
Abstract
Drug resistance remains a major challenge to durable responses in ovarian cancer, the fifth leading cause of cancer-related death among women. In this study, we developed long-term resistant (lt-res, several months) pre-clinical models of two drugs inducing mitotic arrest in TP53-mutated cells: adavosertib (ADA), an investigational WEE1 inhibitor targeting the DNA damage response and currently evaluated in clinical trials, and paclitaxel (PTX), a widely used chemotherapeutic agent in cancer care targeting microtubules. Through integrated multi-omics functional profiling, we identify a shared PI3K/AKT-regulated signaling node that governs drug adaptation across all lt-res models. This node modulates the activity of DNA-damage responses and genotoxic stress to toggle between two adaptive states: activated PI3K/AKT driving a proliferative “fast-bypass” program with sustained cell cycle progression and mitotic evasion, or reduced PI3K/AKT signaling initiating a “slow-repair” state characterized by DNA damage checkpoint engagement, replication slowdown, and increased drug efflux. Notably, upregulation of receptor tyrosine kinases, such as ROR1, was observed in both ADA and PTX lt-res models with activated PI3K/AKT signaling. Targeting ROR1 with zilovertamab-vedotin, a monoclonal antibody-drug conjugate, resulted in enhanced cytotoxicity, demonstrating a new approach against recurrent drug-resistant ovarian cancer.
Similar content being viewed by others


Introduction
Mitotic catastrophe and cell cycle disruption are central vulnerabilities in TP53-mutated cancers, including high-grade serous ovarian carcinoma (HGSOC), the most aggressive subtype of ovarian cancer (OC) accounting for 50–70% of epithelial OCs [1, 2]. HGSOC is characterized by ubiquitous TP53 mutations ( > 90% of tumors), profound genomic instability, and extensive copy number aberrations (CNA), along with defects in homologous recombination and epigenetic alterations [2, 3]. First-line treatment typically combines debulking surgery with platinum-based agents, however, resistance commonly emerges [4, 5].
Loss of TP53 function impairs the G1 checkpoint, forcing reliance on mitotic checkpoints to maintain genomic integrity [6]. This creates a therapeutic window to induce mitotic catastrophe, a form of cell death resulting from aberrant mitosis and chromosome mis-segregation. Two drugs exploiting this vulnerability - adavosertib (ADA), a selective WEE1 kinase inhibitor, and paclitaxel (PTX), a microtubule stabilizer - induce mitotic stress through distinct but convergent mechanisms. ADA disrupts the G2/M checkpoint by inhibiting WEE1, which normally restrains CDK1/2 activity to prevent premature mitosis [7, 8]. In TP53-mutated HGSOC, WEE1 inhibition forces cells with DNA damage into unscheduled mitosis, resulting in replication stress and mitotic catastrophe [9,10,11,12]. WEE1 overexpression correlates with poor chemotherapy response, highlighting its clinical importance [9, 12]. Conversely, PTX stabilizes microtubules, blocking spindle dynamics essential for chromosome segregation and causing metaphase-anaphase arrest followed by mitotic catastrophe [13, 14]. TP53 deficiency sensitizes cells to PTX, as mitotic checkpoints become critical for survival [15,16,17]. However, ADA and PTX resistance is a clinical challenge, as often drug resistance mechanisms are studied under short-term drug exposure, which may not fully recapitulate clinical resistance. Mitotic catastrophe can be circumvented by activation of survival pathways such as PI3K/AKT, altered in ~45% of HGSOC cases, which promotes proliferation and drug resistance by suppressing pro-apoptotic signaling, FOXO3-mediated cell cycle arrest, and enhancing efflux transporter expression [18, 19].
In this study, we investigated the molecular mechanisms underlying the development of long-term resistance (lt-res) to ADA and PTX using models generated through incremental drug exposure - ranging from several weeks in ex vivo primary tumor-derived samples (PDCs) to several months in HGSOC cell lines. A shared resistance mechanism involving a PI3K/AKT signaling switch revealed the adaptability of HGSOC cells to both targeted therapies and chemotherapeutics. Upregulation of receptor tyrosine kinases (RTKs), including ROR1, in PI3K/AKT-activated lt-res models pointed to a therapeutic vulnerability. Treatment with zilovertamab-vedotin, a ROR1-targeting antibody-drug conjugate, demonstrated enhanced efficacy in drug-resistant OC cells.
Materials and methods
Cell culture and creation of resistant cell lines and PDCs
Ovarian cancer cell lines used in this study have been previously characterized by us [20]. All cell lines were authenticated by the Genomics Unit, FIMM Technology Center, University of Helsinki, Finland. The patient-derived cells (PDCs) were acquired upon informed consent and under institutional ethical review board-approved protocols (no. 56/13/03/03/2014) at the University of Helsinki Central Hospital. The PDCs were cultured in 2D as described previously [21]. Cell counting was done with Countess II automated cell counter (Invitrogen™). All cell lines and PDCs were routinely tested for mycoplasma contamination and were found mycoplasma free.
Resistant cell lines and PDCs were established by gradual increase of the Paclitaxel (PTX, #S1150, Selleckchem, Houston, TX, USA) or Adavosertib (ADA, #S1525, Selleckchem), concentrations depending on the EC50 of each parental cell line or PDC. Cells were allowed to rest if extensive cell death was detected, and drug amount was increased once the cells showed recovery. The resistance was tested also after cryopreservation and thawing. Drug resistant cell lines were also authenticated to confirm the original phenotype. To maintain the resistant phenotype, cells were cultured with the maximum concentration of drug added to the culture media. To target ROR1 expression with a monoclonal antibody, the cells were treated with Zilovertamab vedotin (#HY-P99956, MedChemExpress, Monmouth Junction, NJ, USA) for assays as indicated.
Generation of stable shRNA cell lines
Stable shRNA cell lines expressing control and ROR1 shRNA were generated as described previously by using an inducible pLKO-Tet-On lentiviral expression system [22]. The expression of shRNA was induced by 100 ng/ml doxycycline (DOX) treatment refreshed every three days and ROR1 silencing was verified by immunoblot analysis. The ADA resistant shROR1 Kuramochi and JHOS2 cells were created by gradually increasing the drug concentration (ADA/PTX) with DOX maintained in the media.
For ROR1high cells, ROR1 cloned into the pEF-IRES-P vector was transfected using FuGENE HD transfection agent (Promega, Madison, WI, USA) as per manufacturer instructions [23]. ROR1high clones were selected with puromycin and the ROR1 expression was verified with immunoblotting. The resistant JHOS2 and OVCAR8 ROR1high cells were developed by gradually increasing the ADA or PTX concentration, and puromycin was added every week to maintain the selection pressure.
Drug sensitivity and resistance testing (DSRT)
DSRT was performed as described previously [24]. Briefly, 1000–1500 cells were added to wells of 384-well plates with drugs pre-plated over a 10,000-fold concentration range (in five concentrations). After a three-day incubation at 37 °C and 5.0% CO2, CellTiter-Glo (CTG) 2.0 Assay (Promega) was used to measure cell viability according to manufacturer’s instruction. Luminescence signal was detected using Envision plate reader (PerkinElmer, Waltham, MA, USA) or PHERAstar FS (BMG Labtech, Ortenberg, Germany) after timepoints described for each experiment. For each drug, dose response curves were generated and DSS were calculated as previously described [24]. The 85th percentile of all DSS values was used to obtain a cutoff value for effective drugs within each cell strain (parental and corresponding resistant). The DSRT data analyses were performed using BREEZE2 [25] and R (4.3.2). Heatmaps were produced with the R package ComplexHeatmap (2.15.4) [26]. All the DSS values along with the differences between each resistant and parental cell line are available in Source Data.
Immunoblotting
Cells were lysed with Triton-X lysis buffer (50 mM Tris-HCl pH 7.5, 10% glycerol, 150 mM NaCl, 1 mM EDTA, 1% Triton-x-100, 50 mM NaF) supplemented with protease and phosphatase inhibitor cocktails (#B14001 and #B15001, Selleckchem). Protein in the lysates was quantified using Pierce 660 nm Protein Assay reagent and Nanodrop (Thermo Fisher Scientific), then mixed with 4X Laemmli sample buffer (#1610747, Bio-Rad Laboratories, California, USA) and β-mercaptoethanol, separated in SDS-PAGE with 4-20% gradient gels (Bio-Rad Laboratories), and transferred to 0.45 µm nitrocellulose membranes, followed by incubation with the following primary antibodies (all used 1:1,000 dilution, if not stated otherwise): β-tubulin (#2146, Cell Signaling Technology (CST), Danvers, MA, USA), β-actin (#4967, CST), caveolin 1 (#3267, CST), FoxO3a (#99199, CST), c-MYC (#13987, CST), AKT (#2920, CST), AKT-pSer473 (#4060, CST), E-cadherin (#14472S, CST), E-cadherin (#3195, CST), ERK1/2 (#4696, CST), ERK1/2-pThr202/Tyr204 (#9101, CST), GAPDH (#G8795, Sigma-Aldrich, Burlington, MA, USA or #AHP1628, Bio-Rad Laboratories), NF-kB p65 (#6956, CST), NF-kB p65-pSer536 (#3033, CST), STAT3 (#9139, CST), STAT3-pTyr705 (#9145, CST), STAT1 (#65917, CST), STAT1-pTyr701 (#88845, CST), PTK7 (#25618, CST), ROR1 (#16540, CST, 1:500), EGFR (#4267, CST), WEE1 (#13084, CST). 10 µg of protein was loaded in all the immunoblots. Secondary antibodies were IRDye® 800CW Donkey anti-Mouse IgG or IRDye® 680RD Donkey anti-Rabbit IgG (#926-32212 and #926-68073, LI-COR, Lincoln, NE, USA) and blots were scanned with Odyssey® Fc Imaging system (LI-COR). Image analysis was done using the Image Studio Lite software (LI-COR).
