
Abstract
The aberrant expression of the mesenchymal FGFR2c variant in pancreatic ductal adenocarcinoma (PDAC)-derived cells enhances EMT and tumorigenic features, with PKCε-dependent signaling emerging as the main downstream pathway involved. Since lipid rafts are specialized microdomains functioning as signaling hubs and considering their relevance in the induction of EMT and cell invasion in cancer, their potential contribution in FGFR2c-mediated tumorigenesis cannot be excluded. In this study, we aimed to assess whether a possible link exists between lipid raft stability and the oncogenic activity of FGFR2c by analyzing the impact of raft perturbation on the establishment of the aberrant FGFR2c/PKCε axis in PDAC cells. Immunofluorescence and biochemical analyses revealed that ligand-dependent activation of FGFR2c led to an increased localization of the receptor within lipid rafts. Moreover, disruption of lipid rafts by methyl β-cyclodextrin (MβCD) attenuated the FGFR2c downstream signaling, as well as the consequent enhancement of EMT and of MCL1/SRC-mediated cell invasion. In addition, co-immunoprecipitation experiments, coupled to gene silencing approaches, highlighted the cation channel TRPA1 as a potential contributor to FGFR2c oncogenic signaling by regulating its recruitment to cholesterol-enriched signaling platforms. Overall, our findings indicate that FGFR2c, TRPA1 and lipid raft components represent promising targets for the development of novel cancer type-specific therapeutic strategies.
Similar content being viewed by others


Introduction
Fibroblast growth factor receptors (FGFRs) are receptor tyrosine kinases whose aberrant signaling is a widespread event in cancer. This phenomenon is triggered by receptor mutations or the establishment of abnormal autocrine/paracrine loops, resulting from receptor isoform switching [1]. The relevance of FGFRs in cancer is highlighted by the success of target therapies using next-generation small inhibitors or antibodies, which show a low side effect risk and can overcome resistance [1].
The in vitro efficacy of antibodies against FGFs or FGFRs in inhibiting some malignant features of the pancreatic ductal adenocarcinoma (PDAC) suggests their role in tumor/stroma crosstalk, identifying the FGF/FGFR axis as a suitable therapeutic target for combination therapies in this cancer type [2]. However, while anti-FGFR preclinical studies are encouraging, clinical research is still emerging and further investigations are urgently required to identify additional molecular players for more effective target therapies [3]. Among FGFRs, the aberrantly expressed mesenchymal isoforms of FGFR1 and FGFR2 appear to be mainly involved in pancreatic oncogenesis [2]. In this regard, previous studies from our group have proposed that the FGFR2 isoform switch and the consequent expression of the mesenchymal FGFR2c variant, which induces EMT and invasion in human normal keratinocytes [4, 5], can also exacerbate these oncogenic features in human pancreatic ductal adenocarcinoma (PDAC) cells [6]. In both these cancer contexts, an aberrant FGFR2c/PKCε axis was shown to affect the signaling pathway [6,7,8]. More recently, we also demonstrated that the oncogenic role of FGFR2c in PDAC cells is supported by a pore-independent activity of the cation channel TRPA1, whose protein stability is, in turn, dependent on FGFR2c expression and signaling [9]. Our findings are consistent with literature-based evidence, suggesting a potential functional link between FGFR2 and TRPA1. In fact, the expression of FGFR2 and TRPA1 is positively correlated in several cancers [10], and protein–protein interaction studies in lung cancer [11, 12] have revealed a complex mechanistic picture that could underlie their possible interplay. However, although the involvement of TRPA1 in the acquisition of cancer hallmarks is generally accepted, particularly in lung cancer, the possibility of a crosstalk between the channel and FGFR2 remains debated [11, 13, 14], especially in light of recent molecular studies highlighting both positive and negative correlation between them, depending on the tumor contexts [10].
As regards pancreatic cancer, at the moment, the existence of a mechanistic connection between FGFR2 and TRPA1 is poorly investigated. However, their expression profile in PDAC-derived cells appears similarly variable and partially overlapping, with PANC-1 cells expressing high levels of both the receptor and the cation channel [15, 16]. Furthermore, both TRPA1 and FGFR2 have recently been proposed to play a role in regulating PANC-1 cell motility/invasion [16, 17], making it reasonable to suppose that a FGFR2/TRPA1 functional cross-talk could also exist in these cells. Therefore, exploring this possibility and elucidating the underlying molecular mechanisms represents a challenge worth addressing in order to fully understand the contribution of the FGFR2c-mediated oncogenic axis in the exacerbation of EMT and cell invasion.
Lipid rafts are small cholesterol-enriched plasma membrane microdomains enriched in cholesterol, sphingolipids, gangliosides, including GM1, and signaling proteins. They contribute to the modulation of signaling pathways involved in several biological processes [18]. Interestingly, the loss of lipid raft integrity leads to cell signaling dysregulation in a variety of diseases, including cancer [19,20,21]. In fact, in epithelial tumors, intact lipid rafts appeared to be essential for the establishment of multiple and aberrant signaling axes, stemming from the plasma membrane, which in turn results in various oncogenic outcomes, such as dysregulated proliferation, EMT and cell invasion [22,23,24,25]. This is also the case of cell survival in PDAC cells, which is triggered by a sustained EGFR/Akt axis, resulting from lipid raft-mediated inhibition of EGFR endocytosis [26].
The involvement of lipid rafts in the acquisition of EMT traits and cell migration/invasion is further supported by evidence that their key structural proteins, such as flotillin and caveolin, can play a role in promoting these cancer hallmarks in different tumor contexts [27]. However, despite the broad research interest in this field, the role of lipid rafts in cancer remains controversial and may depend on the cell type [28, 29].
Interestingly, under physiological conditions, FGFRs, including FGFR2c, are localized and activated within the lipid rafts [30, 31], and TRPs, including TRPA1, interact with lipid rafts, which in turn affect their function [32, 33]. Therefore, it is reasonable to hypothesize that a complex interplay between FGFRs, TRPs, and lipid rafts may occur, contributing to the establishment of receptor-mediated aberrant signaling during carcinogenesis. Identifying the mechanisms underlying this crosstalk and providing new key molecular targets could be clinically relevant for advancing cancer therapy.
Materials and methods
Cells and treatments
The PANC-1 and MIA PaCa-2 cell lines (ATCC, Manassas, VA, USA) were cultured and subjected to gene silencing approaches to generate FGFR2 and TRPA1 stable depletion, as previously reported [6].
For both TRPA1 and FGFR2c overexpression, cells were transfected with pcDNA3.1+hsTRPA1_myc_dTom (Addgene, Watertown, MA, USA; plasmid 183179) and/or with pCI-neo expression vector containing human FGFR2c (FGFR2c) using JetPei DNA Transfection Reagent (Polyplus Transfection, Illkirch, France; 101-42 N), according to the manufacturer’s protocol.
For growth factor stimulation, cells were left untreated or incubated with fibroblast growth factor 2 (FGF2) (PeproTech, London, UK, BFGF 100-188) 100 ng/mL for 10 min at 37 °C, to trigger receptor activation and signaling, or with FGF2 25 ng/mL for 24 h at 37 °C, to assess the downstream effects. In order to in parallel obtain FGFR2c activation and the inhibition of its internalization, cells were stimulated with FGF2 100 ng/ml for 1 h at 4 °C.
