
Abstract
Endoplasmic reticulum stress (ERS) dynamically regulates cell fate decisions within the tumor microenvironment (TME) through the PERK, IRE1α, and ATF6 pathways of the unfolded protein response (UPR), forming an “ERS-Death Axis” interconnected with apoptosis, autophagy, pyroptosis, and ferroptosis. Its molecular network involves CHOP-mediated apoptotic imbalance, NLRP3 inflammasome-activated pyroptosis, the ATF4–CHAC1 axis-driven ferroptosis, and the dual roles of autophagy (protective or pro-death). Oxidative stress further amplifies the biological functions of this network. The ERS-Death Axis exhibits significant heterogeneity across different tumors. Therapeutic strategies targeting this axis have demonstrated clear potential, including specific modulation of core UPR molecules, pathway activation by natural compounds, synergistic combinations with immune checkpoint inhibitors and metabolic interventions, and enhanced targeting and efficacy through nanodelivery systems. However, clinical translation faces key challenges such as tumor heterogeneity, drug delivery efficiency, and complex resistance mechanisms. In-depth elucidation of the tumor-specific mechanisms underlying the ERS-Death Axis will provide crucial theoretical support for overcoming bottlenecks in cancer therapy and optimizing combination treatment regimens, propelling this axis to become a core target for precision oncology.
Similar content being viewed by others

Facts
-
ERS regulates cell death modalities such as apoptosis and autophagy through the UPR pathway (PERK, IRE1α, ATF6), forming an “ERS–Death Axis” that is closely associated with the tumor microenvironment.
-
Hypoxia, nutrient deprivation, and oxidative stress are major drivers of ERS in the tumor microenvironment, influencing tumor progression and therapy resistance.
-
The ERS–Death Axis exhibits specificity across different tumors (e.g., breast cancer, colorectal cancer, liver cancer), involving distinct molecular mechanisms and regulatory pathways.
-
Therapeutic strategies targeting core UPR pathways and downstream molecules (e.g., CHOP, GRP78) show anti-tumor potential in preclinical studies, and combination therapies (e.g., with immunotherapy or metabolic intervention) can enhance efficacy.
Open questions
-
How can personalized treatment standards be developed based on tumor subtype-specific features of the ERS–Death Axis?
-
How can the activation intensity of ERS be precisely regulated to balance antitumor effects and toxicity to normal tissues?
-
How can ERS-mediated drug resistance mechanisms (e.g., exosome-mediated escape) be effectively blocked?
Introduction
The endoplasmic reticulum (ER), a central organelle responsible for protein folding and calcium homeostasis, undergoes functional disruption that triggers endoplasmic reticulum stress (ERS)—a prevalent stress response in the tumor microenvironment (TME) [1, 2]. Under TME pressures such as hypoxia, nutrient deprivation, and oxidative stress, tumor cells accumulate excessive unfolded or misfolded proteins in the ER lumen, thereby activating the unfolded protein response (UPR). This response initiates adaptive regulation through three core pathways—PERK, IRE1α, and ATF6—to sustain cell survival [3]. However, when ERS persists or becomes overactivated, it surpasses cellular adaptive thresholds and subsequently modulates various forms of programmed cell death (PCD), including apoptosis, autophagy, pyroptosis, and ferroptosis, thereby contributing to tumorigenesis, therapy resistance, and immune microenvironment remodeling [4, 5].
The crosstalk network between ERS and PCD (termed the “ERS-Death Axis”) exhibits high complexity. On one hand, ERS can induce apoptosis via CHOP-mediated Bcl-2 family imbalance and JNK pathway activation [6,7,8,9], trigger pyroptosis through NLRP3 inflammasome activation [10], or promote ferroptosis via the ATF4–CHAC1 axis [11, 12]. On the other hand, ERS-induced autophagy may either exert a protective role by clearing misfolded proteins or switch to a pro-death effect under lysosomal dysfunction [13, 14]. This bidirectional regulation is influenced by tumor type, microenvironmental conditions, and genetic background, as evidenced by the heterogeneous ERS responses across different breast cancer subtypes [15, 16].
A comprehensive understanding of the molecular mechanisms underlying the ERS-Death Axis, along with the identification of its tumor-specific regulatory patterns, is crucial for developing precision therapeutic strategies targeting ERS. This review systematically examines the interplay between ERS and various PCD pathways, the amplifying role of oxidative stress, and immune microenvironment modulation, while summarizing the distinct features of the ERS-Death Axis across tumor types. Furthermore, we discuss its clinical translation potential and challenges, aiming to provide novel theoretical insights and intervention targets for cancer therapy.
Driving factors of ERS in the TME
The drivers of ERS in the TME constitute a complex pathophysiological process involving the synergistic effects of multiple microenvironmental stressors. The rapid proliferation of tumor tissue leads to significant alterations in the local microenvironment, including hypoxia, nutrient deprivation, oxidative stress, and acidosis—harsh conditions that collectively serve as primary inducers of ERS [5] (Fig. 1). Hypoxia, a hallmark feature of the TME, not only directly disrupts ER protein folding by inhibiting disulfide bond formation but also upregulates the expression of redox enzymes such as ERO1α through HIF-1α activation, thereby influencing tumor angiogenesis and immune evasion [17, 18]. This hypoxia-induced ERS plays a pivotal role in tumor progression and therapy resistance, particularly in modulating chemosensitivity and immunotherapy response [19, 20].

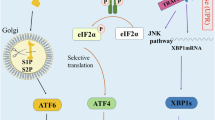
Multiple stress conditions within the TME lead to the accumulation of unfolded/misfolded proteins in the endoplasmic reticulum (ER), triggering ER stress (ERS). This activates three key UPR sensors located on the ER membrane: PERK, IRE1α, and ATF6. The PERK–eIF2α–ATF4 axis primarily induces the expression of the transcription factor CHOP (C/EBP homologous protein), which serves as a central mediator of apoptosis and pyroptosis while also modulating genes involved in ferroptosis. Simultaneously, the IRE1α–XBP1s axis activates adaptive responses through metabolic reprogramming, such as enhanced lipogenesis, and protective autophagy to alleviate cellular stress. The ATF6 pathway contributes to cellular adaptation by upregulating ER chaperone proteins including GRP78 and participates in CHOP-mediated apoptotic signaling. Integration of these signaling cascades, particularly through the molecular hub CHOP, determines the ultimate cellular fate: promoting survival under mild or transient stress conditions, or driving cell death through diverse programmed cell death modalities under severe or prolonged stress. CHOP thus functions as a critical molecular switch balancing pro-survival and pro-death outcomes in response to proteotoxic stress. Created with BioRender.com.
Nutrient deprivation, another critical characteristic of the TME, triggers ERS through multiple mechanisms [5]. The high demand for nutrients by rapidly proliferating tumor cells leads to deficiencies in essential metabolites such as glucose and amino acids, activating the IRE1α/XBP1 signaling pathway to promote lipid synthesis and ER expansion. This metabolic reprogramming not only sustains tumor cell survival but also provides alternative energy sources by regulating autophagy [21,22,23]. Notably, nutrient deprivation-induced ERS also influences the tumor immune microenvironment, transmitting regulatory signals via exosomes and other carriers to suppress T-cell function and promote the polarization of immunosuppressive M2-type macrophages [24].
External therapeutic pressures, including chemotherapy and radiotherapy, are also significant contributors to ERS in tumor cells [25]. Certain targeted therapies, such as immune checkpoint inhibitors (ICIs), may enhance tumor cell immune evasion by upregulating ERS markers like GRP78 [23], highlighting the dual role of ERS in therapy resistance. Therefore, intervention strategies targeting ERS pathways—such as combining ERS inducers with immunotherapies—may represent a novel approach to overcoming treatment resistance.
Core mechanisms of ERS-mediated tumor regulatory networks
The interactive network between ERS and programmed cell death
The ERS-apoptosis axis
Apoptosis, as a crucial form of PCD, plays a central role in maintaining tissue homeostasis and organismal development [26, 27]. This highly conserved physiological process is activated through two major pathways—the intrinsic (mitochondrial) and extrinsic (death receptor) pathways—and is characterized by cellular shrinkage, chromatin condensation, DNA fragmentation, and apoptotic body formation [28,29,30]. In tumor biology, dysregulation of apoptotic pathways has been widely recognized as a hallmark of cancer, with its abnormalities not only promoting tumorigenesis and progression but also closely associated with chemotherapy resistance [31].
The crosstalk between ERS and apoptosis constitutes a critical molecular network in the TME. At the core of this network lies the UPR system, which senses and responds to proteostatic imbalance in the ER through three primary pathways (IRE1, PERK, and ATF6), exhibiting highly pathway-specific regulatory mechanisms (Fig. 2) [32, 33]. Adaptive ERS helps modulate protein synthesis to maintain cellular homeostasis [34]. However, under the sustained ERS conditions of the TME, the UPR system shifts from a pro-survival to a pro-apoptotic mode, a transition involving a cascade of key molecular events [35, 36]. Notably, the expression level of CHOP (C/EBP homologous protein, also known as GADD153) is frequently used as a critical biomarker for assessing ERS-induced apoptosis [37,38,39]. The upregulation of the CHOP transcription factor represents a central event in this process, initiating the caspase cascade by regulating the balance of the Bcl-2 family (promoting BIM/PUMA while suppressing Bcl-2), ultimately triggering apoptosis [6].

ERS triggers the molecular cascade of apoptosis through the PERK, IRE1, and ATF6 pathways. Under ERS conditions, the IRE1 pathway is activated through the kinase domain of IRE1, which recruits and binds TRAF2 to initiate the ASK1-JNK/p38 MAPK signaling cascade. This leads to phosphorylation of Bcl-2 (inhibiting its anti-apoptotic function) and activation of the pro-apoptotic protein Bim. Simultaneously, IRE1 degrades survival factor mRNAs via the RIDD mechanism, collectively promoting apoptosis. In the PERK pathway, ERS induces PERK-mediated phosphorylation of eIF2α, enabling selective translation of ATF4. ATF4 then translocates to the nucleus and upregulates CHOP expression, which promotes pro-apoptotic molecules such as Bim and PUMA while suppressing Bcl-2, ultimately initiating the caspase cascade. The ATF6 pathway involves its proteolytic cleavage (generating ATF6-50), which regulates CHOP and death receptor DR5 (TRAIL Receptor-2) expression, thereby activating the caspase-8-dependent extrinsic apoptotic pathway. These three pathways are not independent but exhibit crosstalk and synergistic regulation through key nodes such as CHOP and apoptosis-related proteins, collectively determining the cellular fate between survival and apoptosis under ERS conditions. ER endoplasmic reticulum; PERK protein kinase RNA-like endoplasmic reticulum kinase, IRE1 inositol requiring enzyme 1, AFT6 transcription factor 6. Created with BioRender.com.
