT1C-positive cancer-associated fibroblast facilitates immunosuppression through promoting IL-6-induced M2-like phenotype of macrophage. Oncoimmunology. 2024;13:2352179.
Lu Y, Li D, Cao Y, Ying L, Tao Q, Xiong F, et al. A genomic signature reflecting fibroblast infiltration into gastric cancer is associated with prognosis and treatment outcomes of immune checkpoint inhibitors. Front Cell Dev Biol. 2022;10:862294.
Peng Q, Zhang P, Liu G, Lu L. Integrated single-cell and bulk RNA sequencing analyses identify an immunotherapy nonresponse-related fibroblast signature in gastric cancer. Anticancer Drugs. 2024;35:952–68.
Zheng H, Liu H, Li H, Dou W, Wang X. Weighted gene co-expression network analysis identifies a cancer-associated fibroblast signature for predicting prognosis and therapeutic responses in gastric cancer. Front Mol Biosci. 2021;8:744677.
Li J, Sun J, Zeng Z, Liu Z, Ma M, Zheng Z, et al. Tumour-associated macrophages in gastric cancer: from function and mechanism to application. Clin Transl Med. 2023;13:e1386.
Shi T, Zhang Y, Wang Y, Song X, Wang H, Zhou X, et al. DKK1 Promotes tumor immune evasion and impedes anti-PD-1 treatment by inducing immunosuppressive macrophages in gastric cancer. Cancer Immunol Res. 2022;10:1506–24.
Pei X, Zhang SL, Qiu BQ, Zhang PF, Liu TS, Wang Y. Cancer cell secreted legumain promotes gastric cancer resistance to anti-PD-1 immunotherapy by enhancing macrophage M2 polarization. Pharmaceuticals. 2024;17:951.
Lv K, Sun M, Fang H, Wang J, Lin C, Liu H, et al. Targeting myeloid checkpoint Siglec-10 reactivates antitumor immunity and improves anti-programmed cell death 1 efficacy in gastric cancer. J Immunother Cancer. 2023;11:e007669.
Tang Y, Zhou C, Li Q, Cheng X, Huang T, Li F, et al. Targeting depletion of myeloid-derived suppressor cells potentiates PD-L1 blockade efficacy in gastric and colon cancers. Oncoimmunology. 2022;11:2131084.
Tsutsumi C, Ohuchida K, Katayama N, Yamada Y, Nakamura S, Okuda S, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells contribute to the development of an immunosuppressive tumor microenvironment in gastric cancer. Gastric Cancer. 2024;27:248–62.
Luo Q, Dong Z, Xie W, Fu X, Lin L, Zeng Q, et al. Apatinib remodels the immunosuppressive tumor ecosystem of gastric cancer enhancing anti-PD-1 immunotherapy. Cell Rep. 2023;42:112437.
Cui JX, Xu XH, He T, Liu JJ, Xie TY, Tian W, et al. L-kynurenine induces NK cell loss in gastric cancer microenvironment via promoting ferroptosis. J Exp Clin Cancer Res. 2023;42:52.
Jiang L, Zhao X, Li Y, Hu Y, Sun Y, Liu S, et al. The tumor immune microenvironment remodeling and response to HER2-targeted therapy in HER2-positive advanced gastric cancer. IUBMB Life. 2024;76:420–36.
Masoumi J, Ghorbaninezhad F, Saeedi H, Safaei S, Khaze Shahgoli V, Ghaffari Jolfayi A, et al. siRNA-mediated B7H7 knockdown in gastric cancer lysate-loaded dendritic cells amplifies expansion and cytokine secretion of autologous T cells. Biomedicines. 2023;11:3212.
Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34.
Yuan Z, Li Y, Zhang S, Wang X, Dou H, Yu X, et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. 2023;22:48.
Li C, Qiu S, Liu X, Guo F, Zhai J, Li Z, et al. Extracellular matrix-derived mechanical force governs breast cancer cell stemness and quiescence transition through integrin-DDR signaling. Signal Transduct Target Ther. 2023;8:247.
Dong S, Zhang S, Zhao P, Lin G, Ma X, Xu J, et al. A combined analysis of bulk and single-cell sequencing data reveals that depleted extracellular matrix and enhanced immune processes co-contribute to fluorouracil beneficial responses in gastric cancer. Front Immunol. 2022;13:999551.
Zhang X, Wu X, Sun Y, Chu Y, Liu F, Chen C. TRIM44 regulates tumor immunity in gastric cancer through LOXL2-dependent extracellular matrix remodeling. Cell Oncol. 2023;46:423–35.
Ohno S, Tachibana M, Fujii T, Ueda S, Kubota H, Nagasue N. Role of stromal collagen in immunomodulation and prognosis of advanced gastric carcinoma. Int J Cancer. 2002;97:770–4.
Ferreira RM, Figueiredo J, Pinto-Ribeiro I, Gullo I, Sgouras DN, Carreto L, et al. Activation of Laminin gamma2 by Helicobacter pylori promotes invasion and survival of gastric cancer cells with E-cadherin defects. J Infect Dis. 2022;226:2226–37.
Pan S, Zhu J, Liu P, Wei Q, Zhang S, An W, et al. FN1 mRNA 3’-UTR supersedes traditional fibronectin 1 in facilitating the invasion and metastasis of gastric cancer through the FN1 3’-UTR-let-7i-5p-THBS1 axis. Theranostics. 2023;13:5130–50.
Jang M, Koh I, Lee JE, Lim JY, Cheong JH, Kim P. Increased extracellular matrix density disrupts E-cadherin/beta-catenin complex in gastric cancer cells. Biomater Sci. 2018;6:2704–13.
Lu Y, Jin Z, Hou J, Wu X, Yu Z, Yao L, et al. Calponin 1 increases cancer-associated fibroblasts-mediated matrix stiffness to promote chemoresistance in gastric cancer. Matrix Biol. 2023;115:1–15.
