
Abstract
Natural killer (NK) cells exhibit remarkable adaptability within the tumour microenvironment (TME), where dynamic shifts in phenotype, function and metabolism govern their dual roles in antitumour immunity and tumour immune evasion. In the TME, NK cells undergo receptor remodelling, which is characterised by upregulated inhibitory signals and suppressed activating receptors, leading to the formation of dysfunctional subsets, such as exhausted TIM-3⁺ NK cells or tissue-resident CD49a⁺ populations. Immunosuppressive factors within the TME drive a transition from cytotoxic activity to regulatory or senescent-like states, impairing tumour surveillance. Metabolic reprogramming further compromises NK cell effector functions, as nutrient deprivation and metabolic byproducts disrupt energy pathways and suppress immune responses. Therapeutic strategies targeting this plasticity include engineered natural killer (NK) cells with enhanced specificity, metabolic restoration approaches and microenvironment-modulating interventions. However, challenges persist because of TME heterogeneity and persistent dysfunctional states. Understanding these adaptive mechanisms provides a framework for developing NK cell-based therapies that leverage plasticity to counteract tumour resistance.
Similar content being viewed by others


FACTS
-
Natural killer cells exhibit remarkable adaptability within the tumour microenvironment, where dynamic shifts in phenotype, function and metabolism govern their dual roles in anti-tumour immunity and tumour immune evasion.
-
Therapeutic strategies targeting this plasticity include engineered NK cells with enhanced specificity, metabolic restoration approaches and microenvironment-modulating interventions.
-
Understanding these adaptive mechanisms provides a framework for developing NK cell-based therapies that leverage plasticity to counteract tumour resistance.
Introduction
Natural killer (NK) cells, which are derived from bone marrow haematopoietic stem cells, play a central role as effector cells in the innate immune system. Studies have shown that under physiological conditions, there are three types of NK cells in human bone marrow: NK1, NK2 and NK3. These three primary NK cell populations can be divided into six subgroups and the NK2 cell population also shows a tumour-specific trend [1,2,3]. NK cells exhibit a rapid response mechanism and rely on germline-encoded receptors such as NKG2D⁺ and DNAM-1⁺ to effectively identify and eliminate both malignant tumour cells and virus-infected cells [4,5,6]. Moreover, different subsets of NK cells are present in the tumour microenvironment (TME), whose expression of different markers differs, resulting in functional differences, as listed in Table 1 [7,8,9,10,11,12]. The cytotoxic potential of NK cells is precisely regulated by a dynamic balance between activating receptors (such as NKp30, NKG2D⁺, NKp44 and NKp46) and inhibitory checkpoint molecules (for instance, KIRs, NKG2A and TIGIT). This regulatory equilibrium allows NK cells to discriminate between healthy cells and stressed target cells, as comprehensively summarised in [13, 14].
Nevertheless, the immunosuppressive TME disrupts this delicate balance through multiple interrelated mechanisms. First, the TME deprives NK cells of metabolic energy through the accumulation of waste products such as lactate, which directly impairs bioenergetic homoeostasis. Simultaneously, energetic reprogramming occurs in NK cells, involving alterations in genetic regulation that induce a functional state characterised by metabolic exhaustion, epigenetic silencing and phenotype remodelling [15, 16]. This synergistic effect of the TME leads to the exhaustion of NK cells, driving multidimensional plasticity in their phenotype, function and metabolic profile.
Recent research has shed light on the dynamic adaptability of NK cells within the TME. For example, sex-specific energetic regulation has been reported: NK cell-specific knockout of UTX significantly inhibited lung metastasis of melanoma in male mice but had no such effect in female mice. These findings are consistent with the documented mechanism by which TME-mediated epigenetic silencing impairs NK cytotoxicity, including hypermethylation of the NKG2D⁺ promoter and histone deacetylation at the DNAM-1⁺ locus [17]. In non-small cell lung cancer (NSCLC), the TME is enriched with myeloid-derived suppressor cells (MDSCs) that exhibit a CD39⁺CD73⁺ phenotype —a feature not observed in infiltrating NK cells, which primarily undergo functional exhaustion via other pathways (e.g. upregulation of the exhaustion marker TIM-3) rather than CD39/CD73 expression [18]. These MDSCs establish an immunosuppressive signalling axis called the ‘adenosine-A2AR axis’ to directly inhibit T cell function, while also impairing NK cell activity indirectly. Specifically, the CD39 protein on MDSCs first hydrolyses extracellular ATP (an energy-providing molecule for cells) into AMP; subsequently, the CD73 protein further converts AMP into adenosine. The adenosine accumulated in the TME then binds to the adenosine A2A receptor (A2AR) expressed on the surface of both T cells and NK cells [19].
For T cells, adenosine-A2AR binding directly suppresses their proliferation and cytotoxicity, weakening their ability to kill tumour cells. For NK cells, this binding does not disrupt oxidative phosphorylation (OXPHOS) or glycolytic pathways— mechanisms that lack support in current NSCLC-focused studies—but instead blocks their maturation process: it reduces the proportion of mature cytotoxic NK cell subsets (e.g. CD27⁻CD11b⁺ in murine models, CD56dimCD16⁺ in humans) and impairs their secretion of interferon-γ (IFN-γ) (a key cytokine that enhances anti-tumour immunity). Consequently, the NK cells show reduced responsiveness to IL-2 (a molecule that stimulates immune cell activation), diminished cytokine secretion and blunted anti-tumour cytotoxicity—ultimately reinforcing the immunosuppressive state of the TME [18, 19]. This metabolic–immune crosstalk establishes a positive feedback loop of immunosuppression, which is further amplified by hypoxic conditions and TGF-β signalling in the TME.
Additional evidence emphasises the role of soluble noninflammatory factors in NK cell polarisation. In sepsis models, activation of the IL-10/IDO axis promotes the differentiation of NK cells into a regulatory phenotype (CD56brightCD16−). Notably, IDO, which is highly expressed in most human tumours, is closely correlated with the loss of NK cell cytotoxicity, highlighting its significance as an immunosuppressive factor secreted by the TME [20, 21]. The spatial heterogeneity of NK cell subsets has been revealed through prognostic modelling in EBV-associated gastric cancer, differentiating between TIM-3⁺ exhausted subsets and CD49a⁺ tissue-resident subsets. Tissue-resident NK cells, which are adapted to specific TMEs, typically exhibit reduced cytotoxic function and variable production of inflammatory cytokines [22,23,24]. Collectively, these findings demonstrate that the TME impairs the antitumour function of NK cells through coordinated energetic, metabolic and phenotypic reprogramming.
In response to these mechanisms, emerging chemotherapeutic strategies adopt combination approaches. Engineered chimeric antigen receptor-NK (CAR-NK) cells, especially fourth-generation constructs incorporating IL-15 or logic-gated antigen recognition modules, increase cell persistence and target specificity. Metabolic interventions, including NAD⁺ supplementation to restore OXPHOS capacity and IDO inhibitors to counteract tryptophan depletion, aim to revitalise the epigenetic and functional responsiveness of NK cells. Environmental remodelling strategies, such as TGF-β siRNA carriers and oncolytic viruses (e.g. oHSV-CXCL10), seek to disrupt the immunosuppression network by modulating cytokine signalling and enhancing immune cell infiltration [17]. Despite these advancements, significant challenges remain, including the spatial heterogeneity of tumour lesions, host immune rejection of engineered cells and complex interactions with the extracellular matrix.
In conclusion, the complex interactions between NK cells and the TME necessitate the development of multidimensional therapeutic strategies. These approaches must integrate the molecular mechanisms of NK cell dysfunction with the unique constraints of the tumour environment, promoting the design of more effective and personalised chemotherapeutic interventions.
Phenotype plasticity: dynamic receptor remodelling and subset heterogeneity
NK cell phenotypic plasticity is operationally defined as receptor network remodelling measurable by flow cytometry (e.g. CD49a⁺ tissue-resident subset enrichment) and epigenetic assays (e.g. NKG2D⁺ promoter hypermethylation). The phenotypic plasticity of NK cells within the TME involves a highly dynamic adaptive process meticulously choreographed by competitive receptor–ligand interactions and profound microenvironmental reprogramming. This biological manifestation originates from the delicate equilibrium maintained between activating receptors—such as NKG2D⁺ and DNAM-1⁺, which trigger cytotoxicity cascades—and inhibitory receptors such as killer immunoglobulin-like receptors (KIRs) and TIGIT. The latter recognises subsequently expressed major histocompatibility complex (MHC) class I molecules on healthy cells. When MHC class I engages with inhibitory KIRs, it transduces dominant negative signals that effectively suppress NK cell effector functions. This process establishes an evolutionarily conserved surveillance mechanism designed to eliminate MHC-I-deficient targets while safeguarding against autoimmunity [25,26,27]. Under normal physiological conditions, this MHC class I-dependent inhibitory signalling efficiently curtails NK cell activation. However, tumours frequently downregulate MHC class I expression as an immune evasion strategy. In theory, this should render them vulnerable to NK cell-mediated dialysis [14, 28]. Paradoxically, advanced tumours subvert this surveillance system by inducing compensatory mechanisms. These mechanisms give rise to heterogeneous NK cell populations with functionally distinct phenotypes, highlighting the intricate nature of tumour–immune interactions [29].