In recent years, ERS-induced apoptotic pathways have demonstrated significant clinical potential in cancer therapy. Multiple studies have shown that targeted modulation of ERS-related pathways can effectively induce tumor cell apoptosis while overcoming resistance to conventional treatments. The Fujian Wan team discovered that knockout of the YTHDF2 gene enhances ERS through the GLI2–JNK pathway, significantly suppressing stemness traits and promoting apoptosis in cervical cancer cells, offering a novel therapeutic target [40]. Diterpenoid tanshinone exhibits potent antitumor effects in lung adenocarcinoma models via the IRE1α/caspase12 pathway [41]. Particularly noteworthy is the synergistic combination of 2-Deoxyglucose and hydroxychloroquine, which induces apoptosis in triple-negative breast cancer through the PERK–ATF4–CHOP axis while simultaneously inhibiting protective autophagy [42]. In drug delivery systems, ER-targeted nanoparticles encapsulating siGRP94 have been developed for hepatocellular carcinoma (HCC), triggering ERS cascades and apoptosis through forced calcium influx, achieving tumor-specific therapy [43].
The ERS-autophagy axis
The interplay between ERS and autophagy constitutes a sophisticated regulatory network that plays a dual role in tumorigenesis and progression. As a crucial homeostatic mechanism, autophagy is markedly activated under ERS conditions, forming a dynamic ERS-autophagy regulatory axis [44]. Similar to the ERS-apoptosis axis, this process is primarily regulated through the three canonical UPR pathways (Fig. 3), yet exhibits distinct molecular signatures and biological consequences [45]. During the initial phase of ERS, acute stress predominantly triggers protective autophagy, facilitating the clearance of misfolded proteins and damaged organelles to maintain cellular viability [46]. However, under persistent ERS conditions, chronic stress leads to impaired autophagic flux and lysosomal dysfunction, potentially switching autophagy toward a pro-death function [13, 14]. This precisely orchestrated network enables cells to mount context-dependent responses based on stress intensity and duration, ultimately determining cellular fate [47].

The IRE1α pathway plays a central regulatory role in the molecular mechanism of the ERS-autophagy axis through its dual functionality. On one hand, its kinase domain activates the JNK signaling pathway, promoting the dissociation of the Bcl-2/Beclin1 complex and initiating autophagosome formation. On the other hand, its nuclease activity selectively degrades mRNAs encoding mitochondrial quality control proteins (such as PINK1) via the RIDD mechanism, thereby regulating selective autophagy. In the PERK-eIF2α-ATF4 pathway, ERS activates PERK, which phosphorylates eIF2α to inhibit global protein synthesis while selectively translating ATF4. As a transcription factor, ATF4 regulates the expression of a series of autophagy-related genes. For instance, it induces CHOP expression, which further promotes the transcription of autophagy-related genes. Additionally, ATF4 directly binds to the promoter regions of autophagy-related genes, enhancing the expression of key autophagy molecules such as Beclin1, ATG5, ATG12, and LC3, thereby inducing autophagy. In the ATF6 pathway, upon ERS activation, ATF6 translocates from the ER to the Golgi apparatus, where it is cleaved and activated. The released cytosolic domain then enters the nucleus to regulate autophagy-related genes. Furthermore, ERS disrupts ER calcium homeostasis, leading to the release of calcium ions into the cytoplasm. This activates calcium-dependent autophagy pathways via PKC and CaMKII signaling. Created with BioRender.com.
The UPR-mediated activation of autophagy has been extensively validated across various tumor models. In HepG2 hepatoblastoma cells, hydrogen peroxide-induced oxidative damage is potentiated by the ERS inducer tunicamycin, accompanied by increased LC3II/LC3-I ratio and p62 degradation - effects that are reversed by the ERS inhibitor salubrinal, unequivocally demonstrating direct ERS-autophagy crosstalk [48]. Colorectal cancer (CRC) studies reveal that excessive proliferation-induced ERS activates protective autophagy through IRE1α and PERK pathways, promoting tumor cell survival and drug resistance [49]. These findings not only elucidate resistance mechanisms but also inform novel combination therapies. Therapeutic modulation of the ERS-autophagy axis has shown promising antitumor efficacy. Sequential treatment strategies - initiating UPR with ERS inducers followed by autophagy inhibitors - demonstrate remarkable synergy in refractory cancers like triple-negative breast cancer [42]. Notably, carbon monoxide-induced moderate ERS activates protective autophagy in T cells, enhancing their antitumor functionality and suggesting novel immunotherapeutic approaches [50]. Furthermore, prognostic models incorporating ERS and mitophagy-related genes exhibit robust predictive value in lung adenocarcinoma, highlighting their clinical potential [51].
The ERS-autophagy interaction displays remarkable tissue specificity. In ovarian cancer, ERS suppresses PI3K/AKT/mTOR signaling to concurrently promote autophagy and apoptosis, increasing chemosensitivity [52]; whereas in photodynamic therapy for cholangiocarcinoma, ERS-induced autophagy initially exerts cytoprotection but transitions to pro-death effects upon lysosomal damage [53]. Such context-dependence underscores the need for personalized therapeutic approaches. Mechanistically, emerging epigenetic regulators like the LINC00963/miR-320a/XBP1 axis [54] and ZNF263-mediated super-enhancer activation [55] provide novel molecular insights into tumor heterogeneity and targeted intervention.
The ERS-pyroptosis axis
Pyroptosis, as a novel form of programmed inflammatory cell death, plays a critical role in infections, inflammatory diseases, and tumor immunity [56, 57]. It is characterized by the formation of pores in the cell membrane, leading to the release of cellular contents and triggering strong inflammatory responses. This process is mediated by key effector proteins of the Gasdermin family, particularly Gasdermin D (GSDMD) and Gasdermin E (GSDME) [58,59,60]. Within the TME, the dual regulatory role of pyroptosis has attracted increasing attention, as it can not only activate anti-tumor immunity by releasing damage-associated molecular patterns (DAMPs) but may also promote inflammation-associated tumor progression [61, 62]. The molecular coupling between ERS and pyroptosis forms a complex regulatory network, with its core mechanism lying in the interaction between the UPR and NLRP3 inflammasome activation [10] (Fig. 4).

In tumors, the ERS-pyroptosis axis involves three core signaling pathways that interconnect ERS and pyroptosis. Activated IRE1α initiates the downstream JNK signaling pathway through its kinase domain, leading to phosphorylation and activation of transcription factors that regulate pyroptosis-related gene expression. Concurrently, its ribonuclease activity modulates mRNA stability via the RIDD pathway, altering the intracellular protein profile and further contributing to pyroptosis regulation. ERS also activates PERK, which phosphorylates eIF2α to selectively enhance ATF4 translation. Upregulated ATF4 increases CHOP expression, which in turn promotes pyroptosis by facilitating the interaction between TXNIP and NLRP3, thereby activating the NLRP3 inflammasome. Additionally, CHOP impairs mitochondrial function, causing mitochondrial damage and the release of mitochondrial components such as mitochondrial DNA, which further amplifies NLRP3 inflammasome activation and drives pyroptosis. The ATF6 pathway modulates pyroptosis indirectly by downregulating P2×7R/NLRP3 or activating sFRP2/NF-κB signaling. Proteolytic cleavage of ATF6 can also trigger caspase-4-dependent NLRP3 inflammasome activation. These collective mechanisms ultimately lead to cleavage of GSDMD and maturation and release of IL-1β/IL-18, resulting in pyroptotic cell death. Created with BioRender.com.
The ERS-pyroptosis axis exhibits dual effects in the TME. On one hand, it can induce immunogenic cell death (ICD) in tumor cells, releasing DAMPs such as ATP, HMGB1, and calreticulin, which promote dendritic cell (DC) maturation and T cell activation, thereby enhancing anti-tumor immune responses [56]. For example, the metabolite trimethylamine N-oxide induces tumor cell pyroptosis by activating the PERK pathway, improving the response to immunotherapy in triple-negative breast cancer [63]. Nanoparticles such as CS-HAP@KAE can convert “cold” tumors into “hot” tumors through the ERS-pyroptosis axis, enhancing the efficacy of ICIs [64]. On the other hand, excessive activation of the ERS-pyroptosis axis may exacerbate tumor-associated inflammation and immune suppression. For instance, in renal cell carcinoma, STING inhibits ERS-mediated pyroptosis to promote immune evasion, while targeted degradation of STING restores PERK/CHOP-dependent pyroptosis and enhances anti-tumor immunity [65]. Additionally, the ERS-pyroptosis axis is closely associated with metabolic reprogramming, such as glucose metabolism disorders and TXNIP/NLRP3 pathway activation, influencing tumor progression [66, 67]. For example, the ZDHHC1 gene suppresses tumor growth by inducing ERS-pyroptosis, while ginsenosides and flavonoids inhibit pyroptosis in diabetes-associated tumors by modulating the ERS-TXNIP/NLRP3 axis [68].
In terms of therapeutic strategies, targeting the ERS-pyroptosis axis has emerged as a promising direction. For instance, baicalin reduces pyroptosis in Mycobacterium tuberculosis-infected macrophages by inhibiting the PERK/TXNIP/NLRP3 pathway [69]. Nanoplatforms such as PCAN enhance anti-tumor immunity by specifically inducing ERS-dependent type II ICD and pyroptosis [70].
ERS-ferroptosis axis
Ferroptosis is a unique mode of cell death driven by iron-dependent phospholipid peroxidation, involving dysregulated iron metabolism, imbalance in the lipid antioxidant system, and accumulation of lipid peroxides [71]. The high metabolic demands and hypoxic microenvironment created by the malignant proliferation of tumor cells can lead to the accumulation of misfolded or unfolded proteins in the ER, thereby triggering ERS [71]. These two processes interact through core signaling pathways such as PERK-eIF2α-ATF4, IRE1α, and ATF6 (Fig. 5).

The PERK-eIF2α-ATF4 signaling axis plays a critical role in ERS-mediated ferroptosis. Activation of PERK leads to phosphorylation of eIF2α, which promotes the translation of ATF4. This in turn upregulates pro-ferroptotic genes such as CHOP and CHAC1. CHAC1 degrades glutathione, thereby weakening the cellular antioxidant capacity, while ATF4 may also modulate HO-1 to increase the labile iron pool, promoting the Fenton reaction and lipid peroxidation. The IRE1α-XBP1 pathway exhibits a dual role in ferroptosis regulation. On one hand, the RNase activity of IRE1α can cleave and downregulate genes involved in glutathione synthesis (e.g., SLC7A11), reducing antioxidant defense and facilitating ferroptosis. On the other hand, IRE1α-XBP1 signaling may indirectly suppress ferroptosis by maintaining endoplasmic reticulum homeostasis. The role of the ATF6 pathway in ferroptosis is relatively less studied, but emerging evidence suggests that ATF6 can transcriptionally regulate TRIM37 to influence ferroptosis. In cervical cancer, ATF6 activation induces TRIM37 expression, which ubiquitinates and degrades ACSL4—a key enzyme promoting lipid peroxidation—thereby inhibiting ferroptosis. Furthermore, the crosstalk between ERS and iron metabolism represents another crucial mechanism in ferroptosis regulation. ERS can promote intracellular iron accumulation through calcium signaling (e.g., the SLC12A5-PNCK pathway) or via upregulation of transferrin. Created with BioRender.com.