Zhang X, Zhao Y, Chen X. Collagen extracellular matrix promotes gastric cancer immune evasion by activating IL4I1-AHR signaling. Transl Oncol. 2024;49:102113.
Honda CK, Kurozumi S, Fujii T, Pourquier D, Khellaf L, Boissiere F, et al. Cancer-associated fibroblast spatial heterogeneity and EMILIN1 expression in the tumor microenvironment modulate TGF-beta activity and CD8(+) T-cell infiltration in breast cancer. Theranostics. 2024;14:1873–85.
Liu X, Li J, Yang X, Li X, Kong J, Qi D, et al. Carcinoma-associated fibroblast-derived lysyl oxidase-rich extracellular vesicles mediate collagen crosslinking and promote epithelial-mesenchymal transition via p-FAK/p-paxillin/YAP signaling. Int J Oral Sci. 2023;15:32.
Sleeboom JJF, van Tienderen GS, Schenke-Layland K, van der Laan LJW, Khalil AA, Verstegen MMA. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci Transl Med. 2024;16:eadg3840.
Bian Y, Xiang Z, Wang Y, Ren Q, Chen G, Xiang B, et al. Immunomodulatory roles of metalloproteinases in rheumatoid arthritis. Front Pharmacol. 2023;14:1285455.
Levin M, Udi Y, Solomonov I, Sagi I. Next generation matrix metalloproteinase inhibitors - novel strategies bring new prospects. Biochim Biophys Acta Mol Cell Res. 2017;1864:1927–39.
Hessmann E, Buchholz SM, Demir IE, Singh SK, Gress TM, Ellenrieder V, et al. Microenvironmental determinants of pancreatic cancer. Physiol Rev. 2020;100:1707–51.
Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122:899–910.
Xing X, Guo J, Ding G, Li B, Dong B, Feng Q, et al. Analysis of PD1, PDL1, PDL2 expression and T cells infiltration in 1014 gastric cancer patients. Oncoimmunology. 2018;7:e1356144.
Huang C, Chen B, Wang X, Xu J, Sun L, Wang D, et al. Gastric cancer mesenchymal stem cells via the CXCR2/HK2/PD-L1 pathway mediate immunosuppression. Gastric Cancer. 2023;26:691–707.
Sun L, Wang Q, Chen B, Zhao Y, Shen B, Wang H, et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018;9:928.
Miliotis C, Slack FJ. miR-105-5p regulates PD-L1 expression and tumor immunogenicity in gastric cancer. Cancer Lett. 2021;518:115–26.
Ma ZL, Ding YL, Jing J, Du LN, Zhang XY, Liu HM, et al. ATRA promotes PD-L1 expression to control gastric cancer immune surveillance. Eur J Pharmacol. 2022;920:174822.
Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71:264–79.
Hu C, Song J, Kwok T, Nguyen EV, Shen X, Daly RJ. Proteome-based molecular subtyping and therapeutic target prediction in gastric cancer. Mol Oncol. 2024;18:1437–59.
Hu X, Wang Z, Wang Q, Chen K, Han Q, Bai S, et al. Molecular classification reveals the diverse genetic and prognostic features of gastric cancer: a multi-omics consensus ensemble clustering. Biomed Pharmacother. 2021;144:112222.
Wu B, Zhang B, Li B, Wu H, Jiang M. Cold and hot tumors: from molecular mechanisms to targeted therapy. Signal Transduct Target Ther. 2024;9:274.
Zheng S, Wang W, Shen L, Yao Y, Xia W, Ni C. Tumor battlefield within inflamed, excluded or desert immune phenotypes: the mechanisms and strategies. Exp Hematol Oncol. 2024;13:80.
Park S, Ock CY, Kim H, Pereira S, Park S, Ma M, et al. Artificial intelligence-powered spatial analysis of tumor-infiltrating lymphocytes as complementary biomarker for immune checkpoint inhibition in non-small-cell lung cancer. J Clin Oncol. 2022;40:1916–28.
Indini A, Massi D, Pirro M, Roila F, Grossi F, Sahebkar A, et al. Targeting inflamed and non-inflamed melanomas: biological background and clinical challenges. Semin Cancer Biol. 2022;86:477–90.
Bai S, Chen L, Yan Y, Li R, Zhou Y, Wang X, et al. Exploration of different hypoxia patterns and construction of a hypoxia-related gene prognostic index in colorectal cancer. Front Immunol. 2022;13:853352.
Broz MT, Ko EY, Ishaya K, Xiao J, De Simone M, Hoi XP, et al. Metabolic targeting of cancer associated fibroblasts overcomes T-cell exclusion and chemoresistance in soft-tissue sarcomas. Nat Commun. 2024;15:2498.
Song D, Wu Y, Li J, Liu J, Yi Z, Wang X, et al. Insulin-like growth factor 2 drives fibroblast-mediated tumor immunoevasion and confers resistance to immunotherapy. J Clin Invest. 2024;134:e183366.
Ilyas SI, Affo S, Goyal L, Lamarca A, Sapisochin G, Yang JD, et al. Cholangiocarcinoma - novel biological insights and therapeutic strategies. Nat Rev Clin Oncol. 2023;20:470–86.
Li X, Gulati M, Larson AC, Solheim JC, Jain M, Kumar S, et al. Immune checkpoint blockade in pancreatic cancer: tTrudging through the immune desert. Semin Cancer Biol. 2022;86:14–27.
Andre T, Tougeron D, Piessen G, de la Fouchardiere C, Louvet C, Adenis A, et al. Neoadjuvant nivolumab plus ipilimumab and adjuvant nivolumab in localized deficient mismatch repair/microsatellite instability-high gastric or esophagogastric junction adenocarcinoma: the GERCOR NEONIPIGA Phase II Study. J Clin Oncol. 2023;41:255–65.
Janjigian YY, Sanchez-Vega F, Jonsson P, Chatila WK, Hechtman JF, Ku GY, et al. Genetic predictors of response to systemic therapy in esophagogastric cancer. Cancer Discov. 2018;8:49–58.