Central to this adaptive process is the bidirectional remodelling of receptor networks. In hepatocellular carcinoma (HCC), the CD155-TIGIT axis serves as a prime example of how tumours exploit inhibitory checkpoints. The high-affinity binding of TIGIT to CD155 inhibits NK cells through two mechanisms: by recruiting SHP-2 adenylyl cyclase to the cell surface to degrade the phosphorylated products of the PI3K/Akt pathway, thereby preventing the endocytosis cycle of NKG2D⁺ or by competing with CD155 for binding, thus preventing the activation of the receptor DNAM-1⁺ to prevent interaction and subsequent triggering of signal transduction. TIGIT, which has the highest binding affinity for CD155, interacts with overexpressed CD155 on tumour cells. This interaction delivers potent inhibitory signals that override concurrent activating receptor stimulation, such as that mediated by NKG2D⁺ [30]. Previous experiments have revealed a new mechanism in liver cancer in which the high expression of CD155 on platelet-adhered circulating tumour cells (CTCs) enables them to evade NK cell death. Blocking the binding of CD155 to TIGIT can effectively increase the clearance of circulating CTCs by NK cells [31]. Clinical investigations have demonstrated that elevated TIGIT expression on NK cells is associated with functional exhaustion and an unfavourable tumour prognosis [32]. As a result, TIGIT blockade has emerged as a promising therapeutic approach to reinstate NK cell cytotoxicity [33]. Simultaneously, CD155 functions as a ligand for the activating receptor DNAM-1⁺ on NK cells [14, 17, 30]. In the TME, DNA damage responses lead to the release of soluble CD155 (sCD155). This sCD155 competes for DNAM-1⁺ binding, promotes receptor degradation and downregulates activating signalling [34]. Mechanistically, TIGIT further disrupts DNAM-1⁺homogenisation, preventing its interaction with membrane-bound CD155. Consequently, this abrogates the costimulatory signals crucial for NK cell activation [35]. Lysosomal-mediated communication adds another layer of regulation. HCC-derived exosomal circHRF1 transfers miRNAs to NK cells, posttranscriptionally regulating PD-1 expression. This exacerbates the exhaustion phenotype beyond imbalances in the receptor expression profile. Maelstrom components further reinforce this suppressive environment. Adipose-derived mesenchymal stem cells (MSCs) secrete TGF-β, whose transcription downregulates NKG2D⁺ and DNAM-1⁺ by reducing DAP10 mRNA and protein levels. This functionally disconnects NK cells from stress ligand recognition [36].
Energetic reprogramming stabilises these dysfunctional phenotypes through chromatin-level modifications that establish heritable gene expression patterns. Recent studies have revealed a critical mechanism of immune escape involving the proteolytic shedding of NKG2D ligands and altered membrane topology. To clarify the biological basis of this mechanism, NKG2D (NK cell lectin-like receptor subfamily D) acts as a key activating receptor on NK cells. The ligands of NKG2D are mainly divided into two families: MHC class I-related molecules A/B (MICA/B) and UL16-binding proteins (ULBPs, e.g. ULBP1-6). Under physiological conditions, these ligands are weakly expressed on healthy cells; however, in tumour cells, stress signals such as DNA damage, oxidative stress or viral infection can significantly upregulate their expression. When NKG2D ligands bind to NKG2D on NK cells, they activate downstream cytotoxic signalling pathways (e.g. PI3K/Akt and MAPK), thereby promoting the release of granzyme B and perforin, as well as the secretion of IFN-γ, which together mediate the killing of tumour cells. Notably, tumour cells have evolved a ‘ligand shedding’ strategy to escape NKG2D-mediated immune surveillance. This process proceeds in two sequential steps: first, endoplasmic reticulum protein 5 mediates the cleavage of disulphide bonds in the α3 domain of MICA/B; then, metalloproteases (e.g. ADAM10/17 and MMP14) further cleave the modified MICA/B, releasing soluble MICA/B (sMICA/B) into the TME. Secreted sMICA/B can bind to NKG2D on the surface of NK cells, inducing receptor internalisation and degradation, which ultimately inhibits NK cell activity. Clinically, elevated serum levels of sMICA/B are observed in patients with melanoma and NSCLC and this elevation is associated with poor survival outcomes [37]. Compared with peripheral or liver-resident NK cells, tumour-infiltrating NK cells exhibit significantly fewer membrane protrusions, as visualised by transmission and scanning electron microscopy. This defect arises from reduced sphingomyelin levels in NK cell membranes, driven by tumour-induced dysregulation of serine metabolism. Mechanistically, serine depletion in the TME impairs SM biosynthesis, leading to disrupted membrane protrusion formation and reduced immune synapse formation [38]. In ovarian cancer, hypermethylation of CpG islands in the NKG2D⁺ promoter region induces long-term transcriptional silencing. In melanoma, histone deacetylase (HDAC)-mediated chromatin compaction at the DNAM-1⁺ locus impedes transcriptional activation [39,40,41]. These epigenetic changes create a long-lasting ‘memory’ of dysfunction. They persist even in the absence of continuous TME signals and synergise with surface receptor modulation to enforce sustained NK cell paralysis.
The spatiotemporal heterogeneity within the TME drives subset specialisation through niche-specific cues. In EBV-associated gastric cancer, the hypoxic tumour core is enriched in TIM-3⁺ exhausted NK cells, characterised by impaired IFN-γ production and reduced cytotoxic potential. Comparative analysis in colorectal cancer reveals distinct CD16 expression profiles. Compared with their peripheral blood counterparts, tumour-infiltrating NK cells display significantly lower CD16 levels, while adjacent normal tissues exhibit intermediate expression. This highlights the phenotypic divergence between tissue-resident and circulating NK cell populations. In contrast, cardiovascular NK cells retain the CD16⁺ cytotoxic phenotype. They mediate antibody-dependent cellular cytotoxicity (ADCC) through immunoreceptor tyrosine-based activation motif (ITAM) signalling in the CD3ζ chain, which is coupled with high-affinity IgE receptors [38, 42, 43]. This compartmentalisation is exemplified in glioblastoma. In contrast to peripheral CD56dimCD16+ cells, which maintain cytotoxic function, tumour-infiltrating CD56brightCD16− NK cells secrete VEGF and IL-10 to promote pathogenesis and immunosuppression [44]. Hypoxia-inducible factor 1α (HIF-1α) and TGF-β gradients drive this specialisation. HIF-1α stabilises CD73⁺ expression to facilitate adenine-mediated immunosuppression, while TGF-β induces differentiation into tissue-resident CD49a⁺⁺CD103⁺ NK cells at the expense of migratory cytotoxic subsets [12]. Notably, this plasticity is environmentally dependent. Ectopic Eomes expression in innate lymphoid cells (ILC1s) can convert them into NK-like cells with restored cytotoxicity in murine models. This challenges the traditional concept of irreversible lineage commitment in tissue-resident populations [45]. Clinical correlations include chronic hepatitis B-induced chemokine receptor reprogramming (CCR1 downregulation/CXCR6 regulation) in circulating NK cells, which impairs their homing to infected hepatocytes. Additionally, extranodal NK/T-cell lymphoma drives PD-1/TIM-3⁺coexpression on NK cells, which is associated with shortened progression-free survival [46, 47].
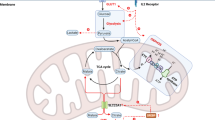
The convergence of receptor dynamics and subset specialisation constitutes a ‘double-hit’ immunosuppression mechanism. Transcriptional silencing of activating receptors (e.g. NKp30 promoter hypermethylation) works in tandem with checkpoint expression (e.g. TIGIT and PD-1) to disrupt both target recognition and effector function. Transcript analyses underscore the microenvironmental regulation of receptor plasticity. Compared with peripheral blood cells, bone marrow-derived NK cells exhibit distinct inhibitory receptor profiles (e.g. KIR2DL1 predominance), as depicted in Fig. 1. Figure 1 further delineates the multidimensional regulatory network underlying this receptor plasticity, highlighting three key regulatory axes: first, the activating–inhibitory receptor balance: NKG2E (an activating receptor) recognises stress ligands such as MICA/B to enhance NK cell killing, while NKG2A (an inhibitory receptor) binds HLA-E (e.g. CD155) to transmit suppressive signals—this balance is disrupted in the TME, where tumours upregulate NKG2A ligands to silence NK cells [30]; second, CD155-mediated bidirectional regulation: CD155 on tumour cells acts as a ‘molecular switch’: it activates NK cells via DNAM-1⁺ binding but inhibits them via CD96/TIGIT interaction. In HCC, platelet-adhered CTCs overexpress CD155, which preferentially binds TIGIT on NK cells to block NKG2D⁺ endocytosis and PI3K/Akt signalling, enabling CTC escape [31]. Third, cytokine‒transcription factor crosstalk occurs: IL-2R mediates JAK‒STAT5 signalling to promote NK cell proliferation, but this pathway is countered by CISH (a negative regulator that degrades STAT5). In the TME, reduced IL-2 availability and increased CISH expression synergise to weaken activating signals, while upregulated PD-L1 and TIM-3⁺ (exhaustion markers) further lock NK cells into dysfunction [29]. Collectively, Fig. 1 illustrates how TME-driven perturbations in these axes converge to remodel NK cell receptors, laying the foundation for subset heterogeneity (e.g. TIM-3⁺ exhausted cells and CD49a⁺ tissue-resident cells), as discussed earlier. These findings reflect the plasticity of the NK cell phenotype in the TME and these cell surface proteins affect the different states of NK cell activation and inhibition. Overcoming these layered regulatory mechanisms requires integrated therapeutic strategies. These strategies should target both cell-intrinsic plasticity—through epigenetic modulators and receptor-engineered CAR-NK cells—and extrinsic TME signals—such as TGF-β inhibitors and hypoxia-targeting agents—to restore NK cell anti-immunosuppression competence.