Spheroid formation assay
Cells were stained with 2 µM CellTracker Green (#C7025, Thermo Fisher Scientific), according to manufacturer´s instructions. The stained cells were cultured for three days in their corresponding media on ultra-low attachment 384-well plates (#3830 Corning Inc., NY, USA). An hour prior imaging, 0.9 µM of Hoechst 33342 (#62249 Thermo Scientific, US) was added to the cells. Images were taken with Opera Phenix High-Content Imager (PerkinElmer) and analysis of the spheroid size and shape was done with Harmony High-Content Imaging and Analysis Software (PerkinElmer).
Patient-derived cell 3D organoid assay
The patient-derived drug-resistant cells and their parental controls were cultured over six weeks in the conditions described above. The EC50 values of resistant cells were obtained by dose-response experiment with five concentrations of the drugs. 1500 cells per well were seeded on a 384-well plate, and drugs were diluted into culture media. CellTiter-Glo (CTG) 2.0 Assay was performed as in DSRT and luminescence signal was detected using Tecan Spark (Tecan, Männedorf, Switzerland).
For the 3D assay, 1500 cells were embedded in 66% VitroGel ORGANOID-4 hydrogel (#VHM04-4, TheWell Bioscience, NJ, USA) on ultra-low attachment 384-well plates (Corning) in volume 10 µl. Seven replicates were used. The hydrogel was allowed to solidify 15 min at 37°C prior addition of 25 µl of top medium. Drugs were diluted in top media and refreshed at day 7. 3D organoid development was monitored by capturing images with Leica DMi1 microscope equipped with a Flexacam C1 camera. On day 14, the top media was removed and CTG assay was performed for bulk viability as described above.
For live cell immunofluorescence imaging, on day 14, organoids were stained with 100 nM TMRE (tetramethylrhodamine, methyl ester) for 1 h at 37°C, followed by PBS wash and imaging with LSM780 confocal microscope (Zeiss, Oberkochen, Germany). Images were taken as 300 µm z-stack with 10 µm interval, with 10X objective, using the same exposure time, and light power across all sample conditions. Image analysis was performed with FiJi (ImageJ, 10.2023) [27]. Images were filtered with 3D Gaussian blur using x = 2 y = 2, and z = 2. Then, the 3D Object Counter plugin [28] was used to segment the images, count the 3D objects, and measure their volumes.
Cell Painting sample preparation
Cells (10,000/well) were plated to a 96-well plate (#3631, Corning) and let grow to near confluence for 48 h. Next, cells were fixed with 4% paraformaldehyde and stored in PBS. Fixed cells were permeabilized with Triton-X buffer and washed with PBS prior to adding dyes. The following dyes were used in 1% BSA, 0,1% Tween20, PBS: WGA (#W32464, Wheat Germ Agglutinin, Thermo Fisher Scientific, 1:500), ConA (#C11252, Concanacalin A, Thermo Fisher Scientific, 1:50), Phalloidin 568 (#A12380, Thermo Fisher Scientific, 1:50), SYTO14 (#S7576, Thermo Fisher Scientific, 1:500) and Hoechst 33342 (#62249, Thermo Fisher Scientific, 1:5000). Images were taken with Opera Phenix (PerkinElmer) at 40X. Figures are representative from several fields from a single well.
Image analysis
Raw images were preprocessed with illumination correction using the CIDRE method [29], followed by nuclei segmentation using the nucleAlzer deep learning model [30] in the BIAS software. Subsequent feature extraction was performed in CellProfiler (4.2.5) [31]. After enhancement, the image was masked using the MaskImage module. Nucleolei were then segmented using an adaptive Otsu threshold (IdentifyPrimaryObjects). The average number of nucleolei per cell was calculated at a field of view (FOV) level. Cells were segmented using the IdentifySecondaryObjects module. A global propagation strategy was employed to segment the cytoplasm using the previously identified nuclei as seed objects. Neighbor cells were determined using the MeasureObjectNeighbors module, whereby all cells within a 20-pixel distance were considered as neighbors. Intensity, size, shape and texture features were computed for cells, nuclei and nucleolei. For the actin analysis, actin neurites were enhanced with the EnhanceOrSuppressFeatures module based with the tubeness filter, that has been optimized for detecting tubular structures. A smoothing scale of 5 was applied to adjust the sensitivity of the filter, before a global threshold was applied using a minimum cross-entropy algorithm and a threshold correction factor of 3.5. Actin objects were further filtered based on their shape features, specifically their eccentricity (restricted to be between 0.95 and 1.0 to ensure elongated structures). Additionally, only fibers with a major axis length between 50 and 500 pixels and a minor axis length below 75 pixels were retained for further analysis. The downstream analysis was performed in Python (3.11) using the SciPy and scikit-learn python libraries. Features were selected based on their mean values of each field-of-view to determine the features with the highest variance. Both uniform manifold approximation and projection embeddings (UMAPs) and violin plots represent feature distributions at the FOV level.
Single-cell RNA sequencing (scRNA-seq) sample preparation
The sample preparation was done as described by Dini et al. [19]. In brief, cells were plated to 6-well plates three days prior to scRNA-seq. For short term samples, parental JHOS2, Kuramochi and Ovsaho were treated with 500 nM of ADA (JHOS2, Kuramochi, and Ovsaho) or 100 nM of PTX (JHOS2 and COV318), to get 72 h treatment of the drugs. For scRNA analysis, 10,000 cells were collected for each sample.
Library chemistry, sequencing, and preprocessing of raw FASTQ files
Single-cell gene expression profiling was conducted using the 10x Genomics Chromium Single Cell 3’RNAseq platform (10x Genomics, #CG000317). The Chromium Single Cell 3’RNAseq workflow, including library preparation, were done using the Chromium Next GEM Single Cell 3’ Gene expression version 3.1 Dual Index chemistry with Feature Barcoding technology. Sequencing of libraries was performed on an Illumina NovaSeq 6000 system (Illumina). Preprocessing of FASTQ files was conducted using the Cell Ranger pipeline (version 7.1.0, 10x Genomics), where cellranger mkfastq was used to produce FASTQ files, and cellranger count was employed to perform alignment and UMI quantification. Alignments were made against the GRCh38 (GENCODE v32/Ensembl 98) reference genome assembly.
scRNA-seq data analysis
The scRNA-seq bioinformatic analyses were performed as previously described by Dini et al. [19] using R (4.3.2). Briefly, the pre-processing was done separately for each cell line utilizing the R package Seurat (5.0.3) [32]. Initially, the hashtag-oligo (HTO) data from the experimental batches was normalized using the NormalizeData function. Cells were then demultiplexed with the HTODemux function using default parameters, and negative and doublet cells were excluded. The remaining cells classified by HTO were subjected to mRNA quality control, assessing the number of unique molecular identifiers (UMIs) and the percentage of UMIs derived from mitochondrial genes. Normalization and variance stabilization of the scRNA-seq data was performed using NormalizeData, and highly variable features were identified with FindVariableFeatures using default settings. Following this, the layers were combined, and cell cycle heterogeneity was scored using CellCycleScoring, using the cell cycle phase references provided by Tirosh et al. [33]. The Seurat object was subsequently re-split into the original layers, and the data was scaled using the ScaleData function, regressing out G2M and S phase scores. Principal component analysis (PCA) was performed on the RNA data using RunPCA with default parameters, followed by Harmony [34] integration of the experimental batches using the IntegrateLayers function. UMAP was computed with RunUMAP using the reduction from the Harmony integration. To quantify differences in cell cycle phase distribution across parental and lt-res cell lines, the cell cycle phases were extracted from the Seurat object metadata. For each cell line, cell counts per phase were converted to percentages. The resulting distributions were visualized as stacked bar plots using ggplot2 (3.5.1) [35].
Pathway activity scores at the single-cell level were computed using decoupleR (2.8.0) [36] and the PROGENy [37] model pathway weights for target genes. The human interaction weights and the top 500 responsive genes, ranked by p-value, were used for inference using a multivariate linear model (run_mlm) following the package’s vignette. The results were scaled and visualized in heatmaps using the package ComplexHeatmap.
The gene module analysis was performed as previously described by França et al. [38] with minor adaptations. First, the signatures were created by average linkage hierarchical clustering of the cells based on the gene expression profiles of the top 1000 most variable genes on the centered dataset using one minus the Pearson correlation as the distance metric. To identify potential gene expression signatures, clusters without a clear differential expression signal, with fewer than 30 cells and where less than 20% of the cells expressed the gene were excluded. Each cluster was evaluated for differential expression relative to all other cells using the FindMarkers function and the MAST test, whereafter the mitochondrial and ribosomal genes were filtered out. Clusters were retained if they showed a signature of differentially expressed genes with an average fold change greater than 1.5 and an adjusted p-value < 0.001, and at least 50 genes meeting these criteria. Gene signatures were then hierarchically clustered based on their Jaccard index, and for pairs with a similarity above 0.75, the signature containing the largest number of genes was retained.