The ERS-ferroptosis axis exhibits a dual role in tumors. On one hand, it provides new therapeutic targets. Tanshinone IIA can exert anti-tumor effects by downregulating the PERK–ATF4–HSPA5 pathway-mediated activation of ferroptosis [72]. The irreversible proteasome inhibitor carfilzomib, combined with 125I brachytherapy, synergistically induces apoptosis, necroptosis, and ferroptosis by enhancing ERS and the UPR, producing significant anti-tumor effects in esophageal squamous cell carcinoma [73]. Nanodrug delivery systems, such as pH-sensitive nanoplatforms [74] and iron-based metal-organic framework nanocages [75], can achieve tumor-specific therapy by inducing ferroptosis. Combining the regulation of the ERS-ferroptosis axis with ICIs to induce ferroptosis in tumor cells, or the synergistic effect of proteasome inhibitors with ferroptosis signaling suppression, offers new strategies to enhance anti-tumor efficacy [76, 77]. On the other hand, tumor cells may exploit this axis to evade death [78]. In non-small cell lung cancer, GPER1 can promote the activation of PI3K/AKT/mTOR signaling, induce SCD1 expression, and thereby prevent ferroptosis in tumor cells, facilitating tumor progression [79]. The TME components, such as immune cells and extracellular matrix, engage in complex signaling crosstalk with the ERS-ferroptosis axis.
CHOP: a central integrator of ERS-mediated cell death
CHOP belongs to the CCAAT/enhancer-binding protein (C/EBPs) family. It is a 29 kD protein consisting of 169 amino acid residues in humans (168 in rodents). Its structure comprises two functional domains: an N-terminal transcriptional activation domain and a C-terminal basic leucine zipper (bZIP) domain [35]. Under normal physiological conditions, CHOP is ubiquitously expressed at very low levels [80]. However, under persistent ERS conditions, it is significantly transcriptionally upregulated through all three branches of the UPR—PERK–eIF2α–ATF4, IRE1α–XBP1, and ATF6—making it a reliable marker of severe ERS [3].
As a core effector downstream of ERS, CHOP orchestrates multiple forms of PCD by regulating downstream target genes. In apoptosis, CHOP initiates the caspase cascade by altering the balance of Bcl-2 family proteins (promoting BIM/PUMA expression and suppressing Bcl-2) [6,7,8,9]; in autophagy, it modulates autophagic flux through the transcriptional regulation of key autophagy-related genes [44]; in pyroptosis, CHOP interacts with inflammatory pathways such as the NLRP3 inflammasome, participating in the regulation of PERK/CHOP-dependent pyroptosis [10, 65]; in ferroptosis, it promotes iron-dependent cell death by regulating genes involved in redox and iron metabolism (e.g., CHAC1 and SAT1) [11, 12]. Notably, as a member of the C/EBPs family, CHOP itself is involved in regulating genes encoding proteins related to proliferation, differentiation, and energy metabolism, playing an important role in determining cell fate [71].
The central regulatory role of CHOP in these death pathways establishes its function as a molecular “switch” that determines cell survival or death under stress conditions. This multifaceted regulatory capability highlights the important therapeutic significance of CHOP in targeting ERS-driven cell death in cancer. For example, the synergistic combination of 2-deoxyglucose and hydroxychloroquine induces apoptosis in triple-negative breast cancer cells via the PERK–ATF4–CHOP axis [42], while targeted degradation of STING enhances antitumor immunity by restoring PERK/CHOP-dependent pyroptosis [65]. These findings indicate that CHOP is not only a key biomarker of ERS but also a promising target for therapeutic intervention, providing an important theoretical basis for developing novel anticancer strategies.
The amplifier role of oxidative stress
Oxidative stress and ERS exhibit a tightly coordinated “amplification” relationship during tumorigenesis and progression. As a key driver, oxidative stress can exacerbate ERS through multiple pathways, thereby reshaping the biological behavior of tumor cells and the TME. The large amounts of reactive oxygen species (ROS) generated by oxidative stress directly disrupt the redox homeostasis of the ER, leading to protein misfolding and aggregation—a core trigger of ERS. For example, in glioblastoma, glutamine deficiency reduces glutathione levels, significantly elevating ROS and causing the accumulation of unfolded proteins in the ER lumen, which activates ERS sensor proteins such as PERK, IRE1α, and ATF6, initiating the UPR [81]. Meanwhile, the activation of ERS signaling pathways can, in turn, promote ROS generation, forming a vicious cycle. For instance, oridonin-induced ERS in CRC cells disrupts ER calcium homeostasis, releasing calcium ions into the cytoplasm and activating mitochondrial ROS production, further intensifying oxidative stress and driving tumor cells toward death [82].
The “amplification” regulation of ERS-related signaling pathways by oxidative stress profoundly influences tumor cell metabolic reprogramming and survival adaptability. In M2 tumor-associated macrophages, oxidative stress synergistically activates ERS via the IRE1-XBP1 pathway, suppressing glycolysis while promoting oxidative phosphorylation and lipid accumulation, thereby maintaining the immunosuppressive M2 phenotype and fostering a favorable environment for tumor growth [83]. In cancer cells, sustained oxidative stress-ERS conditions drive metabolic shifts that favor tumor proliferation, such as upregulating key glycolytic enzymes to enhance glucose uptake and utilization while inhibiting fatty acid oxidation to ensure biosynthetic precursor supply [84, 85]. Moreover, the mutual amplification of oxidative stress and ERS significantly affects tumor cell sensitivity and resistance to therapy. In chemodynamic therapy, H₂O₂ self-supplying nanocomposites (e.g., (Cu₂SeCa₂) @LA) generate ROS under near-infrared light, triggering ERS and upregulating the PERK-mediated eIF2α phosphorylation pathway to induce ICD, thereby killing tumors [85]. However, tumor cells can also exploit the oxidative stress-ERS interplay to develop resistance—for example, by activating the ER chaperone protein GRP78 to repair misfolded proteins while enhancing the antioxidant defense system to reduce ROS levels and diminish therapeutic efficacy [86]. Additionally, ERS induced by oxidative stress can release DAMPs, such as calreticulin and high-mobility group box 1 (HMGB1), activating dendritic cells and modulating the TME to influence immunotherapy outcomes [87,88,89].
Regulation of the immune microenvironment
The ERS in the TME plays a pivotal role in tumor immune escape by modulating immune cell functions. Research indicates that the UPR induced by ERS exerts dual regulatory effects in both tumor cells and immune cells—sustaining tumor cell survival while also reshaping the immune microenvironment through non-cell-autonomous mechanisms. In tumor cells, activation of key UPR molecules such as the IRE1α/XBP1 and PERK/ATF4 pathways promotes the formation of an immunosuppressive TME. For instance, XBP1 upregulates cholesterol synthesis and secretion, activating myeloid-derived suppressor cells and thereby inhibiting CD8(+)T cell function [76, 90]. Additionally, ERO1A influences immunogenic cell death in tumor cells by regulating the balance between IRE1α and PERK signaling, attenuating the therapeutic efficacy of PD-1 blockade [91]. Clinical data further reveal that lung cancer patients with high ERO1A expression exhibit poor responses to neoadjuvant immunotherapy, suggesting its potential as a biomarker for immunotherapy resistance [91].
ERS also modulates immune checkpoint molecule expression to affect T cell function. For example, metformin activates AMPK to phosphorylate PD-L1 at the S195 site, promoting its degradation via the ER-associated degradation pathway and thereby enhancing cytotoxic T lymphocyte activity [92]. Similarly, ERS-induced ATF4 can deliver the long non-coding RNA SNHG6 through small extracellular vesicles, suppressing T cell-mediated immune responses and promoting M2 macrophage polarization to establish an immunosuppressive TME [93]. In HCC, ERS-related gene signatures significantly correlate with immune cell infiltration and poor prognosis, with low GP6 expression strongly linked to immunotherapy resistance and reduced survival rates [94].
ERS exerts particularly notable effects on myeloid cell function. In glioblastoma, IRE1α recruits monocytes and neutrophils via the UBE2D3/NF-κB axis, fostering a pro-tumor immune microenvironment [95]. Moreover, the PERK pathway upregulates phosphoserine aminotransferase 1 and serine metabolism to enhance the immunosuppressive function of M2 macrophages, while PERK inhibition improves the efficacy of PD-1 blockade [96]. In melanoma models, the PERK inhibitor GSK2656157 combined with an mRNA vaccine promotes M1 macrophage polarization and enhances CD8(+) T cell infiltration, significantly suppressing tumor growth and metastasis [97]. ERS also influences antitumor immune responses by regulating ICD. For instance, the photosensitizer ER-Cy triggers NLRP3 inflammasome activation and pyroptosis via ERS induction, releasing DAMPs to promote dendritic cell maturation and T cell activation [98]. Similarly, nanoparticle-mediated ERS activation enhances the ICD effect of chemotherapeutic drugs by releasing ATP and HMGB1, which activate antigen-presenting cells and reverse the immunosuppressive TME [99].
Characteristics of the ERS-death axis in different tumor types
Breast cancer (BC)
BC exhibits a multi-layered and dynamically evolving interaction between ERS and cell death. At the molecular level, ERS regulates cell fate through three major signaling pathways, with the PERK/eIF2α/ATF4/CHOP pathway playing a pivotal role in determining cell survival or death [100]. Notably, different BC subtypes display significant heterogeneity in ERS responses: in estrogen receptor-positive (ER+) breast cancer, moderate ERS activation promotes cellular adaptation, whereas sustained activation induces apoptosis through mitochondria-associated endoplasmic reticulum membrane-mediated calcium signaling and ROS production [15, 16]. In contrast, triple-negative breast cancer, which exhibits higher basal ERS levels, is more sensitive to ERS inducers but also develops resistance mechanisms through anti-apoptotic proteins such as FLIP [101].
At the TME level, stress factors such as hypoxia, acidosis, and nutrient deprivation persistently activate the UPR, while ERS, in turn, remodels the TME [102, 103]. This bidirectional regulation manifests in two ways: on one hand, ERS induces ICD through calreticulin exposure, HMGB1 release, and ATP secretion, enhancing anti-tumor immunity [104]; on the other hand, it promotes immune suppression via mechanisms such as PD-L1 glycosylation [103]. Particularly noteworthy is the discovery that ERS-induced exosomal miR-27a-3p upregulates macrophage PD-L1 expression through the MAGI2/PTEN/PI3K axis [103], providing a novel perspective for understanding tumor immune escape. In terms of therapeutic translation, ERS-targeting strategies can be broadly categorized into two approaches: first, direct modulation of core ERS molecules, such as combining the PERK agonist CCT020312 with taxanes to overcome drug resistance [105, 106]; second, the use of natural compounds (e.g., γ- and δ-tocotrienols [107], oleandrin [104]) to specifically activate the ERS-death axis.
