
Abstract
Therapeutic cancer vaccines aim to expand and activate antigen-specific T cells for the targeted elimination of cancer cells. While early clinical trials faced challenges due to suboptimal antigen-specific T-cell activation, recent advancements in antigen discovery and vaccine platform engineering have revitalized the field. This review provides a comprehensive overview of key tumor antigens, including tumor-associated antigens, viral oncoprotein antigens, neoantigens, and cryptic antigens, with a focus on their immunogenicity and therapeutic potential. Advances in our understanding of traditional cancer vaccination targets, in conjunction with the timely identification of novel antigen epitopes, have facilitated the strategic selection of vaccination targets. We also discuss the evolution of cancer vaccine platforms—spanning peptide-based formulations to advanced mRNA vectors—emphasizing innovative strategies to optimize antigen delivery efficiency and adjuvant effects. Efficient antigen delivery and adjuvant selection overcome immune tolerance and tumor-induced immunosuppression. Furthermore, we examine recent clinical trial data and emerging combination approaches that integrate cancer vaccines with other immunotherapies to increase efficacy. While significant progress has been made, challenges remain in improving vaccine-induced T-cell responses, overcoming immune suppression, and translating these advances into effective clinical interventions. Addressing these hurdles will be critical for realizing the full potential of cancer vaccines in immunotherapy.
Similar content being viewed by others
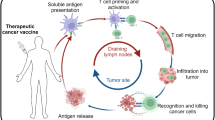

Introduction
Immunotherapy has emerged as a strategy to reprogram the immune system to control tumors, fundamentally changing cancer treatment paradigms. Immune checkpoint inhibitors (ICIs), which block cancer-derived immune-suppressive signals, represent the most successful immunotherapy approach to date [1]. Long-term complete response has been achieved in specific subsets of patients with certain cancers [2]. However, most cancer patients either fail to respond or experience acquired resistance, underscoring the urgent need for new cancer immunotherapy strategies [3].
The tumor microenvironment (TME) involves multiple immune-suppressive mechanisms that inhibit antitumor immunity, allowing cancer cells to evade immune detection [4]. The density of tumor-infiltrating T cells is correlated with patient prognosis. Primary and acquired resistance to ICIs often results from insufficient antitumor T cells [3]. To address this problem, numerous immunotherapy strategies have been developed to expand preexisting and prime de novo tumor-specific T cells to reactivate antitumor immune responses. A promising approach for providing more de novo antitumor T cells is therapeutic cancer vaccination [5]. This strategy focuses on identifying tumor antigens and designing potent vaccine formulations capable of delivering both antigens and adjuvant signals to activate antigen-presenting cells (APCs) for efficient T-cell priming and reactivation inside lymphoid tissues [6]. Earlier clinical trials of cancer vaccines relied on tumor-associated antigens (TAAs) that were overexpressed in cancer cells and utilized traditional vaccine platforms, including proteins, DNA, viral vectors, and whole cancer cells [7]. However, these trials largely failed because of self-tolerance to TAAs, poor adjuvants, toxicity, and the suppressive nature of the tumor microenvironment. Recent technological advancements have revolutionized cancer vaccination by targeting tumor-specific antigens (TSAs).
Cancer is characterized by genomic instability, with numerous point mutations and chromosomal alterations accumulating during tumor progression [8]. These genetic mutations can give rise to altered proteins, which are recognized by the immune system as “nonself” antigens or neoantigens, potentially triggering cellular immune responses that counteract tumor growth [9]. Tumor mutation burdens (TMBs) are generally correlated with the ICI response rate. With rapid advances in genomic sequencing and antigen prediction across diverse HLA complexes, potential immunogenic neoantigens can now be identified with certain precision [10]. Artificial intelligence (AI) may further accelerate the identification of genuine immunogenic neoantigens. These epitopes are tumor-specific and nontolerant to the host immune system. Furthermore, novel vaccine platforms have been developed to enhance antitumor T-cell responses. This review explores tumor antigens and adjuvants, detailing the development of diverse vaccination platforms, including peptide and mRNA vaccines. Optimizing antigen selection and vaccine formulation holds the potential to revitalize cancer vaccination as a treatment strategy capable of stimulating immune responses in ICI-resistant patients.