The signatures were further refined by excluding genes with very low expression, and by only retaining signatures containing more than 50 genes. To assess similarity between the remaining signatures, pairwise comparisons were performed using the Jaccard similarity index. The results were converted into a distance matrix for hierarchical clustering using the average linkage method and visualized as a dendrogram. To integrate the signatures into gene modules, clusters of signatures that were too small or too large were excluded. The genes were grouped into modules based on their expression across the clusters of signatures as defined by the hierarchical clustering dendrogram. Genes were included in the modules only if they appeared in a sufficient number of signatures within the module. The top 50–100 genes from each signature in a cluster were merged and ranked by their average expression ratio in cells between that cluster and the other clusters. The data was scaled for each module separately for visualization using ComplexHeatmap and key genes were visualized in each module. The gene modules allow for overlap, enabling genes to be associated with multiple modules.
To identify pathways enriched in genes associated with long-term drug resistance, gene sets from modules predominantly associated with each resistant cell line were selected (JHOS2 ADA: module A; Kuramochi ADA: modules A, B, C; Ovsaho ADA: module A; JHOS2 PTX: modules B, C; COV318 PTX: modules C, D). Gene set enrichment analysis was performed for each model using the gost() function of the gprofiler2 package (0.2.3) [39], querying the Reactome database and applying an FDR significance threshold of 0.05. To identify shared pathway-level alterations across models, enriched terms from each cell line were compiled, and selected significant terms were visualized using ggplot2.
To quantify the degree of transcriptomic perturbation within each untreated and treated cell line, energy-distance (E-distance) was calculated using the scPerturbR package (0.1.0) [40] separately for ADA and PTX. The Seurat object was split by cell line, and each subset underwent normalization, variable feature identification, scaling, and dimensionality reduction using RunPCA. E-test statistics were computed for each group using the edist() function, grouping by cell line and using the PCA reduction. The results were visualized using ComplexHeatmap.
Differential gene expression analysis between the parental and long-term drug-resistant cell lines as well as between parental and short-term or intermediate drug-resistant cell lines was conducted using the FindMarkers function with the log-normalized counts and default settings. The results were filtered to contain only protein coding genes, and the p-value was adjusted based on the Bonferroni correction. The gene expression fold-changes and adjusted p-values were visualized in volcano plots using the EnhancedVolcano package (1.20.0) [41]. Significant genes (log2FC > 0.25 or < -0.25, adjusted p-value < 0.05) were colored, and selected genes were labeled.
For long-term treated cells, over-representation analysis was conducted using the enrichGO function of the clusterProfiler package (4.10.1) [42]. The analysis was applied separately on the list of significantly upregulated and downregulated genes derived from the differential gene expression analysis. The analysis included all available GO ontologies, and the org.Hs.eg.db database (3.18.0) was used as the reference for human gene annotations. P-values were adjusted using the Benjamini-Hochberg method, with a significance threshold of p-value < 0.05 and q-value < 0.2. Selected pathways were included based on relevance and significance (FDR < 0.05) and visualized in a dot plot using ggplot2. For short-term treated cells, differentially expressed genes were filtered for average logFC ≥ |0.5| and q-value ≤ 0.1. Fisher’s exact test results was performed against the MSigDB database, and results were filtered for entries including the words “REACTOME”, “KEGG”, “PID”, and “HALLMARK”. Finally, the 10 most significant pathways retained in the set were retained and plotted. P-values were adjusted based on the Bonferroni correction. Finally, only pathways whose q-value is ≤ 0.1 are returned.
Copy number alterations in the long-term drug-resistant cell lines were analyzed using the InferCNV package (1.20.0) [43], using the parental cell lines as references. The analysis was performed by running InferCNV::run with a cutoff of 0.1, analysis_mode set to ‘sample’, and HMM_type set to ‘i3’. The results were visualized using the ComplexHeatmap and the circlize (0.4.16) [44] packages.
Three publicly available ovarian cancer scRNA-seq datasets (Olbrecht et al., Qian et al., and Zhang et al.) [45,46,47] were preprocessed and integrated using Harmony. Cells with more than 100,000 total counts or more than 25% mitochondrial gene expression were excluded. Doublets were identified and excluded using DoubletFinder (2.0.4) [48], using the tutorial at https://biostatsquid.com/doubletfinder-tutorial/. Cell type annotation was performed using scType [49] with a custom marker gene list. ROR1 and WEE1 normalized expression levels of treatment-naïve epithelial tumor cells were visualized as violin plots.
Clinical samples in the DECIDER cohort
The sample collection and analysis are part of the DECIDER cohort (https://www.deciderproject.eu/). All patients participating in the study provided written informed consent. The study and the use of all clinical material have been approved by The Ethics Committee of the Hospital District of Southwest Finland (ETMK) under decision number EMTK: 145/1801/2015. DECIDER [18] raw bulk RNA-Seq count data for 329 samples was quantified to gene level counts with Kallisto [50]. scRNA-seq count data were downloaded for 43,945 ovary cells with associated metadata annotations, including cell type (endothelium, epithelium, erythrocyte, fibroblast, lymphocyte, myeloid, plasma or undefined) and sample location (normal, tumor, or tumor-adjacent) [46, 51]. DECIDER bulk RNA-seq count data were then deconvoluted using the R package BayesPrism (2.0) [52] and the scRNA-seq count and annotation data were used to identify overall cell type composition and gene expression by cell type for all samples. Deconvoluted raw counts were normalized with the ‘vst’ function of the R package DESeq2 (1.40) [53]. Tumor epithelial expression of WEE1 and ROR1 was analyzed in treatment-naïve and post-NACT samples. Samples were stratified into high and low expression groups based on the median vst normalized expression of the reciprocal gene and visualized as box plots using ggplot2. Statistical comparisons were performed using a two-tailed Wilcoxon test with the ggpubr package (0.6.0) [54]. Additonally, vst normalized expression of samples from relevant tissues (ascites, omentum, ovary, peritoneum) were visualized in box plots as described above.
LINCS analysis
Level 5 Library of Integrated Network-Based Cellular Signatures (LINCS) [55] cell line chemical perturbation data was filtered to samples with an origin of ovary and drug treatment of ADA. To compare the LINCS data to the single-cell data, the ADA lt-res samples and their corresponding parentals were divided into two groups, and pseudobulk expression data was generated for each of them using the AggregateExpression Seurat function and the RNA assay. The resulting counts were normalized using DESeq2 (1.42.1) size factor normalization, and z-scores were generated for all genes in each pseudobulk sample. MODZ were derived by subtracting the parental z-scores from the lt-res z-scores for each gene. The top 100 positive and negative common genes between ADA lt-res and the LINCS dataset were visualized in heatmaps using ComplexHeatmap, and relevant genes were labeled.
Results
Generation of ADA- and PTX lt-res models in HGSOC
To investigate the molecular mechanisms of drug resistance linked to cell cycle inhibition in clinically relevant settings, we developed multiple preclinical models of ADA- and PTX lt-res cell lines that were selected to cover the genomic and tissue-of-origin representability and heterogeneity characteristic of HGSOC (Fig. 1a, Table 1) [56]. The sensitivity of drug-naïve cell lines to ADA or PTX varied across models (Supplementary Fig. 1a). Among HGSOC cell lines and normal ovarian surface epithelial cells (HOSE1C and HOSE2C), WEE1 expression was relatively higher in Ovsaho and Kuramochi. Given that WEE1 localization is regulated by phosphorylation, we also observed elevated levels of pWEE1 (Ser642) in Ovsaho and Kuramochi cells suggesting cytoplasmic localization driven by AKT-mediated phosphorylation (Supplementary Fig. 1b) [57].

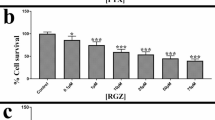
a Schematic representation of the mechanisms by which adavosertib (ADA) and paclitaxel (PTX) affect the cell cycle in high-grade serous ovarian cancer (HGSOC). Right, clinical trials where ADA and PTX were used in HGSOC. b Representative brightfield microscopy images of drug-naïve (parental) and long-term resistant (lt-res) HGSOC cell models. Scale bar: 100 µm. Below, dose–response curves for ADA and PTX in drug-naïve and lt-res cells following 3-day treatment. Cell viability was assessed using the CellTiter-Glo (CTG) assay. Data are presented as relative proliferation (normalized to untreated control = 1). c Half-maximal effective concentration (EC50) calculated from dose–response curves, with fold changes comparing lt-res to drug-naïve cells. d Baseline proliferation rates determined from luminescence values at day 0 and day 6 (CTG assay). Fold change was calculated relative to day 0. Data represent mean ± SD; n = 4 replicates. Statistical analysis was performed using unpaired two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001).
Drug-naïve HGSOC cell lines were subjected to escalating doses of either ADA or PTX over a period of at least four months. This process yielded various drug-resistant models, with some cell lines showing continuous resistance development over four months, while others undergoing senescence at early stages of drug exposure (Table 1). We obtained three ADA- and two PTX- long-term resistant cell models (ADA lt-res/PTX lt-res, 18 weeks drug exposure), while the COV362 cell line failed to develop resistance to both drugs. ADA resistance increased 1.5- to 109-fold, while PTX resistance rose 18- to 27-fold, based on EC50 values compared to their drug-naïve counterparts (Fig. 1b,c). We hypothesize that this gradual dose escalation led to a continuum of resistant phenotypes, reflecting enhanced drug tolerance at higher concentrations [38]. Baseline proliferation assays showed significantly reduced growth rates in lt-res models, reflecting changes in the phenotype (Fig. 1d).