For lipid raft perturbation, cells were incubated with 5 µM or 2.5 µM methyl-β-cyclodextrin (MβCD) (Sigma-Aldrich; St. Louis, Missouri, USA; C4555) for 20 min and 24 h at 37 °C, respectively.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay protocol was previously described [34]. Briefly, cells were incubated for 3 h with MTT solution 5 mg/ml in DMEM, washed with PBS and then incubated with 100 µl DMSO, gently shaking for 5 min. Absorbance was read on the GloMax plate reader.
Cholesterol extraction by high-performance thin-layer chromatography (HPTLC)
Cells, either treated or untreated with MβCD, were lysed in a lysis buffer containing 1% Triton X-100 for 20 min at 4 °C. Following protein concentration assessment, the lysate was subjected to cholesterol analysis. Neutral lipid extracts were separated by HPTLC, using a solvent system of hexane/diethyl ether/acetic acid (70:30:1, v/v/v) and were detected by staining with 2% copper acetate solution in 8% phosphoric acid and subsequent heating at 120 °C for 15 min. Quantification was carried out using NIH Image 1.62 (Mac OS X, Apple Computer International). (Mac OS X, Apple Computer International).
Immunofluorescence
Cells were grown on coverslips and processed as previously described [6]. Cells were then incubated with the following primary antibodies: monoclonal antibody anti-vimentin (1:50 in PBS; Dako, Glostrup, Denmark; M0725) Cholera Toxin B subunit FITC conjugate (CTxB-FITC) (1:50 in PBS, Sigma-Aldrich; St. Louis, Missouri, USA; C1655) for 1 h at 25 °C; polyclonal antibodies anti-Bek (H-80) (1:25 in PBS, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; sc-20735) for 1 h at 4 °C.
The primary antibodies were visualized, and images were taken, as previously described [6]. Quantitative analysis of vimentin fluorescence intensity and that of FGFR2 colocalization with cholera toxin-FITC was performed using dedicated functions of the Axiovision software (Zeiss), delimiting single cell areas in 5 fields from three independent experiments. For Fluorescence intensity analysis, the results were expressed as mean values ± SD and reported in a graph as fold change with respect to the control value. For colocalization analysis, the results were expressed as mean percentage ± SD.
Isolation of Triton X-100 soluble and insoluble fractions
Fractions were isolated according to Skibbens et al [35]. Briefly, cells, both treated and untreated, were lysed with 1 ml of extraction buffer (25 mM HEPES, pH 7.5, 0.15 NaCl, 1% Triton X-100 and 100 kallikrein U/ml aprotinin) for 20 min on ice. Lysates were collected and centrifuged for 2 min in a Brinkmann microfuge at 12,000 rpm at 4 °C. Sovranatants, containing Triton X-100 soluble material, were collected; pellets were undertaken to a second centrifugation (30 s) in order to remove the remaining soluble material. The pellets were then solubilized in 100 ml of buffer containing 50 mM Tris-HCl, pH 8.8, 5 mM EDTA and 1% SDS. DNA was sheared by passage through a 22 ga. needle. The insoluble- and soluble-Triton X-100 fractions were considered as raft fractions and non-raft fractions, respectively, as checked by Western blotting, using anti-Flotillin pAb.
Immunoprecipitation
Cells were lysed as previously reported [36]. The lysates were mixed with protein A/G-acrylic beads (Sigma-Aldrich, St. Louis, Missouri, USA; P3296) and stirred by a rotary shaker for 2 h at 4 °C to preclear nonspecific binding. After centrifugation (500 × g for 1 min), the supernatant was recovered and immunoprecipitated with mouse anti-FGFR2 antibodies plus protein A/G-acrylic beads. A mouse IgG isotypic control (Sigma-Aldrich; St. Louis, Missouri, USA; I5006) was used as a negative control. The immunoprecipitates (IPs) were split into two aliquots. The first one was subjected to Western blot analysis for FGFR2 or TRPA1 detection; the second one was checked by dot blot for GM1 detection.
Western blot analysis
Cells were lysed, and total lysates were resolved and blotted as previously reported [17]. The membranes were incubated with anti-Flotillin (Abcam Cambridge, UK; ab13493) anti-E-cadherin (GT311 GeneTex, Irvine, California, USA), anti-vimentin (M0725, Dako, Glostrup, Denmark), anti-phospho-Fibroblast Growth Factor Receptor Substrate 2 α (FRS2-α) (Tyr196) (Cell Signaling Technology, Beverly, MA, USA, 3864), the anti-phospho-Sarcoma kinase (Src) Family (Tyr416, D49G4) (Cell Signaling Technology, Beverly, MA; USA;6943), monoclonal antibodies anti-p-MTOR (Ser 2448; Cell Signaling; Beverly, MA, USA; 5536S), anti-p-p44/42 mitogen-activated protein kinase (MAPK) (p-ERK1/2) (Thr202/Tyr204; Cell Signaling; 9101S), anti-Bek (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; C17, sc-122), anti-p-S6K (ser 371, Cell Signaling, Beverly, MA, USA; 9208), and anti p-PKCε (Ser729, Abcam, Cambridge, UK; ab63387), anti-Myeloid Cell Leukemia 1 (Mcl-1) (D35A5) (Cell Signaling; Beverly, MA, USA; 5453), anti-TRPA1 (Abcam; Abcam Cambridge, UK; ab62053),. The membranes were stripped as reported [6] and probed again with anti-p44/42 MAPK (ERK1/2) (Cell Signaling; Beverly, MA, USA; 4695S), anti-S6K (Cell Signaling; Beverly, MA, USA;9202), anti-PKCε (Abcam; Abcam Cambridge, UK;ab124806), anti-FRS2 (H-91) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; sc-8318), anti-HSP90 (Proteintech Inc., Rosemont, IL, USA, 13171-1-AP), anti-MTOR (Cell Signaling; Beverly, MA, USA; 2983S) polyclonal antibodies or anti-Src (Cell Signaling; Beverly, MA, USA;36D10), anti-ACTB (Sigma-Aldrich; St. Louis, Missouri, USA; A5441) monoclonal antibodies for protein equal loading. Densitometric analysis was performed as previously described [17]. Mean values (± SD) from three different experiments were normalized and expressed as fold increase with respect to the control. All uncropped gels are reported as Supplementary material.
Dot blot analysis
Briefly, aliquots of FGFR IPs, prepared as described above, were spotted onto nitrocellulose strips. The strips were blocked for 1 h with 5% BSA in TBS/T (Bio-Rad Laboratories, Hercules, CA, USA) to block the residual binding sites on the paper. The strips were rinsed for 10 min in TBS/T and then incubated with Cholera Toxin B Subunit-Peroxidase conjugate (Sigma-Aldrich; St. Louis, Missouri, USA; C3741) for 1 h at 25 °C, or with anti-FGFR pAb or with anti-TRPA1 pAb and further incubated for 1 h at 37 °C with HRP-conjugated anti-rabbit IgG. Immunoreactivity was assessed by chemiluminescence reaction, using the ECL Western detection system.