Antigen selection for the design of cancer vaccines
The development of an effective vaccine for tumor rejection requires the identification of reliable tumor antigens that are highly expressed in tumor tissues and capable of eliciting robust antigen-specific T-cell responses. On the basis of parental gene expression, tumor antigens are broadly categorized into tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) (Fig. 1A). TAAs are self-proteins that are overexpressed in cancer cells, including tissue-specific antigens and developmentally associated germline/cancer testis antigens. TSAs are antigens that are restricted to tumors and are rarely found in normal tissues. They can arise from genetic alterations (neoantigens), oncogenic viruses involved in oncogenic transformation (oncoviral antigens), expression of endogenous retroviral elements, or presumably noncoding sequences within tumors (cryptic antigens).

Cancer Antigen Categories and Personalized Neoantigen Vaccine Design. A Tumor antigens can be roughly classified into five categories: tumor-associated antigens (TAAs), cancer/testis antigens, oncoviral antigens, neoantigens, and cryptic antigens. The table summarizes the key properties of each category, including tumor specificity, central tolerance, patient prevalence, clonality among cancer cells, and clinical exploration. Notable examples are also provided. B Clinical procedure for designing personalized neoantigen vaccines. Tumor and normal tissue (e.g., PBMC) biopsies are obtained from patients and sent for next-generation sequencing (NGS) and mutation calling. The identified nonsynonymous mutations are evaluated for MHC binding, antigen presentation, transcript levels, self-dissimilarity, and clonality. Selected neoantigen epitopes are synthesized into peptide or mRNA vaccines for patient treatment
Some tumor antigens, referred to as shared or universal antigens, are present in multiple patients and serve as promising targets for universal vaccines. In contrast, personalized antigens are unique to specific cancer cells in individual patients and have garnered attention with advances in the rapid identification of neoantigens. In the following sections, we summarize the characteristics of different tumor antigen types, outline key experimental pipelines for formulating neoantigen vaccines in the clinic, and discuss critical considerations for identifying and selecting neoantigens for individualized therapeutic cancer vaccines.
Tumor-associated antigens
Some tumor types highly express tissue-specific antigens, which have been proven to be immunogenic and tested in clinical trials [7, 11]. Human epidermal growth factor receptor 2 (HER2/Neu), a member of the EGFR kinase family, is overexpressed in approximately 20% of breast cancers and some gastrointestinal and ovarian tumors [12]. This antigen is commonly targeted with therapeutic anti-HER2 antibodies or antibody‒drug conjugates (ADCs) [13]. A peptide vaccine comprising both HLA-I- and HLA-II-binding HER2 peptides has induced durable CD8 + T-cell responses in patients, demonstrating the immunogenicity of this antigen [14]. Gp100, which is expressed in melanoma, has been extensively studied as a target for both vaccine and adoptive cell transfer (ACT) therapy [15, 16]. Phase III clinical trials have shown that gp100 peptide vaccination can improve the overall response rate (ORR) when it is combined with high-dose IL-2 but not with ipilimumab [15, 17]. However, gp100 vaccine monotherapy has limited therapeutic efficacy, possibly because of central tolerance mechanisms that eliminate high-affinity gp100-recognizing T cells. In contrast, ACT of engineered T cells with high affinity for gp100 has demonstrated superior antitumor immunity in melanoma models but is associated with autoimmune side effects [18]. Prostatic acid phosphatase (PAP) and prostate-specific antigen (PSA) are expressed on prostate epithelial cells [19]. A phase III trial of sipuleucel-T, an autologous GM-CSF-stimulated monocyte mixture pulsed with PAP, demonstrated a 4-month survival benefit over control vaccination, leading to FDA approval for prostate cancer [20]. However, peptide- or virus-based PAP and PSA vaccines failed to demonstrate a survival benefit in clinical trials, highlighting the need to improve vaccination platforms to increase T-cell activation [21]. Other antigens, such as p53, MUC-1, IDO1, survivin, and PD-L1, are overexpressed in various tumor types and have been evaluated in clinical trials for their ability to induce antigen-specific T-cell responses [22,23,24,25,26].