Cell Painting reveals distinct organelle changes in lt-res models
Next, we used Cell Painting [58] and high-content imaging to examine morphological changes in drug-naïve and lt-res HGSOC cells. Five stains were employed to label cellular components: Hoechst 33342 (DNA), wheat germ agglutinin (WGA, Golgi and plasma membrane), concanavalin A (ConA, endoplasmic reticulum), SYTO 14 (nucleoli and cytoplasmic RNA), and phalloidin (actin) (Fig. 2a,b, Supplementary Fig. 2a). Microscopic imaging revealed visually distinct phenotypic differences in lt-res cells, such as multi-nuclei formation in JHOS2 and Kuramochi ADA lt-res, and elongated actin filaments in multiple models. Uniform Manifold Approximation and Projection (UMAP) of all morphological features showed clear separation between lt-res and drug-naïve cells, particularly in JHOS2 ADA lt-res, JHOS2 PTX lt-res, COV318 PTX lt-res, and Ovsaho ADA lt-res (Fig. 2a, b). Quantitative analysis of actin filaments revealed an increase in length but a decrease in filament number in PTX lt-res models (Fig. 2c) [59]. Our analysis of neighboring cells also revealed distinct differences between drug-naïve and lt-res cell lines in both PTX lt-res models, with PTX lt-res models showing substantially more neighboring cells compared to their drug-naïve cells, a trend also observed in Ovsaho ADA lt-res cells (Fig. 2d). Since neighboring cell counts are influenced by overall cell density, averages were calculated using regression slope parameters. Consistently, PTX lt-res cells had an increased number of neighboring cells, suggesting an enhanced cell adhesion phenotype, a trait previously shown in multiple PTX-treated cancers.

a, b Representative merged images from the Cell Painting assay and Uniform Manifold Approximation and Projection (UMAP) plots based on morphological profiles of drug-naïve, ADA- and PTX lt-res HGSOC cells. Scale bar: 100 µm. Individual fluorescence channels are shown in Supplementary Fig. 2. c Quantification of actin filament length (pixels) and number per cell (phalloidin staining). d Scatter plots showing the average number of neighboring cells per cell in each field of view (FOV), with 25 FOVs per condition. Analysis was performed across 25 FOVs per condition. Regression lines were calculated at fixed cell counts (x = 40 and x = 60) to enable comparison across varying densities. e, f Representative images of 3D spheroids from drug-naïve, ADA lt-res e, and PTX lt-res f cells cultured in ultra-low attachment plates. Live cells were stained with CellTracker Green; nuclei were counterstained with Hoechst after 3 days. Spheroid volume (×10⁵ µm³) and intact object count were quantified. Each data point represents an individual well. Bars indicate mean ± SD (n = 4–6). Statistical analysis was performed using two-tailed Student’s t-test (*P < 0.05, **P < 0.01).
Additionally, TMRE (active mitochondria) and Calcein AM (viable cells) staining revealed a distinct morphological alteration between parental and lt-res models in JHOS2 and COV318 (Supplementary Fig. 2b). We also assessed 3D growth and spheroid formation of lt-res models (Fig. 2e,f). Both ADA and PTX lt-res models produced more spheroids, but they were smaller and more dispersed than in drug-naïve cells, indicating that lt-res models have undergone EMT changes [60] compared to drug-naïve cells.
Single-cell transcriptomics uncovers a PI3K/AKT-driven convergence in mitotic escape and stress adaptation
To investigate transcriptional adaptations in long-term resistance, we performed single-cell RNA sequencing (scRNA-seq) of parental and lt-res HGSOC cell lines (Fig. 3a). UMAP clustering segregated cells by treatment and lineage, with most lt-res models showing marked transcriptional shifts, except for Kuramochi ADA lt-res, which retained a more parental-like profile. Cell cycle phase analysis revealed G2/M enrichment in all lt-res populations—indicative of checkpoint engagement- except in COV318 PTX lt-res, which maintained more G1-phase cells consistent with their lower proliferation (Fig. 3b).

a UMAP plots showing the distribution of drug-naïve and long-term resistant cells for each HGSOC cell line. Colors denote cell line and treatment condition. b Bar charts showing the proportion of cells (%) in each cell cycle phase across cell lines and treatment conditions. c Heatmap illustrating the scaled signaling pathway activity scores of PROGENy pathways in PTX lt-res and ADA lt-res cell lines and parental cells. Red color indicates higher pathway activity, and blue color indicates lower pathway activity. d Heatmaps of gene modules for each cell line. Letters A-D indicate the gene modules identified within each cell line. Columns represent signatures (gene sets derived from cell clusters), and rows represent the genes within each module. Color indicates z-scored expression of module genes across signatures (capped at ± 3). The stacked bars below each heatmap show the percentage of cells from each condition present in each signature. e Dot plot showing shared significantly enriched pathways among the gene modules identified in panel d (JHOS2 ADA module A, Kuramochi ADA modules A, B, C, Ovsaho ADA module A, JHOS2 PTX modules B, C and COV318 PTX modules C, D). Dot size represents the gene ratio, and dot color corresponds to the adjusted p-value. f Immunoblot analysis for signaling pathways of interest in drug-naïve and lt-res models. β-actin was used as loading control. Below, a schematic representation of the two PI3K/AKT-regulated drug resistance mechanisms, with background colors matching the lt-res models where the mechanisms were active. Left (PI3K/AKT OFF): Nuclear FOXO3 represses MYC, activates DNA Damage Response (DDR, ATR/CHK1), and together with active WEE1 stalls cells in G2, leading to replication stress responses. Right (PI3K/AKT ON): AKT phosphorylates FOXO3 (pFOXO3) causing its cytoplasmic retention, induction of FOXM1/MYC, reduced WEE1 function, and RTK upregulation (e.g., ROR1), enabling mitotic escape and survival.
Inference of copy number alterations (CNAs) from scRNA-seq data revealed heterogeneous genetic evolution across lt-res models (Supplementary Fig. 3a). Each cell line exhibited distinct CNA profiles; for instance, COV318 PTX lt-res had gains on chromosomes 4, 6, 8, and 15, while Ovsaho ADA lt-res showed gains on chromosomes 11, 12, and 16. These diverse CNA patterns underscore the genomic heterogeneity of resistance in HGSOC [38, 61].
PROGENy analysis of pathway activity revealed upregulation of PI3K/AKTsignaling across lt-res models, often together with activated MAPK, and NF-κB signaling, indicating cytoplasmic pathway rewiring (Fig. 3c) [37]. Module-based clustering of transcriptomes identified conserved and model-specific transcriptional programs driving resistance (Fig. 3d). ADA lt-res cells shared a mitotic module (Module A) enriched for genes regulating mitosis and checkpoint progression (e.g., CDK1, PLK1, AURKA, CDC20). Kuramochi ADA lt-res also showed stress-related modules involving immediate early response (FOS, STAT3, HIF1) and cytoskeletal adaptation (SLIT2/3, ROBO3, PVT1). In PTX lt-res models, JHOS2 adopted a proliferative program driven by FOXM1 and homologous recombination repair (HR), while COV318 displayed stress-adaptive signatures involving oxidative stress (TXNIP, NQO1), DNA damage response (GADD45A), EMT, and drug efflux (ABCB1).
Gene set enrichment analysis (GSEA) confirmed these divergent adaptations (Fig. 3e). ADA and PTX lt-res models (Kuramochi ADA, Ovsaho ADA, JHOS2 PTX) were enriched for cell cycle and checkpoint programs, whereas COV318 PTX and JHOS2 ADA showed activation of MAPK4/6, NOTCH, and ABCB pathways—suggesting stress-adaptive strategies. These patterns were also evident from differential gene expression and over-representation analyses (Supplementary Fig. 3b, c), with ADA lt-res models favoring mitotic programs, while PTX lt-res models displayed metabolic and proteasome-related signatures.
Immunoblot validation supported the transcriptional findings (Fig. 3f). Concurring with the pathway analysis, an increase in pAKT (PI3K pathway) and pERK (MAPK pathway) were observed in the resistant models. The PI3K/AKT activation and downstream FOXO3 phosphorylation were evident in proliferative models (Kuramochi ADA, Ovsaho ADA, JHOS2 PTX), while COV318 PTX and JHOS2 ADA showed low pAKT and active FOXO3, consistent with a stress-adaptive phenotype. Notably, RTKs including ROR1, EGFR, and PTK7 were upregulated across several lt-res models, with ROR1 correlating with PI3K/AKT activation, reinforcing its role in resistance [62,63,64]. Collectively, these findings reveal that despite distinct transcriptional strategies, all lt-res models converge on a PI3K/AKT-centered axis that modulates FOXO3 to balance proliferative escape and stress adaptation, thusforming a unifying mechanism of resistance across diverse genetic contexts and drug pressures.
PI3K/AKT activation distinguishes short-term signaling bursts from long-term adaptive remodeling in HGSOC drug resistance
To investigate whether HGSOC cells respond differently to acute versus prolonged drug pressure, we also compared the transcriptomic profiles of short-term resistant (st-res; 3-day treatment) and lt-res models. For JHOS2, we additionally included an intermediate resistant (i-res) population exposed to submaximal (SubMAX) doses of ADA or PTX (Table 1, Fig. 4a), representing a transitional adaptation state. Dimensionality reduction of the transcriptomic data revealed distinct clustering patterns: lt-res cells formed clearly separated clusters, while st-res and drug-naïve populations clustered closely together (Fig. 4b).