Colorectal cancer (CRC)
The interaction between ERS and cell death in CRC exhibits complex and dynamic regulatory characteristics. Studies have shown that the ferroptosis inducer RSL3 significantly activates all three major pathways of the UPR, with the PERK pathway negatively regulating ferroptosis by modulating ATF4’s binding ability to the SLC7A11 promoter [108]. This discovery highlights the critical role of ERS pathways in regulating ferroptosis sensitivity, offering a novel approach to overcoming apoptosis resistance.
In terms of the interplay between ERS and the immune microenvironment, research has revealed that ERS-induced ICD exhibits dual regulatory properties. Macrocarpal I, by activating the PERK/eIF2α/ATF4/CHOP pathway, not only triggers classic ICD markers such as calreticulin exposure and HMGB1 release but also induces ferroptosis. This dual-death mechanism significantly enhances the immune response to anti-PD-1 therapy [109]. However, sustained ERS may also promote immune escape through exosomal miR-27a-mediated PD-L1 upregulation [110], underscoring the importance of temporally controlled therapeutic strategies.
Various ERS-targeting treatment approaches have shown promising translational potential. Natural compounds such as curcumin and gambogenic acid induce ERS-dependent apoptosis by specifically activating the ATF6 and IRE1α pathways [111, 112], while α-hederin causes irreversible proteostasis collapse by simultaneously blocking ERAD and autophagic flux [113]. Notably, cetuximab-resistant cells exhibit sensitivity to the proteasome inhibitor carfilzomib [114], revealing a new strategy for overcoming targeted therapy resistance through ERS modulation. Additionally, the SPARC protein competitively binds to GRP78, relieving its anti-apoptotic effects and providing a novel approach to restoring chemotherapy sensitivity [115].
Hepatocellular carcinoma (HCC)
In HCC, the regulation of the ERS-death axis primarily revolves around the three core UPR pathways, influencing tumor progression and therapeutic response through apoptosis, autophagy, and specialized forms of cell death. Various natural compounds and drugs can induce HCC cell death by activating ERS pathways. For instance, Icaritin triggers ERS and mitochondrial dysfunction by targeting BHLHE40, leading to caspase-independent cytoplasmic vacuolization [116]. Fisetin induces intracellular Ca²⁺ release, activating the PERK–ATF4–CHOP pathway while promoting GRP78 exosome secretion. It also enhances ERS-mediated apoptosis in radiation-resistant HCC cells and suppresses radiation-induced epithelial-mesenchymal transition to overcome radioresistance [117]. Compounds like (-)-agelasidine A and prodigiosin upregulate UPR components (PERK, IRE1α) and pro-apoptotic proteins (CHOP, Bax, caspases) while downregulating anti-apoptotic Bcl-2, synergistically activating intrinsic and extrinsic apoptotic pathways—effects reversible by the ERS inhibitor 4-PBA [94, 118]. Other agents, including Annona muricata L and Xanthatin, induce ERS via the PERK-eIF2α-ATF4-CHOP axis, with Xanthatin specifically promoting ATF4 nuclear translocation to amplify CHOP-dependent apoptosis, an effect abolished by CHOP knockdown [119, 120].
Under therapeutic pressure, the interplay between ERS and autophagy becomes a critical determinant of resistance. Sorafenib induces ERS but concurrently activates protective autophagy via the PERK–ATF4–Beclin1 pathway to counteract apoptosis, while low-dose melatonin inhibits this axis to restore sorafenib sensitivity [121]. In 5-fluorouracil (5-FU)-resistant cells, konjac glucomannan reverses resistance by downregulating TLR4 to reactivate the PERK/ATF4/CHOP pathway [122]. The ATP citrate lyase inhibitor BMS-303141 triggers the p-eIF2α/ATF4/CHOP axis, synergizing with sorafenib to suppress tumor growth [123], whereas 125I brachytherapy combined with lobaplatin amplifies apoptosis via the same pathway [124]. Additionally, ubiquitin-specific peptidase 18 attenuates ERS-mediated apoptosis by inhibiting PERK phosphorylation and CHOP expression [125], whereas total flavonoids of Oldenlandia diffusa simultaneously induce apoptosis and autophagy via PERK-eIF2α-ATF4 activation [126].
ERS-related molecules also govern HCC metastasis and microenvironment remodeling. The histidine-rich calcium-binding protein enhances ERS adaptation through the PERK-eIF2α-ATF4-CHOP axis, fostering anoikis resistance and metastasis, with its expression correlating with tumor size and TNM stage [127]. ERS-associated gene signatures link to increased immune cell infiltration in HCC, where PPP1R16A may remodel the TME via the MIF/CD74+CXCR4 signaling axis [128]. Melatonin selectively blocks ATF6, suppressing COX-2 to augment ERS-induced apoptosis, while the lncRNA RMRP regulates PERK-mediated apoptosis, with its knockdown enhancing cell death [121].
Glioblastoma (GBM)
Therapeutic resistance in GBM is closely associated with dysregulation of ERS and the UPR. Research demonstrates that various natural compounds and targeted agents induce GBM cell death while overcoming treatment resistance by modulating ERS/UPR pathways. Sulforaphane selectively triggers apoptosis in GBM cells through activation of the ATF4–CHOP axis [129], whereas the proteasome inhibitor marizomib upregulates ERS markers such as GRP78, IRE1α, and CHOP to induce caspase-3-dependent apoptosis, independent of ROS and autophagy [130]. Notably, remdesivir exerts antitumor effects via PERK-mediated UPR, demonstrating superior efficacy to temozolomide (TMZ) while maintaining a favorable safety profile [131].
The dynamic regulation of ERS pathways plays a dual role in GBM treatment resistance. TMZ-resistant cells exhibit suppressed ERS and enhanced proteasomal activity, with PSMC2 overexpression inhibiting pro-death autophagy via the JNK-Bcl-2-Beclin1 pathway—an effect reversible by PSMC2 targeting [132]. Additionally, therapy-induced senescent GBM cells rely on the PERK-ATF4-CHOP axis for survival, and PERK inhibition promotes apoptosis while delaying recurrence [133]. Integrin α3 silencing disrupts β1 subunit maturation, leading to immature β1 accumulation and ERS-dependent DR5 upregulation, resensitizing resistant cells to TRAIL [134]. Combination strategies targeting key ERS nodes show significant therapeutic potential. The UBA1 inhibitor TAK-243 induces irreversible UPR by disrupting protein ubiquitination, with efficacy correlating with GRP78 expression, while the GRP78 inhibitor HA15 synergistically enhances its effects [135]. The PDI inhibitor CCF642 amplifies TMZ efficacy through irreversible UPR, and its nanoformulation markedly suppresses tumor growth in vivo [136]. Furthermore, metabolic interventions such as nutrient deprivation effectively eliminate glioma stem cells via ERS-mitochondrial coupling pathways, highlighting the therapeutic value of microenvironment modulation [134].
Lung cancer (LC)
The progression and therapeutic resistance of lung cancer are closely linked to dysregulated ERS within the TME. Studies reveal that ERO1A shapes an immunosuppressive TME by balancing IRE1α and PERK signaling, and its ablation enhances PD-1 blockade efficacy while promoting ICD [91]. Similarly, Derlin-3 drives M2 macrophage polarization via the Hrd1/p38/PRDM1 pathway, fostering immune suppression [137]. Photodynamic therapy, through ROS-dependent mechanisms, induces ERS and DNA damage, significantly boosting tumor immunogenicity [138].
ERS-related molecular markers hold prognostic value in lung cancer. Risk signatures based on key ERS-related genes accurately predict patient survival, with high-risk groups exhibiting reduced immune infiltration and diminished treatment sensitivity [139]. Among ERS-associated lncRNAs, RP11-295G20.2 promotes tumor progression by regulating cell migration and ERS [140]. TBL2, a novel driver gene, aids tumor adaptation to ERS by upregulating ATF4 and serves as an independent poor prognostic factor in LUAD [141]. Therapeutic strategies targeting the ERS-death axis demonstrate antitumor effects. Natural compounds like Icariside II [142] and Dihydroartemisinin [143] enhance chemosensitivity by activating ERS pathways, while the Gefitinib derivative L1Au2 induces ERS-dependent immunogenic death by dual-targeting TrxR and EGFR [144]. Narciclasine upregulates NOXA via the IRE1α-JNK/p38 axis, synergizing with cisplatin to amplify apoptosis [145]. Fascaplysin triggers ferroptosis and apoptosis through ROS-mediated ERS while upregulating PD-L1 to improve immunotherapy [146].
The ERS regulatory network intersects with metabolic reprogramming. 3,3’-Diselenodipropionic acid induces ERS-associated apoptosis via reductive stress, with confirmed in vivo antitumor activity [147]. The novel PPARɣ ligand PPZ023 provokes ERS-dependent death through the PPARγ/ROS/PERK axis, and its exosomes transmit death signals to overcome radioresistance [148].
Pancreatic ductal adenocarcinoma (PDAC)
In PDAC, ERS drives disease progression and therapeutic resistance through multiple mechanisms within the TME. Single-cell analyzes reveal heterogeneity in tumor-associated neutrophils (TANs), with terminally differentiated TAN-1 subsets exhibiting enhanced glycolysis regulated by the transcription factor BHLHE40 [149]. These TANs foster an immunosuppressive TME via CCL5 secretion and upregulation of the immune checkpoint Nectin2, while targeted interventions can reverse T-cell exhaustion [150].
ERS plays a central role in PDAC treatment resistance. RUNX1 activates the BiP/PERK/eIF2α pathway to promote gemcitabine resistance, an effect reversible by RUNX1 inhibition [151]. Innovative nanodelivery systems, such as CB-5083/miR-142 co-loaded nanoparticles, induce immunogenic cell death by multitargeting ERS and immune checkpoints [152]. The GRP78 inhibitor YUM70-PROTAC triggers irreversible ERS-dependent apoptosis, demonstrating strong synergy [153]. Metabolic reprogramming intersects with ERS regulation. BZW1, a PERK adaptor protein, enhances glycolysis via HIF1α/c-Myc, and its suppression overcomes metabolic stress resistance [154]. Natural compounds like secoemestrin C disrupt ER proteostasis to induce aberrant YAP degradation, while 2-hydroxy nervonic acid provokes ERS-mediated apoptosis through metabolic derivatives [155].
Therapeutically, anlotinib activates ERS via the ROS-PERK/eIF2α/ATF4 axis, with Nrf2 inhibition augmenting efficacy [156]. Curcumin/gelatin-blended nanofibrous mats sustain ERS induction to suppress STAT3 signaling and promote apoptosis [157]. Multifunctional nanoplatforms amplify immunogenic death through ERS, synergizing powerfully with immunotherapy [158].