Scratch assay
Cells were seeded at 2.5 × 105 cells on 35 mm plates and grown until confluence. Confluent cells were serum starved for 12 h, and then a standardized cell-free area was introduced by scraping the monolayer with a sterile tip. Plates were photographed immediately after scratching (T0 = 100% Open Area) and then left untreated or stimulated with FGF2 for 24 h in the presence or not of MβCD. Phase contrast images were taken along the scratch at low magnification (10×) using an Axiovert 200 inverted microscope (Zeiss). Cell migration was quantified as previously described [17]. The results are expressed as the mean values of three independent experiments ± SD. p-values were calculated using Student’s t test, and the significance level has been defined as p > 0.05.
In vitro invasion assay
The in vitro assay and its quantitative analysis were assessed as previously reported [6]. Briefly, cells were seeded on Matrigel pre-coated Transwell Boyden chamber filters and left unstimulated or stimulated with FGF2 in the presence or not of MβCD for 24 h. FGF2 was added to the medium of the bottom chamber to stimulate cell chemotaxis.
Primers
Oligonucleotide primers were designed with Primer-BLAST [37] and purchased from Invitrogen. The following primers were used: for the Snail1 target gene: 5′-GCTGCAGGACTCTAATCCAGA-3′ (sense), 5′- ATCTCCGGAGGTGGGATG-3′ (antisense); for the STAT3 target gene: 5′-CAGAGATGTGGGAATGGGGG- 3′ (sense), 5′-TGGCAAGGAG TGGGTCTCTA-3′ (antisense); for the FRA1 target gene: 5′-GCAGGCGGAGACTGACAAA-3′ (sense), 5′- GATGGGTCGGTGGGCTTC- 3′, MMP2 target gene: 5′-AGAAGGCTGTGTTCTTTGCAG-3′ (sense), 5′-AGGCTGGTCAGTGGCTTG-3′ (antisense), MMP9 target gene: 5′- TGACAGCGACAAGAAGTG-3′ (sense), 5′-CAGTGAAGCGGTACATAGG-3′ (antisense). For the 18S rRNA, housekeeping sequences were previously reported [6].
RNA extraction and cDNA synthesis
Total RNA was obtained using TRIzol (Invitrogen, Waltham, MA, USA) and prepared as reported [6]. The total RNA concentration was evaluated by spectrophotometry; the cDNA was obtained with iScriptTM cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA; 170-8891) according to the manufacturer’s protocol.
PCR amplification and real-time quantitation
Real-time PCR and gene expression quantitation were performed as previously described [6]. Results are reported as mean values ± SD from three different experiments in triplicate.
Statistics
Statistical analysis was performed using GraphPad Prism software Inc. version 9.5.0 (San Diego, CA, USA). The p-values for all graphs were determined using the Student’s t-test, as indicated in the figure legends. Significance levels were denoted by asterisks: * p < 0.05, ** p < 0.005, *** p < 0.001, and **** p < 0.0001, corresponding to increasing levels of significance.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Ethical approval and informed consent were not necessary. All experiments were performed exclusively using commercially available immortalized cell lines.
Results
Ligand-dependent signaling of FGFR2c requires intact lipid rafts
FGFR2c is highly and variably expressed in PDAC-derived cells and tissues, where its expression correlates with clinical malignant features. FGFR2c expression also correlates with tumor growth and metastasis in in vivo mouse models, as well as cell growth, stemness and invasion in in vitro models [15, 38]. In line with this evidence, we previously selected PANC-1 and MIA PaCa-2 cell lines, which express divergent levels of FGFR2c (high and very low, respectively), to demonstrate that this receptor is a key player in the exacerbation of EMT profile, anchorage-independent growth and cell invasion in PDAC cells [6, 17]. In the present study, we took advantage of the same two cell lines to investigate the possible involvement of lipid rafts in FGFR2c oncogenic function.
We first investigated the receptor distribution on the cell surface and its possible relocation following its activation. To this aim, PANC-1 cells transiently overexpressing FGFR2c (PANC-1/FGFR2c) were stimulated with FGF2 for 1 h at 4 °C, to trigger receptor activation at the plasma membrane, blocking its internalization. The efficiency of FGFR2c transient transfection was assessed by Western blot analysis (Fig. 1A). In immunofluorescence assays, cell surface FGFR2c was checked using an antibody recognizing the extracellular portion of the receptor, while lipid rafts were detected using CTxB-FITC, which labels monosialoganglioside type 1 (GM1), a widely recognized marker of lipid rafts [39]. Quantitative immunofluorescence analysis showed that, although most of the receptors did not colocalize with CTxB-FITC in unstimulated cells, the rate of colocalization was significantly increased following ligand stimulation (Fig.1B). Receptor relocalization into lipid rafts was further supported by changes in plasma membrane distribution, which became predominantly dotted after FGF2 stimulation (Fig. 1B, right panels). This redistribution of FGFR2c was also assessed by biochemical experiments using Triton X-100 fractions. Western blot analysis showed that, in the TX-100 insoluble fractions containing lipid rafts, the band corresponding to FGFR2c at 120 KDa was significantly increased by FGF2. On the contrary, the corresponding band in TX-100 soluble fractions, although more intense, appeared unaffected by ligand stimulation (Fig. 1C).

PANC-1 cells transiently transfected with pCI-neo FGFR2c expression plasmid (PANC-1 FGFR2c) were left untreated or stimulated with FGF2 for 1 h 4 °C, to trigger receptor activation, impairing its internalization. A Western blot analysis, performed using, shows that the 120 KDa band corresponding to the molecular weight of FGFR2 is more evident in cells transfected with the FGFR2c variant. The equal loading was assessed using anti-beta actin (ACTB) antibody. For densitometric analysis, the values from three different experiments (n = 3) were normalized, expressed as mean values ± SD and reported in a graph as fold increase with respect to the control value. Student t test was performed, and significance levels have been defined as p < 0.05: **p < 0.01. B Quantitative immunofluorescence analysis was performed using anti-FGFR2 bek H80 antibody, which recognizes the extracellular portion of the receptor, and CTxB-FITC, which binds the monosialic ganglioside type 1 (GM1). Nuclei were stained with DAPI. Colocalization analysis was performed by scanning cells with an ApoTome System (Zeiss) and analyzing the images from two independent experiments (n = 2) with the Axiovision software (Zeiss). The treatment with FGF2 increases the colocalization of FGFR2c with GM1 and changes the plasma membrane distribution of the receptor from continuous to dotted. The colocalization was expressed as mean percentage ± SD. Student t test was performed, and significance levels have been defined as p < 0.05: ***p < 0.001. Bars: 10 μm C PANC-1 FGFR2c cells were lysed to obtain TX100 soluble and insoluble fractions as reported in Materials and Methods. The efficiency of the fraction extraction was assessed by checking the distribution of the lipid raft marker flotillin. Western blot analysis shows a significant increase of FGFR2c amount in the insoluble fraction after FGF2 stimulation. Densitometric analysis was performed as reported: *p < 0.05.