NKG2D (NK cell lectin-like receptor subfamily D): Activating receptor that recognises stress ligands including MICA, MICB and ULBPs and enhances the killing activity of NK cells against tumours; NKG2A (NK cell lectin-like receptor subfamily A): inhibitory receptors that bind HLA-E (such as CD155) to transmit inhibitory signals and mediate tumour immune escape; CD96 (T-cell activation inhibitory receptor): immune checkpoint molecule that binds to CD155 to inhibit NK cytotoxicity; CD155 (poliovirus receptor): tumour ligand that bidirectionally regulates NK cell function (activated by DNAM-1⁺ and inhibited by CD96/TIGIT); IL-2R (interleukin-2 receptor): mediates IL-2 signalling and drives NK cell proliferation and effector function through the JAK-STAT pathway (JAK, STAT5); CISH (cytokine-induced SH2 protein): negatively regulates IL-2 signalling and degrades STAT5 to prevent overactivation; PD-L1 (programmed death ligand 1): an immune checkpoint ligand that binds to PD-1 on NK cells to inhibit their function and mark the exhaustion state; TIM-3⁺ (T cellular immunoglobulin mucin 3): a depletion marker that binds to galectin-9 to inhibit NK cell activity. This figure was created with BioRender.com.
Functional plasticity: from cytotoxicity to carcinogenicity
Functional plasticity—quantified by cytotoxic assays (e.g. reductions in granzyme B secretion) and cytokine profiling (e.g. upregulation of IL-10 expression)—reflects the hierarchical molecular reprogramming that drives NK cells from cytotoxic effectors to regulatory states under TME stress. This intricate phenotype transition is meticulously orchestrated by hierarchical molecular reprogramming spanning receptor signalling, energetic modification and metabolic adaptation. At the heart of this process lies the dynamic disequilibrium between activating and inhibitory receptor networks (Fig. 2) [48, 49]. The molecular circuits that govern NK cell functional shifts are shown in Fig. 2, with clear delineation of activating cues, inhibitory signals and functional readouts: activating pathways: Proinflammatory cytokines (IL-2, IL-15 and IL-18) and activating receptors (NKp46, NKp44 and NKG2D) drive the secretion of pro-tumouricidal factors (IFN-γ, TNF-α and GM-CSF) and degranulation (marked by CD107a). For example, IL-15 sustains NK cell survival and cytotoxicity via mTOR activation, whereas the chemokines CXCL10 and CXCL11 recruit NK cells to tumour margins [50, 51]. Inhibitory perturbations: the TME disrupts these pathways through multiple mechanisms: (1) tumour-derived TGF-β and IL-10 downregulate activating receptors (NKG2D, NKp30) and reduce cytokine secretion (IFN-γ↓, CCL3↓/CCL5↓); (2) immune checkpoint ligands (PD-L1) and stromal cells (Treg, TAM) reinforce suppression—Tregs secrete TGF-β to block granzyme B expression, whereas TAMs increase adenosine signalling to inhibit NK cell activation [52, 53]; and (3) hypoxia (marked by HIF-1α↑) induces VEGF secretion, shifting NK cells towards a pro-angiogenic state rather than a cytotoxic state. Functional readouts: the chart links these signals to measurable functional changes: exhausted NK cells show reduced CD107a (degranulation) and ATP levels, whereas regulatory NK cells upregulate IL-10 and lose antitumour cytokine production. This finding aligns with our earlier observation in triple-negative breast cancer, where TGF-β induces a TOMM20-low mitochondrial phenotype that impairs granzyme B exocytosis [54]. This network map (Fig. 2) explains why NK cells transition from cytotoxic effectors to immunosuppressive regulators in the TME: inhibitory signals overwhelm activating cues, leading to coordinated loss of cytotoxic function and increased pro-tumour activity. There is complex crosstalk between NK cells and various TMEs. NK cells and TMEs sometimes cooperate with each other, but more often, they fight against each other, thus eliciting different antitumour responses [54]. Moreover, mutual interference occurs between NK cells and other immune cells in the TME [55]. Previous studies have shown that NK cells under IL-12 or IL-2 conditions can initiate DC maturation of Th1 cells that produce IFN-γ. NK cells under IL-18 conditions induce Th1 polarisation only when they are cocultured with DCs and T cells, whereas the release of IL-2 by T cells promotes the production of IFN-γ. When NK cells are under IL-4 conditions, they produce nonpolarized T cells that release only low levels of IL-2 [56]. Treg cells in the TME can also interfere with NK cells because of their sensitivity to immunosuppressive agents [57]. Previous studies have shown that Tregs directly inhibit NK cell responses through the activity of transforming growth factor (TGF)-β, thereby affecting their antitumour activity [52].

Proinflammatory cytokines (IFN-γ, TNF-α and GM-CSF) and chemokines (CCL3, CCL5 and CXCL10/11) drive immune activation, whereas CD107a is involved in NK cell degranulation. NK cell surface receptors (NKp46, NKp44, TIM) and inhibitory checkpoints (PD-1, NKG2A) reflect functional modulation. Immunosuppressive factors (IL-10, TGF-β and PD-L1) and hypoxia-related markers (HIF-1α and VEGF) highlight tumour-driven immune evasion. This figure was created with BioRender.com.
Prolonged exposure to tumour-derived factors promotes receptor desensitisation through the transcriptional downregulation of pivotal activating receptors such as NKG2D, DNAM-1 and NKp30. Intriguingly, this occurs despite the intact preservation of the core effector machinery, effectively muting the cytotoxic potential and pushing NK cells into a paradoxical state of functional tolerance. This context-dependent plasticity manifests diversely in different biological scenarios. For instance, Yishen-Jiangu pills alleviate osteoarthritis inflammation by suppressing synovial NK cell activation via inhibition of the ERK/NK-1 receptor pathway. Conversely, in breast cancer models, the epigenetic silencing of cytotoxicity receptors mediated by TGF-β/SMAD3 is achieved through histone deacetylation at the TBX21 locus, underscoring the tissue-specific nature of these adaptive mechanisms [58].
TGF-β orchestrates NK cell functional plasticity through a hierarchical triad of mechanisms. In vitro studies have shown that TGF-β treatment blocks IL-15-induced mTOR phosphorylation of S6 kinase (S6K) and 4EBP1, reducing NK cell metabolic activity (e.g. glucose uptake and OXPHOS) by 40–60%. This inhibition is comparable to the effect of the mechanistic target of rapamycin complex 1 (mTORC1) inhibitor rapamycin, as both TGF-β and rapamycin reduce NK cell proliferation, granzyme B expression and cytotoxicity against tumour cells. Mechanistically, TGF-β signalling via SMAD2/3 represses mTOR activity, uncoupling IL-15-mediated metabolic reprogramming and impairing effector function. Additionally, the mRNA endonuclease Regnase-1 acts as a negative regulator of IFN-γ production. Its deletion relieves the transcriptional repression of OCT2 (Pou2f2) and IkBζ (Nfkbiz), which form a complex with NF-κB to bind a distal enhancer (DI + 55) 55 kb downstream of the IFN-γ promoter. This interaction increases IFN-γ transcription by 4.6-fold, as confirmed by ChIP-seq, which revealed increased H3K27 acetylation at the DI + 55 locus in Reg1ΔNK-NK cells [50].