Conclusions and future perspectives
Therapeutic strategies targeting the ERS-death axis through precise modulation of UPR core pathways (PERK, IRE1α, ATF6) and downstream molecules (CHOP, GRP78, XBP1, etc.) have demonstrated significant antitumor potential in preclinical studies. As comprehensively summarized in Small Molecule Compounds (Table 1) and Biomacromolecules, Natural Extracts, and Novel Formulations (Table 2), these preclinical evidences not only validate the therapeutic potential across diverse cancer types but also provide a roadmap for future drug development by systematically categorizing agents based on their specificity for UPR components and translational status.
For the PERK pathway, inhibitors (e.g., GSK2656157) that block protective autophagy or agonists (e.g., CCT020312) that enhance pro-death signals can reverse resistance in non-small cell lung cancer and triple-negative breast cancer, respectively [17, 18]. IRE1α-targeting agents (e.g., STF-083010) suppress kinase or nuclease activity to induce apoptosis in pancreatic cancer while mitigating mitochondrial and autolysosomal alterations [159]. ATF6 modulators (e.g., curcumin and melatonin) selectively regulate the ERS-death axis in colorectal and liver cancers by activating or inhibiting its transcriptional function [20, 21]. Downstream targeting approaches—such as GRP78 inhibitors (HA15, YUM70) triggering irreversible ERS in pancreatic cancer [22, 23] and CHOP enhancers (e.g., oleandrin) inducing subtype-specific apoptosis in breast cancer [123]—highlight the advantages of precision modulation. Combination strategies further expand therapeutic potential: PERK inhibitors paired with mRNA vaccines remodel the immune microenvironment in glioblastoma [24]; nanocarrier-mediated ERS inducers synergize with PD-1 inhibitors to convert “cold” tumors to “hot” [37]; and metabolic interventions (e.g., 2-deoxyglucose] combined with autophagy inhibitors amplify lethal ERS effects in triple-negative breast cancer [25].
Despite these promising preclinical results, translating ERS-death axis targeting strategies into clinical practice faces several critical challenges. Tumor heterogeneity leads to markedly divergent ERS-death axis characteristics (e.g., differential responses between ER⁺ and triple-negative breast cancers) [15, 16, 26], complicating the establishment of unified treatment protocols. The double-edged nature of ERS modulation necessitates precise control over activation intensity to avoid toxicity in normal tissues. Current drug delivery systems still lack sufficient targeting specificity and tissue penetration, as exemplified by the fibrotic stroma in pancreatic cancer that impedes nanoparticle delivery [27, 28]. Additionally, complex resistance mechanisms (e.g., FLIP upregulation and exosomal miRNA-mediated immune evasion) [26] further hinder therapeutic efficacy.
To overcome these barriers, future research must focus on [160, 161]: utilizing multi-omics technologies to identify ERS-specific biomarkers for improved patient stratification; developing spatiotemporally controllable ERS modulation systems that balance pro-death effects with adaptive resistance; engineering intelligent nanocarriers with enhanced targeting capabilities; and exploring triplet combination strategies integrating ERS modulators, ICIs, and metabolic interventions for multidimensional synergy against resistance. Breakthroughs in these areas will facilitate the transition of ERS-death axis research from fundamental theory to personalized clinical applications, ultimately providing innovative approaches for precision cancer therapy.
References
Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71–88.
Yan T, Ma X, Guo L, Lu R. Targeting endoplasmic reticulum stress signaling in ovarian cancer therapy. Cancer Biol Med. 2023;20:748–64.
Salvagno C, Mandula JK, Rodriguez PC, Cubillos-Ruiz JR. Decoding endoplasmic reticulum stress signals in cancer cells and antitumor immunity. Trends Cancer. 2022;8:930–43.
Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692–706.
Urra H, Aravena R, González-Johnson L, Hetz C. The UPRising connection between endoplasmic reticulum stress and the tumor microenvironment. Trends Cancer. 2024;10:1161–73.
Zhang W, Shi Y, Oyang L, Cui S, Li S, Li J, et al. Endoplasmic reticulum stress-a key guardian in cancer. Cell Death Discov. 2024;10:343.
Sari FR, Watanabe K, Widyantoro B, Thandavarayan RA, Harima M, Zhang S, et al. Partial inactivation of cardiac 14-3-3 protein in vivo elicits endoplasmic reticulum stress (ERS) and activates ERS-initiated apoptosis in ERS-induced mice. Cell Physiol Biochem. 2010;26:167–78.
Sari FR, Watanabe K, Thandavarayan RA, Harima M, Zhang S, Muslin AJ, et al. 14-3-3 protein protects against cardiac endoplasmic reticulum stress (ERS) and ERS-initiated apoptosis in experimental diabetes. J Pharmacol Sci. 2010;113:325–34.
Gao FF, Quan JH, Lee MA, Ye W, Yuk JM, Cha GH, et al. Trichomonas vaginalis induces apoptosis via ROS and ER stress response through ER-mitochondria crosstalk in SiHa cells. Parasites Vectors. 2021;14:603.
Cheng SB, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, et al. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019;10:927.
Chen J, Liu D, Lei L, Liu T, Pan S, Wang H. et al. CNPY2 aggravates renal tubular cell ferroptosis in diabetic nephropathy by regulating PERK/ATF4/CHAC1 pathway and MAM integrity. Adv Sci. Adv Sci. 2025;12:e2416441.
Zhao Y, Zheng Y, Li H, Li Y, Wang R, Cai Y, et al. Protein folding dependence on selenoprotein M contributes to steady cartilage extracellular matrix repressing ferroptosis via PERK/ATF4/CHAC1 axis. Osteoarthr Cartil. 2025;33:261–75.
Habshi T, Shelke V, Kale A, Anders HJ, Gaikwad AB. Role of endoplasmic reticulum stress and autophagy in the transition from acute kidney injury to chronic kidney disease. J Cell Physiol. 2023;238:82–93.
Nakashima A, Cheng SB, Kusabiraki T, Motomura K, Aoki A, Ushijima A, et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci Rep. 2019;9:11466.
Fan P, Jordan VC. Estrogen receptor and the unfolded protein response: double-edged swords in therapy for estrogen receptor-positive breast cancer. Target Oncol. 2022;17:111–24.
Fan P, Jordan VC. PERK, beyond an unfolded protein response sensor in estrogen-induced apoptosis in endocrine-resistant breast cancer. Mol Cancer Res. 2022;20:193–201.
Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16:533–44.
Zito E, Guarrera L, Janssen-Heininger YMW. Fingerprint of the oxido-reductase ERO1: a protein disulfide bond producer and supporter of cancer. Biochim Biophys Acta Rev cancer. 2024;1879:189027.
Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4:423–36.
He J, Zhou Y, Sun L. Emerging mechanisms of the unfolded protein response in therapeutic resistance: from chemotherapy to Immunotherapy. Cell Commun Signal. 2024;22:89.
Peng C, Wang J, Wang S, Zhao Y, Wang H, Wang Y, et al. Endoplasmic reticulum stress: triggers microenvironmental regulation and drives tumor evolution. Cancer Med. 2025;14:e70684.
Guerra L, Bonetti L, Brenner D. Metabolic modulation of immunity: a new concept in cancer immunotherapy. Cell Rep. 2020;32:107848.
Madhavan S, Nagarajan S. GRP78 and next generation cancer hallmarks: an underexplored molecular target in cancer chemoprevention research. Biochimie. 2020;175:69–76.
Li Y, You J, Zou Z, Sun G, Shi Y, Sun Y, et al. Decoding the tumor microenvironment: exosome-mediated macrophage polarization and therapeutic frontiers. Int J Biol Sci. 2025;21:4187–214.
Nie Z, Chen M, Wen X, Gao Y, Huang D, Cao H, et al. Endoplasmic reticulum stress and tumor microenvironment in bladder cancer: the missing link. Front Cell Dev Biol. 2021;9:683940.
Fujita K, Iwama H, Oura K, Tadokoro T, Samukawa E, Sakamoto T. et al. Cancer therapy due to apoptosis: Galectin-9. Int J Mol Sci. 2017;18:74.
Balaji S, Terrero D, Tiwari AK, Ashby CR Jr, Raman D. Alternative approaches to overcome chemoresistance to apoptosis in cancer. Adv Protein Chem Struct Biol. 2021;126:91–122.
Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13:847345.
Guchelaar HJ, Vermes A, Vermes I, Haanen C. Apoptosis: molecular mechanisms and implications for cancer chemotherapy. Pharm World Sci. 1997;19:119–25.
Dallaporta B, Pablo M, Maisse C, Daugas E, Loeffler M, Zamzami N, et al. Proteasome activation as a critical event of thymocyte apoptosis. Cell Death Differ. 2000;7:368–73.
Kashyap D, Garg VK, Goel N. Intrinsic and extrinsic pathways of apoptosis: role in cancer development and prognosis. Adv Protein Chem Struct Biol. 2021;125:73–120.
Sims SG, Cisney RN, Lipscomb MM, Meares GP. The role of endoplasmic reticulum stress in astrocytes. Glia. 2022;70:5–19.
Wu H, Zheng S, Zhang J, Xu S, Miao Z. Cadmium induces endoplasmic reticulum stress-mediated apoptosis in pig pancreas via the increase of Th1 cells. Toxicology. 2021;457:152790.
Sun M, Zhang X, Tan B, Zhang Q, Zhao X, Dong D. Potential role of endoplasmic reticulum stress in doxorubicin-induced cardiotoxicity-an update. Front Pharmacol. 2024;15:1415108.
Hu H, Tian M, Ding C, Yu S. The C/EBP Homologous Protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083.
Wang P, Li J, Tao J, Sha B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J Biol Chem. 2018;293:4110–21.
Lin R, Ma M, Han B, Zheng Y, Wang Y, Zhou Y. Esophageal cancer stem cells reduce hypoxia-induced apoptosis by inhibiting the GRP78-perk-eIF2α-ATF4-CHOP pathway in vitro. J Gastrointest Oncol. 2023;14:1669–93.
Chen J, Fan W, Fan J, Xie J, Wang Y, Wang Y, et al. Tetrahydrocurcumin attenuates polymyxin B sulfate-induced HK-2 cells apoptosis by inhibiting endoplasmic reticulum stress-mediated PERK/eIF2α/ATF4/CHOP signaling pathway axis. Environ Toxicol. 2024;39:4995–5007.
Shi M, Liu K, Li X, Zeng XL, Liu XJ. Melatonin ameliorates PM2.5-induced airway inflammation and apoptosis by PERK/eIF2α/ATF4/CHOP in chronic obstructive pulmonary disease mice. Toxicol Appl Pharmacol. 2025;499:117314.
Wan F, Qiu F, Deng Y, Hu H, Zhang Y, Zhang JY, et al. Knockdown of YTHDF2 initiates ERS-induced apoptosis and cancer stemness suppression by sustaining GLI2 stability in cervical cancer. Transl Oncol. 2024;46:101994.