Germline/testis antigens, which are expressed primarily in fetal tissues and testes, can be re-expressed in malignant tissues, contributing to carcinogenesis and immune evasion. Wilms’ tumor protein (WT1), a developmental transcription factor involved in oncogenesis, is overexpressed in most acute myeloid leukemias (AMLs), breast cancers, and Wilms’ tumors [27]. The National Cancer Institute (NCI) has ranked WT1 as a top candidate for cancer vaccination [28]. Vaccination with a heteroclitic WT1 peptide (Galinpepimut-S) with enhanced HLA affinity induced T-cell responses in most AML patients in a phase II trial, supporting further exploration of this antigen [29]. New York-esophageal cancer 1 (NY-ESO-1), a cancer-testis antigen, is highly expressed in synovial sarcomas and variably expressed in melanoma, ovarian and esophageal cancers [30]. Spontaneous antigen-specific T-cell responses against NY-ESO-1 are frequently observed, indicating its immunogenicity [31, 32]. However, multiple clinical trials have failed, likely because of suboptimal vaccination designs and heterogeneous antigen expression in cancers [33,34,35]. In synovial sarcoma, where NY-ESO-1 expression is homogeneous, adoptive transfer of NY-ESO-1-specific T cells has yielded impressive clinical responses [36]. A state-of-the-art RNA vaccine encoding NY-ESO-1 and three other TAAs has demonstrated promising clinical outcomes, suggesting that potent vaccination platforms targeting multiple antigens can improve efficacy and prevent antigen escape [37]. Melanoma-associated antigen 3 (MAGE-A3), a cancer-testis antigen with antiapoptotic functions, is preferentially expressed in melanoma, non-small cell lung cancer (NSCLC), and breast cancer [38]. A TLR9 agonist-adjuvanted MAGE-A3 vaccine induced CD4+ T-cell and antibody responses in a phase III trial but failed to improve disease-free survival [39, 40]. In a large randomized trial, a multivalent melanoma vaccine containing MAGE-A3, Melan-A, gp100, and tyrosinase (seviprotimut-L) improved outcomes in subsets of younger patients and those with ulcerated primary melanomas, emphasizing the importance of patient selection for vaccination [41].
Central tolerance mechanisms may delete high-affinity TCR clones against TAAs or induce regulatory T cells toward tissue-specific antigens. Thus, while TAAs are potential candidates for patients with a low tumor mutational burden (TMB), their efficacy is limited by immune tolerance, poor immunogenicity, and tumor heterogeneity. Potent adjuvants may overcome tolerance but can cause detrimental autoimmunity due to the expression of the antigen in normal tissues. Therefore, the development of TAA-based vaccines must balance tumor-specific T-cell responses with autoimmune toxicity.
Tumor-specific antigens
Currently, more efforts are focused on developing cancer vaccines targeting shared TSAs or personalized neoantigens, which could circumvent immune tolerance and elicit potent tumor-specific T-cell responses with improved safety.
Oncoviral antigens
Some oncogenic viruses, such as Epstein–Barr virus (EBV) and human papillomavirus (HPV), can drive oncogenic transformation [42]. These viral antigens are not subject to central tolerance, making them ideal targets for vaccination. EBV-encoded latent membrane proteins (LMP1 and LMP2) are expressed in natural killer (NK)–T-cell lymphoma, nasopharyngeal carcinoma, and other malignancies [43]. Adoptive transfer of autologous cytotoxic T cells targeting LMPs has resulted in durable clinical responses in lymphoma patients, spurring interest in clinical trials for EBV vaccines [44]. An mRNA vaccine encoding truncated domains with multiple T-cell epitopes from EBV proteins has demonstrated cellular immunity and prolonged survival in preclinical models [45]. Additionally, a modified vaccinia Ankara (MVA) virus expressing an Epstein–Barr nuclear antigen (EBNA)–LMP2 fusion protein increased CD4+ and CD8+ T-cell responses in EBV-positive patients, encouraging further large-scale clinical studies [46].
HPV E6 and E7 are viral TSAs that inhibit the tumor suppressors p53 and retinoblastoma protein (pRb), thereby promoting proliferation and tumorigenesis in squamous epithelial cells [47]. Nearly 60% of oropharyngeal cancers and almost all cervical cancers are positive for E6 and E7 antigens, making them highly attractive targets for vaccination [48]. Both mRNA-based and peptide-based vaccines have elicited robust antigen-specific T-cell responses in preclinical HPV mouse tumor models [49, 50]. In clinical studies, synthetic long peptides (SLPs), DNA, and viral-vector vaccines have demonstrated promising efficacy in treating cervical intraepithelial neoplasia, an early stage of carcinogenesis [51,52,53]. However, further research is needed to evaluate the effectiveness of E6/E7-based vaccines in more advanced-stage tumors, potentially in combination with other therapies to achieve clinical benefit.