Fuca G, Cohen R, Lonardi S, Shitara K, Elez ME, Fakih M, et al. Ascites and resistance to immune checkpoint inhibition in dMMR/MSI-H metastatic colorectal and gastric cancers. J Immunother Cancer. 2022;10:e004001.
Wang Z, Wang X, Xu Y, Li J, Zhang X, Peng Z, et al. Mutations of PI3K-AKT-mTOR pathway as predictors for immune cell infiltration and immunotherapy efficacy in dMMR/MSI-H gastric adenocarcinoma. BMC Med. 2022;20:133.
Chen KH, Yuan CT, Tseng LH, Shun CT, Yeh KH. Case report: mismatch repair proficiency and microsatellite stability in gastric cancer may not predict programmed death-1 blockade resistance. J Hematol Oncol. 2016;9:29.
Liang H, Li Z, Huang Z, Wu C, Qiu Y, Liang Y, et al. Prognostic characteristics and clinical response to immunotherapy targeting programmed cell death 1 for patients with advanced gastric cancer with liver metastases. Front Immunol. 2022;13:1015549.
Han Z, Cheng S, Dai D, Kou Y, Zhang X, Li F, et al. The gut microbiome affects response of treatments in HER2-negative advanced gastric cancer. Clin Transl Med. 2023;13:e1312.
Weng S, Li M, Deng J, Xu H, Ren Y, Zhou Z, et al. Epigenetically regulated gene expression profiles decipher four molecular subtypes with prognostic and therapeutic implications in gastric cancer. Clin Epigenet. 2023;15:64.
Shao J, Zhang W, Li Y, Tang Y, Fan L. Metabolic and immune-related gene signatures: predictive stratification and prognostic implications in gastric cancer. J Gene Med. 2024;26:e3635.
Wang H, Wu J, Ling R, Li F, Yang Q, He J, et al. Fibroblast-derived LPP as a biomarker for treatment response and therapeutic target in gastric cancer. Mol Ther Oncolytics. 2022;24:547–60.
Sundar R, Huang KK, Qamra A, Kim KM, Kim ST, Kang WK, et al. Epigenomic promoter alterations predict for benefit from immune checkpoint inhibition in metastatic gastric cancer. Ann Oncol. 2019;30:424–30.
Wang J, Feng J, Chen X, Weng Y, Wang T, Wei J, et al. Integrated multi-omics analysis and machine learning identify hub genes and potential mechanisms of resistance to immunotherapy in gastric cancer. Aging. 2024;16:7331–56.
Gu L, Ding D, Wei C, Zhou D. Cancer-associated fibroblasts refine the classifications of gastric cancer with distinct prognosis and tumor microenvironment characteristics. Front Oncol. 2023;13:1158863.
Wu X, Zhou F, Cheng B, Tong G, Chen M, He L, et al. Immune activity score to assess the prognosis, immunotherapy and chemotherapy response in gastric cancer and experimental validation. PeerJ. 2023;11:e16317.
Jiang Q, Chen L, Chen H, Tang Z, Liu F, Sun Y. Integrated analysis of stemness-related LncRNAs Helps predict the immunotherapy responsiveness of gastric cancer patients. Front Cell Dev Biol. 2021;9:739509.
Yang H, Gou X, Feng C, Zhang Y, Chai F, Hong N, et al. Computed tomography-detected extramural venous invasion-related gene signature: a potential negative biomarker of immune checkpoint inhibitor treatment in patients with gastric cancer. J Transl Med. 2023;21:4.
Cao LL, Lu H, Soutto M, Bhat N, Chen Z, Peng D, et al. Multivalent tyrosine kinase inhibition promotes T cell recruitment to immune-desert gastric cancers by restricting epithelial-mesenchymal transition via tumour-intrinsic IFN-gamma signalling. Gut. 2023;72:2038–50.
Xu H, Fu X, Liu B, Weng S, Guo C, Quan L, et al. Immune perturbation network identifies an EMT subtype with chromosomal instability and tumor immune-desert microenvironment. iScience. 2023;26:107871.
Fu Y, Wang B, Fu P, Zhang L, Bao Y, Gao ZZ. Delineation of fatty acid metabolism in gastric cancer: therapeutic implications. World J Clin Cases. 2023;11:4800–13.
Liu R, Chu W, Liu X, Hong J, Wang H. Establishment of Golgi apparatus-related genes signature to predict the prognosis and immunotherapy response in gastric cancer patients. Medicine. 2024;103:e37439.
Kovacs SA, Fekete JT, Gyorffy B. Predictive biomarkers of immunotherapy response with pharmacological applications in solid tumors. Acta Pharmacol Sin. 2023;44:1879–89.
Yuan C, Zhang J, Deng C, Xia Y, Li B, Meng S, et al. Crosstalk of histone and RNA modifications identified a stromal-activated subtype with poor survival and resistance to immunotherapy in gastric cancer. Front Pharmacol. 2022;13:868830.
Liu Z, Sun L, Peng X, Liu S, Zhu Z, Huang C. An immunogenic cell death-related signature predicts prognosis and immunotherapy response in stomach adenocarcinoma. Apoptosis. 2023;28:1564–83.
Lou S, Wang Y, Zhang J, Yin X, Zhang Y, Wang Y, et al. Patient-level DNA damage repair pathway profiles and anti-tumor immunity for gastric cancer. Front Immunol. 2021;12:806324.
Jiang Q, Sun J, Chen H, Ding C, Tang Z, Ruan Y, et al. Establishment of an immune cell infiltration score to help predict the prognosis and chemotherapy responsiveness of gastric cancer patients. Front Oncol. 2021;11:650673.
Ren Q, Zhu P, Zhang H, Ye T, Liu D, Gong Z, et al. Identification and validation of stromal-tumor microenvironment-based subtypes tightly associated with PD-1/PD-L1 immunotherapy and outcomes in patients with gastric cancer. Cancer Cell Int. 2020;20:92.