Lou ZH, Xia RM, Li XJ, Cheng RB, Shao KD, Zhang GJ. Anti-lung cancer mechanisms of diterpenoid tanshinone viaendoplasmic reticulum stress-mediated apoptosis signal pathway. Zhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi =China J Chin Mater Med. 2018;43:4900.
Zhou N, Liu Q, Wang X, He L, Zhang T, Zhou H, et al. The combination of hydroxychloroquine and 2-deoxyglucose enhances apoptosis in breast cancer cells by blocking protective autophagy and sustaining endoplasmic reticulum stress. Cell Death Discov. 2022;8:286.
Bian X, Fan N, Li M, Han D, Li J, Fan L, et al. An ER-Horse detonating stress cascade for hepatocellular carcinoma nanotherapy. ACS Nano. 2023;17:4896–912.
Lu Z, Peng Q, Hu R, Wang Y, Fan K, Zhang T. Naringin attenuates inflammatory injury to the bovine endometrium by regulating the endoplasmic reticulum stress-PI3K/AKT-autophagy axis. Front Pharmacol. 2024;15:1424511.
Chipurupalli S, Samavedam U, Robinson N. Crosstalk between ER stress, autophagy and inflammation. Front Med. 2021;8:758311.
Jin H, Yang Y, Zhu X, Zhou Y, Xu Y, Li J, et al. DDRGK1-mediated ER-phagy attenuates acute kidney injury through ER-stress and apoptosis. Cell Death Dis. 2024;15:63.
Gao LJ, Li P, Ma T, Zhong ZQ, Xu SJ. Ligustilide alleviates neurotoxicity in SH-SY5Y cells induced by Aβ(25-35) via regulating endoplasmic reticulum stress and autophagy. Phytother Res. 2021;35:1572–84.
Wu Z, Wang H, Fang S, Xu C. Roles of endoplasmic reticulum stress and autophagy on H2O2‑induced oxidative stress injury in HepG2 cells. Mol Med Rep. 2018;18:4163–74.
Shi Y, Jiang B, Zhao J. Induction mechanisms of autophagy and endoplasmic reticulum stress in intestinal ischemia-reperfusion injury, inflammatory bowel disease, and colorectal cancer. Biomed Pharmacother. 2024;170:115984.
Chakraborty P, Parikh RY, Choi S, Tran D, Gooz M, Hedley ZT, et al. Carbon monoxide activates PERK-regulated autophagy to induce immunometabolic reprogramming and boost antitumor T-cell function. Cancer Res. 2022;82:1969–90.
Lin X, Yang M, Huang Y, Huang X, Shi H, Chen B, et al. Gene signatures of endoplasmic reticulum stress and mitophagy for prognostic risk prediction in lung adenocarcinoma. IET Syst Biol. 2024;18:103–17.
Hu JL, Hu XL, Guo AY, Wang CJ, Wen YY, Cang SD. Endoplasmic reticulum stress promotes autophagy and apoptosis and reverses chemoresistance in human ovarian cancer cells. Oncotarget. 2017;8:49380–94.
He C, Xia J, Gao Y, Chen Z, Wan X. Chlorin A-mediated photodynamic therapy induced apoptosis in human cholangiocarcinoma cells via impaired autophagy flux. Am J Transl Res. 2020;12:5080–94.
Cui Y, Xu H, Yang Y, Zhao D, Wen Y, Lv C, et al. The regulation of miR-320a/XBP1 axis through LINC00963 for endoplasmic reticulum stress and autophagy in diffuse large B-cell lymphoma. Cancer Cell Int. 2021;21:305.
Cui J, Liu J, Fan L, Zhu Y, Zhou B, Wang Y, et al. A zinc finger family protein, ZNF263, promotes hepatocellular carcinoma resistance to apoptosis via activation of ER stress-dependent autophagy. Transl Oncol. 2020;13:100851.
Zhaoyun L, Wang H, Yang C, Zhao X, Hui L, Song J. et al. Enhancing antitumor immunity via ROS-ERS and pyroptosis-induced immunogenic cell death in multiple myeloma. J Immunother Cancer. 2025;13:e011717
Wang F, Wang J, Liang X, Wu Z, Xue J, Yin L, et al. Ghrelin inhibits myocardial pyroptosis in diabetic cardiomyopathy by regulating ERS and NLRP3 inflammasome crosstalk through the PI3K/AKT pathway. J Drug Target. 2024;32:148–58.
Han J, Cheng C, Zhang J, Fang J, Yao W, Zhu Y, et al. Myricetin activates the Caspase-3/GSDME pathway via ER stress induction of pyroptosis in lung cancer cells. Front Pharmacol. 2022;13:959938.
Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781.
Wright SS, Kumari P, Fraile-Ágreda V, Wang C, Shivcharan S, Kappelhoff S, et al. Transplantation of gasdermin pores by extracellular vesicles propagates pyroptosis to bystander cells. Cell. 2025;188:280–91.e17.
Xiao Y, Zhang T, Ma X, Yang QC, Yang LL, Yang SC, et al. Microenvironment-responsive prodrug-induced pyroptosis boosts cancer immunotherapy. Adv Sci. 2021;8:e2101840.
Wang H, Wang T, Yan S, Tang J, Zhang Y, Wang L, et al. Crosstalk of pyroptosis and cytokine in the tumor microenvironment: from mechanisms to clinical implication. Mol Cancer. 2024;23:268.
Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab. 2022;34:581–94.e8.
Chen Q, Peng B, Lin L, Chen J, Jiang Z, Luo Y, et al. Chondroitin sulfate-modified hydroxyapatite for caspase-1 activated induced pyroptosis through Ca overload/ER Stress/STING/IRF3 pathway in colorectal cancer. Small. 2024;20:e2403201.
Wu S, Wang B, Li H, Wang H, Du S, Huang X, et al. Targeting STING elicits GSDMD-dependent pyroptosis and boosts anti-tumor immunity in renal cell carcinoma. Oncogene. 2024;43:1534–48.
Le X, Mu J, Peng W, Tang J, Xiang Q, Tian S, et al. DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics. 2020;10:9495–511.
Lv W, Wu X, Dou Y, Yan Y, Chen L, Fei Z. et al. Homer1 protects against retinal ganglion cell pyroptosis by inhibiting endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation after middle cerebral artery occlusion-induced retinal ischemia. Int J Mol Sci. 2023;24:16811.
Li K, Wang YJ, Chen C, Wang XJ, Li W. Targeting pyroptosis: a novel strategy of ginseng for the treatment of diabetes and its chronic complications. Phytomed Int J Phytother Phytopharmacol. 2025;138:156430.
Fu Y, Shen J, Li Y, Liu F, Ning B, Zheng Y, et al. Inhibition of the PERK/TXNIP/NLRP3 axis by baicalin reduces NLRP3 inflammasome-mediated pyroptosis in macrophages infected with Mycobacterium tuberculosis. Mediat Inflamm. 2021;2021:1805147.
Zhang Y, Yan Y, Liu J, Xia H, Zhou J, Cui Y. et al. An endoplasmic reticulum stress-specific nanoinducer selectively evokes type-II immunogenic cell death for pyroptotic cancer immunotherapy. Adv Mater.2025;37:e2501953.
Yang J, Wang Y, Liu F, Zhang Y, Han F. Crosstalk between ferroptosis and endoplasmic reticulum stress: a potential target for ovarian cancer therapy (Review). Int J Mol Med. 2025;55:97.
Guo C, Zhao W, Wang W, Yao Z, Chen W, Feng X.Study on the antitumor mechanism of tanshinone IIA in vivo and in vitro through the regulation of PERK-ATF4-HSPA5 pathway-mediated ferroptosis.in vivo and in vitro through the regulation of PERK-ATF4-HSPA5 pathway-mediated ferroptosis. Molecules. 2024;29:1557.
Wang C, Zha YL, Wang H, Sun B, Qiang WG, Yuan Y, et al. Carfilzomib promotes Iodine-125 seed radiation-induced apoptosis, paraptosis, and ferroptosis in esophageal squamous cell carcinoma by aggravating endoplasmic reticulum stress. Transl Oncol. 2025;57:102393.
Fu F, Wang W, Wu L, Wang W, Huang Z, Huang Y, et al. Inhalable biomineralized liposomes for cyclic Ca(2+)-burst-centered endoplasmic reticulum stress enhanced lung cancer ferroptosis therapy. ACS Nano. 2023;17:5486–502.
Zhao Z, Wu Y, Liang X, Liu J, Luo Y, Zhang Y, et al. Sonodynamic therapy of NRP2 monoclonal antibody-guided MOFs@COF targeted disruption of mitochondrial and endoplasmic reticulum homeostasis to induce autophagy-dependent ferroptosis. Adv Sci. 2023;10:e2303872.
Lou X, Gao D, Yang L, Wang Y, Hou Y. Endoplasmic reticulum stress mediates the myeloid-derived immune suppression associated with cancer and infectious disease. J Transl Med. 2023;21:1.
Yang J, Xu H, Wu W, Huang H, Zhang C, Tang W, et al. Ferroptosis signaling promotes the release of misfolded proteins via exosomes to rescue ER stress in hepatocellular carcinoma. Free Radic Biol Med. 2023;202:110–20.
Zhang X, Li W, Ma Y, Zhao X, He L, Sun P, et al. High-fat diet aggravates colitis-associated carcinogenesis by evading ferroptosis in the ER stress-mediated pathway. Free Radic Biol Med. 2021;177:156–66.
Chen J, Zhao R, Wang Y, Xiao H, Lin W, Diao M, et al. G protein-coupled estrogen receptor activates PI3K/AKT/mTOR signaling to suppress ferroptosis via SREBP1/SCD1-mediated lipogenesis. Mol Med. 2024;30:28.
Ren Q, Liu Z, Wu L, Yin G, Xie X, Kong W, et al. C/EBPβ: The structure, regulation, and its roles in inflammation-related diseases. Biomed Pharmacother. 2023;169:115938.
Yin H, Liu Y, Dong Q, Wang H, Yan Y, Wang X, et al. The mechanism of extracellular CypB promotes glioblastoma adaptation to glutamine deprivation microenvironment. Cancer Lett. 2024;597:216862.
Zhou F, Gao H, Shang L, Li J, Zhang M, Wang S, et al. Oridonin promotes endoplasmic reticulum stress via TP53-repressed TCF4 transactivation in colorectal cancer. J Exp Clin Cancer Res. 2023;42:150.
Jiang M, Li X, Zhang J, Lu Y, Shi Y, Zhu C, et al. Dual inhibition of endoplasmic reticulum stress and oxidation stress manipulates the polarization of macrophages under hypoxia to sensitize immunotherapy. ACS Nano. 2021;15:14522–34.
Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
Feng X, Lin T, Chen D, Li Z, Yang Q, Tian H, et al. Mitochondria-associated ER stress evokes immunogenic cell death through the ROS-PERK-eIF2α pathway under PTT/CDT combined therapy. Acta Biomater. 2023;160:211–24.
Zhang Z, Shen C, Zhou F, Zhang Y. Shikonin potentiates therapeutic efficacy of oxaliplatin through reactive oxygen species-mediated intrinsic apoptosis and endoplasmic reticulum stress in oxaliplatin-resistant colorectal cancer cells. Drug Dev Res. 2023;84:542–55.
Xu Z, Xu J, Sun S, Lin W, Li Y, Lu Q, et al. Mecheliolide elicits ROS-mediated ERS driven immunogenic cell death in hepatocellular carcinoma. Redox Biol. 2022;54:102351.
Li W, Yang J, Luo L, Jiang M, Qin B, Yin H, et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat Commun. 2019;10:3349.
Yang Z, Teng Y, Lin M, Peng Y, Du Y, Sun Q, et al. Reinforced immunogenic endoplasmic reticulum stress and oxidative stress via an orchestrated nanophotoinducer to boost cancer photoimmunotherapy. ACS Nano. 2024;18:7267–86.
Yang Z, Huo Y, Zhou S, Guo J, Ma X, Li T, et al. Cancer cell-intrinsic XBP1 drives immunosuppressive reprogramming of intratumoral myeloid cells by promoting cholesterol production. Cell Metab. 2022;34:2018–35.e8.
Liu L, Li S, Qu Y, Bai H, Pan X, Wang J, et al. Ablation of ERO1A induces lethal endoplasmic reticulum stress responses and immunogenic cell death to activate anti-tumor immunity. Cell Rep Med. 2023;4:101206.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–20.e7.
Liu C, Zhou X, Zeng H, Yu J, Li W, Zhang W, et al. Endoplasmic reticulum stress potentiates the immunosuppressive microenvironment in hepatocellular carcinoma by promoting the release of SNHG6-enriched small extracellular vesicles. Cancer Immunol Res. 2024;12:1184–201.
Zhan K, Yang X, Li S, Bai Y. Correlation of endoplasmic reticulum stress patterns with the immune microenvironment in hepatocellular carcinoma: a prognostic signature analysis. Front Immunol. 2023;14:1270774.
Obacz J, Archambeau J, Lafont E, Nivet M, Martin S, Aubry M, et al. IRE1 endoribonuclease signaling promotes myeloid cell infiltration in glioblastoma. Neuro-Oncology. 2024;26:858–71.
Raines LN, Zhao H, Wang Y, Chen HY, Gallart-Ayala H, Hsueh PC, et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat Immunol. 2022;23:431–45.
Li X, Ma L, Guo J, Wei Y, Ma S, Mai Y, et al. Synergistic anti-tumor effects of mRNA vaccine and PERK inhibitor combination in melanoma treatment. Colloids Surf B Biointerfaces. 2025;254:114808.
Wu C, Gao M, Xiao W, Huang X, Yang X, Wu Z, et al. Light-activatable manganese carbonate nanocubes elicit robust immunotherapy by amplifying endoplasmic reticulum stress-meditated pyroptotic cell death. J Exp Clin Cancer Res. 2025;44:147.
Zhu MY, Wang T, Wang HD, Wang HZ, Chen HY, Zhang S, et al. LW-213 induces immunogenic tumor cell death via ER stress mediated by lysosomal TRPML1. Cancer Lett. 2023;577:216435.
Jin Y, Huynh DTN, Heo KS. Ginsenoside Rh1 inhibits tumor growth in MDA-MB-231 breast cancer cells via mitochondrial ROS and ER stress-mediated signaling pathway. Arch Pharmacal Res. 2022;45:174–84.
Cano-González A, Mauro-Lizcano M, Iglesias-Serret D, Gil J, López-Rivas A. Involvement of both caspase-8 and Noxa-activated pathways in endoplasmic reticulum stress-induced apoptosis in triple-negative breast tumor cells. Cell Death Dis. 2018;9:134.
Adamczyk-Grochala J, Bloniarz D, Zielinska K, Lewinska A, Wnuk M. DNMT2/TRDMT1 gene knockout compromises doxorubicin-induced unfolded protein response and sensitizes cancer cells to ER stress-induced apoptosis. Apoptosis. 2023;28:166–85.
Yao X, Tu Y, Xu Y, Guo Y, Yao F, Zhang X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J Cell Mol Med. 2020;24:9560–73.
Li X, Zheng J, Chen S, Meng FD, Ning J, Sun SL. Oleandrin, a cardiac glycoside, induces immunogenic cell death via the PERK/elF2α/ATF4/CHOP pathway in breast cancer. Cell Death Dis. 2021;12:314.
Li X, Yu X, Zhou D, Chen B, Li W, Zheng X, et al. CCT020312 inhibits triple-negative breast cancer through PERK pathway-mediated G1 phase cell cycle arrest and apoptosis. Front Pharmacol. 2020;11:737.
Cai W, Rong D, Ding J, Zhang X, Wang Y, Fang Y, et al. Activation of the PERK/eIF2α axis is a pivotal prerequisite of taxanes to cancer cell apoptosis and renders synergism to overcome paclitaxel resistance in breast cancer cells. Cancer Cell Int. 2024;24:249.
Comitato R, Guantario B, Leoni G, Nesaretnam K, Ronci MB, Canali R, et al. Tocotrienols induce endoplasmic reticulum stress and apoptosis in cervical cancer cells. Genes Nutr. 2016;11:32.
Saini KK, Chaturvedi P, Sinha A, Singh MP, Khan MA, Verma A, et al. Loss of PERK function promotes ferroptosis by downregulating SLC7A11 (System Xc⁻) in colorectal cancer. Redox Biol. 2023;65:102833.
Zhang Y, Li H, Zhao Y, Liu L, Zhou Y, Pan X, et al. Macrocarpal I induces immunogenic cell death and synergizes with immune checkpoint inhibition by targeting tubulin and PARP1 in colorectal cancer. Cell Death Discov. 2025;11:73.
Colangelo T, Polcaro G, Ziccardi P, Muccillo L, Galgani M, Pucci B, et al. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2016;7:e2108.
Xu W, Shen Y. Curcumin affects apoptosis of colorectal cancer cells through ATF6-mediated endoplasmic reticulum stress. Chem Biol Drug Des. 2024;103:e14433.
Liu C, Xu J, Guo C, Chen X, Qian C, Zhang X, et al. Gambogenic acid induces endoplasmic reticulum stress in colorectal cancer via the aurora A pathway. Front Cell Dev Biol. 2021;9:736350.
Wang Q, Feng H, Li Z, Wu Q, Li L, Sun D, et al. α-Hederin induces human colorectal cancer cells apoptosis through disturbing protein homeostasis. Chem Biol Interact. 2023;386:110785.
Zulkifli A, Tan FH, Areeb Z, Stuart SF, Gomez J, Paradiso L. et al. Carfilzomib promotes the unfolded protein response and apoptosis in cetuximab-resistant colorectal cancer. Int J Mol Sci. 2021;22:7114.
Chern YJ, Wong JCT, Cheng GSW, Yu A, Yin Y, Schaeffer DF, et al. The interaction between SPARC and GRP78 interferes with ER stress signaling and potentiates apoptosis via PERK/eIF2α and IRE1α/XBP-1 in colorectal cancer. Cell Death Dis. 2019;10:504.
Wei W, Wang H, Fu L, Liu H. Icaritin induces paraptosis in hepatocellular carcinoma cells by targeting BHLHE40 via endoplasmic reticulum stress and mitochondrial dysfunction. Phytomedicine. 2025;143:156870.
Kim TW. Fisetin, an anti-inflammatory agent, overcomes radioresistance by activating the PERK-ATF4-CHOP axis in liver cancer. Int J Mol Sci. 2023;24:9076.
Wang J, Liu H, Zhu L, Wang J, Luo X, Liu W. et al. Prodigiosin from Serratia Marcescens in cockroach inhibits the proliferation of hepatocellular carcinoma cells through endoplasmic reticulum stress-induced apoptosis. Molecules. 2022;27:7281.
Liu N, Yang HL, Wang P, Lu YC, Yang YJ, Wang L, et al. Functional proteomic analysis revels that the ethanol extract of Annona muricata L. induces liver cancer cell apoptosis through endoplasmic reticulum stress pathway. J Ethnopharmacol. 2016;189:210–7.
Shi TL, Zhang L, Cheng QY, Yu JS, Liu J, Shen YJ, et al. Xanthatin induces apoptosis by activating endoplasmic reticulum stress in hepatoma cells. Eur J Pharmacol. 2019;843:1–11.
Zhou B, Lu Q, Liu J, Fan L, Wang Y, Wei W, et al. Melatonin increases the sensitivity of hepatocellular carcinoma to sorafenib through the PERK-ATF4-Beclin1 pathway. Int J Biol Sci. 2019;15:1905–20.
Shi Y, Ma J, Chen KE, Chen B. Konjac glucomannan enhances 5-FU-induced cytotoxicity of hepatocellular carcinoma cells via TLR4/PERK/CHOP signaling to induce endoplasmic reticulum stress. Oncol Res. 2022;30:201–10.
Zheng Y, Zhou Q, Zhao C, Li J, Yu Z, Zhu Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2α/ATF4/CHOP axis. J Cell Mol Med. 2021;25:1468–79.
Li D, Wang WJ, Wang YZ, Wang YB, Li YL. Lobaplatin promotes (125)I-induced apoptosis and inhibition of proliferation in hepatocellular carcinoma by upregulating PERK-eIF2α-ATF4-CHOP pathway. Cell Death Dis. 2019;10:744.
Li L, Liu N, Yang P, Rao C, Kong L, Huang Y, et al. USP18 attenuates endoplasmic reticulum stress via the PERK-eIF2α-ATF4 axis to reduce apoptosis in hepatocellular carcinoma cells. Sci Rep. 2025;15:15659.
Chen H, Shang X, Yuan H, Niu Q, Chen J, Luo S, et al. Total flavonoids of Oldenlandia diffusa (Willd.) Roxb. suppresses the growth of hepatocellular carcinoma through endoplasmic reticulum stress-mediated autophagy and apoptosis. Front Pharmacol. 2022;13:1019670.
Xia S, Wu J, Zhou W, Zhang M, Zhao K, Tian D, et al. HRC promotes anoikis resistance and metastasis by suppressing endoplasmic reticulum stress in hepatocellular carcinoma. Int J Med Sci. 2021;18:3112–24.
Cheng Z, Li S, Yang S, Long H, Wu H, Chen X, et al. Endoplasmic reticulum stress promotes hepatocellular carcinoma by modulating immunity: a study based on artificial neural networks and single-cell sequencing. J Transl Med. 2024;22:658.
Li N, Jiang Y, Wang A, Jiang T, Dai H, Xia C, et al. Sulforaphane induces cell morphology change and cell apoptosis by activating endoplasmic reticulum stress in glioblastoma. BMC Cancer. 2025;25:1050.