Neoantigens
Neoantigens are a subset of TSAs arising from tumor-specific genetic mutations. Neoantigen epitopes are absent in normal tissues, making them exempt from immune tolerance and more recognizable by T cells [6, 10, 54,55,56,57]. Single-cell RNA sequencing (scRNA-seq) across various cancers has suggested that most intratumoral T cells recognize neoantigen epitopes rather than TAAs [58,59,60,61,62], underscoring their superior immunogenicity and potential for therapeutic vaccination.
Molecular characterization of neoantigens
Genomic mutations, such as single-nucleotide variants (SNVs), insertions and deletions (indels), and gene fusions, can generate immunogenic neoantigens [10]. SNVs involve the substitution of single nucleotides within the genome and are the most abundant mutation type in cancers [63]. The SNV burden is correlated with the clinical efficacy of ICI therapies [64, 65]. Early studies focused on SNV-derived neoantigens, demonstrating primary clinical efficacy in various tumor models. Some common SNVs, such as the KRAS G12 and the IDH1 R132H mutations, are immunogenic and are shared across cancer patients [66, 67]. However, most SNVs are patient-specific and limited by tumor heterogeneity [68]. Indels can introduce frameshift sequences unrelated to the original protein with potential immunogenicity [69]. Clinical studies have shown that indel-derived neoantigens exhibit higher HLA-binding affinity than SNV-derived epitopes do. Moreover, shared immunogenic polyepitopes from indels have been observed in microsatellite-unstable tumors [70]. Gene fusions occur through chromosomal rearrangements, creating new sequences. Although gene fusion-derived antigens are rare compared with SNVs and indels [71], a well-known example is the BCR–ABL1 fusion gene, which is found in ~90% of patients with chronic myelogenous leukemia (CML) [72]. In addition to genomic mutations, neoantigens can also arise from transcriptional and posttranscriptional alterations, such as alternative splicing and A-to-I RNA editing [73,74,75,76,77,78,79,80]. However, these neoantigens require further validation in clinical trials.
Although some neoantigens are shared among a small subset of patients, most neoantigens are highly individualized [68, 69, 81]. The development of personalized neoantigen vaccines is therefore necessary. However, this approach faces significant challenges, as the immunogenicity of neoepitopes varies considerably, with only 1–2% of identified nonsynonymous mutations in tumors recognized by T cells [82]. Advancing neoantigen vaccines requires sophisticated experimental methods, AI-driven analysis, and computational pipelines to identify tumor-expressed mutated peptides and assess their vaccine potential, offering a promising yet complex and resource-intensive approach for cancer immunotherapy.
Identification of neoantigen epitopes
Identifying mutated neoantigen epitopes is a fundamental step in developing personalized cancer vaccines. This process begins with next-generation sequencing (NGS) of malignant tissues to detect tumor-specific genetic mutations (Fig. 1B). Sequencing reads are aligned to a reference genome from the patient’s normal tissue, enabling the detection of nonsynonymous mutations through mutation-calling software [83]. Detection reliability varies among SNVs, indels, and gene fusions, with SNVs exhibiting the highest sensitivity and specificity [81, 84,85,86]. To increase accuracy, researchers often select consensus mutations identified by at least two independent mutation-calling software programs for downstream analysis.
Another approach combines NGS with mass spectrometry to profile the HLA immunopeptidome [87,88,89]. Procedurally, HLA-peptide complexes are immunoprecipitated from tumor cells, and the eluted peptides are analyzed via mass spectrometry to identify HLA-bound epitopes. These analyses reveal not only the mutated neoantigens presented on HLA molecules but also their relative abundance, providing valuable insights into their immunogenic potential. Additionally, this approach enables the discovery of nonmutational antigens, such as cryptic antigens. However, sensitivity limitations and the need for large tumor samples hinder the clinical application of mass spectrometry. Despite these challenges, mass spectrometry-derived datasets are instrumental in refining bioinformatics tools that predict peptide-HLA binding affinity and antigen processing efficiency, thereby aiding in the prioritization of neoantigens for therapeutic use [90, 91].