The first aspect is epigenetic silencing mediated by TGF-β. SMAD3 recruits HDACs to TBX21/PRF1 promoters, inducing chromatin compaction that silences T-bet and granzyme B. Lactate in the TME further inhibits HDAC activity, promoting NKG2D promoter methylation—a type of metabolic‒epigenetic cross-talk that exacerbates receptor downregulation [59, 60]. The second aspect is receptor signalling network perturbation. In HCC, TGF-β1 upregulates CD96, disrupting the CD226-CD96-TIGIT axis. This receptor perturbation, combined with NKG2D downregulation, forms a dual blockade of activation signals, reducing NK cell cytotoxicity [61]. The last aspect is mitochondrial metabolic dysfunction. In triple-negative breast cancer, TGF-β1 induces a TOMM20-low phenotype, disrupting mitochondrial protein import and OXPHOS. The resulting ATP depletion impairs granzyme B exocytosis, directly compromising cytotoxicity. TGF-β-coordinated immunosuppressive pathways play a role in this process. TGF-β‘s triad of mechanisms involving TGF-β synergises with canonical immunosuppression pathways. For example, activation of the IL-10/IDO axis polarises NK cells to a CD56brightCD16− regulatory phenotype. Moreover, paradoxical IFN-γ-JAK1/STAT1 signalling upregulates PD-L1 expression on NK cells, establishing a self-perpetuating loop of immunosuppression through the inhibition of PD-1+ T cells. The interaction between PD-1 and PD-L1 results in the recruitment of protein tyrosine phosphatase 2 (SHP-2), which in turn downregulates PI3K/Akt signalling to maintain immune tolerance [62, 63].
Spatial cues within the TME further determine the functional polarisation of NK cells. A reduction in membrane protrusions impairs the formation of lytic immunological synapses, as demonstrated by reduced tight junction formation and F-actin polarisation in tumour-infiltrating NK cells. Coculture experiments revealed that compared with >90% of peripheral NK cells, only 21–50% of intratumoural NK cells bind to tumour cells. This defect is correlated with decreased granzyme B polarisation and cytotoxicity, as measured by flow cytometry and real-time cell index assays [38]. Single-cell sequencing of EBV+ gastric cancer reveals spatial segregation of the following NK subsets: IFN-γ-high cells at vascularises margins and PD-L1+ cells in hypoxic cores. This correlates with local lactate gradients (>10 mM in cores), directly linking metabolic stress to phenotypic polarisation [64]. Adipose-derived MSCs secrete TGF-β and prostaglandin E2 (PGE2). These factors downregulate the production of perforin and granzyme B, regulate PD-L1 expression, disrupt IL-15-mediated survival signalling in NK cells and paradoxically increase NKG2D⁺ surface expression [50].
Lactate accumulation, a characteristic feature of TME acidosis, induces metabolic paralysis through dual mechanisms. Tumour acidosis (pH 6.2–6.5) impairs cytokine signalling by reducing the binding of IL-2 to IL-2Rα (EC₅₀ ↑2.3-fold at pH 6.5). This results from histidine protonation at the IL-2:IL-2Rα interface, disrupting receptor dimerisation. In B16 melanoma, acidosis suppresses IL-2-induced STAT5 phosphorylation by 40–60%, a defect reversed by bicarbonate neutralisation [65].
In CRLM, tumour-derived lactate (11.5 ± 2.8 mmol/L, pH 6.6) induces liver-resident NK cell apoptosis via mitochondrial ROS accumulation, a process rescued by MitoTempo [66]. Additionally, the immune checkpoint receptor TIM-3 plays a pivotal role in NK cell dysfunction via ligand-mediated signalling. Among its four ligands, galectin-9 has emerged as the most potent suppressor of NK cell cytotoxicity and proliferation. In head and neck squamous cell carcinoma (HNSCC), galectin-9 binding to TIM-3 impairs NK cell-mediated killing by disrupting PI3K signalling while concurrently promoting IFN-γ release in a TIM-3-dependent manner. This dual effect highlights the context-dependent role of TIM-3 in modulating NK cell effector functions. Mechanistically, galectin-9 also engages CD44 to suppress the proliferation of NK cells, a pathway that is resistant to conventional TIM-3 blockade [67]. This acidification impairs intracellular pH regulation in NK cells, causing mitochondrial ROS accumulation and ATP depletion. Clinically, CRLM patients with lower intratumoural NK cell numbers have higher recurrence rates, highlighting lactate as a key mediator of immune evasion [66]. Hypoxia exacerbates this state through DRP1 (S616)-mediated mitochondrial fragmentation. This fragmentation disrupts citrate-malate shuttling and redistributes H3K27ac histone marks from cytotoxic gene promoters (PRF1 and GZMB) to tolerance-associated loci (LAG-3 and TIM-3⁺), epigenetically locking NK cells into immunosuppressive states [68, 69].
Therapeutic strategies aimed at targeting this plasticity necessitate integrated interventions across multiple dimensions. PD-1/PD-L1 blockade disrupts immunoinhibitory ligand–receptor interactions, thereby restoring cytotoxic effector functions, as demonstrated in clinical models [53, 70,71,72]. The inhibition of lactate dehydrogenase reverses mTORC1 suppression, reinstating nuclear factor of activated T cells 1 (NFATc1)/IFN-γ signalling and bioenergetic homeostasis [50, 64]. Epigenetic modulators, such as HDAC inhibitors, prevent T-bet silencing and reactivate NKG2D⁺/NKp30 expression. These modulators synergise with TGF-β receptor antagonists to counteract MSC-mediated suppressive signalling. Crucially, these approaches must take into account the spatial heterogeneity observed in EBV+ gastric cancer, where cytotoxic and regulatory NK subsets occupy distinct tumour niches. This may require the development of spatially targeted delivery systems to optimise therapeutic efficacy [64].
This integrative framework defines NK cell functional plasticity as a multidimensional adaptation process. Shifts in the receptor expression profile initiate the tolerance state, energetic modifications stabilise the dysfunctional phenotype and metabolic reprogramming enforces bioenergetic constraints. Overcoming these multilayered barriers demands combination strategies that simultaneously target cell-intrinsic reprogramming drivers, such as TBX21 genetic and mitochondrial function and extrinsic TME signals, including TGF-β, lactate and hypoxia. By doing so, the reinstatement of NK cell-mediated immunosuppression across anatomical and molecular gradients becomes achievable. The epigenetic silencing induced by TGF-β converges with metabolic reprogramming in the TME, as discussed in Section ‘Metabolic plasticity: adaptation versus dysfunction’.
Metabolic plasticity: adaptation versus dysfunction
The metabolic plasticity of NK cells within the TME presents a paradoxical adaptive mechanism. Metabolic reprogramming driven by the imperative for survival in the face of nutrient deprivation ultimately compromises the cytotoxic effector functions of NK cells. Hypoxia-induced lactate accumulation and acidic pH impose substantial bioenergetic stress. This forces NK cells to prioritise the maintenance of cellular homeostasis over their immune surveillance role, thereby triggering a complex cascade of metabolic rewiring events. In breast cancer TMEs, this dualistic adaptation is clearly manifested. Hypoxia also inhibits the proliferation of NK cells and reduces their cytotoxicity [73]. Elevated levels of 27-hydroxycholesterol, a cholesterol metabolite that inhibits the transcriptional regulation mediated by sterol regulatory element-binding protein (SREBP), disrupt glucose metabolism in tumour-infiltrating NK cells [73,74,75,76]. Simultaneously, glucose-deprived NK cells activate compensatory glutamine dependency pathways. This metabolic vulnerability can be exploited by glutamine inhibitors such as CB-839, which selectively deplete the energy of tumour cells while safeguarding the viability of NK cells. The metabolic flexibility of NK cells extends to lipid metabolism as well. In HCC models, cholesterol enrichment enhances the antitumour capacity of NK cells through lipid raft-mediated receptor clustering. In contrast, in sepsis, TGF-β suppresses carnitine palmitoyl transferase 1A (CPT1A)-dependent fatty acid oxidation (FAO), leading to mitochondrial fragmentation and a subsequent loss of cytotoxicity [77, 78].
The interaction between nutrient sensing and effector function in NK cells reveals an intricate network of regulatory mechanisms. Tumour-derived kynurenine activates the aryl hydrocarbon receptor (AHR) pathway, which in turn suppresses glycolysis. Conversely, β-hydroxybutyrate restores mitochondrial efficiency and promotes the expression of granzyme B, demonstrating the metabolite-specific modulation of NK cell activity [79, 80]. These adaptations come with functional trade-offs. Although glutamine dependency enables NK cells to survive in glucose-limited niches in the short term, chronic exposure to 27-hydroxycholesterol induces epigenetic silencing of cytotoxicity genes through SREBP-mediated chromatin remodelling. This results in persistent dysfunction of NK cells even after the resolution of metabolic stress [77, 81]. Similarly, lactate accumulation not only inhibits the mTORC1, thereby blocking the nuclear translocation of NFATc1 and the production of IFN-γ, but also redistributes H3K27ac histone marks from the promoters of cytotoxicity genes to tolerance-associated loci. This energetic modification firmly entrenches the immunosuppressive state of NK cells [50, 64].