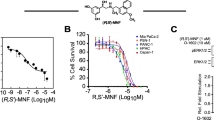
We then investigated the relevance of the lipid raft integrity in FGFR2c oncogenic signaling using the lipid raft-disrupting agent MβCD [40]. To determine suitable conditions for both short-term and long-term cholesterol depletion, PANC-1 cells were treated with different concentrations of MβCD for different times (Fig. 2A, B). The results confirmed that the treatment with 5 μM MβCD for 20 min, as previously used [41], induced a significant depletion of cholesterol (Fig. 2A), without affecting cell viability (Fig. 2B). For long-term treatments (24 h), the concentration of 2.5 μM MβCD was selected based on its effects on cholesterol efflux and cell viability (Fig. 2A, B). To analyse the effects of MβCD on early FGFR2c signaling, we compared PANC-1 cells, which express high levels of FGFR2c, with MIA PaCa-2 cells, which express very low levels of FGFR2c, as expected [6]. Biochemical approaches showed that the difference in FGFR2 expression is not affected by lipid raft perturbation (Fig. 2C). In contrast, lipid raft perturbation significantly inhibited the FGF2-mediated phosphorylation of the FRS2, a widely recognized sensor of FGFR2c activation, as well as the phosphorylation of the hub signaling molecule PKCε and downstream substrates ERK1/2 (Fig. 2C) in PDAC-1 cells. A comparable impairment of phosphorylation/activation was also observed for MTOR and its downstream substrate S6K (Fig. 2C), indicating a general inhibition of FGFR2c signaling. As expected, MIA PaCa-2 cells displayed no significant signaling response to FGF2 treatment, nor any effect as a consequence of lipid raft perturbation (Fig. 2C). The absence of an appreciable response to FGF2 in these cells suggested that they express very low levels of all FGFRs able to bind FGF2, including FGFR3c and FGFR4 [15]. Our molecular analysis, performed by real-time RT-PCR, confirmed the FGFR expression profile as previously described for PANC-1 and MIA PaCa-2 cells (Supplementary Fig. 1A) [15]. However, to confirm that the stronger signaling response to FGF2 treatment in PANC-1 cells is primarily due to high expression of FGFR2c, we checked ERK1/2 phosphorylation in stable FGFR2 shRNA clones. We previously demonstrated that, since both PANC-1 and MIA PaCa-2 cells do not express appreciable levels of FGFR2b, the effect of FGFR2 shRNA is entirely at the expense of FGFR2c expression [6]. Western blot analysis showed that the strong enhancement of ERK signaling, detected in PANC-1 cells, was significantly dampened by FGFR2 depletion, while the more modest response in MIA PaCa-2 cells remained unaffected (Supplementary Fig. 1B). This evidence confirmed that the signaling response of PANC-1 cells can be ascribed to FGFR2c, whereas the modest response in MIA PaCa-2 does not. In addition, our results also suggest that, regardless of the variability of FGFR profile, in both cell lines, the endogenous expression of other FGFRs able to recognize FGF2 (such as FGFR3c and FGFR4) is not sufficient to generate an appreciable signaling response.

A, B PANC-1 cells were treated with the reported concentrations of methyl-β-cyclodextrin (MβCD) for the different reported times. Cells were then processed for free cholesterol (CHOL) analysis (A) or for MTT assay (B), as reported in materials and methods. A For densitometric analysis of cholesterol levels, the resulting values from two different experiments (n = 2) were expressed as mean values ± SD and reported in graph as fold change respect to the control value. B The cell survival rate was expressed as mean optical density (O.D.) values from three independent experiments (n = 3) ± SD. The concentration of 5 µM for 20 min and 2.5 µM for 24 h were chosen for short-term and long-term experiments, respectively. Student t test was performed and significance levels have been defined as p < 0.05: *p < 0.05; ** p < 0.01; *** p < 0.001. C PANC1 and MIA PaCa-2 cells treated with 5 mM MβCD for 20 min and left untreated or stimulated with FGF2 for 10 min at 37 °C, to induce receptor activation and signaling. Western blot analysis shows the difference in FGFR2 expression, which is not affected by lipid raft perturbation. In addition, only in PDAC-1 cells, lipid raft perturbation represses the phosphorylation of the FGFR2c signaling platform FRS2, that of PKCε and- ERK1/2, as well as that of MTOR and its substrate S6K. MIA PaCa-2 cells, which express very low levels of FGFR2c, display no significant changes in substrate phosphorylation/activation, neither in response to FGF2 or lipid raft perturbation. Densitometric analysis was performed as reported in Fig. 1: *p < 0.05; ** p < 0.01.
Lipid raft perturbation counteracts FGFR2c-mediated enhancement of EMT phenotype and cell invasion
To investigate the downstream events triggered by FGFR2c signaling and their dependence on lipid raft integrity, PANC-1 and MIA PaCa-2 cells were treated for 24 h with FGF2 in the presence of 2.5 mM MβCD. Real−time RT−PCR showed that, only in PANC-1 cells, the previously described increase in EMT−related transcription factors Snail1, STAT3 and FRA1 [6] was significantly impaired by MβCD (Fig. 3A). These results suggested the lipid raft disruption interferes with the enhancement of the EMT program controlled by FGFR2c.

PANC-1 and MIA PaCa-2 cells were treated with FGF2 for 24 h at 37 °C in the presence or not of 2.5 mM MβCD. A Real−time RT − PCR shows that the induction of the EMT−related transcription factors Snail1, STAT3 and FRA1 in response to FGF2 stimulation is only visible in PANC−1 cells and is reversed by the treatment with MβCD. The mRNA levels are expressed as mean value ± SD (n = 3) and reported in the graph as fold increase with respect to the control. Student’s t-test was performed, with significance levels defined as p values < 0.05. p < 0.05: *p < 0.05; ** p < 0.01; *** p < 0.001. B Western blot analysis shows that, only in PANC-1 cells, the decrease of the epithelial marker E-cadherin and the increase of the mesenchymal marker vimentin induced by FGF2 stimulation are counteracted by lipid raft perturbation. The expression of EMT-related genes, E-cadherin and vimentin do not significantly change in MIA PaCa-2 cells. Densitometric analysis was performed as reported above: *p < 0.05; *** p < 0.001. C Quantitative immunofluorescence analysis shows that the increase in the immunostaining corresponding to the mesenchymal marker vimentin, visible after FGF2 stimulation only in PANC-1 cells, was inhibited by MβCD. In MIA PaCa-2 cells the vimentin immunostaining does not significantly change, independently from FGF2 and/or MβCD treatment. Quantitative analysis of the fluorescence intensity was performed as reported in Materials and Methods. Results were expressed as mean values ± SD and reported in the graph as fold change with respect to the control value. Student’s t test was performed, with significance levels defined as p values < 0.05: p < 0.05. *p < 0.05; *** p < 0.001. Bars: 20 μm.