While hundreds of nonsynonymous mutations might be identified within a tumor, only a small subset qualifies as viable neoantigens for vaccine development [82]. Selecting optimal candidates is critical to ensuring a robust immune response, minimizing antigen escape, and effectively targeting tumor cells. Several parameters are considered during this process, including binding affinity to the patient’s HLA molecules, immunogenicity, transcript expression levels, and clonality. A promising neoantigen should bind to HLA molecules and be presented on APCs or tumor cells for efficient T-cell priming and cytotoxicity. Algorithms trained on datasets derived from mass spectrometry and peptide-binding assays are now widely used to predict peptide-HLA binding affinities, filtering out candidates with poor presentation potential [91,92,93,94]. The sufficient presentation of neoantigens on tumor cells is partly determined by the abundance of neoantigen-encoding RNA transcripts, as high transcript levels correlate with increased pMHC complex density on tumor cells [95]. RNA sequencing data analysis helps quantify transcript levels, ensuring that selected neoantigens are sufficiently expressed to trigger immune responses [96, 97].
Immunogenicity, the ability of a neoantigen to provoke a strong immune response, is another key criterion. Neoantigens that highly discriminate from self-peptides are less immune tolerant and more likely to be recognized by T cells [98, 99]. Bioinformatic algorithms assess peptide similarity to the normal self-proteome, prioritizing those with the highest likelihood of immune recognition [98, 100]. Additionally, some neoantigens exhibit sequence similarity to pathogen-derived antigens, which can trigger cross-reactivity with preexisting T-cell populations primed against infectious agents [101, 102]. These neoantigens may benefit from this preformed immune memory, enhancing their therapeutic efficacy [99].
Clonality is another critical consideration in neoantigen selection. Tumors are genetically heterogeneous, with subclonal mutations arising at different stages or sites during cancer progression [103]. Clonal neoantigens, which occur early in tumorigenesis and are present in most tumor cells, are preferred over subclonal mutations. These clonal neoantigens reduce the risk of tumor escape. Mutations in genes that promote oncogenic transformation or tumor cell survival are desirable targets for vaccine development, as they are less likely to be eliminated by selective pressure [104]. Despite these advances, neoantigen selection remains challenging due to the sheer number of potential epitopes, difficulties in defining immunogenicity, and the complexity of tumor biology. Most selected mutated antigens might not be able to stimulate effective T-cell responses, and 20–30 neoantigen candidates are commonly included during the development of peptide- or mRNA-based cancer vaccines. Integrating experimental data, clinical insights, and advanced bioinformatics—particularly artificial intelligence and machine learning—will streamline the selection of neoantigen candidates. Optimized computational pipelines incorporating HLA-binding affinity, transcript expression, self-dissimilarity, and clonality will enhance the efficacy of personalized cancer vaccines. However, further technological advancements are needed to simplify and expedite individualized vaccine production for broader clinical applications.
Cryptic antigens
Emerging evidence has shown that noncoding gene elements can encode antigen peptides presented by HLAs to induce T-cell responses. These antigens are referred to as cryptic antigens [105]. Cancer-associated epigenetic dysregulation, splicing aberrations, and cellular stress may promote the generation of tumor-specific or tumor-associated cryptic antigens, which may be immunogenic [106,107,108]. Human endogenous retroviral elements (HERVs) are remnants of ancient retrovirus integration events and are located throughout the human genome. These elements are usually silent in normal tissues via epigenetic modifications [109]. However, under pathological conditions such as cancer, HERVs can be reactivated, leading to the expression of viral antigens that serve as potential targets for cancer vaccination [110,111,112,113,114]. Circular RNAs (circRNAs) constitute another class of noncoding RNAs that play crucial roles in cancers [115]. Recent studies have revealed that circRNAs are translatable and can produce distinct peptides, making them valuable sources of TSAs. CircFAM53B is a circRNA expressed in breast cancer and melanoma that translates immunogenic peptides and elicits a T-cell response for antitumor effects [87]. Cryptic antigens are shared across multiple cancer types and are potential targets for cancer vaccines, warranting further exploration in clinical settings.