Kusaczuk M, Tyszka N, Kretowski R, Cechowska-Pasko M. The proteasome inhibitor marizomib evokes endoplasmic reticulum stress and promotes apoptosis in human glioblastoma cells. Pharmaceuticals. 2024;17:1089.
Chen Y, Guo Y, Li S, Xu J, Ning W, Zhao C, et al. Remdesivir inhibits the progression of glioblastoma by enhancing endoplasmic reticulum stress. Biomed Pharmacother. 2023;157:114037.
Roy PK, Deepak K, Das CK, Das A, Biswas A, Jena BC, et al. PSMC2 promotes resistance against temozolomide in glioblastoma via suppressing JNK-mediated autophagic cell death. Biochem Pharmacol. 2025;233:116755.
Ketkar M, Desai S, Rana P, Thorat R, Epari S, Dutt A, et al. Inhibition of PERK-mediated unfolded protein response acts as a switch for reversal of residual senescence and as senolytic therapy in glioblastoma. Neuro-Oncology. 2024;26:2027–43.
Kuranaga Y, Yu B, Osuka S, Zhang H, Devi NS, Bae S. et al. Targeting integrin α3 blocks β1 maturation, triggers endoplasmic reticulum stress, and sensitizes glioblastoma cells to TRAIL-mediated apoptosis. Cells. 2024;13:753.
Zhang X, Wu R, Tian C, Wang W, Zhou L, Guo T, et al. GRP78 blockade overcomes intrinsic resistance to UBA1 inhibitor TAK-243 in glioblastoma. Cell Death Discov. 2022;8:133.
Kiang KM, Tang W, Song Q, Liu J, Li N, Lam TL, et al. Targeting unfolded protein response using albumin-encapsulated nanoparticles attenuates temozolomide resistance in glioblastoma. Br J Cancer. 2023;128:1955–63.
Lin L, Chen L, Lin G, Chen X, Huang L, Yang J. Derlin-3 manipulates the endoplasmic reticulum stress and IgG4 secretion of plasma cells in lung adenocarcinoma. Oncogene. 2025;44:2620–33.
Yu TT, Hu J, Li QR, Peng XC, Xu HZ, Han N, et al. Chlorin e6-induced photodynamic effect facilitates immunogenic cell death of lung cancer as a result of oxidative endoplasmic reticulum stress and DNA damage. Int Immunopharmacol. 2023;115:109661.
Wan L, Chen Z, Yang J, Wu G, Xu Y, Cui J, et al. Identification of endoplasmic reticulum stress-related signature characterizes the tumor microenvironment and predicts prognosis in lung adenocarcinoma. Sci Rep. 2023;13:19462.
Yu L, Zhou S, Hong W, Lin N, Wang Q, Liang P. Characterization of an endoplasmic reticulum stress-associated lncRNA prognostic signature and the tumor-suppressive role of RP11-295G20.2 knockdown in lung adenocarcinoma. Sci Rep. 2024;14:12283.
Kosai K, Masuda T, Kitagawa A, Tobo T, Ono Y, Ando Y, et al. Transducin beta-like 2 is a potential driver gene that adapts to endoplasmic reticulum stress to promote tumor growth of lung adenocarcinoma. Ann Surg Oncol. 2023;30:7538–48.
Tang Z, Du W, Xu F, Sun X, Chen W, Cui J, et al. Icariside II enhances cisplatin-induced apoptosis by promoting endoplasmic reticulum stress signalling in non-small cell lung cancer cells. Int J Biol Sci. 2022;18:2060–74.
Han N, Yang ZY, Xie ZX, Xu HZ, Yu TT, Li QR, et al. Dihydroartemisinin elicits immunogenic death through ferroptosis-triggered ER stress and DNA damage for lung cancer immunotherapy. Phytomedicine. 2023;112:154682.
Wang L, Ma X, Zhou L, Luo M, Lu Y, Wang Y, et al. Dual-targeting TrxR-EGFR alkynyl-Au(I) gefitinib complex induces ferroptosis in gefitinib-resistant lung cancer via degradation of GPX4. J Med Chem. 2025;68:5275–91.
Lee JH, Seo SH, Shim J, Kim YN, Yoon K. Narciclasine enhances cisplatin-induced apoptotic cell death by inducing unfolded protein response-mediated regulation of NOXA and MCL1. Cell Mol Biol Lett. 2025;30:59.
Luo L, Xu G. Fascaplysin induces apoptosis and ferroptosis, and enhances Anti-PD-1 immunotherapy in non-small cell lung cancer (NSCLC) by promoting PD-L1 expression. Int J Mol Sci. 2022;23:13774.
Gandhi VV, Gandhi KA, Kumbhare LB, Goda JS, Gota V, Priyadarsini KI, et al. 3,3’-Diselenodipropionic acid (DSePA) induces reductive stress in A549 cells triggering p53-independent apoptosis: A novel mechanism for diselenides. Free Radic Biol Med. 2021;175:1–17.
Kim TW, Hong DW, Kang CM, Hong SH. A novel PPARɣ ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells. Exp Mol Med. 2020;52:1730–43.
Wang L, Liu Y, Dai Y, Tang X, Yin T, Wang C, et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut. 2023;72:958–71.
Luo H, Ikenaga N, Nakata K, Higashijima N, Zhong P, Kubo A, et al. Tumor-associated neutrophils upregulate Nectin2 expression, creating the immunosuppressive microenvironment in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2024;43:258.
She C, Wu C, Guo W, Xie Y, Li S, Liu W, et al. Combination of RUNX1 inhibitor and gemcitabine mitigates chemo-resistance in pancreatic ductal adenocarcinoma by modulating BiP/PERK/eIF2α-axis-mediated endoplasmic reticulum stress. J Exp Clin Cancer Res. 2023;42:238.
Li CY, Chou TF, Lo YL. An innovative nanoformulation utilizing tumor microenvironment-responsive PEG-polyglutamic coating and dynamic charge adjustment for specific targeting of ER stress inducer, microRNA, and immunoadjuvant in pancreatic cancer: In vitro investigations. Int J Biol Macromol. 2024;254:127905.
Samanta S, Yang S, Debnath B, Xue D, Kuang Y, Ramkumar K, et al. The hydroxyquinoline analogue YUM70 inhibits GRP78 to induce ER stress-mediated apoptosis in pancreatic cancer. Cancer Res. 2021;81:1883–95.
Li Z, Ge Y, Dong J, Wang H, Zhao T, Wang X, et al. BZW1 facilitates glycolysis and promotes tumor growth in pancreatic ductal adenocarcinoma through potentiating eIF2α phosphorylation. Gastroenterology. 2022;162:1256–71.e14.
Wang J, Chen M, Wang M, Zhao W, Zhang C, Liu X, et al. The novel ER stress inducer Sec C triggers apoptosis by sulfating ER cysteine residues and degrading YAP via ER stress in pancreatic cancer cells. Acta Pharm Sin B. 2022;12:210–27.
Yang L, Zhou X, Sun J, Lei Q, Wang Q, Pan D, et al. Reactive oxygen species mediate anlotinib-induced apoptosis via activation of endoplasmic reticulum stress in pancreatic cancer. Cell Death Dis. 2020;11:766.
Cheng T, Zhang Z, Shen H, Jian Z, Li J, Chen Y, et al. Topically applicated curcumin/gelatin-blended nanofibrous mat inhibits pancreatic adenocarcinoma by increasing ROS production and endoplasmic reticulum stress mediated apoptosis. J Nanobiotechnol. 2020;18:126.
Lo YL, Li CY, Chou TF, Yang CP, Wu LL, Chen CJ, et al. Exploring in vivo combinatorial chemo-immunotherapy: Addressing p97 suppression and immune reinvigoration in pancreatic cancer with tumor microenvironment-responsive nanoformulation. Biomed Pharmacother. 2024;175:116660.
Blanc M, Habbouche L, Xiao P, Lebeaupin C, Janona M, Vaillant N, et al. Bax Inhibitor-1 preserves pancreatic β-cell proteostasis by limiting proinsulin misfolding and programmed cell death. Cell Death Dis. 2024;15:334.
Shi W, Han H, Zou J, Zhang Y, Li H, Zhou H, et al. Identification of dihydrotanshinone I as an ERp57 inhibitor with anti-breast cancer properties via the UPR pathway. Biochem Pharmacol. 2021;190:114637.
Zhou W, Fang H, Wu Q, Wang X, Liu R, Li F, et al. Ilamycin E, a natural product of marine actinomycete, inhibits triple-negative breast cancer partially through ER stress-CHOP-Bcl-2. Int J Biol Sci. 2019;15:1723–32.
Kim SY, Hwang S, Choi MK, Park S, Nam KY, Kim I. Molecular mechanisms underlying the effects of the small molecule AMC-04 on apoptosis: roles of the activating transcription factor 4-C/EBP homologous protein-death receptor 5 pathway. Chem-Biol Interact. 2020;332:109277.
Qu J, Zeng C, Zou T, Chen X, Yang X, Lin Z. Autophagy induction by trichodermic acid attenuates endoplasmic reticulum stress-mediated apoptosis in colon cancer cells. Int J Mol Sci. 2021;22:5566.
Qu C, Ma J, Liu X, Xue Y, Zheng J, Liu L, et al. Dihydroartemisinin exerts anti-tumor activity by inducing mitochondrion and endoplasmic reticulum apoptosis and autophagic cell death in human glioblastoma cells. Front Cell Neurosci. 2017;11:310.
Zhu J, Tian S, Li KT, Chen Q, Jiang Y, Lin HD, et al. Inhibition of breast cancer cell growth by methyl pyropheophenylchlorin photodynamic therapy is mediated though endoplasmic reticulum stress-induced autophagy in vitro and vivo. Cancer Med. 2018;7:1908–20.
Wang Q, Wang P, Xiao Z. Resistant starch prevents tumorigenesis of dimethylhydrazine-induced colon tumors via regulation of an ER stress-mediated mitochondrial apoptosis pathway. Int J Mol Med. 2018;41:1887–98.
Acknowledgements
We would like to express our gratitude to Biorender for providing us with beautiful templates for our scientific illustrations and supporting us in the publication process (https://www.biorender.com/).
Funding
The Basic Research Projects of Science and Technology Department of Guizhou Province (Qian Ke He-zk [2022]-659). The healthcare Commission (Healthcare Service and Security Capacity Enhancement Grant Funding) JZ-2024-11D.
Author information
Authors and Affiliations
Contributions
Hongyu Chai drafted the manuscript; Qian Hu, Shun Yao and Shuoguo Ma participated in the data investigation and analysis; Wei Su revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chai, H., Hu, Q., Yao, S. et al. Endoplasmic reticulum stress-mediated programmed cell death in the tumor microenvironment. Cell Death Discov. 11, 559 (2025). https://doi.org/10.1038/s41420-025-02862-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02862-6