Wong CC, Yu J. Gut microbiota in colorectal cancer development and therapy. Nat Rev Clin Oncol. 2023;20:429–52.
Lu Y, Yuan X, Wang M, He Z, Li H, Wang J, et al. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J Hematol Oncol. 2022;15:47.
Wang Y, Han W, Wang N, Han M, Ban M, Dai J, et al. The role of microbiota in the development and treatment of gastric cancer. Front Oncol. 2023;13:1224669.
Raoul P, De Gaetano V, Sciaraffia G, Ormea G, Cintoni M, Pozzo C, et al. Gastric cancer, immunotherapy, and nutrition: the role of microbiota. Pathogens. 2024;13:357.
Simpson RC, Shanahan ER, Batten M, Reijers ILM, Read M, Silva IP, et al. Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiome. Nat Med. 2022;28:2344–52.
Cameron G, Nguyen T, Ciula M, Williams SJ, Godfrey DI. Glycolipids from the gut symbiont Bacteroides fragilis are agonists for natural killer T cells and induce their regulatory differentiation. Chem Sci. 2023;14:7887–96.
Guo J, Meng F, Hu R, Chen L, Chang J, Zhao K, et al. Inhibition of the NF-kappaB/HIF-1alpha signaling pathway in colorectal cancer by tyrosol: a gut microbiota-derived metabolite. J Immunother Cancer. 2024;12:e008831.
de Almeida CV, Taddei A, Amedei A. The controversial role of Enterococcus faecalis in colorectal cancer. Therap Adv Gastroenterol. 2018;11:1756284818783606.
Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol. 2019;17:156–66.
Maleki Kakelar H, Barzegari A, Dehghani J, Hanifian S, Saeedi N, Barar J, et al. Pathogenicity of Helicobacter pylori in cancer development and impacts of vaccination. Gastric Cancer. 2019;22:23–36.
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176:3000–9.
Wu YY, Lin CW, Cheng KS, Lin C, Wang YM, Lin IT, et al. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol. 2010;161:551–9.
Satoh-Takayama N, Kato T, Motomura Y, Kageyama T, Taguchi-Atarashi N, Kinoshita-Daitoku R, et al. Bacteria-induced group 2 innate lymphoid cells in the stomach provide immune protection through induction of IgA. Immunity. 2020;52:635–49.e4.
Peng R, Liu S, You W, Huang Y, Hu C, Gao Y, et al. Gastric microbiome alterations are associated with decreased CD8+ tissue-resident memory T cells in the tumor microenvironment of gastric cancer. Cancer Immunol Res. 2022;10:1224–40.
Ling Z, Shao L, Liu X, Cheng Y, Yan C, Mei Y, et al. Regulatory T cells and plasmacytoid dendritic cells within the tumor microenvironment in gastric cancer are correlated with gastric microbiota dysbiosis: a preliminary study. Front Immunol. 2019;10:533.
Qiu Q, Lin Y, Ma Y, Li X, Liang J, Chen Z, et al. Exploring the emerging role of the gut microbiota and tumor microenvironment in cancer immunotherapy. Front Immunol. 2020;11:612202.
McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019;20:e77–91.
Sun M, Wu W, Chen L, Yang W, Huang X, Ma C, et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat Commun. 2018;9:3555.
Parada Venegas D, De la Fuente MK, Landskron G, Gonzalez MJ, Quera R, Dijkstra G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277.
Chen L, Sun M, Wu W, Yang W, Huang X, Xiao Y, et al. Microbiota metabolite butyrate differentially regulates Th1 and Th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis. 2019;25:1450–61.
Natarajan N, Pluznick JL. From microbe to man: the role of microbial short chain fatty acid metabolites in host cell biology. Am J Physiol Cell Physiol. 2014;307:C979–85.
Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. 2020;11:4457.
Choi Y, Lichterman JN, Coughlin LA, Poulides N, Li W, Del Valle P, et al. Immune checkpoint blockade induces gut microbiota translocation that augments extraintestinal antitumor immunity. Sci Immunol. 2023;8:eabo2003.
Schupack DA, Mars RAT, Voelker DH, Abeykoon JP, Kashyap PC. The promise of the gut microbiome as part of individualized treatment strategies. Nat Rev Gastroenterol Hepatol. 2022;19:7–25.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–9.
Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9.
Abate M, Vos E, Gonen M, Janjigian YY, Schattner M, Laszkowska M, et al. A novel microbiome signature in gastric cancer: a two independent cohort retrospective analysis. Ann Surg. 2022;276:605–15.
Oster P, Vaillant L, Riva E, McMillan B, Begka C, Truntzer C, et al. Helicobacter pylori infection has a detrimental impact on the efficacy of cancer immunotherapies. Gut. 2022;71:457–66.
Cao Y, Yu K, Zhang Z, Gu Y, Gu Y, Li W, et al. Blockade of V-domain immunoglobulin suppressor of T-cell activation reprograms tumour-associated macrophages and improves efficacy of PD-1 inhibitor in gastric cancer. Clin Transl Med. 2024;14:e1578.
An HW, Seok SH, Kwon JW, Choudhury AD, Oh JS, Voon DC, et al. The loss of epithelial Smad4 drives immune evasion via CXCL1 while displaying vulnerability to combinatorial immunotherapy in gastric cancer. Cell Rep. 2022;41:111878.
Song J, Wei R, Huo S, Liu C, Liu X. Versican enrichment predicts poor prognosis and response to adjuvant therapy and immunotherapy in gastric cancer. Front Immunol. 2022;13:960570.
Zhang D, Sun R, Di C, Li L, Zhao F, Han Y, et al. Microdissection of cancer-associated fibroblast infiltration subtypes unveils the secreted SERPINE2 contributing to immunosuppressive microenvironment and immuotherapeutic resistance in gastric cancer: a large-scale study integrating bulk and single-cell transcriptome profiling. Comput Biol Med. 2023;166:107406.