Indeed, parallel biochemical and immunofluorescence experiments showed that the decrease of the epithelial marker E-cadherin and the increase of the mesenchymal marker vimentin, enhanced by FGF2 stimulation only in PANC-1 cells, were significantly counteracted by MβCD pre-treatment (Fig. 3B, C). Otherwise, the expression of these EMT markers did not significantly change in MIA PaCa-2 cells, confirming that their modulations are closely dependent on FGFR2c expression and activation (Fig. 3B, C).
Since the acquisition of EMT phenotype is linked to increased cell motility, and we previously demonstrated that FGF2 increases cell motility in the PANC-1 cell line [17], we first analyzed the impact of lipid raft perturbation on this event. The in vitro scratch assay confirmed that, only in PANC-1 cells, FGF2 stimulation induced an increase in the migratory response, leading to cell repopulation of the scratch area (Supplementary Fig. 2, arrows), which was significantly impaired by MβCD treatment (Supplementary Fig. 2). The inhibition of FGF2-induced cell motility in response to lipid raft disruption was comparable to that obtained following stable depletion of FGFR2c by shRNA. On the other hand, cell migration in MIAPaCa-2 cells appeared independent of both FGF2 stimulation and MβCD presence (Supplementary Fig. 2). These results confirmed that the increased cell motility in response to FGF2 can be attributed to the elevated expression of FGFR2c, which in turn requires lipid raft integrity.
EMT and cell motility are single elements of a more complex cell reprogramming that leads to cell invasion. We have previously proposed that, in PDAC cells, FGFR2c-mediated cell invasion is linked to MCL-1 induction and SRC phosphorylation/activation [17]. Interestingly, it has been found that the activation of SRC family members, which are involved in EMT in several cancers [42, 43], including PDAC [44], can take place in the lipid rafts, conferring invasive potential to epithelial cancer cells [45]. In line with this observation, here we found that the increase of MCL-1 protein expression and of SRC phosphorylation, induced by FGF2 stimulation in PANC-1 cells (Fig. 4A), was significantly dampened by MβCD (Fig. 4A), suggesting its dependence on intact lipid rafts.

PANC-1 and MIA PaCa-2 cells were treated with FGF2 for 24 h at 37 °C in the presence or not of MβCD 2.5 mM. A Western blot analysis shows that the increase of either MCL-1 expression or of SRC phosphorylation (tyr416 site), observed only in PANC-1 cells in response to FGF2, was significantly inhibited by lipid raft perturbation. Densitometric analysis was performed as reported above: *p < 0.05. B Real-time RT-PCR analysis shows that the increase of MMP-2 and MMP-9 mRNA levels, induced by FGF2 stimulation only in PANC-1 cells, is significantly repressed by MβCD treatment. The mRNA levels are expressed as mean value ± SD (n = 3) and reported in the graph as fold increase with respect to the control. Student’s t test was performed, with significance levels defined as p-values < 0.05. p < 0.05: *p < 0.05; ** p < 0.01. C In vitro invasion assay performed in PANC-1 and MIA PaCa-2 cells stably transfected with an unrelated short harpin RNA (PANC-1 Cx shRNA; MIA PaCa-2 Cx shRNA) or with FGFR2c shRNA (PANC-1 FGFR2c shRNA; MIA PaCa-2 FGFR2c shRNA) to obtain FGFR2c depletion. Cells were stimulated with FGF2 in the presence or not of MβCD, as above. The increase of cell invasion in response to FGF2, visible only in PANC-1 cells, was significantly dampened by lipid raft perturbation. The effect of MβCD is comparable to that observed after FGFR2c depletion. No significant effects are visible in MIA PaCa-2 in response to FGF2 stimulation, independently of lipid raft perturbation or FGFR2c depletion. Quantitative analysis was assessed by counting for each sample the migrated cells in 10 microscopic fields from two independent experiments (n = 2). Results were expressed as mean values ± SD. Student’s t test was performed, with significance levels defined as p-values < 0.05. p < 0.05. *p < 0.05. Bar: 20 μm.
The secretion of MMP-2 and MMP-9 is a crucial event connecting the EMT program to tumor cell invasion in several carcinomas, including PDAC [46, 47]. Therefore, to further assess the relevance of the interplay between FGFR2c signaling and lipid rafts in the functional link between EMT and invasion, we monitored the impact of receptor activation and of raft perturbation on the expression of MMP-2 and MMP-9, which are the MMPs mainly represented in PDAC [48]. Real-time RT-PCR analysis highlighted that, only in PANC-1 cells, FGF2 stimulation induced an increase of both MMP-2 and MMP-9 mRNA levels, which was significantly repressed by MβCD treatment (Fig. 4B).
To assess if the effects induced by the lipid raft perturbation could actually result in an effective impairment of cell-invasive ability, we moved on to the functional in vitro invasion assay of the transwell Boyden chambers pre-coated with Matrigel. Upon seeding, cells were serum starved overnight, and then FGF2 was added in the bottom chamber for 48 h in order to stimulate cell chemotaxis. The results showed that the number of PANC- 1 cells invading the bottom side of the filter was increased by FGF2 stimulation, but significantly reduced by the presence of MβCD (Fig. 4C). The effects of lipid raft disruption in these cells were comparable to that observed in response to the stable depletion of FGFR2c by shRNA (Fig. 4C). No significant changes were detected in MIAPaCa-2 cells in terms of MCL1/SRC signaling, MMPs expression and cell invasion, independently from lipid rafts perturbation or FGFR2c shRNA stable transduction (Fig. 4A, B, C), further confirming their dependence on FGFR2c high expression.
The enhanced targeting of active FGFR2c to lipid rafts requires TRPA1 expression
We recently proposed that the expression of the cation channel TRPA1 is required for FGFR2c-mediated oncogenic outcomes in PDAC cells [9]. However, although the role of this channel appeared to be relevant and independent of its pore function [9], the molecular mechanism underlying TRPA1/FGFR2c crosstalk remains unknown.
Since TRP channels contribute to the compartmentalization of signaling molecules into plasma membrane domains, including lipid rafts [33], we investigated the possible involvement of TRPA1 in FGFR2c targeting to lipid rafts. PANC-1 cells stably transduced with TRPA1 shRNA or with an unrelated shRNA were transiently transfected with pCI-neo FGFR2c (PANC-1 TRPA1 shRNA/FGFR2c; PANC-1 Cx shRNA /FGFR2c), stimulated with FGF2 for 1 h at 4 °C and then subjected to sucrose-gradient fractionation, as reported above. The efficiency of either stable TRPA1 depletion and FGFR2c overexpression was assessed by Western blot analysis performed using whole lysates (Fig. 5A). The subsequent biochemical analysis performed on the extracted fractions showed that TRPA1 depletion impaired the increase of FGFR2c amount in the TX-100 insoluble fractions after FGF2 stimulation (Fig. 5B). Furthermore, HPTLC analysis to evaluate the free cholesterol (CHOL) content in PANC-1 TRPA1 shRNA and PANC-1 Cx shRNA, treated or not with the TRPA1 antagonist A907079, showed not significant changes in cholesterol levels (Supplementary Fig. 3), confirming that TRPA1 depletion or inactivation did not affect the stability of lipid rafts. Therefore, the results obtained by sucrose-gradient fractions suggest that TRPA1 is required for the recruitment of FGFR2c within lipid rafts after its ligand-dependent activation.