a Schematic overview of the experimental workflow for generating long-term (lt-res), intermediate (i-res), and short-term (st-res) drug-resistant models. b UMAP plots showing the distribution of drug-naïve and resistant cells for each HGSOC cell line. Colors denote cell line and treatment condition. c Energy-distance (E-distance) heatmaps illustrating the degree of transcriptomic perturbation in parental, ADA- and PTX- resistant cells. Color intensity reflects the degree of perturbation, with darker shades indicating higher perturbation and lighter shades indicating lower perturbation. Color annotations correspond to identities as shown in panel b. d Heatmaps illustrating the scaled signaling pathway activity scores of PROGENy pathways in PTX-res and ADA-res cell lines and the parental cells. Red color indicates higher pathway activity scores, and blue color indicates lower pathway activity scores. e Enrichment analysis by Fisher’s exact test of differentially expressed genes (logFC ≥ |0.5 | , adjusted p-value ≤ 0.1) in all ADA st-res and PTX st-res cells, compared to drug-naive cells. The top 10 most significantly enriched pathways were reported for both upregulated and downregulated genes in st-res cells, from REACTOME, MSigDB HALLMARKS, PID, and KEGG terms. Dot size represents gene count, color intensity reflects adjusted p-value, and odds ratio is reported on the x-axis.
To quantify these dynamics, we employed scPerturb, a benchmark framework for assessing transcriptional divergence in single-cell data of perturbed cells [40]. Interestingly, the E-distance between drug-naïve and st-res cells was greater than that between drug-naïve and lt-res cells in all models except COV318, indicating a strong initial perturbation followed by partial transcriptomic convergence or adaptation over time (Fig. 4c). These trends were further supported by PROGENy pathway activity analysis which revealed distinct signaling profiles between st- and lt-resistance states (Fig. 4d). Notably, PI3K/AKT signaling emerged as the most strongly activated pathway across all st-res models, underscoring its role as a conserved early response to mitotic stress in HGSOC. Pathway enrichment analysis of differentially expressed genes in ADA and PTX st-res cells revealed consistent enrichment for proliferative programs including E2F and MYC targets, DNA replication, and checkpoint pathways, while downregulated terms were associated with protein biosynthesis and metabolic processes (Fig. 4e). Together, these findings suggest that early drug exposure induces a pronounced PI3K/AKT-driven signaling burst, while long-term resistance arises through progressive transcriptional adaptation and remodeling. In addition, while short term treatment with ADA at 100 nM did not significantly alter ROR1 or PTK7 expression, a higher dose of 500 nM resulted in ROR1 downregulation (Supplementary Fig. 4a), showing that RTKs modulation is also different in st- versus lt-res models, despite PI3K/AKT activation.
Analysis of JHOS2 i-res models identified early adaptive genes. ADA i-res cells upregulated MET, EGFR, ROR1, CDK6, and AKT3, while PTX i-res cells expressed BRCA2, RAD51, and MCM2, consistent with PTX lt-res Module C (Supplementary Fig. 4b), supporting a stepwise acquisition of resistance traits where key pathways emerge “at middle way” during adaptation.
Finally, LINCS-based [65] comparison confirmed significant overlap between ADA lt-res gene signatures and canonical mitotic regulators (AURKA, CDC20, CDK1, GTSE1) from our scRNA-seq modules (Supplementary Fig 4c), reinforcing the relevance of mitotic drive in acquired resistance.
Drug sensitivity and resistance profiles defined by ADA and PTX resistance in HGSOC
We assessed global drug sensitivity and resistance profiles in ADA-(and PTX lt-res models using a DSRT screen that included 526 drugs (Supplementary Fig. 5a), each tested at five concentrations over 10,000-fold dilutions, using cell viability as the readout. The drug sensitivity score (DSS) was used as a single metric to quantify drug responses [66], with an efficacy threshold between 5 and 8.8 for different cell lines, representing the 85th percentile of DSSs distribution (Supplementary Fig. 5b). Hierarchical clustering of DSS values revealed four distinct, mostly cell line-specific, groups: JHOS2 (drug-naïve and ADA/PTX lt-res), Kuramochi and Ovsaho parental, Ovsaho ADA lt-res, and COV318 (drug-naïve and PTX lt-res) together with Kuramochi ADA lt-res, as confirmed by principal component analysis (PCA) (Fig. 5a,b). We also examined the percentage of effective drugs within each mechanism of action (MOA) (Fig. 5c). Kuramochi and Ovsaho ADA lt-res models showed a loss of drug sensitivity, while JHOS2 lt-res acquired sensitivity, particularly to metabolic modifiers and apoptotic drugs. JHOS2 PTX lt-res cells exhibited increased sensitivity compared to drug-naïve cells, a trend not observed in COV318 likely due to its inherently resistant profile.

a Cladogram showing hierarchical clustering of HGSOC cell lines based on drug sensitivity scores (DSS) across a 526-compound drug screen. b Principal component analysis (PCA) of DSS profiles illustrating similarity in drug responses among cell lines. c Class-level analysis of drug hits. Bars represent the percentage of compounds per class with DSS values above the 85th percentile threshold (see Supplementary Fig. 5 for details). d DSS values for selected compound categories, including SMAC mimetics, apoptotic modulators, and standard chemotherapeutics, in ADA- and PTX-resistant versus drug-naïve models. e Validation of drug sensitivity screening using CellTiter-Glo (CTG) assay. Cells were treated with indicated drug concentrations for 3 days. Data represent mean ± SD (n = 4). Statistical significance was assessed using two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001).
Next, our focus was to identify drugs with increased DSS (potency) in ADA- or PTX-resistant models to explore potential co-targeting strategies for overcoming drug resistance. Among MOAs, several SMAC mimetics, Bcl-XL (apoptotic modifiers) and topoisomerase I inhibitors (chemotherapeutics) showed increased potency in various ADA- and/or PTX-resistant models (Fig. 5d,e, Supplementary Fig. 5c).
In JHOS2 ADA lt-res, drugs such as SN-38 (a metabolic modifier), UCN-01, and omipalisib (kinase inhibitor) showed enhanced potency. In Kuramochi and Ovsaho lt-res proteasome inhibitor bortezomib gained significant activity. In PTX lt-res models, chemotherapeutics like cisplatin, cabazitaxel, vinorelbine, and proteasome inhibitors (VLX-1570, tosedostat) exhibited higher DSSs compared to drug-naïve cells. Overall, the drug response profiles of ADA lt-res and PTX lt-res models were highly cell-specific, underscoring the impact of cellular heterogeneity on drug resistance in HGSOC.
ROR1 modulates ADA and PTX long-term resistance in HGSOC
Transcriptomic analysis identified PI3K/AKT as a central signaling switch in ADA and PTX lt-res HGSOC models. Among its upstream activators, ROR1, a Wnt5a/b receptor with lower expression in healthy adult tissues, emerged as a promising therapeutic target due to its potential for reduced off-target toxicity [67]. Given that ADA mediated the upregulation of ROR1 in PI3K/AKT activated lt-res models, we sought to explore the mechanistic link between ROR1 and WEE1 by assessing their expression correlations in clinical datasets. In the DECIDER cohort (n = 316), WEE1 and ROR1 showed strong co-expression across treatment phases (Fig. 6a, Supplementary Fig. 6a, b). This correlation was also evident in HGSOC scRNA-seq datasets from Olbrecht [45], Qian [46], and Zhang [47], supporting the clinical relevance of this axis (Supplementary Fig. 6c).

a Box plots illustrating WEE1 and ROR1 expression (variance-stabilizing transformed, VST) in DECIDER tissue samples across treatment phases: diagnostic (n = 158), post-NACT (n = 69), and relapse (n = 31). Statistical comparisons between treatment phases were performed using a two-tailed Wilcoxon test, with asterisks indicating significance levels (****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05). b Immunoblot analysis of ROR1 in HGSOC cell lines, where β-actin was used as a loading control. c Immunoblot analysis of ROR1 and WEE1 in parental, control plasmid-transfected, and HA-tagged ROR1-plasmid transfected (ROR1high) JHOS2 and OVCAR8 cells. GAPDH served as a loading control. ROR1-HA indicates the HA-tag present in the plasmid. d ROR1 and WEE1 protein levels in doxycycline-induced shROR1 knockdown (KD) and parental cells. Protein expression was normalized to GAPDH and to untreated controls (set to 1). e Proliferation assay comparing parental, ROR1high, and the ADA or PTX-res cells. The parental, ROR1high and their ADA- or PTX-res counterparts were treated with increasing concentrations of ADA or PTX for 3 days. Significance was calculated using two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001), n = 3 or 4 replicates. f Proliferation assay comparing ROR1 knockdown (shROR1) and parental cells, and their respective ADA-res models. The parental, shROR1 and their ADA-res counterparts were treated with increasing concentrations of ADA for 3 days. Significance was calculated using two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001), n = 3 or 4 replicates. g Proliferation assay of JHOS2, Kuramochi, and Ovsaho parental and ADA lt-res cells treated with zilovertamab vedotin (Zilo-VT; 25 µg/mL), ADA (250 nM for JHOS2 and Ovsaho, 500 nM for Kuramochi), or their combination. Cell viability was measured after 3 days using the CTG assay. Data represent mean ± SD (n = 3 or 4 replicates). Statistical significance was determined using two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001).
To investigate causality, we modulated ROR1 expression in HGSOC cell lines. Immunoblotting confirmed variable endogenous ROR1 levels, with high expression in COV318 and Ovsaho (Fig. 6b). Stable overexpression of ROR1 in ROR1-low JHOS2 and ROR1-negative OVCAR8 did not alter WEE1 levels (Fig. 6c), whereas inducible shRNA ROR1 knockdown reduced WEE1 in JHOS2 and Kuramochi but not in Ovsaho (Fig. 6d). The lack of effect in Ovsaho may result from WEE1 cytoplasmic retention via Ser642 phosphorylation or RB loss-driven constitutive WEE1 expression [68].