Wei R, Song J, Liu X, Huo S, Liu C, Liu X. Immunosuppressive MFAP2(+) cancer associated fibroblasts conferred unfavorable prognosis and therapeutic resistance in gastric cancer. Cell Oncol. 2024;47:55–68.
Wang SY, Yang XQ, Wang YX, Shen A, Liang CC, Huang RJ, et al. Overexpression of COX7A1 promotes the resistance of gastric cancer to oxaliplatin and weakens the efficacy of immunotherapy. Lab Invest. 2024;104:102090.
Yin Y, Liu Y, Wang Y, Li J, Liang S, Zhang W, et al. DZIP1 expressed in fibroblasts and tumor cells may affect immunosuppression and metastatic potential in gastric cancer. Int Immunopharmacol. 2023;117:109886.
Ge Y, Wei F, Du G, Fei G, Li W, Li X, et al. The association of sex-biased ATRX mutation in female gastric cancer patients with enhanced immunotherapy-related anticancer immunity. BMC Cancer. 2021;21:240.
Rizvi N, Ademuyiwa FO, Cao ZA, Chen HX, Ferris RL, Goldberg SB, et al. Society for Immunotherapy of Cancer (SITC) consensus definitions for resistance to combinations of immune checkpoint inhibitors with chemotherapy. J Immunother Cancer. 2023;11:e005920.
Guo J, Yu Z, Sun D, Zou Y, Liu Y, Huang L. Two nanoformulations induce reactive oxygen species and immunogenetic cell death for synergistic chemo-immunotherapy eradicating colorectal cancer and hepatocellular carcinoma. Mol Cancer. 2021;20:10.
Li Z, Lai X, Fu S, Ren L, Cai H, Zhang H, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci. 2022;9:e2201734.
Wang M, He M, Zhang M, Xue S, Xu T, Zhao Y, et al. Controllable hypoxia-activated chemotherapy as a dual enhancer for synergistic cancer photodynamic immunotherapy. Biomaterials. 2023;301:122257.
Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383:2207–18.
Zhang TQ, Geng ZJ, Zuo MX, Li JB, Huang JH, Huang ZL, et al. Camrelizumab (a PD-1 inhibitor) plus apatinib (an VEGFR-2 inhibitor) and hepatic artery infusion chemotherapy for hepatocellular carcinoma in Barcelona Clinic Liver Cancer stage C (TRIPLET): a phase II study. Signal Transduct Target Ther. 2023;8:413.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14:356–65.
Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618:144–50.
Wang M, Wisniewski CA, Xiong C, Chhoy P, Goel HL, Kumar A, et al. Therapeutic blocking of VEGF binding to neuropilin-2 diminishes PD-L1 expression to activate antitumor immunity in prostate cancer. Sci Transl Med. 2023;15:eade5855.
Jin ZC, Chen JJ, Zhu XL, Duan XH, Xin YJ, Zhong BY, et al. Immune checkpoint inhibitors and anti-vascular endothelial growth factor antibody/tyrosine kinase inhibitors with or without transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma (CHANCE2201): a target trial emulation study. EClinicalMedicine. 2024;72:102622.
Mansouri A, Padmanaban V, Aregawi D, Glantz M. VEGF and immune checkpoint inhibition for prevention of brain metastases: systematic review and meta-analysis. Neurology. 2021;97:e1484–92.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38:2053–61.
Nishiga Y, Drainas AP, Baron M, Bhattacharya D, Barkal AA, Ahrari Y, et al. Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat Cancer. 2022;3:1351–66.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Lynch E, Duffy AG, Kelly RJ. Immune checkpoint inhibitors: changing the treatment landscape in esophagogastric adenocarcinoma. Pharmaceuticals. 2023;16:102.
Sasaki A, Nakamura Y, Togashi Y, Kuno H, Hojo H, Kageyama S, et al. Enhanced tumor response to radiotherapy after PD-1 blockade in metastatic gastric cancer. Gastric Cancer. 2020;23:893–903.
Liu Z, Wang F, Zhang Y, Lu J, Yang Y. Combination treatment with anti-HER2 therapeutic antibody RC48, PD-1 inhibitor, radiotherapy, and granulocyte macrophage-colony stimulating factor (GM-CSF) in patient with metastatic gastric cancer: a case report. Front Immunol. 2024;15:1321946.
Cornel AM, Dunnebach E, Hofman DA, Das S, Sengupta S, van den Ham F, et al. Epigenetic modulation of neuroblastoma enhances T cell and NK cell immunogenicity by inducing a tumor-cell lineage switch. J Immunother Cancer. 2022;10:e005002.
West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013;73:7265–76.
Zhang Y, Zhao H, Deng W, Lai J, Sang K, Chen Q. Zebularine potentiates anti-tumor immunity by inducing tumor immunogenicity and improving antigen processing through cGAS-STING pathway. Commun Biol. 2024;7:587.
Dupuis J, Bachy E, Morschhauser F, Cartron G, Fukuhara N, Daguindau N, et al. Oral azacitidine compared with standard therapy in patients with relapsed or refractory follicular helper T-cell lymphoma (ORACLE): an open-label randomised, phase 3 study. Lancet Haematol. 2024;11:e406–14.
Godfrey J, Mei M, Chen L, Song JY, Bedell V, Budde E, et al. Results from a phase I trial of pembrolizumab plus vorinostat in relapsed/refractory B-cell non-Hodgkin lymphoma. Haematologica. 2024;109:533–42.
Walsh RJ, Sundar R, Lim JSJ. Immune checkpoint inhibitor combinations-current and emerging strategies. Br J Cancer. 2023;128:1415–7.
Du WY, Masuda H, Nagaoka K, Yasuda T, Kuge K, Seto Y, et al. Janus kinase inhibitor overcomes resistance to immune checkpoint inhibitor treatment in peritoneal dissemination of gastric cancer in C57BL/6 J mice. Gastric Cancer. 2024;27:971–85.