Emerging therapeutic strategies aim to target these key metabolic nodes through engineered interventions and modulation of the microenvironment. Supplementation with nicotinamide adenine dinucleotide (NAD+) enhances the mitochondrial membrane potential in CAR-NK cells. This improvement in mitochondrial function contributes to better cell persistence and a reduction in relapse rates in acute myeloid leukaemia (AML) patients. This approach is paralleled by preclinical studies that use ketone body supplementation to increase mitochondrial efficiency [82]. Moreover, coculturing NK cells with metformin-pretreated MSCs rescues glycolysis in NK cells by upregulating glucose transporter 1 and hexokinase 2. This effectively counteracts the suppressive effects of adipose-derived MSCs, which secrete TGF-β and PGE2 to downregulate the expression of perforin and granzyme B [50]. These strategies highlight the potential of decoupling the bioenergetics of NK cells from the constraints of the TME. However, significant challenges remain in achieving a balance between metabolic rewiring and the preservation of NK cell function.
Spatiotemporal heterogeneity further complicates the targeting of therapeutic interventions. In hypoxic tumour cores, OXPHOS is suppressed through lactate-mediated inhibition of mTORC1. In contrast, normoxic margins allow residual glycolytic activity in NK cells, creating anatomically distinct functional niches. Single-cell analyses confirm this metabolic zonation. In EBV+ gastric cancer, IFN-γhigh cytotoxic NK subsets are found mainly in the vascularises tumour margins, whereas lipid-laden immunosuppressive NK cells dominate the hypoxic zones [64]. This metabolic compartmentalisation reflects the dichotomous role of cholesterol. In HCC, cholesterol promotes lipid raft formation and the clustering of activating receptors. However, in atherosclerosis models, excessive cholesterol induces oxysterol cytotoxicity, which exacerbates NK cell dysfunction. Such context-dependent outcomes emphasise the need for precision medicine. AML patients may benefit from NAD+-enhanced CAR-NK therapies, while solid tumours may require a combined approach targeting lactate transporters (MCT1/4) and AHR signalling to overcome metabolic immunosuppression.
In summary, the metabolic plasticity of NK cells represents a dynamic equilibrium between adaptive survival and functional exhaustion. Therapeutic interventions must carefully navigate the fine line between restoring the bioenergetic competence of NK cells and avoiding iatrogenic metabolic stress. Future strategies that integrate real-time metabolic imaging with spatially resolved omics techniques hold great promise. These strategies could identify niche-specific vulnerabilities, thereby transforming the metabolic adaptation mechanism, which currently promotes tumour survival, into a viable therapeutic target.
Harnessing plasticity for immunotherapy
Receptor engineering: iterative innovation and functional optimisation of CAR-NK cells
Cellular immunotherapy encompasses a wide array of modalities, among which chimeric antigen receptor T (CAR-T) cell therapy has achieved extensive clinical implementation. However, recent research breakthroughs have spotlighted CAR-NK cells as a promising alternative, offering several distinct advantages over CAR-T cells. These advantages include better safety, multiple cytotoxic mechanisms, reduced risk of allogeneic reactions and clinical complete remission [83]. A characteristic feature of CAR-NK technology is gradually overcoming the immunosuppressive hurdles of the solid TME through innovative molecular engineering approaches. These cells are predominantly derived from CD56− human cord blood (CB) NK progenitor cells, with the intracellular domains of most CARs relying on the CD3ζ chain signalling module [84]. First-generation CAR-NK constructs solely contain the CD3ζ signalling domain. In contrast, second- and third-generation designs incorporate costimulatory domains, such as the 4-1BB-CD3ζ chimeric structure, which significantly enhance sustained activation and the antitumour memory response. For example, anti-CD19 CAR-NK cells engineered with CD3ζ exhibit a remarkable increase in cytotoxicity against target cells [84, 85].
The development of CAR-NK molecules has led to the emergence of fourth-generation ‘armed’ CAR systems, which represent a significant departure from earlier designs that relied on native immune receptor domains. These advanced constructs integrate modular molecular payloads, endowing CAR-engineered cells with enhanced functionality and addressing the inherent limitations of conventional immune cell therapies, as depicted in Fig. 3. The iterative evolution of CAR-NK technology across four generations is shown in Fig. 3, highlighting how structural modifications address key clinical challenges. First-generation CAR-NK cells: as shown in the figure, these constructs contain only the CD3ζ signalling domain, enabling basic cytotoxicity but lacking sustained activation—this limits their in vivo persistence, as observed in early CD19-targeted CAR-NK trials for B-cell malignancies [83]. Second/third-generation CAR-NK cells: the addition of costimulatory domains (e.g. 4-1BB-CD3ζ in the second generation, dual costimulatory domains in the third generation) resolves this issue. For example, 4-1BB signalling enhances antitumour memory responses, leading to prolonged CAR-NK survival and 66.7% response rates in diffuse large B-cell lymphoma [86]; fourth-generation ‘armed’ CAR-NK cells: the chart emphasises two key innovations: (1) integration of cytokine genes (e.g. IL-15) to support autonomous persistence—CLDN6-targeted CAR-NK cells with IL-15 modules achieve deep tumour infiltration in pancreatic cancer models [87]; (2) logic-gated antigen recognition (e.g. CD19/CD22, EGFR/IL13Rα2) to reduce off-target toxicity, as exemplified by Senti-401 (CEA-targeted), which spares healthy epithelial cells [88]; notably, Fig. 3 also highlights the shift from ‘native receptor-dependent’ to ‘modular payload-dependent’ design, which is critical for overcoming solid TME immunosuppression—for instance, TGF-βRII mutants in fourth-generation CAR-NK cells block TGF-β-mediated inhibition, a major barrier in glioblastoma [89]; this evolutionary timeline (Fig. 3) underscores how each generation of CAR-NK cells addresses the limitations of the previous generation, providing a framework for current clinical strategies targeting NK cell receptor plasticity. These fourth-generation innovations translate to clinical efficacy in patients with haematological malignancies. In B-cell malignant haematological tumours, CD19-targeted CAR-NK cells, when cocultured with B-cell non-Hodgkin lymphoma cells, increase the secretion of cytotoxic factors and enhance killing activity [85, 90]. The allogeneic logic-gated therapy Senti-401 exemplifies this precision, selectively eliminating carcinoembryonic antigen (CEA)+ tumour cells while sparing CEA+ healthy epithelial cells, as shown in Table 2 [89, 91,92,93,94,95,96,97,98]. An experiment revealed the role of the endonuclease Regnase-1 in the antitumour activity of NK cells. NK cell-specific deletion of Regnase-1 (Reg1ΔNK) increased cytolytic activity and interferon-gamma (IFN-γ) production in vitro and increased intratumoural accumulation of Reg1ΔNK-NK cells in vivo, reducing tumour growth in an IFN-γ-dependent manner. It was also proposed that targeting Regnase-1 could increase the persistence of active NK cells, which could be utilised in cancer immunotherapy [99].

Development of adoptive cell transfer therapy.
Clinical evidence validates the durability of CAR-NK responses. In a study involving 37 CD19+ B-cell malignancy patients treated with cord blood-derived CAR19/IL-15 NK cells, the overall response rate reached 48.6% at both Day 30 and Day 100. The 1-year overall survival and progression-free survival rates were 68% and 32%, respectively. Complete remission was associated with increased CAR-NK cell levels and prolonged persistence. In diffuse large B-cell lymphoma, 4-1BB-stimulated CD19 CAR-NK cells achieved a 66.7% response rate, highlighting the therapeutic potential of integrating costimulatory domains [86, 97, 100].
CRISPR-Cas9-based genome editing has emerged as a potent tool for enhancing CAR-NK functionality. By knocking out inhibitory receptors, such as TIGIT and CD96, it synergises with CAR signalling to reverse TME-induced exhaustion [101, 102]. In AML models, PD-1-knockdown CD33-targeting CAR-NK cells significantly enhance responses to anti-PD-L1 checkpoint inhibitors, highlighting the synthetic lethality manifested between genetic editing and immunotherapy. Translating NKG2D⁺-based activation into CAR designs, Jin et al. developed dual-targeted CD123/NKG2D ligand CAR-T cells that eliminate AML cells and selectively target immunosuppressive subsets in preclinical models. There are also experiments indicating the potential advantages of targeting TIM-3/CD44 or galectin-9 in the treatment of HPV-positive HNSCC [103, 104]. Unlike T cells, NK cells exhibit minimal adverse reactions and lack the risk of cytokine storms; thus, enhancing the localisation of NKG2D⁺ is a promising strategy for improving the safety of cell therapy [105,106,107].