PANC-1 cells stably transduced with TRPA1 (PANC-1 TRPA1 shRNA) or with an unrelated shRNA (PANC-1 Cx shRNA) were transiently transfected with pCI-neo FGFR2c (PANC-1 Cx shRNA /FGFR2c; PANC-1 TRPA1 shRNA/FGFR2c), stimulated with FGF2 for 1 h 4 °C and then processed for fraction extraction or for immunoprecipitation as reported in materials and methods. A Western blot analysis using anti-Bek and anti-TRPA1 antibodies, performed in the whole cell lysates, shows the efficiency of both FGFR2c overexpression and TRPA1 depletion. Densitometric analysis was performed as reported above: *p < 0.05. B Western blot analysis performed on cell fractions shows that the increase of the FGFR2c amount in the total TX-100 insoluble fraction induced by ligand activation is not visible in cells in which TRPA1 has been depleted. C Western blot analysis of cell immunoprecipitates, obtained with anti-FGFR2 antibody bek-C8, shows that the band corresponding to TRPA1 is increased after FGF2 stimulation. D Dot blot analysis performed using Cholera Toxin B Subunit-Peroxidase conjugate, anti-TRPA1 antibody or anti-FGFR2 antibody, bek C8 shows that both GM1 and TRPA1 were consistently enriched in the FGF2-stimulated sample. STD = pure standard GM1. Densitometric analysis was performed as reported above: *p < 0.05; ****p < 0.0001.
To elucidate the molecular mechanisms driving this interplay, we investigated the possibility that the FGFR2c might interact with TRPA1. To assess this, PANC-1 cells overexpressing both FGFR2c and TRPA1 by transient transfection were stimulated with FGF2 for 1 h at 4 °C and then subjected to immunoprecipitation, using the anti-FGFR2 antibody (anti-Bek C8). Western blot analysis of cellular immunoprecipitates revealed that the band corresponding to the molecular weight of TRPA1 was significantly increased after FGF2 stimulation (Fig. 5C). Therefore, the interaction between FGFR2c and TRPA1 was enhanced by the receptor activation. The cellular immunoprecipitates were then spotted onto nitrocellulose strips and analyzed by dot blot, using CTxB -Peroxidase, to detect the lipid-raft component ganglioside GM1. The results showed that both GM1 and TRPA1 were consistently enriched in the FGF2-stimulated sample (Fig. 5D). Overall, the coimmunoprecipitation assays suggested that FGFR2c physically interacts with TRPA1, that this interaction was significantly enhanced by the receptor activation, and that it may be required for the receptor targeting to GM1-enriched lipid rafts.
Discussion
Cholesterol-rich membrane domains, termed lipid rafts, play a key role in the physical compartmentalization of plasma membrane proteins, including receptor tyrosine kinases, and intracellular substrates, serving as pivotal platforms for signal transduction [24, 49, 50]. Therefore, it is not surprising that loss of their integrity can somehow lead to the dysregulation of the cell signaling that occurs in several diseases, including cancer. However, despite the large body of evidence in the field, the specific role of cholesterol metabolism and of lipid rafts in the different tumor contexts remains controversial [28, 29]. For instance, although several reports have highlighted their involvement in tumor cell survival [24, 26], other studies have implicated these plasma membrane domains as death-promoting platforms [24, 27].
Nevertheless, the relevance of lipid rafts in the establishment of EMT and cell invasion has been widely recognized in several tumors [22,23,24,25]. Therefore, in light of our previous findings indicating the role of FGFR2c in the enhancement of these specific oncogenic hallmarks in PDAC, in this work, we explored the possible synergic involvement of the lipid rafts.
Using immunofluorescence, as well as biochemical approaches, we found that, although FGFR2c is mainly distributed outside of lipid rafts, the receptor was recruited within these plasma membrane domains, following its ligand-dependent activation. Interestingly, the disruption of lipid rafts by MβCD-induced cholesterol efflux resulted in a strong inhibition of the oncogenic FGFR2c-PKCε-ERK1/2 axis, which is heavily activated by FGF2 stimulation only in those PDAC cells highly expressing FGFR2c. Considering that ERK1/2 is also a well-established pathway contributing to the establishment of the EMT program in PDAC [51, 52], we have previously demonstrated its involvement in the enhancement of FGFR2c-mediatement EMT signature [6]. In line with our previous observations, in the present study, we show that disruption of lipid rafts by MβCD led to repression of the induction of the EMT-related transcription factors, Snail1, STAT3 and FRA1, as well as to a reversion of the modulation of the epithelial/mesenchymal markers, compatible with the recovery of an epithelial phenotype. Thus, lipid raft integrity appears to be required for FGFR2c signaling and for the enhancement of FGFR2c-mediated EMT. This evidence is in line with previous data obtained in prostate cancer, indicating lipid rafts as platforms for sustained EGFR/ERK1-2 axis, leading to EMT [23]. The evidence that lipid rafts significantly contribute to EMT is also supported by the evidence of the involvement of structural proteins, such as flotillin-1 and -2 and caveolin-1, in the induction of this cancer hallmark; [27, 53, 54].
Another crucial tumor signature recently correlated to lipid raft integrity is cell invasion, which, in PDAC, we found to be enhanced following the activation of the MCL-1/SRC pathway downstream of FGFR2c [17]. Exploring the impact of lipid raft dysregulation, in this work, we found that this signaling axis and the resulting increase in cell invasion, detected only in cells overexpressing FGFR2c after FGF2 stimulation, were inhibited by MβCD. Therefore, the FGFR2c-mediated enhancement in cell migration/invasion is also dependent on lipid raft integrity. These findings are in line with several previous studies, demonstrating a close interplay between lipid rafts and the dynamics of cytoskeletal filaments and focal adhesion components [55, 56]. This is particularly true for SRC family kinases, whose activation within lipid rafts has been found to confer invasive potential to epithelial cancer cells [45]. In this perspective, our data support the possibility that SRC protein may represent a hub signaling molecule, linking lipid rafts to oncogenic FGFR2c signaling and cell motility/invasion. At this point, we concluded that the recruitment of FGFR2c within lipid rafts is a crucial event for its oncogenic activity in PDAC cells, although the molecular mechanisms regulating this sorting remain unclear.
In this regard, we focused our attention on TRPA1, which we recently found to play a pore-independent role in FGFR2c-mediated oncogenic outcomes [9]. The possible contribution of TRPA1 in the acquisition of cancer hallmarks has been proposed in various tumors, including PDAC [16], even if the possible crosstalk of this cation channel with FGFR2 is still a widely debated topic [11, 13, 14]. TRPs channels, including TRPA1, localize in lipid rafts [33], where they can functionally interact with several receptors and signaling substrates, including SRC family members [33]. Furthermore, raft disruption can significantly impair TRP function [32, 57]. Therefore, we explored the possibility that TRPA1 could contribute to the oncogenic outcome of FGFR2c through receptor targeting to lipid rafts. Biochemical approaches in PDAC cells highly expressing FGFR2c showed that the depletion of TRPA1 impaired the FGF2-mediated increase in receptor expression in lipid raft-enriched cell fractions. Since parallel analysis of free cholesterol by HPTLC confirmed that TRPA1 depletion did not alter lipid raft stability, our data indicated that TRPA1 is required for the ligand-dependent sorting of FGFR2c into these plasma membrane domains. Finally, immunoprecipitation approaches, combined with Western blot and Dot blot analysis, highlighted that FGF2 stimulation results in increased FGFR2c interaction with both TRPA1 and the lipid raft marker GM1.