Overall, TSAs are promising targets for cancer vaccines. Technology advancements permit the timely design of personalized vaccines, although they are costly. Identifying appropriate antigens is the crucial first step in vaccine design. Once identified, antigens must be efficiently delivered to APCs and processed for T-cell priming in the lymphoid organ. In addition to TCR signaling, costimulatory signaling and cytokines are required for successful T-cell priming by APCs. Owing to the immune-suppressive nature of the cancer microenvironment, potent adjuvants and delivery systems are necessary to efficiently activate APCs and facilitate T-cell activation and differentiation (Fig. 2). In subsequent sections, we examine current vaccine platforms, focusing on peptide and mRNA vaccines, tracing their development through preclinical and clinical research.

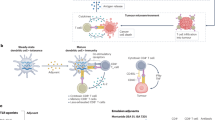
Antigen presentation of cancer vaccines. Peptide vaccines: Synthetic long peptides (SLPs) and adjuvants, such as Toll-like receptor (TLR) agonists, are injected to stimulate an immune response. Antigen-presenting cells (APCs) take up SLPs, process them via cross-presentation pathways, and present peptide‒MHC complexes to T cells. Adjuvants activate APCs, increasing the expression of costimulatory markers such as CD80/86 and promoting their migration to lymph nodes, where they prime and activate T cells. mRNA vaccines: mRNA vaccines encoding tumor antigens are internalized by dendritic cells (DCs) via endocytosis or macropinocytosis, followed by endosomal escape and cytosolic release of mRNA facilitated by ionizable lipids. Within the cytoplasm, mRNAs are translated into antigens, which are subsequently processed into epitopes and presented on the cell surface through interactions with MHC molecules. During this process, the mRNA vaccine might activate innate immune pathways, including TLR7, MDA5, or RIG-1, thereby stimulating innate immune responses. These antigen-presenting DCs then prime antigen-specific T cells, which differentiate into either memory T cells or effector T cells, thereby inducing tumor cell death
Peptide vaccine for cancer therapy
The identification of precise TCR-recognizing epitopes enables the use of synthetic peptides instead of whole tumor cell lysates or proteins in cancer vaccination [116]. Synthetic peptides encompass the minimal immunogenic regions of protein antigens, allowing for the precise modulation of immune responses. The safe and operable manufacture of neoantigen peptides ensures their clinical feasibility; however, their inherently low immunogenicity necessitates additional delivery systems and adjuvants to elicit an effective antigen-specific immune response [117]. Research on peptide-based cancer vaccines has focused on designing peptide sequences to enhance their processing and presentation by APCs [118], as well as combining them with various adjuvant systems to improve their immunogenicity [119]. Furthermore, next-generation peptide vaccines focused on developing delivery systems such as lipid-based carriers to specifically target both peptides and adjuvants to dendritic cells, thereby increasing their activation and antigen presentation for T-cell priming [120].
Design of peptide
Initial studies identified minimal cytotoxic T-cell-recognizing epitopes, typically 8--10 amino acids in length, for vaccination purposes [117]. However, this approach results in limited antigen-specific T-cell responses and unsatisfactory clinical outcomes [121,122,123,124,125]. Subsequent investigations revealed that vaccination with minimal MHC-I binding epitopes could lead to T-cell anergy, possibly due to peptide binding to nonprofessional APCs, which primes T cells in the absence of costimulatory signals [126,127,128]. To address these challenges, synthetic long peptides (SLPs) have been proposed for vaccination. SLPs, which are usually 15--25 amino acids in length, comprise well-defined CD8 epitopes and extension sequences that may contain potential CD4 epitopes. The inclusion of CD4 epitopes enhances CD8+ T-cell priming, and antigen-specific CD4+ T cells may perform antitumor functions in the TME via various mechanisms [129,130,131]. Studies in mice have shown that the intracellular presentation of SLPs is more efficient than that of full-length antigens, resulting in antigen presentation to both CD4+ T cells and CD8+ T cells [132, 133].
The flanking amino acids and the linker regions between the CD4 and CD8 epitopes can significantly impact antigen presentation efficiency. One study using a DNA-based HBV vaccine in HLA-A*0201 transgenic mice revealed that the processing efficacy of an epitope strongly correlated with the presence of specific amino acid residues at the C + 1 position following the C-terminus of the CD8 epitope. The presence of an amide or positively charged amino acid residue contributes to optimal processing, whereas aromatic, negatively charged, or aliphatic residues are associated with suboptimal processing [134]. Another study utilizing the model antigen MELOE-1 screened cathepsin-sensitive linkers to join CD8 and CD4 epitopes. Cathepsin is one of the key proteases in the endosome of dendritic cells (DCs) and significantly impacts antigen processing and presentation. This research reported several candidate linkers that could enhance the cross-presentation of class I epitopes to CD8-specific T cells by up to 100-fold compared with native SLPs, with minimal impact on CD4 epitope presentation [130].