Metabolic reprogramming: resolving TME energy restrictions
The metabolic adaptability of NK cells within the TME has become a focal point of research, with interventions aiming to restore their bioenergetic competence. Mitophagy inducers, such as urolithin A, eliminate dysfunctional mitochondria, thereby restoring OXPHOS efficiency and granzyme B trafficking in NK cells. With respect to lactic acid-mediated NK cell apoptosis, the mitochondrial-targeted antioxidant MitoTempo can effectively block this pathway; under acidic conditions (pH 6.4), MitoTempo reduced the apoptosis rate of liver-resident NK cells from 32.9% to 18.1% (P = 0.0013), providing new ideas for clinical translation. Combinations of lactate transporter inhibitors (such as AZD3965) or microenvironmental buffers (bicarbonate) may further enhance the therapeutic effect [66]. IDO inhibitors, such as epacadostat, block tryptophan depletion, preserving mTORC1 activity and the proliferative potential of NK cells [108]. Single-cell metabolomics studies have revealed that the lipid peroxidation microenvironment in the TME upregulates carnitine palmitoyl transferase 1A (CPT1A)-dependent FAO, reshaping the metabolic resilience of NK cells. This provides a mechanistic basis for lipid metabolism-targeted combination therapies [109, 110].
Cytokine engineering: spatiotemporal control of functional polarisation
Cytokine-based strategies capitalise on the crucial role of soluble factors in regulating NK cells. Next-generation cytokine fusion proteins, such as the IL-15/IL-21 heterodimer, promote NK cell expansion and functional polarisation in melanoma and head and neck cancer models when delivered via spatiotemporally controlled hydrogel systems. Integrating membrane-anchored IL-18 expression into CAR-NK platforms results in the establishment of an autocrine STAT3 activation loop, overcoming TGF-β-mediated immunosuppression [111, 112]. Preclinical data from colorectal cancer liver metastasis models have shown that such engineered NK cells can penetrate the fibrotic stroma, induce immunogenic cell death and stimulate systemic antitumour immunity.
Microenvironment remodelling: multimodal synergistic interventions
A targeted delivery system composed of a cationic liposome-encapsulated TGF-β siRNA combined with galectin-9 inhibitors disrupted adenosine-dependent inhibition by tumour-associated macrophages (TAMs), significantly increasing the tumour infiltration density of CAR-NK cells. In breast cancer models, this strategy enhances both infiltration and killing activity, demonstrating the potential of nanotechnology to reprogramme the immunosuppressive niche [113,114,115].
Oncolytic herpes simplex virus expressing CXCL10 (oHSV-CXCL10) utilises chemokine gradients to recruit peripheral NK cells while inducing tumour cells to release damage-associated molecular patterns, thereby amplifying ADCC. In triple-negative breast cancer models, this combination achieves more than threefold greater tumour regression than monotherapy does, highlighting the synergistic potential of viroimmunotherapy, as shown in Table 3 [15, 66, 73, 86, 112].
Challenges and future directions
We have compiled a table summarising the three types of plasticity of NK cells and their interactions, highlighting the importance of NK cells in immunotherapy (Table 4). Despite significant progress, multiple challenges remain in maximising the potential of CAR-NK cell therapy. Single-cell sequencing research has mapped the spatiotemporal heterogeneity of NK cells in EBV-related gastric cancer, revealing distinct transcriptomic profiles between hypoxic core-residing TIM-3⁺ exhausted subsets and marginal CD49a⁺ tissue-resident NK populations [12, 48]. These results emphasize the crucial need for spatial transcriptomics to guide precision treatment strategies, as noted in recent reviews. The host immune rejection of allogeneic CAR-NK cells, along with the metabolic limitations within the TME—which are characterised by nutrient deficiency and lactate build-up—restricts cell persistence. This has led to the exploration of combination therapies with antiangiogenic drugs (such as bevacizumab) to improve tumour infiltration. Moreover, owing to the dysfunction of the patient’s immune system, there is a scarcity of autologous NK cells, which further increases the reliance on allogeneic sources. However, the poor in vitro expandability of NK cells still presents a major obstacle to large-scale production [87]. Additionally, the circulation route of CAR-NK cells poses difficulties. Prolonged exposure to peripheral blood reduces its effective contact with tumour lesions, thereby weakening therapeutic effectiveness.
Another layer of complexity comes from stromal cell interactions. Adipose-derived MSCs secrete TGF-β and PGE2. These substances work together to induce NK cell dysfunction through receptor downregulation and metabolic reprogramming. Given that TGF-β regulates the phenotype, function and metabolic plasticity of NK cells by inhibiting the mTOR pathway, intervention strategies targeting this axis may become key to overcoming the limitations of the TME. For example, the combined use of TGF-β receptor antagonists (such as SB-431542) and mTOR activators can simultaneously restore the expression of cytotoxic receptors (such as NKG2D), mitochondrial metabolic efficiency and IFN-γ secretion ability of NK cells. This highlights the need for strategies that simultaneously target both tumour cells and immunosuppressive stromal components.
Innovative methods are emerging to address these challenges. The circadian regulation of NK cell metabolism has become a promising approach for timed therapy. Preclinical studies have shown enhanced efficacy by aligning drug delivery with NK cell metabolic peaks, such as the morning glycolysis peak [116,117,118,119,120]. Building on the pH sensitivity of cytokines in the TME, future efforts could focus on developing microenvironment-adaptable IL-2 variants. For instance, the combination of Switch-2 with immune checkpoint inhibitors (e.g. anti-PD-L1) can restore NK cell function while reversing T-cell exhaustion, demonstrating superior antitumour efficacy compared with monotherapy in the B16.SIY model. Additionally, combining IL-2 variants with metabolic modulators (e.g. lactate transporter inhibitors) could further increase their activity by ameliorating the acidic TME, offering a multidimensional solution to overcome NK cell dysfunction in solid tumours [65]. Integrated multiomics analyses (transcriptomic, metabolomic and epigenomic) hold promise for predicting patient-specific NK cell vulnerabilities, enabling the targeted delivery of miR-155 or NKG2D⁺ agonists via nanoparticles. The synergistic combination of NK cell therapy and antiangiogenic agents has already been demonstrated to be effective in renal cell carcinoma, highlighting the potential of multimodal interventions. Emerging technologies such as spatial proteomics and organoid-based TME modelling will deepen our understanding of NK cell plasticity through the replication of tumour niche complexity. However, standardising NK cell sources is still crucial for clinical translation. Currently, batch differences between manually and machine-separated NK cells affect treatment consistency, making robust quality control frameworks essential.
Conclusion
The multidimensional plasticity of NK cells—including phenotypic, functional and metabolic adaptations—is not only a core feature of their immune surveillance ability but also a key mechanism for tumour immune escape within the TME. Phenotypic plasticity creates a ‘double-hit’ immunosuppressive pattern through dynamic receptor network remodelling (for example, imbalance of the TIGIT-CD155 axis), epigenetic silencing (such as NKG2D⁺ promoter hypermethylation) and microenvironment-driven subpopulation heterogeneity (e.g. TIM-3⁺ exhausted NK cells in hypoxic niches). Functionally, under the influence of TGF-β/SMAD3/HDAC-mediated epigenetic reprogramming, metabolic stress (such as lactate-induced mTORC1 inhibition) and spatial cues (for instance, the accumulation of PD-L1+ NK cells in the cores of EBV+ gastric cancer), NK cells shift from cytotoxic effectors to immunosuppressive regulators. Metabolic plasticity reveals a survival paradox for NK cells in the TME: while adaptations such as glutamine dependency or lipid metabolism allow NK cells to tolerate TME stress, these reprogramming events often result in reduced cytotoxic capacity.
In response, emerging immunotherapies use multilevel strategies to overcome TME barriers: receptor engineering (such as combining CAR-NK cells with TGF-βRII mutants), metabolic interventions (for example, NAD+ supplementation to restore OXPHOS) and microenvironment remodelling (such as the use of CD73⁺ inhibitors to disrupt adenosine signalling). In the future, technological innovations integrating single-cell spatiotemporal omics, circadian rhythm regulation (such as optimising the CRY/BMAL1 pathway) and precision metabolic imaging will drive a paradigm shift in NK cell therapy, evolving from ‘passive adaptation’ to ‘active remodelling’ of the TME. These advancements offer the hope of achieving breakthroughs in personalised immunotherapy by systematically addressing the complex layers of NK cell plasticity in cancer.
References
Crinier A, Dumas PY, Escalière B, Piperoglou C, Gil L, Villacreces A, et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell Mol Immunol. 2021;18:1290–304.
Rebuffet L, Melsen JE, Escalière B, Basurto-Lozada D, Bhandoola A, Björkström NK, et al. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat Immunol. 2024;25:1474–88.
Peng L, Renauer PA, Sferruzza G, Yang L, Zou Y, Fang Z, et al. In vivo AAV-SB-CRISPR screens of tumor-infiltrating primary NK cells identify genetic checkpoints of CAR-NK therapy. Nat Biotechnol. 2025;43:752–61.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10.
Mujal AM, Delconte RB, Sun JC. Natural killer cells: from innate to adaptive features. Annu Rev Immunol. 2021;39:417–47.
Coënon L, Geindreau M, Ghiringhelli F, Villalba M, Bruchard M. Natural killer cells at the frontline in the fight against cancer. Cell Death Dis. 2024;15:614.
Judge SJ, Murphy WJ, Canter RJ. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol. 2020;10:49.
Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74:7250–9.