Overall, our data indicate that, at least in the case of PDAC cells, lipid raft integrity is required for FGFR2c-mediated oncogenic outcomes in terms of exacerbation of both EMT and invasive behavior, further supporting the growing evidence pointing to these plasma membrane domains as promising therapeutic targets in cancer [20, 24, 58]. In addition, we highlighted for the first time a pivotal role played by TRPA1/FGFR2c physical interaction in determining the targeting of the receptor to suitable cholesterol-enriched signaling platforms.
Currently, given the multiple proposed functions of TRPs in different physiological and pathological contexts, including cancer, identifying the specific roles of each TRP channel, particularly in tumorigenesis, is a key goal for the development of selective anticancer drugs [59].
Although PDAC exhibits extensive genetic heterogeneity, which represents a major obstacle to understanding the biological behavior of this tumor, and taking into account the limitations of the present study (such as the small sample size and the lack of validations in primary cultures), our data could represent a preliminary advance towards the identification of promising new targets for future combined and cancer-specific therapeutic approaches.
From this perspective, FGFR2c, TRPA1 and lipid rafts could be included in the list of new suitable targets, whose validation will require further analyses to be performed on tissue samples and in vivo models.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Material.
References
Katoh M, Loriot Y, Brandi G, Tavolari S, Wainberg ZA, Katoh M. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol. 2024;21:312–29.
Carter EP, Coetzee AS, Tomas Bort E, Wang Q, Kocher HM, Grose RP. Dissecting FGF signalling to target cellular crosstalk in pancreatic cancer. Cells. 2021;10:847.
Orlandi E, Guasconi M, Vecchia S, Trubini S, Giuffrida M, Proietto M, et al. Exploring the horizon: anti-fibroblast growth factor receptor therapy in pancreatic cancer with aberrant fibroblast growth factor receptor expression–a scoping review. Cancers. 2024;16:2912.
Ranieri D, Rosato B, Nanni M, Magenta A, Belleudi F, Torrisi MR. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget. 2016;7:5440.
Ranieri D, Rosato B, Nanni M, Belleudi F, Torrisi MR. Expression of the FGFR2c mesenchymal splicing variant in human keratinocytes inhibits differentiation and promotes invasion. Mol Carcinog. 2018;57:272–28.
Ranieri D, Guttieri L, Raffa S, Torrisi MR, Belleudi F. Role of FGFR2c and its PKCε downstream signaling in the control of EMT and autophagy in pancreatic ductal adenocarcinoma cells. Cancers. 2021;13:4993.
Ranieri D, Nanni M, Persechino F, Torrisi MR, Belleudi F. Role of PKCε in the epithelial-mesenchymal transition induced by FGFR2 isoform switch. Cell Commun Signal. 2020;18:1–13.
Ranieri D, French D, Raffa S, Guttieri L, Torrisi MR, Belleudi F. Expression of the E5 oncoprotein of HPV16 impacts on the molecular profiles of EMT-related and differentiation genes in ectocervical low-grade lesions. Int J Mol Sci. 2021;22:6534.
Mancini V, Raffa S, Fiorio Pla A, French D, Torrisi MR, Ranieri D, et al. TRPA1 contributes to FGFR2c signaling and to its oncogenic outcomes in pancreatic ductal adenocarcinoma-derived cell lines. Cancers. 2024;16:609.
Cucu D. The potential of TRPA1 as a therapeutic target in cancer–a study using bioinformatic tools. Pharmaceuticals. 2024;17:1657.
Berrout J, Kyriakopoulou E, Moparthi L, Hogea AS, Berrout L, Ivan C, et al. TRPA1-FGFR2 binding event is a regulatory oncogenic driver modulated by miRNA-142-3p. Nat Commun. 2017;8:947.
Kyriakopoulou E, Nikita G. FGFR2 and TRPA1 Interaction in Lung Cancer. FASEB J. 2019;33:lb266–lb266.
Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, et al. Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Cancer Cell. 2018;33:985–1003.e7.
O’Connor D, Finn SP, Gray SG. The role of TRPA1 in lung cancer. Transl Lung Cancer Res. 2025;14:4604–17.
Ishiwata T, Matsuda Y, Yamamoto T, Uchida E, Korc M, Naito Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am J Pathol. 2012;180:1928–41.
Cojocaru F, Şelescu T, Domocoş D, Măruţescu L, Chiritoiu G, Chelaru NR, et al. Functional expression of the transient receptor potential ankyrin type 1 channel in pancreatic adenocarcinoma cells. Sci Rep. 2021;11:2018.
Ranieri D, French D, Persechino F, Guttieri L, Torrisi MR, Belleudi F. The FGFR2c/PKCε axis controls MCL-1-mediated invasion in pancreatic ductal adenocarcinoma cells: perspectives for innovative target therapies. Biomedicines. 2022;10:1652.
Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18:361–74.
Codini M, Garcia-Gil M, Albi E. Cholesterol and sphingolipid-enriched lipid rafts as therapeutic targets in cancer. Int J Mol Sci. 2021;22:726.
Vona R, Iessi E, Matarrese P. Role of cholesterol and lipid rafts in cancer signaling: a promising therapeutic opportunity? Front Cell Dev Biol. 2021;9:622908.
Duranti C, Iorio J, Manganelli V, Bagni G, Colasurdo R, Lottini T, et al. Targeting the hERG1/β1 integrin complex in lipid rafts potentiates statins anti-cancer activity in pancreatic cancer. Cell Death Discov. 2025;11:39.
Hryniewicz-Jankowska A, Augoff K, Sikorski AF. The role of cholesterol and cholesterol-driven membrane raft domains in prostate cancer. Exp Biol Med. 2019;244:1053–61.
Jiang S, Wang X, Song D, Liu X, Gu Y, Xu Z, et al. Cholesterol induces epithelial-to-mesenchymal transition of prostate cancer cells by suppressing degradation of EGFR through APMAP. Cancer Res. 2019;79:3063–75.
Mollinedo F, Gajate C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: implications in tumor progression and therapy: thematic review series: biology of lipid rafts. J Lipid Res. 2020;61:611–35.
Jin H, Koh M, Lim H, Yong HY, Kim ES, Kim SY, et al. Lipid raft protein flotillin-1 is important for the interaction between SOS1 and H-Ras/K-Ras, leading to Ras activation. Int J Cancer. 2023;152:1933–46.
Gyoten M, Luo Y, Fujiwara-Tani R, Mori S, Ogata R, Kishi S, et al. Lovastatin treatment inducing apoptosis in human pancreatic cancer cells by inhibiting cholesterol rafts in plasma membrane and mitochondria. Int J Mol Sci. 2023;24:16814.