Peng J, Zhu Q, Peng Z, Chen Z, Liu Y, Liu B. Patients with positive HER-2 amplification advanced gastroesophageal junction cancer achieved complete response with combined chemotherapy of AK104/cadonilimab (PD-1/CTLA-4 bispecific): A case report. Front Immunol. 2022;13:1049518.
Chiriaco C, Donini C, Cortese M, Ughetto S, Modica C, Martinelli I, et al. Efficacy of CAR-T immunotherapy in MET overexpressing tumors not eligible for anti-MET targeted therapy. J Exp Clin Cancer Res. 2022;41:309.
Chen C, Gu YM, Zhang F, Zhang ZC, Zhang YT, He YD, et al. Construction of PD1/CD28 chimeric-switch receptor enhances anti-tumor ability of c-Met CAR-T in gastric cancer. Oncoimmunology. 2021;10:1901434.
Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022;28:1189–98.
Qi C, Liu C, Gong J, Liu D, Wang X, Zhang P, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial final results. Nat Med. 2024;30:2224–34.
Cao W, Xing H, Li Y, Tian W, Song Y, Jiang Z, et al. Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark Res. 2022;10:38.
Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. 2021;17:3850–61.
Mylod E, Lysaght J, Conroy MJ. Natural killer cell therapy: a new frontier for obesity-associated cancer. Cancer Lett. 2022;535:215620.
Tian M, Zhang S, Tseng Y, Shen X, Dong L, Xue R. Gut microbiota and immune checkpoint inhibitors-based immunotherapy. Anticancer Agents Med Chem. 2022;22:1244–56.
Simpson RC, Shanahan ER, Scolyer RA, Long GV. Towards modulating the gut microbiota to enhance the efficacy of immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2023;20:697–715.
Kim CG, Koh JY, Shin SJ, Shin JH, Hong M, Chung HC, et al. Prior antibiotic administration disrupts anti-PD-1 responses in advanced gastric cancer by altering the gut microbiome and systemic immune response. Cell Rep Med. 2023;4:101251.
Marashi A, Hasany S, Moghimi S, Kiani R, Mehran Asl S, Dareghlou YA, et al. Targeting gut-microbiota for gastric cancer treatment: a systematic review. Front Med. 2024;11:1412709.
Lee SY, Jhun J, Woo JS, Lee KH, Hwang SH, Moon J, et al. Gut microbiome-derived butyrate inhibits the immunosuppressive factors PD-L1 and IL-10 in tumor-associated macrophages in gastric cancer. Gut Microbes. 2024;16:2300846.
Xu X, Ying J. Gut Microbiota and immunotherapy. Front Microbiol. 2022;13:945887.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Routy B, Lenehan JG, Miller WH Jr, Jamal R, Messaoudene M, Daisley BA, et al. Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat Med. 2023;29:2121–32.
Loke P, Orsini F, Lozinsky AC, Gold M, O’Sullivan MD, Quinn P, et al. Probiotic peanut oral immunotherapy versus oral immunotherapy and placebo in children with peanut allergy in Australia (PPOIT-003): a multicentre, randomised, phase 2b trial. Lancet Child Adolesc Health. 2022;6:171–84.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603–15.
Fang E, Liu X, Li M, Zhang Z, Song L, Zhu B, et al. Advances in COVID-19 mRNA vaccine development. Signal Transduct Target Ther. 2022;7:94.
Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer. 2021;20:41.
Li Y, Wang M, Peng X, Yang Y, Chen Q, Liu J, et al. mRNA vaccine in cancer therapy: Current advance and future outlook. Clin Transl Med. 2023;13:e1384.
Sun C, Nagaoka K, Kobayashi Y, Maejima K, Nakagawa H, Nakajima J, et al. Immunotherapies targeting neoantigens are effective in PD-1 blockade-resistant tumors. Int J Cancer. 2023;152:1463–75.
Gao D, Li C, Xie X, Zhao P, Wei X, Sun W, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy with cytokine-induced killer cells improves survival in gastric and colorectal cancer patients. PLoS ONE. 2014;9:e93886.
Han R, Wang Y, Lu L. Sensitizing the efficiency of ICIs by neoantigen mRNA vaccines for HCC treatment. Pharmaceutics. 2023;16:59.
Liu Y, Yan Q, Zeng Z, Fan C, Xiong W. Advances and prospects of mRNA vaccines in cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2024;1879:189068.
Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4(+) T cell metabolic rewiring. Cell Metab. 2019;30:1055–74.e8.
Cai X, Ng CP, Jones O, Fung TS, Ryu KW, Li D, et al. Lactate activates the mitochondrial electron transport chain independently of its metabolism. Mol Cell. 2023;83:3904–20.e7.
Chowdhury S, Kar A, Bhowmik D, Gautam A, Basak D, Sarkar I, et al. Intracellular Acetyl CoA potentiates the therapeutic efficacy of antitumor CD8+ T cells. Cancer Res. 2022;82:2640–55.
Zhang L, Zhao J, Hu X, Wang C, Jia Y, Zhu C, et al. A Peritumorally injected immunomodulating adjuvant elicits robust and safe metalloimmunotherapy against solid tumors. Adv Mater. 2022;34:e2206915.
Wu L, Jin Y, Zhao X, Tang K, Zhao Y, Tong L, et al. Tumor aerobic glycolysis confers immune evasion through modulating sensitivity to T cell-mediated bystander killing via TNF-alpha. Cell Metab. 2023;35:1580–96.e9.
Jin J, Yuan P, Yu W, Lin J, Xu A, Xu X, et al. Mitochondria-targeting polymer micelle of dichloroacetate induced pyroptosis to enhance osteosarcoma immunotherapy. ACS Nano. 2022;16:10327–40.
Yang JF, Xing X, Luo L, Zhou XW, Feng JX, Huang KB, et al. Mitochondria-ER contact mediated by MFN2-SERCA2 interaction supports CD8(+) T cell metabolic fitness and function in tumors. Sci Immunol. 2023;8:eabq2424.
Su S, Zou Z, Chen F, Ding N, Du J, Shao J, et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. 2017;6:e1249558.