Franklin M, Connolly E, Hussell T. Recruited and tissue-resident natural killer cells in the lung during infection and cancer. Front Immunol. 2022;13:887503.
Hashemi E, Malarkannan S. Tissue-resident NK cells: development, maturation, and clinical relevance. Cancers. 2020;12:1553.
Gustafson CE, Qi Q, Hutter-Saunders J, Gupta S, Jadhav R, Newell E, et al. Immune checkpoint function of CD85j in CD8 T cell differentiation and aging. Front Immunol. 2017;8:692.
Corvino D, Kumar A, Bald T. Plasticity of NK cells in cancer. Front Immunol. 2022;13:888313.
Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74.
Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. 2019;16:430–41.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18:85–100.
Liu J, Bai Y, Li Y, Li X, Luo K. Reprogramming the immunosuppressive tumor microenvironment through nanomedicine: an immunometabolism perspective. EBioMedicine. 2024;107:105301.
de Andrade LF, Smyth MJ, Martinet L. DNAM-1 control of natural killer cell functions through nectin and nectin-like proteins. Immunol Cell Biol. 2014;92:237–44.
Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6:e1320011.
Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018;78:1003–16.
Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108:4118–25.
Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47:820–33.
Harmon C, Robinson MW, Fahey R, Whelan S, Houlihan DD, Geoghegan J, et al. Tissue-resident Eomes(hi) T-bet(lo) CD56(bright) NK cells with reduced proinflammatory potential are enriched in the adult human liver. Eur J Immunol. 2016;46:2111–20.
Burt BM, Plitas G, Zhao Z, Bamboat ZM, Nguyen HM, Dupont B, et al. The lytic potential of human liver NK cells is restricted by their limited expression of inhibitory killer Ig-like receptors. J Immunol. 2009;183:1789–96.
Greppi M, De Franco F, Obino V, Rebaudi F, Goda R, Frumento D, et al. NK cell receptors in anti-tumor and healthy tissue protection: mechanisms and therapeutic advances. Immunol Lett. 2024;270:106932.
Zhang C, Liu Y. Targeting NK cell checkpoint receptors or molecules for cancer immunotherapy. Front Immunol. 2020;11:1295.
Myers JA, Schirm D, Bendzick L, Hopps R, Selleck C, Hinderlie P, et al. Balanced engagement of activating and inhibitory receptors mitigates human NK cell exhaustion. JCI Insight. 2022;7:e155585.
Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst. 2014;106:dju200.
Yu L, Liu X, Wang X, Yan F, Wang P, Jiang Y, et al. TIGIT(+) TIM-3(+) NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virus‑related hepatocellular carcinoma. Oncoimmunology. 2021;10:1942673.
Portale F, Di Mitri D. NK cells in cancer: mechanisms of dysfunction and therapeutic potential. Int J Mol Sci. 2023;24:4636.
Sun Y, Li T, Ding L, Wang J, Chen C, Liu T, et al. Platelet-mediated circulating tumor cell evasion from natural killer cell killing through immune checkpoint CD155-TIGIT. Hepatology. 2025;81:791–807.
Zhang W, Zhao Z, Li F. Natural killer cell dysfunction in cancer and new strategies to utilize NK cell potential for cancer immunotherapy. Mol Immunol. 2022;144:58–70.
Yahata T, Mizoguchi M, Kimura A, Orimo T, Toujima S, Kuninaka Y, et al. Programmed cell death ligand 1 disruption by clustered regularly interspaced short palindromic repeats/Cas9-genome editing promotes antitumor immunity and suppresses ovarian cancer progression. Cancer Sci. 2019;110:1279–92.
Molfetta R, Zitti B, Lecce M, Milito ND, Stabile H, Fionda C, et al. CD155: A Multi-Functional Molecule in Tumor Progression. Int J Mol Sci. 2020;21:9584.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110.
Xing S, Ferrari de Andrade L. NKG2D and MICA/B shedding: a ‘tag game’ between NK cells and malignant cells. Clin Transl Immunol. 2020;9:e1230.
Zheng X, Hou Z, Qian Y, Zhang Y, Cui Q, Wang X, et al. Tumors evade immune cytotoxicity by altering the surface topology of NK cells. Nat Immunol. 2023;24:802–13.
Watts GS, Futscher BW, Holtan N, DeGeest K, Domann FE, Rose SL. DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med Genom. 2008;1:47.
Hornig E, Heppt MV, Graf SA, Ruzicka T, Berking C. Inhibition of histone deacetylases in melanoma—a perspective from bench to bedside. Exp Dermatol. 2016;25:831–8.
Koukoura O, Spandidos DA, Daponte A, Sifakis S. DNA methylation profiles in ovarian cancer: implication in diagnosis and therapy (Review). Mol Med Rep. 2014;10:3–9.
Tang F, Li J, Qi L, Liu D, Bo Y, Qin S, et al. A pan-cancer single-cell panorama of human natural killer cells. Cell. 2023;186:4235–4251.e4220.
Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene. 2020;39:428–42.
Montaldo E, Del Zotto G, Della Chiesa M, Mingari MC, Moretta A, De Maria A, et al. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A. 2013;83:702–13.
Lim AI, Verrier T, Vosshenrich CAJ, Di Santo JP. Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol. 2017;44:61–8.
Mirzaee V, Shahriari J, Hajghani M. CCR5 on the NK cells and its ligand (RANTES) expressions are disrupted in south-eastern Iranian patients with chronic hepatitis B infection. Iran Red Crescent Med J. 2014;16:e12458.
Feng Y, Zhong M, Liu Y, Wang L, Tang Y. Expression of TIM-3 and LAG-3 in extranodal NK/T cell lymphoma, nasal type. Histol Histopath. 2018;33:307–15.
Picant V, Revol-Bauz L, Tonon L, Casini T, Voissière A, Poujol D, et al. Interleukin-35 impairs human NK cell effector functions and induces their ILC1-like conversion with tissue residency features. Nat Commun. 2025;16:6135.
Pfefferle A, Jacobs B, Netskar H, Ask EH, Lorenz S, Clancy T, et al. Intra-lineage plasticity and functional reprogramming maintain natural killer cell repertoire diversity. Cell Rep. 2019;29:2284–2294.e2284.
Viel S, Marçais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9:ra19.
Shen J, Zhang Y, Tang W, Yang M, Cheng T, Chen Y, et al. Short IL-18 generated by caspase-3 cleavage mobilizes NK cells to suppress tumor growth. Nat Immunol. 2025;26:416–28.
Liu W, Wei X, Li L, Wu X, Yan J, Yang H, et al. CCR4-mediated chemotaxis of regulatory T cells suppress the activation of T cells and NK cells via TGF-β pathway in human non-small cell lung cancer. Biochem Biophys Res Commun. 2017;488:196–203.
Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M, et al. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J Immunother Cancer. 2021;9:e77458.
Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. 2019;10:2278.
Zhou Y, Cheng L, Liu L, Li X. NK cells are never alone: crosstalk and communication in tumour microenvironments. Mol Cancer. 2023;22:34.
Agaugué S, Marcenaro E, Ferranti B, Moretta L, Moretta A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood. 2008;112:1776–83.
Bozward AG, Warricker F, Oo YH, Khakoo SI. Natural killer cells and regulatory T cells cross-talk in hepatocellular carcinoma: exploring therapeutic options for the next decade. Front Immunol. 2021;12:643310.
Johansson MH, Höglund P. The dynamics of natural killer cell tolerance. Semin Cancer Biol. 2006;16:393–403.
Naik A, Thakur N. Epigenetic regulation of TGF-β and vice versa in cancers—a review on recent developments. Biochim Biophys Acta Rev Cancer. 2024;1879:189219.
Tang X, Tu G, Yang G, Wang X, Kang L, Yang L, et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 2019;452:79–89.
Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. 2019;70:168–83.
Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong W, et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. 2024;23:108.
Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, et al. PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy. J Cell Physiol. 2019;234:16824–37.
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24:657–71.
Gaggero S, Martinez-Fabregas J, Cozzani A, Fyfe PK, Leprohon M, Yang J, et al. IL-2 is inactivated by the acidic pH environment of tumors, enabling engineering of a pH-selective mutein. Sci Immunol. 2022;7:eade5686.
Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol Res. 2019;7:335–46.
Wang J, Li H, Kulkarni A, Anderson JL, Upadhyay P, Onyekachi OV, et al. Differential impact of TIM-3 ligands on NK cell function. J Immunother Cancer. 2025;13:e010618.
Zhang D, Liu Y, Tang Y, Wang X, Li Z, Li R, et al. Increased mitochondrial fission is critical for hypoxia-induced pancreatic beta cell death. PLoS ONE. 2018;13:e0197266.
Pouikli A, Maleszewska M, Parekh S, Yang M, Nikopoulou C, Bonfiglio JJ, et al. Hypoxia promotes osteogenesis by facilitating acetyl-CoA-mediated mitochondrial-nuclear communication. EMBO J. 2022;41:e111239.
Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE. 2018;13:e0191358.