Next, we assessed whether ROR1 modulates resistance acquisition. ROR1 overexpression in JHOS2 did not increase ADA resistance (Fig. 6e), consistent with its unchanged ROR1-PI3K/AKT activation (Fig. 3d). However, it enhanced PTX resistance, aligning with ROR1 upregulation in JHOS2 PTX lt-res cells. In contrast, OVCAR8 ROR1high cells showed enhanced ADA resistance (Supplementary Fig. 6d), while ROR1 knockdown in JHOS2 and Kuramochi reduced ADA resistance (Fig. 6f). These findings confirm that ROR1 promotes ADA or PTX resistance in a context-dependent manner.
To evaluate therapeutic potential, we tested zilovertamab-vedotin, an anti-ROR1 antibody-drug conjugate in clinical trials for ROR1 positive tumors and preclinically active in HGSOC [69, 70]. Its cytotoxicity correlated with ROR1 expression in both parental and lt-res models (Fig. 6g, Supplementary Fig. 6e). Ovsaho and Kuramochi lt-res cells, with higher ROR1 levels, were more sensitive to zilovertamab-vedotin than JHOS2 lt-res. Co-treatment with ADA or PTX had an additive effect in parental but not lt-res cells, although zilovertamab-vedotin was less effective in PTX lt-res cells possibly due to ABCB1-mediated efflux (based on the upregulated ABCB1 levels seen in the transcriptomic data) or impaired ADC internalization.
Targeting ROR1 to overcome ADA and PTX resistance in primary tumor-derived HGSOC organoid cultures
To further validate the clinical relevance of our findings, we examined the development of ADA and PTX resistance in ex vivo PDCs from Murumägi et al. [21] (Table 1). A strong correlation between ROR1 and WEE1 expression was observed across the PDCs, with PDC#2 and PDC#3 exhibiting high levels of both proteins along with elevated pWEE1 Ser642 (Fig. 7a). PDCs were then subjected to prolonged ex vivo treatment with escalating concentrations of ADA or PTX to induce resistance (ADA or PTX res). Drug exposure was feasible for up to 4-5 weeks, after which the cells entered senescence, likely due to limitations of the ex vivo culture environment. Continuous ADA or PTX treatment elicited pronounced morphological alterations in all PDCs (Fig. 7b), indicative of drug-induced phenotypic remodeling. Functional assessment via cell viability assays confirmed an on-going intermediate resistance (i-res) development to both drugs, particularly in PDC#2 and PDC#3, although bulk cell viability measurements may underestimate underlying phenotypic changes (Fig. 7c). Immunoblotting revealed dynamic modulation of ROR1 expression in all PDC-res models underscoring its association with ADA and PTX resistance. Notably, ROR1 was upregulated in PDC#3 ADA res and PDC#1 PTX res (Supplementary Fig. 7a).

a Immunoblot analysis of ROR1, WEE1 and pWEE1 expression in three primary tumor-derived HGSOC cultures (PDC#1-3). β-actin was used as a loading control. b Representative brightfield images of drug-naïve (parental) and intermediate-resistant ADA (ADA i-res) or PTX (PTX i-res) PDCs. Scale bar: 100 µm. c Proliferation assay of parental and ADA or PTX i-res PDCs treated with ADA or PTX, respectively. Data represent mean ± SD (n = 3 or 4 replicates). Significance was determined with one-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001). d Representative images of tetramethylrhodamine ethyl ester (TMRE)-stained parental, ADA res or PTX i-res PDCs organoids embedded in VitroGel® 3D matrix and cultured for 14 days with zilovertamab vedotin (25 µg/mL), ADA (100 nM/PDC1, 2000nM/PDC2 and 500 nM/PDC3) PTX (10 nM/PDC1, 50 nM/PDC2 and 10 nM/PDC3), or their combination as indicated. Grid square width: 100 µm. e Bar charts illustrating the mean volume in pixels (px) across n = 3-4 replicates, where error bars correspond to standard deviation. Significance was determined with one-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001).
We next evaluated the efficacy of zilovertamab-vedotin (zilo-VT) in long-term 3D organoid cultures derived from drug-naïve and i-res PDCs. Robust organoid growth was achieved in PDC#1 and PDC#3, while PDC#2 produced smaller and dispersed structures (Fig. 7d, e). Zilo-VT induced potent cytotoxicity in a ROR1-dependent fashion. Both drug-naïve and ADA i-res PDC#3 (a ROR1 high sample) exhibited strong sensitivity to zilo-VT, with combinatorial ADA treatment further enhancing the response. Conversely, despite ROR1 upregulation, PDC#1 PTX i-res responded only modestly, mirroring observations in PTX lt-res cell lines that display reduced sensitivity to anti-ROR1 ADCs despite increased ROR1 expression.
Together, these findings suggest that ROR1 upregulation in ADA-resistant models confers vulnerability to zilovertamab-vedotin, although therapeutic efficacy appears to depend not solely on ROR1 levels, but also on downstream factors affecting ADC processing and cellular susceptibility.
Discussion
The development of resistance to chemotherapy and targeted therapies is a major challenge in HGSOC treatment [71, 72]. Our study provides crucial insights into ADA and PTX resistance and potential therapeutic strategies for HGSOC treatment.
Cell Painting analysis identified morphological changes, such as multinucleation, cytoskeletal remodeling, and increased cell adhesion, all linked to (de-)differentiation and partial EMT [70, 73]. The changes of the actin filament length and number suggest an inhibition of the actin nucleation, concurring with the previous data showing that PTX can affect acting cytoskeleton organization [59]. These physical alterations, alongside molecular changes, highlight the role of cytoskeletal restructuring and enhanced mitotic control in driving drug resistance in HGSOC.
A key finding of this study is the emergence of the PI3K/AKT signaling axis as a central hub governing resistance to both ADA and PTX in HGSOC. Through integrative scRNA-seq analyses, we identified distinct co-expression modules in lt-res models that converge on PI3K/AKT pathway activation. Our data show that in models like Kuramochi ADA, Ovsaho ADA, and JHOS2 PTX, this PI3K/AKT-pFOXO3 axis is highly activated leading to mitotic escape and drug adaptation. In contrast, other models (e.g., JHOS2 ADA, COV318 PTX lt-res) adopt a “slow-repair” mode, with sustained nuclear FOXO3 activity enforcing replication stress responses and drug efflux mechanisms. This dynamic PI3K/AKT-FOXO3 switch serves as a molecular bifurcation point: either driving resistance via proliferation (FOXO3 suppression upregulation) or promoting tolerance via stress checkpoints (FOXO3 activation). Our data also reveal that while all lt-res models converge on a PI3K/AKT–FOXO3 signaling axis, the upstream adaptive mechanisms differ between resistance types and cellular backgrounds. Kuramochi and Ovsaho cells both exhibit elevated pSer642-WEE1 levels (Supplementary Fig 1b), indicative of constitutive AKT activation likely driven by their respective genomic aberrations - KRAS amplification and RB loss, respectively. Consequently, PI3K/AKT signaling remains active in the ADA lt-res models derived from these lines. In contrast, JHOS2 cells display drug-specific modulation of PI3K/AKT–FOXO3, with ADA and PTX alternately toggling pathway activity according to their MOA.
These findings are consistent with prior studies showing that PI3K/AKT activation is a frequent resistance mechanism in ovarian cancer [74], and that FOXO3 inactivation plays a central role in tumor progression and therapy escape [75]. By connecting upstream RTK signaling with the PI3K/AKT-FOXO3 axis, our study uncovers a shared, targetable vulnerability across mitotic drug-resistant states in HGSOC.
Our DSRT screens also revealed cell-specific drug response profiles highlighting the heterogeneity in therapeutic vulnerabilities in HGSOC. Kuramochi and Ovsaho ADA lt-res models exhibited a general loss of drug sensitivity, while JHOS2 ADA lt-res models gained sensitivity to metabolic modifiers and apoptotic drugs. In PTX lt-res models, agents like cisplatin and cabazitaxel showed increased potency, suggesting that these drugs may be effective in overcoming PTX resistance, as shown in clinical settings in OC [76, 77].
We also discovered a feedback loop via ROR1 upregulation in ADA and PTX lt-res models. ROR1 has gained attention for anti-cancer therapy due to its minimal expression in healthy adult tissues [78, 79]. Our previous data showed that ROR1 is upregulated by neoadjuvant chemotherapy in HGSOC, suggesting that this receptor is particularly important in drug resistance in OC [64]. Here, we show that ROR1 was significantly upregulated in PI3K/AKT-activated ADA- and PTX- HGSOC resistant models, both in cell lines and ex vivo PDCs. Although ROR1 is typically activated through non-canonical Wnt5a/b signaling, we found no correlation between ROR1 and Wnt5a/b expression in lt-res HGSOC models (data not shown). Instead, ROR1–PI3K/AKT activation appears Wnt-independent, suggesting a pathway switch from Wnt-mediated to PI3K/AKT-driven signaling under chronic drug pressure. This indicates that, rather than engaging dual signaling, resistant cells reprogram survival pathways to favor ROR1-mediated PI3K/AKT activation.