Greenlee JD, Subramanian T, Liu K, King MR. Rafting down the metastatic cascade: the role of lipid rafts in cancer metastasis, cell death, and clinical outcomes. Cancer Res. 2021;81:5–17.
Ding X, Zhang W, Li S, Yang H. The role of cholesterol metabolism in cancer. Am J Cancer Res. 2019;9:219.
DuBroff R, de Lorgeril M. Cholesterol confusion and statin controversy. World J Cardiol. 2015;7:404.
Dufour, Guenou C, Kaabeche H, Bouvard K, Sanjay D, Marie A, et al. FGFR2-Cbl interaction in lipid rafts triggers attenuation of PI3K/Akt signaling and osteoblast survival. Bone. 2008;42:1032–9.
Bryant MR, Marta CB, Kim FS, Bansal R. Phosphorylation and lipid raft association of fibroblast growth factor receptor-2 in oligodendrocytes. Glia. 2009;57:935–46.
Horváth Á, Payrits M, Steib A, Kántás B, Biró-Süt T, Erostyák J, et al. Analgesic effects of lipid raft disruption by sphingomyelinase and myriocin via transient receptor potential vanilloid 1 and transient receptor potential ankyrin 1 ion channel modulation. Front Pharmacol. 2021;11:593319.
Bobkov D, Semenova S. Impact of lipid rafts on transient receptor potential channel activities. J Cell Physiol. 2022;237:2034–44.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63.
Skibbens JE, Roth MG, Matlin KS. Differential extractability of influenza virus hemagglutinin during intracellular transport in polarized epithelial cells and nonpolar fibroblasts. J Cell Biol. 1989;108:821–32.
Manganelli V, Recalchi S, Capozzi A, Riitano G, Mattei V, Longo A, et al. Autophagy induces protein carbamylation in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Rheumatology. 2018;57:2032–41.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134.
Ueda J, Matsuda Y, Yamahatsu K, Uchida E, Nait Z, Korc M, et al. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene. 2014;33:4485–95.
Kenworthy AK, Schmieder SS, Raghunathan K, Tiwari A, Wang T, Kelly CV, et al. Cholera toxin as a probe for membrane biology. Toxins. 2021;13:543.
Galbiati F, Razani B, Lisanti MP. Emerging themes in lipid rafts and caveolae. Cell. 2001;106:403–11.
Garofalo T, Ferri A, Sorice M, Azmoon P, Grasso M, Mattei V, et al. Neuroglobin overexpression plays a pivotal role in neuroprotection through mitochondrial raft-like microdomains in neuroblastoma SK-N-BE2 cells. Mol Cell Neurosci. 2018;88:167–76.
Young AI, Timpson P, Gallego-Ortega D, Ormandy CJ, Oakes SR. Myeloid cell leukemia 1 (MCL-1), an unexpected modulator of protein kinase signaling during invasion. Cell Adh Migr. 2018;12:513–23.
Ortiz MA, Mikhailova T, Li X, Porter BA, Bah A, Kotula L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun Signal. 2021;19:67.
Castillo L, Young AIJ, Mawson A, Schafranek P, Steinmann AM, Nessem D, et al. MCL-1 antagonism enhances the anti-invasive effects of dasatinib in pancreatic adenocarcinoma. Oncogene. 2020;39:1821–9.
Kajiwara K, Chen PK, Abe Y, Okuda S, Kon S, Adachi J, et al. Src activation in lipid rafts confers epithelial cells with invasive potential to escape from apical extrusion during cell competition. Curr Biol. 2022;32:3460–76.
Khalili-Tanha G, Radisky ES, Radisky DC, Shoari A. Matrix metalloproteinase-driven epithelial-mesenchymal transition: implications in health and disease. J Transl Med. 2025;23:436.
Joshi VB, Gutierrez Ruiz OL, Razidlo GL. The cell biology of metastatic invasion in pancreatic cancer: updates and mechanistic insights. Cancers. 2023;15:2169.
Slapak EJ, Duitman J, Tekin C, Bijlsma MF, Spek CA. Matrix metalloproteases in pancreatic ductal adenocarcinoma: key drivers of disease progression? Biology. 2020;9:80.
Barbat C, Trucy M, Sorice M, Garofalo T, Manganelli V, Fischer A, et al. p56lck, LFA-1 and PI3K but not SHP-2 interact with GM1-or GM3-enriched microdomains in a CD4–p56lck association-dependent manner. Biochem J. 2007;402:471–81.
Ruzzi F, Cappello C, Semprini MS, Scalambra L, Angelicola S, et al. Lipid rafts, caveolae, and epidermal growth factor receptor family: friends or foes? Cell Cell Commun Signal. 2024;22:489.
Huang L, Chen S, Fan H, Ji D, Chen C, Sheng W. GINS2 promotes EMT in pancreatic cancer via specifically stimulating ERK/MAPK signaling. Cancer Gene Ther. 2021;28:839–49.
Safa AR. Epithelial-mesenchymal transition: a hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance. J Cancer Metastasis Treat. 2020;6:36.
Liang W, Hao Z, Han JL, Zhu DJ, Jin ZF & Xie WL. CAV-1 contributes to bladder cancer progression by inducing epithelial-to-mesenchymal transition. Urol Oncol. 2014;32:855–63.
Li Y, Shan F, Chen J. Lipid raft-mediated miR-3908 inhibition of migration of breast cancer cell line MCF-7 by regulating the interactions between AdipoR1 and Flotillin-1. World J Surg Oncol. 2017;15:1–10.
Huang Y, Guo Y, Xu Y, Liu F, Dai S. Flotillin-1 promotes EMT of gastric cancer via stabilizing Snail. PeerJ. 2022;10:e13901.
Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014;1838:532–45.
Startek JB, Talavera K. Lipid raft destabilization impairs mouse TRPA1 responses to cold and bacterial lipopolysaccharides. Int J Mol Sci. 2020;21:3826.
Mollinedo F, Janssen H, de la Iglesia-Vicente J, Villa-Pulgarin JA, Calafat J. Selective fusion of azurophilic granules with Leishmania-containing phagosomes in human neutrophils. J Biol Chem. 2010;285:34528–36.
Marini M, Titiz M, Souza Monteiro de Araújo D, Geppetti P, Nassini R, De Logu F. TRP channels in cancer: signaling mechanisms and translational approaches. Biomolecules. 2023;13:1557.
Author information
Authors and Affiliations
Contributions
Study design: FB and DR; experimental work and data analysis: VM, SR, V Mang, TG and DS. Manuscript writing: FB, VM, and DR. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Ethical approval and informed consent were not necessary. All experiments were performed exclusively using commercially available immortalized cell lines.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Stephen Tait
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mancini, V., Manganelli, V., Garofalo, T. et al. Role of lipid rafts in the FGFR2c-mediated oncogenic signaling by involvement of TRPA1 channel in pancreatic ductal adenocarcinoma cells. Cell Death Dis 17, 259 (2026). https://doi.org/10.1038/s41419-026-08513-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-026-08513-7