Ma X, Jia S, Wang G, Liang M, Guo T, Du H, et al. TRIM28 promotes the escape of gastric cancer cells from immune surveillance by increasing PD-L1 abundance. Signal Transduct Target Ther. 2023;8:246.
Fuchs CS, Ozguroglu M, Bang YJ, Di Bartolomeo M, Mandala M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated PD-L1-positive advanced gastric or gastroesophageal junction cancer: 2-year update of the randomized phase 3 KEYNOTE-061 trial. Gastric Cancer. 2022;25:197–206.
Kwon M, An M, Klempner SJ, Lee H, Kim KM, Sa JK, et al. Determinants of response and intrinsic resistance to PD-1 blockade in microsatellite instability-high gastric cancer. Cancer Discov. 2021;11:2168–85.
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 clinical KEYNOTE-059 trial. JAMA Oncol. 2018;4:e180013.
Shitara K, Di Bartolomeo M, Mandala M, Ryu MH, Caglevic C, Olesinski T, et al. Association between gene expression signatures and clinical outcomes of pembrolizumab versus paclitaxel in advanced gastric cancer: exploratory analysis from the randomized, controlled, phase III KEYNOTE-061 trial. J Immunother Cancer. 2023;11:e006920.
Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016;17:717–26.
Van Cutsem E, Valderrama A, Bang YJ, Fuchs CS, Shitara K, Janjigian YY, et al. Quality of life with first-line pembrolizumab for PD-L1-positive advanced gastric/gastroesophageal junction adenocarcinoma: results from the randomised phase III KEYNOTE-062 study. ESMO Open. 2021;6:100189.
Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449–58.
Klempner SJ, Bendell JC, Villaflor VM, Tenner LL, Stein SM, Rottman JB, et al. Safety, efficacy, and biomarker results from a phase Ib study of the anti-DKK1 antibody DKN-01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther. 2021;20:2240–9.
Kawazoe A, Fukuoka S, Nakamura Y, Kuboki Y, Wakabayashi M, Nomura S, et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): an open-label, single-arm, phase 2 trial. Lancet Oncol. 2020;21:1057–65.
Chung HC, Kang YK, Chen Z, Bai Y, Wan Ishak WZ, Shim BY, et al. Pembrolizumab versus paclitaxel for previously treated advanced gastric or gastroesophageal junction cancer (KEYNOTE-063): as randomized, open-label, phase 3 trial in Asian patients. Cancer. 2022;128:995–1003.
Van Cutsem E, Amonkar M, Fuchs CS, Alsina M, Ozguroglu M, Bang YJ, et al. Health-related quality of life in advanced gastric/gastroesophageal junction cancer with second-line pembrolizumab in KEYNOTE-061. Gastric Cancer. 2021;24:1330–40.
Janjigian YY, Kawazoe A, Yanez P, Li N, Lonardi S, Kolesnik O, et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature. 2021;600:727–30.
Yamaguchi K, Minashi K, Sakai D, Nishina T, Omuro Y, Tsuda M, et al. Phase IIb study of pembrolizumab combined with S-1 + oxaliplatin or S-1 + cisplatin as first-line chemotherapy for gastric cancer. Cancer Sci. 2022;113:2814–27.
Chen LT, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer. 2020;23:510–9.
Lee CK, Kim HS, Jung M, Kim H, Bae WK, Koo DH, et al. Open-label, multicenter, randomized, biomarker-integrated umbrella trial for second-line treatment of advanced gastric cancer: K-Umbrella Gastric Cancer Study. J Clin Oncol. 2024;42:348–57.
Hasegawa H, Shitara K, Takiguchi S, Takiguchi N, Ito S, Kochi M, et al. A multicenter, open-label, single-arm phase I trial of neoadjuvant nivolumab monotherapy for resectable gastric cancer. Gastric Cancer. 2022;25:619–28.
Moehler M, Xiao H, Blum SI, Elimova E, Cella D, Shitara K, et al. Health-related quality of life with nivolumab plus chemotherapy versus chemotherapy in patients with advanced gastric/gastroesophageal junction cancer or esophageal adenocarcinoma from CheckMate 649. J Clin Oncol. 2023;41:5388–99.
Kawazoe A, Yamamoto N, Sugimoto N, Kawakami H, Oshima T, Yamaguchi K, et al. Phase II study of the liposomal formulation of eribulin (E7389-LF) in combination with nivolumab: results from the gastric cancer cohort. Clin Cancer Res. 2024;30:1264–72.
Li S, Yu W, Xie F, Luo H, Liu Z, Lv W, et al. Neoadjuvant therapy with immune checkpoint blockade, antiangiogenesis, and chemotherapy for locally advanced gastric cancer. Nat Commun. 2023;14:8.
Lin JX, Tang YH, Zheng HL, Ye K, Cai JC, Cai LS, et al. Neoadjuvant camrelizumab and apatinib combined with chemotherapy versus chemotherapy alone for locally advanced gastric cancer: a multicenter randomized phase 2 trial. Nat Commun. 2024;15:41.
Jing C, Wang J, Zhu M, Bai Z, Zhao B, Zhang J, et al. Camrelizumab combined with apatinib and S-1 as second-line treatment for patients with advanced gastric or gastroesophageal junction adenocarcinoma: a phase 2, single-arm, prospective study. Cancer Immunol Immunother. 2022;71:2597–608.
Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64.
Li J, Bai Y, Chen Z, Ying J, Guo Y, Fang W, et al. SAFFRON-104: a phase Ib/II study of sitravatinib alone or with tislelizumab in advanced hepatocellular carcinoma and gastric cancer/gastroesophageal junction cancer. Cancer Immunol Immunother. 2024;73:219.
Shi JW, Zhou Y, Wu S. Clinical efficacy and safety of adjuvant immunotherapy (Tislelizumab) plus chemotherapy vs. adjuvant chemotherapy alone in lymph node-positive patients with gastric cancer after D2 radical resection: a prospective, 2-arm, phase II study. Eur Rev Med Pharmacol Sci. 2023;27:10472–80.
Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol. 2019;30:1479–86.
Ko AH, Kim KP, Siveke JT, Lopez CD, Lacy J, O’Reilly EM, et al. Atezolizumab Plus PEGPH20 versus chemotherapy in advanced pancreatic ductal adenocarcinoma and gastric cancer: MORPHEUS Phase Ib/II Umbrella Randomized Study Platform. Oncologist. 2023;28:553–e472.
Li J, Deng Y, Zhang W, Zhou AP, Guo W, Yang J, et al. Subcutaneous envafolimab monotherapy in patients with advanced defective mismatch repair/microsatellite instability high solid tumors. J Hematol Oncol. 2021;14:95.
Doi T, Iwasa S, Muro K, Satoh T, Hironaka S, Esaki T, et al. Phase 1 trial of avelumab (anti-PD-L1) in Japanese patients with advanced solid tumors, including dose expansion in patients with gastric or gastroesophageal junction cancer: the JAVELIN Solid Tumor JPN trial. Gastric Cancer. 2019;22:817–27.
Gou M, Zhang Y, Wang Z, Qian N, Dai G. PD-1 inhibitor combined with albumin paclitaxel and apatinib as second-line treatment for patients with metastatic gastric cancer: a single-center, single-arm, phase II study. Invest New Drugs. 2024;42:171–8.
Kang YK, Bang YJ, Kondo S, Chung HC, Muro K, Dussault I, et al. Safety and Tolerability of Bintrafusp Alfa, a bifunctional fusion protein targeting TGFbeta and PD-L1, in Asian patients with pretreated recurrent or refractory gastric cancer. Clin Cancer Res. 2020;26:3202–10.
Liu D, Zhou J, Wang Y, Li M, Jiang H, Liu Y, et al. Bifunctional anti-PD-L1/TGF-betaRII agent SHR-1701 in advanced solid tumors: a dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med. 2022;20:408.
Kim TH, Lee D, Oh HJ, Ham IH, Lee DM, Lee Y, et al. Targeting GAS6/AXL signaling improves the response to immunotherapy by restoring the anti-immunogenic tumor microenvironment in gastric cancer. Life Sci. 2023;335:122230.
Mao D, Zhou Z, Chen H, Liu X, Li D, Chen X, et al. Pleckstrin-2 promotes tumour immune escape from NK cells by activating the MT1-MMP-MICA signalling axis in gastric cancer. Cancer Lett. 2023;572:216351.
Zhang J, Dong Y, Yu S, Hu K, Zhang L, Xiong M, et al. IL-4/IL-4R axis signaling drives resistance to immunotherapy by inducing the upregulation of Fcgamma receptor IIB in M2 macrophages. Cell Death Dis. 2024;15:500.
Zhang P, Gu Y, Fang H, Cao Y, Wang J, Liu H, et al. Intratumoral IL-1R1 expression delineates a distinctive molecular subset with therapeutic resistance in patients with gastric cancer. J Immunother Cancer. 2022;10:e004047.
Wang Q, Cao T, Zhang X, Hui J, Wang C, Zhang W, et al. ATXN2-mediated PI3K/AKT activation confers gastric cancer chemoresistance and attenuates CD8(+) T cell cytotoxicity. J Immunol Res. 2022;2022:6863240.
Song J, Yang R, Wei R, Du Y, He P, Liu X. Pan-cancer analysis reveals RIPK2 predicts prognosis and promotes immune therapy resistance via triggering cytotoxic T lymphocytes dysfunction. Mol Med. 2022;28:47.
Sundar R, Huang KK, Kumar V, Ramnarayanan K, Demircioglu D, Her Z, et al. Epigenetic promoter alterations in GI tumour immune-editing and resistance to immune checkpoint inhibition. Gut. 2022;71:1277–88.
Cui Y, Li Q, Li W, Wang Y, Lv F, Shi X, et al. NOTCH3 is a prognostic factor and is correlated with immune tolerance in gastric cancer. Front Oncol. 2020;10:574937.
Huang X, Zhang J, Zheng Y. ANTXR1 Is a prognostic biomarker and correlates with stromal and immune cell infiltration in gastric cancer. Front Mol Biosci. 2020;7:598221.
Wang S, Zhang S, Li X, Li X, Zhao S, Guo J, et al. HIGD1B, as a novel prognostic biomarker, is involved in regulating the tumor microenvironment and immune cell infiltration; its overexpression leads to poor prognosis in gastric cancer patients. Front Immunol. 2024;15:1415148.
Li Y, Qiu J, Meng Z, Yin S, Ruan M, Zhang W, et al. MFG-E8 promotes M2 polarization of macrophages and is associated with poor prognosis in patients with gastric cancer. Heliyon. 2024;10:e23917.
Acknowledgements
This work was supported by the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2022KY1246), and the Science and Technology Bureau of Jiaxing City (2023AZ31002 and 2022AZ10009).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Hans-Uwe Simon
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, D., Zhou, J., Ruan, S. et al. Overcoming immunotherapy resistance in gastric cancer: insights into mechanisms and emerging strategies. Cell Death Dis 16, 75 (2025). https://doi.org/10.1038/s41419-025-07385-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-07385-7
This article is cited by
-
Tumor microenvironment dynamics in gastric cancer pathogenesis and therapeutic resistance
Molecular Cancer (2026)
-
Metastatic organotropism in peritoneal metastasis: Paget’s hypothesis revisited
Clinical and Experimental Medicine (2026)
-
Exploring the role of miR-221/222 in gastrointestinal cancers: implications for innovative therapeutic strategies
Cancer Cell International (2025)
-
Spatially resolved endothelial signaling via nampt-itga5 drives immune evasion in stem-like gastric cancer
Cancer Immunology, Immunotherapy (2025)
-
SPTSSA facilitates gastric cancer progression with modulating PD-L1 in immunomicroenvironment through Wnt/β-catenin pathway
Cellular Oncology (2025)