Functionally, ROR1 acts as a context-dependent activator of the PI3K/AKT–FOXO3 axis, supporting mitotic escape and adaptive survival. These findings provide a rationale for combining ROR1-targeted ADCs with ADA or PTX, where co-inhibition of the ROR1–PI3K/AKT pathway could prevent checkpoint evasion and enhance cytotoxicity. Such combinations may effectively counteract adaptive signaling particularly in ROR1high/PI3K/AKThigh tumors, offering a translational strategy to overcome mitotic resistance in HGSOC. However, the efficacy of ROR1-targeted ADCs likely depends on ROR1 surface density, internalization efficiency, and drug retention, parameters that may vary across resistance states. Consistent with this, PTX-resistant models showed limited sensitivity to zilovertamab-vedotin, likely due to drug-efflux mechanisms and impaired ADC internalization.
In summary, our study highlights the complexity of drug resistance in HGSOC, a representative model among the TP53 mutated cancers, and identifies PI3K/AKT signaling as a central adaptive hub that orchestrates divergent resistance trajectories to ADA and PTX. These findings underscore the value of targeting PI3K/AKT-driven resistance mechanisms such as ROR1 upregulation and reinforce the need for personalized therapeutic strategies in recurrent HGSOC.
Data availability
The datasets generated and analyzed during this study are available in Mendeley Data (https://doi.org/10.17632/dd6bvtrv9b.1). Here, the following files have been deposited: raw counts data for all samples; corresponding metadata associated with the samples; Cell Painting features extracted with CellProfiler; all supplementary source data. Raw and processed scRNA-seq data from this manuscript were deposited to Gene Expression Ontology (GEO) under accession number GSE297836. The DECIDER data is available from Lahtinen et al. [18]. All original code has been deposited at https://github.com/ungureanulab/OC-resistance-profiling and https://github.com/FranziskaBentz/CP-drug-res-HGSOC.
References
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299–309. https://doi.org/10.1038/S41586-019-1730-1.
Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. https://doi.org/10.1038/nature10166.
Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–94. https://doi.org/10.1038/nature14410.
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018;379:2495–505. https://doi.org/10.1056/NEJMOA1810858.
McNeish IA, Ledermann JA, Webber L, James L, Kaye SB, Hall M, et al. A randomised, placebo-controlled trial of weekly paclitaxel and saracatinib (AZD0530) in platinum-resistant ovarian, fallopian tube or primary peritoneal cancer. Ann Oncol. 2014;25:1988–95. https://doi.org/10.1093/annonc/mdu363.
Wang H, Guo M, Wei H, Chen Y Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transd Targeted Therapy. 2023;8. https://doi.org/10.1038/S41392-023-01347-1.
Moore KN, Chambers SK, Hamilton EP, Chen LM, Oza AM, Ghamande SA, et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin Cancer Res. 2022;28:36–44. https://doi.org/10.1158/1078-0432.CCR-21-0158.
Leijen S, Van Geel RMJM, Sonke GS, De Jong D, Rosenberg EH, Marchetti S, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patientswith tp53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34:4354–61. https://doi.org/10.1200/JCO.2016.67.5942.
Slipicevic A, Holth A, Hellesylt E, Tropé CG, Davidson B, Flørenes VA. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol Oncol. 2014;135:118–24. https://doi.org/10.1016/j.ygyno.2014.07.102.
Westin SN, Coleman RL, Fellman BM, Yuan Y, Sood AK, Soliman PT, et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J Clin Oncol. 2021;19:14–15. https://doi.org/10.1200/JCO.2021.39.15_SUPPL.5505.
Embaby A, Kutzera J, Geenen JJ, Pluim D, Hofland I, Sanders J, et al. WEE1 inhibitor adavosertib in combination with carboplatin in advanced TP53 mutated ovarian cancer: A biomarker-enriched phase II study. Gynecol Oncol. 2023;174:239–46. https://doi.org/10.1016/j.ygyno.2023.05.063.
Mahajan K, Mahajan NP. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet. 2013;29:394–402. https://doi.org/10.1016/J.TIG.2013.02.003.
Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–81. https://doi.org/10.1091/mbc.E14-04-0916.
Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–7. https://doi.org/10.1038/277665A0.
Ferlini C, Cicchillitti L, Raspaglio G, Bartollino S, Cimitan S, Bertucci C, et al. Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res. 2009;69:6906–14. https://doi.org/10.1158/0008-5472.CAN-09-0540.
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–98. https://doi.org/10.1146/ANNUREV.PHARMTOX.39.1.361.
Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JTM, Fojo T, et al. Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 1997;272:17118–25. https://doi.org/10.1074/JBC.272.27.17118.
Lahtinen A, Lavikka K, Virtanen A, Li Y, Jamalzadeh S, Skorda A, et al. Evolutionary states and trajectories characterized by distinct pathways stratify patients with ovarian high grade serous carcinoma. Cancer Cell. 2023;41:1103–1117.e12. https://doi.org/10.1016/J.CCELL.2023.04.017.
Dini A, Barker H, Piki E, Sharma S, Raivola J, Murumägi A, et al. A multiplex single-cell RNA-Seq pharmacotranscriptomics pipeline for drug discovery. Nat Chem Biol. 2024;21:432–42. https://doi.org/10.1038/s41589-024-01761-8.
Piki E, Dini A, Rantanen F, Bentz F, Paavolainen L, Barker H, et al. Molecular and functional profiling of primary normal ovarian cells defines insights into cancer development and drug responses. Mol Ther Oncol. 2024;32:200903. https://doi.org/10.1016/j.omton.2024.200903.
Murumägi A, Ungureanu D, Khan S, Arjama M, Välimäki K, Ianevski A, et al. Drug response profiles in patient-derived cancer cells across histological subtypes of ovarian cancer: real-time therapy tailoring for a patient with low-grade serous carcinoma. Br J Cancer. 2023;128:678–90. https://doi.org/10.1038/S41416-022-02067-Z.
Karvonen H, Niininen W, Murumägi A, Ungureanu D. Targeting ROR1 identifies new treatment strategies in hematological cancers. Biochem Soc Trans. 2017;45:457–64. https://doi.org/10.1042/BST20160272.
Hobbs S, Jitrapakdee S, Wallace JC. Development of a Bicistronic Vector Driven by the Human Polypeptide Chain Elongation Factor 1α Promoter for Creation of Stable Mammalian Cell Lines That Express Very High Levels of Recombinant Proteins. Biochem Biophys Res Commun. 1998;252:368–72. https://doi.org/10.1006/BBRC.1998.9646.
Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3:1416–29. https://doi.org/10.1158/2159-8290.CD-13-0350.
Potdar S, Ianevski F, Ianevski A, Tanoli Z, Wennerberg K, Seashore-Ludlow B, et al. Breeze 2.0: an interactive web-tool for visual analysis and comparison of drug response data. Nucleic Acids Res. 2023;51:W57–W61. https://doi.org/10.1093/NAR/GKAD390.
Gu Z. Complex heatmap visualization. iMeta. 2022;1:e43. https://doi.org/10.1002/imt2.43.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. https://doi.org/10.1038/NMETH.2019.
Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. https://doi.org/10.1111/J.1365-2818.2006.01706.X.
Smith K, Li Y, Piccinini F, Csucs G, Balazs C, Bevilacqua A, et al. CIDRE: An illumination-correction method for optical microscopy. Nat Methods. 2015;12:404–6. https://doi.org/10.1038/NMETH.3323.
Hollandi R, Szkalisity A, Toth T, Tasnadi E, Molnar C, Mathe B, et al. nucleAIzer: A Parameter-free Deep Learning Framework for Nucleus Segmentation Using Image Style Transfer. Cell Syst. 2020;10:453–458.e6. https://doi.org/10.1016/J.CELS.2020.04.003.
Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 2021;22:1–11. https://doi.org/10.1186/s12859-021-04344-9.
Hao Y, Stuart T, Kowalski MH, Choudhary S, Hoffman P, Hartman A, et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. 2024;42:293–304. https://doi.org/10.1038/S41587-023-01767-Y.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189 https://doi.org/10.1126/SCIENCE.AAD0501.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–96. https://doi.org/10.1038/s41592-019-0619-0.
Wickham H ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016.
Badia-IMompel P, Vélez Santiago J, Braunger J, Geiss C, Dimitrov D, Müller-Dott S, et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinf Adv. 2022;2. https://doi.org/10.1093/BIOADV/VBAC016.
Schubert M, Klinger B, Klünemann M, Sieber A, Uhlitz F, Sauer S, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun. 2018;9:1–11. https://doi.org/10.1038/s41467-017-02391-6.
França GS, Baron M, King BR, Bossowski JP, Bjornberg A, Pour M, et al. Cellular adaptation to cancer therapy along a resistance continuum. Nature. 2024;631:876–83. https://doi.org/10.1038/S41586-024-07690-9.
Peterson H, Kolberg L, Raudvere U, Kuzmin I, Vilo J gprofiler2 -- an R package for gene list functional enrichment analysis and namespace conversion toolset g: Profiler. F1000Research. 2020;9. https://doi.org/10.12688/f1000research.24956.2.
Peidli S, Green TD, Shen C, Gross T, Min J, Garda S, et al. scPerturb: harmonized single-cell perturbation data. Nat Methods. 2024;21:531–40. https://doi.org/10.1038/S41592-023-02144-Y.
Blighe K, Rana S, Emir T, Ostendorf B, Grioni A, Lewis M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R Package Vers. 2021;1:0.
Xu S, Hu E, Cai Y, Xie Z, Luo X, Zhan L, et al. Using clusterProfiler to characterize multiomics data. Nature Protocols. 2024;19. https://doi.org/10.1038/S41596-024-01020-Z.