Bejarano L, Jordāo MJC, Joyce JA. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11:933–59.
Gardiner CM. NK cell metabolism. J Leukoc Biol. 2019;105:1235–42.
Kennedy PR, Arvindam US, Phung SK, Ettestad B, Feng X, Li Y, et al. Metabolic programs drive the function of therapeutic NK cells in hypoxic tumor environments. Sci Adv. 2024;10:eadn1849.
Li D, Long W, Huang R, Chen Y, Xia M. 27-Hydroxycholesterol inhibits sterol regulatory element-binding protein 1 activation and hepatic lipid accumulation in mice. Obesity. 2018;26:713–22.
Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem. 2004;279:52772–80.
Liu W, Chakraborty B, Safi R, Kazmin D, Chang CY, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis, increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021;12:5103.
Presnell SR, Spear HK, Durham J, Riddle T, Applegate A, Lutz CT. Differential fuel requirements of human NK cells and human CD8 T cells: glutamine regulates glucose uptake in strongly activated CD8 T cells. ImmunoHorizons. 2020;4:231–44.
Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology. 2020;158:1713–27.
Tang M, Tu Y, Gong Y, Yang Q, Wang J, Zhang Z, et al. β-hydroxybutyrate facilitates mitochondrial-derived vesicle biogenesis and improves mitochondrial functions. Mol Cell. 2025;85:1395–1410.e1395.
Finlay DK. Metabolic regulation of natural killer cells. Biochem Soc Trans. 2015;43:758–62.
Ying D, Zhang G, Huang H, Tan WS, Cai H. Optimizing glutamine concentration enhances ex vivo expansion of natural killer cells through improved redox status. Biotechnol Prog. 2024;40:e3464.
Llorente-Folch I, Düssmann H, Watters O, Connolly NMC, Prehn JHM. Ketone body β-hydroxybutyrate (BHB) preserves mitochondrial bioenergetics. Sci Rep. 2023;13:19664.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–53.
Strecker MI, Wlotzka K, Strassheimer F, Roller B, Ludmirski G, König S, et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology. 2022;11:2127508.
Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat Med. 2024;30:772–84.
Wu X, Matosevic S. Gene-edited and CAR-NK cells: opportunities and challenges with engineering of NK cells for immunotherapy. Mol Ther Oncolytics. 2022;27:224–38.
Luo J, Guo M, Huang M, Liu Y, Qian Y, Liu Q, et al. Neoleukin-2/15-armored CAR-NK cells sustain superior therapeutic efficacy in solid tumors via c-Myc/NRF1 activation. Signal Transduct Target Ther. 2025;10:78.
Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell. 2019;35:489–503.e488.
Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Investig. 2021;131:e142116.
Yang N, Zhang C, Zhang Y, Fan Y, Zhang J, Lin X, et al. CD19/CD20 dual-targeted chimeric antigen receptor-engineered natural killer cells exhibit improved cytotoxicity against acute lymphoblastic leukemia. J Transl Med. 2024;22:274.
Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med. 2022;28:2133–44.
Marofi F, Saleh MM, Rahman HS, Suksatan W, Al-Gazally ME, Abdelbasset WK, et al. CAR-engineered NK cells: a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res Ther. 2021;12:374.
Biederstädt A, Rezvani K. Engineering the next generation of CAR-NK immunotherapies. Int J Hematol. 2021;114:554–71.
Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14:73.
Burger MC, Forster MT, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro-Oncology. 2023;25:2058–71.
Asadi M, Kiani R, Razban V, Faraji SN, Ahmadi A, Fallahi J, et al. Harnessing the power of CAR-NK cells: a promising off-the-shelf therapeutic strategy for CD38-positive malignancies. Iran J Immunol. 2023;20:410–26.
Lei W, Liu H, Deng W, Chen W, Liang Y, Gao W, et al. Safety and feasibility of 4-1BB co-stimulated CD19-specific CAR-NK cell therapy in refractory/relapsed large B cell lymphoma: a phase 1 trial. Nat Cancer. 2025;6:786–800.
Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci USA. 2022;119:e2122379119.
Sun X, Nagahama Y, Singh SK, Kozakai Y, Nabeshima H, Fukushima K, et al. Deletion of the mRNA endonuclease Regnase-1 promotes NK cell anti-tumor activity via OCT2-dependent transcription of Ifng. Immunity. 2024;57:1360–1377.e1313.
Shapiro RM, Romee R. iPSC-derived CD19 CAR NK cells for relapsed or refractory lymphoma. Lancet. 2025;405:98–9.
Wang M, Krueger JB, Gilkey AK, Stelljes EM, Kluesner MG, Pomeroy EJ, et al. Precision enhancement of CAR-NK cells through non-viral engineering and highly multiplexed base editing. J Immunother Cancer. 2025;13:e009560.
Bexte T, Albinger N, Al Ajami A, Wendel P, Buchinger L, Gessner A, et al. CRISPR/Cas9 editing of NKG2A improves the efficacy of primary CD33-directed chimeric antigen receptor natural killer cells. Nat Commun. 2024;15:8439.
Ge J, Meng Y, Guo J, Chen P, Wang J, Shi L, et al. Human papillomavirus-encoded circular RNA circE7 promotes immune evasion in head and neck squamous cell carcinoma. Nat Commun. 2024;15:8609.
Li J, Hu H, Lian K, Zhang D, Hu P, He Z, et al. CAR-NK cells in combination therapy against cancer: a potential paradigm. Heliyon. 2024;10:e27196.
Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021;11:146.
Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. 2015;212:307–17.
Tang S, Zhang Y, Wang P, Tang Q, Liu Y, Lu F, et al. NKG2D-CAR-targeted iPSC-derived MSCs efficiently target solid tumors expressing NKG2D ligand. iScience. 2025;28:112343.
Wang M, Liu Y, Li Y, Lu T, Wang M, Cheng Z, et al. Tumor microenvironment-responsive nanoparticles enhance IDO1 blockade immunotherapy by remodeling metabolic immunosuppression. Adv Sci. 2025;12:e2405845.
Sheppard S, Srpan K, Lin W, Lee M, Delconte RB, Owyong M, et al. Fatty acid oxidation fuels natural killer cell responses against infection and cancer. Proc Natl Acad Sci USA. 2024;121:e2319254121.
Delconte RB, Owyong M, Santosa EK, Srpan K, Sheppard S, McGuire TJ, et al. Fasting reshapes tissue-specific niches to improve NK cell-mediated anti-tumor immunity. Immunity. 2024;57:1923–1938.e1927.
Yang M, Qin C, Tao L, Cheng G, Li J, Lv F, et al. Synchronous targeted delivery of TGF-β siRNA to stromal and tumor cells elicits robust antitumor immunity against triple-negative breast cancer by comprehensively remodeling the tumor microenvironment. Biomaterials. 2023;301:122253.
Möller AM, Vettermann S, Baumann F, Pütter M, Müller D. Trifunctional antibody-cytokine fusion protein formats for tumor-targeted combination of IL-15 with IL-7 or IL-21. Front Immunol. 2025;16:1498697.
Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23:556–67.
Shobaki N, Sato Y, Suzuki Y, Okabe N, Harashima H. Manipulating the function of tumor-associated macrophages by siRNA-loaded lipid nanoparticles for cancer immunotherapy. J Control Release. 2020;325:235–48.
Xu Z, Wang Y, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636–45.
Tomar MS, Mohit, Kumar A, Shrivastava A. Circadian immunometabolism: a future insight for targeted therapy in cancer. Sleep Med Rev. 2025;80:102031.
Zeng Y, Guo Z, Wu M, Chen F, Chen L. Circadian rhythm regulates the function of immune cells and participates in the development of tumors. Cell Death Discov. 2024;10:199.
Zlacká J, Zeman M. Glycolysis under circadian control. Int J Mol Sci. 2021;22:13666.
Arjona A, Sarkar DK. Circadian oscillations of clock genes, cytolytic factors, and cytokines in rat NK cells. J Immunol. 2005;174:7618–24.
Arjona A, Sarkar DK. Evidence supporting a circadian control of natural killer cell function. Brain Behav Immun. 2006;20:469–76.
Acknowledgements
Figures were created with BioRender.com.
Funding
This work was supported by the National Natural Science Foundation of China (No.82373368, No. 82502069), the Natural Science Basic Research Plan of Shaanxi Province (No. 2024JC-YBQN-0834), the Innovation Capacity Support Plan of Shaanxi Province (No. 2024ZC-KJXX-110), and the Xi’an Science and Technology Plan Project (No. 25YXYJYB00001).
Author information
Authors and Affiliations
Contributions
Xinya Yang and Zhaoyang Song: writing- original draft preparation. Ligang Chen, Qingge Jia and Mingyang Li: writing— reviewing and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Francesca Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, X., Song, Z., Chen, L. et al. Multidimensional plasticity of natural killer cells in tumours. Cell Death Dis 16, 916 (2025). https://doi.org/10.1038/s41419-025-08361-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-08361-x