Tickle T, Tirosh I, Georgescu C, Brown M, Haas B inferCNV of the Trinity CTAT Project. GitHub. https://github.com/broadinstitute/inferCNV [Accessed 14th January 2025].
Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2. https://doi.org/10.1093/BIOINFORMATICS/BTU393.
Olbrecht S, Busschaert P, Qian J, Vanderstichele A, Loverix L, Van Gorp T, et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 2021;13:1–30. https://doi.org/10.1186/S13073-021-00922-X.
Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30:745–62. https://doi.org/10.1038/S41422-020-0355-0.
Zhang K, Erkan EP, Jamalzadeh S, Dai J, Andersson N, Kaipio K, et al. Longitudinal single-cell RNA-seq analysis reveals stress-promoted chemoresistance in metastatic ovarian cancer. Sci Adv. 2022;8. https://doi.org/10.1126/SCIADV.ABM1831.
McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. https://doi.org/10.1016/J.CELS.2019.03.003.
Ianevski A, Giri AK, Aittokallio T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat Commun. 2022;13:1–10. https://doi.org/10.1038/s41467-022-28803-w.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7. https://doi.org/10.1038/NBT.3519.
Luo H, Xia X, Huang LBin, An H, Cao M, Kim GD, et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun. 2022;13:1–17. https://doi.org/10.1038/s41467-022-34395-2.
Chu T, Wang Z, Pe’er D, Danko CG. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat Cancer. 2022;3:505–17. https://doi.org/10.1038/s43018-022-00356-3.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. https://doi.org/10.1186/S13059-014-0550-8.
Kassambara A. ggpubr: ‘ggplot2’. Based Publication Ready Plots. 2023.
Evangelista JE, Clarke DJB, Xie Z, Lachmann A, Jeon M, Chen K, et al. SigCom LINCS: data and metadata search engine for a million gene expression signatures. Nucleic acids Res. 2022;50:W697–W709. https://doi.org/10.1093/NAR/GKAC328.
Jin H, Zhang C, Zwahlen M, von Feilitzen K, Karlsson M, Shi M, et al. Systematic transcriptional analysis of human cell lines for gene expression landscape and tumor representation. Nat Commun. 2023;14:1–15. https://doi.org/10.1038/s41467-023-41132-w.
Katayama K, Fujita N, Tsuruo T. Akt/Protein Kinase B-Dependent Phosphorylation and Inactivation of WEE1Hu Promote Cell Cycle Progression at G2/M Transition. Mol Cell Biol. 2005;25:5725–37. https://doi.org/10.1128/MCB.25.13.5725-5737.2005.
Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc. 2016;11:1757–74. https://doi.org/10.1038/nprot.2016.105.
Klimaszewska-Wiśniewska A, Hałas-Wiśniewska M, Grzanka A, Grzanka D. Evaluation of Anti-Metastatic Potential of the Combination of Fisetin with Paclitaxel on A549 Non-Small Cell Lung Cancer Cells. Int J Mol Sci. 2018;19:661 https://doi.org/10.3390/IJMS19030661.
Santamaría PG, Moreno-Bueno G, Cano A. Contribution of Epithelial Plasticity to Therapy Resistance. J Clin Med. 2019;8:676 https://doi.org/10.3390/JCM8050676.
Dai J, Zheng S, Falco MM, Bao J, Eriksson J, Pikkusaari S, et al. Tracing back primed resistance in cancer via sister cells. Nat Commun. 2024;15:1–14. https://doi.org/10.1038/s41467-024-45478-7.
Raivola J, Dini A, Karvonen H, Piki E, Salokas K, Niininen W, et al. Multiomics characterization implicates PTK7 in ovarian cancer EMT and cell plasticity and offers strategies for therapeutic intervention. Cell Death Dis. 2022;13:1–12. https://doi.org/10.1038/s41419-022-05161-5.
Sheetz JB, Mathea S, Karvonen H, Malhotra K, Chatterjee D, Niininen W, et al. Structural Insights into Pseudokinase Domains of Receptor Tyrosine Kinases. Mol Cell. 2020;79:390–405.e7. https://doi.org/10.1016/J.MOLCEL.2020.06.018.
Piki E, Dini A, Raivola J, Salokas K, Zhang K, Varjosalo M, et al. ROR1-STAT3 signaling contributes to ovarian cancer intra-tumor heterogeneity. Cell Death Discov. 2023;9:1–10. https://doi.org/10.1038/S41420-023-01527-6.
Niepel M, Hafner M, Duan Q, Wang Z, Paull EO, Chung M, et al. Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat Commun. 2017;8:1–11. https://doi.org/10.1038/S41467-017-01383-W.
Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep. 2014;4:1–10. https://doi.org/10.1038/srep05193.
Peng H. Perspectives on the development of antibody-drug conjugates targeting ROR1 for hematological and solid cancers. Antib Therapeut. 2021;4:222–7. https://doi.org/10.1093/ABT/TBAB023.
Kumarasamy V, Nambiar R, Wang J, Rosenheck H, Witkiewicz AK, Knudsen ES. RB loss determines selective resistance and novel vulnerabilities in ER-positive breast cancer models. Oncogene. 2022;41:3524–38. https://doi.org/10.1038/S41388-022-02362-2.
Maloney SM, Hoover CA, Morejon-Lasso LV, Prosperi JR. Mechanisms of taxane resistance. Cancers. 2020;12:1–57. https://doi.org/10.3390/CANCERS12113323.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29. https://doi.org/10.1038/NRCLINONC.2017.44.
Wang L, Wang X, Zhu X, Zhong L, Jiang Q, Wang Y, et al. Drug resistance in ovarian cancer: from mechanism to clinical trial. Mol Cancer. 2024;23:1–26. https://doi.org/10.1186/S12943-024-01967-3.
Ortiz M, Wabel E, Mitchell K, Horibata S. Mechanisms of chemotherapy resistance in ovarian cancer. Cancer Drug Resistance. 2022;5:304–16. https://doi.org/10.20517/CDR.2021.147.
Debaugnies M, Rodríguez-Acebes S, Blondeau J, Parent MA, Zocco M, Song Y, et al. RHOJ controls EMT-associated resistance to chemotherapy. Nature. 2023;616:168–75. https://doi.org/10.1038/s41586-023-05838-7.
Aliyuda F, Moschetta M, Ghose A, Sofia Rallis K, Sheriff M, Sanchez E, et al. Advances in Ovarian Cancer Treatment Beyond PARP Inhibitors. Curr Cancer Drug Targets. 2023;23:433–46. https://doi.org/10.2174/1568009623666230209121732.
Liu Y, Wang Y, Li X, Jia Y, Wang J, Ao X. FOXO3a in cancer drug resistance. Cancer Lett. 2022;540:215724. https://doi.org/10.1016/J.CANLET.2022.215724.
Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer. N Engl J Med. 2006;354:34–43. https://doi.org/10.1056/NEJMOA052985.
Goldberg JM, Piver MS, Hempling RE, Recio FO. Paclitaxel and cisplatin combination chemotherapy in recurrent epithelial ovarian cancer. Gynecol Oncol. 1996;63:312–7. https://doi.org/10.1006/gyno.1996.0328.
Zhang H, Qiu J, Ye C, Yang D, Gao L, Su Y, et al. ROR1 expression correlated with poor clinical outcome in human ovarian cancer. Sci Rep. 2014;4. https://doi.org/10.1038/SREP05811.
Quezada MJ, Lopez-Bergami P The signaling pathways activated by ROR1 in cancer. Cell Signall. 2023;104. https://doi.org/10.1016/j.cellsig.2023.110588.
Acknowledgements
We thank prof. Lauri Aaltonen lab for supporting the study, prof. Sampsa Hautaniemi for providing the DECIDER clinical dataset, Dr. Susanna Säisä, Anna Näätänen, Salla Hyyppä and Dr. Antti Hassinen for excellent technical assistance. We thank Biocenter Finland-supported FIMM High-Throughput Biomedicine Unit for providing pre-plated drug plates, FIMM High Content Imaging and Analysis Unit for providing the high-content confocal imaging services and FIMM Single Cell Analytics Unit for providing the sequencing services. Furthermore, these operations thank the Biocenter Finland for instrument funding. This work was supported by the University of Oulu and funded by Research Council of Finland (Profi6 #336449, #333583, #360437 to DU, #349787 to JR, and #340273 to LP), Sigrid Juselius Foundation (to DU and AM), and Finnish Cancer Foundation (to DU and AM). Doctoral research of EP was funded by Oulu University UniOGS/HBS-DP, and FB by University of Helsinki DPBM doctoral programme. The authors wish to acknowledge the CSC-IT Center for Science (Finland) for computational resources.
Author information
Authors and Affiliations
Contributions
JR generated lt-res and i-res HGSOC cell lines. JR, EM, PS, AMK, AD, AM, and EP performed experiments. DU, FR, AD, EM and JR performed scRNA-seq experiments. FR and AD conducted the scRNA-seq data analysis. HB and FR analyzed the clinical data provided by SH. FB and LP analyzed the Cell Painting data. DU supervised the research, and wrote the manuscript with JR, and FR. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical statement
All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Angelo Peschiaroli
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raivola, J., Rantanen, F., Dini, A. et al. ROR1-PI3K/AKT signaling drives adaptive resistance to cell cycle blockade in TP53 mutated ovarian cancer. Cell Death Dis 17, 276 (2026). https://doi.org/10.1038/s41419-026-08501-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-026-08501-x

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}