The epitope sequences that recognized by CD8+ T cells can be modified to enhance antigen-specific responses [117]. Heteroclitic peptides, which introduce specific residue substitutions to improve the immunogenicity of original peptides, are beneficial for tumor-associated antigens, which often exhibit poor immunogenicity due to their central tolerance [116, 135]. An important strategy for designing heteroclitic peptides is to increase the binding affinity of cytotoxic T-cell epitopes to MHC-I molecules, as this correlates with improved immunogenicity [136,137,138]. Simple modifications to MHC anchor residues, which substitute for suboptimal anchor residues, have proven effective in enhancing the immunogenicity of tumor-associated antigens, including gp100, p53, Trp-1, and survivin [135, 136, 139,140,141].
Overall, modifying the TCR-recognizing region and incorporating specific enzymatic cleavage sites can enhance the pMHC-TCR interaction and antigen presentation, thereby increasing immune responses. However, SLPs alone cannot efficiently activate T cells without providing costimulatory signals or cytokine signals. Potent immune adjuvants are required to activate APCs and provide costimulatory signals and cytokines.
Adjuvant system
Aluminum-based adjuvants have been widely used in prophylactic vaccinations that induce an antibody response [142]. Although they are safe, aluminum salts induce an enhanced Th2 immune response with suppressed CD8+ T-cell activation, which has hindered their application in therapeutic cancer vaccinations [143, 144]. Thus, other adjuvants have been explored to enhance CTL responses in peptide-based cancer vaccines.
Innate immune sensors, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), and cytosolic DNA sensors, detect pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPs) and activate APCs to initiate adaptive immune responses [145]. Various agonists targeting these sensors have been synthesized and employed as adjuvants in peptide-based cancer vaccines [144, 146]. The structural characteristics of these immune sensors and downstream signaling pathways have been extensively characterized and reviewed elsewhere [147]. Here, we briefly describe the key immune cells and molecules that are activated by several commonly used adjuvants to enhance peptide vaccination-induced adaptive immunity.
Poly-ICLC, derived from polyinosinic–polycytidylic acid (poly I:C) and stabilized with carboxymethylcellulose and polylysine, is a commonly used adjuvant for neoantigen-based peptide vaccines [148,149,150]. Following endocytosis by APCs, poly-ICLC engages TLR3 on endosomal membranes and subsequently activates melanoma differentiation-associated protein 5 (MDA5) in the cytosol [151]. Preclinical studies indicate that TLR3 signaling upregulates CD80 and CD86 on DCs, enhancing interleukin-12 (IL-12) secretion and thereby facilitating T-cell priming. Concurrently, MDA5 activation induces robust type I interferon (IFN) production, which drives IL-15α/IL-15 complex formation on myeloid cells and promotes CTL expansion. Genetic ablation of the IFNα/β receptor in non–T-cell hematopoietic populations or blockade of IL-15 abrogated CTL expansion induced by poly–ICLC-adjuvanted vaccination. These findings illustrate how poly-ICLC simultaneously engages multiple innate sensors to modulate cytokine profiles and enhance CTL responses [152]. In addition to immune-stimulating activity, cationic stabilizers can induce a “proton sponge” effect that destabilizes endosomal membranes and promotes antigen cross-presentation.
Saponins are plant-derived natural products that can be formulated in liposomes with additional adjuvants, such as monophosphoryl lipid A (MPL), a TLR4 agonist. Saponin-based adjuvant systems elicit potent Th1-biased immune responses and have been approved for the development of vaccines against infectious diseases, including shingles (Shingrix) and respiratory syncytial virus (Arexvy) [153]. Upon uptake by APCs via a cholesterol-dependent mechanism, saponins destabilize endosomal membranes to facilitate antigen leakage and cross-presentation [154]. Preclinical studies have demonstrated that saponins also induce lipid body formation in CD11b+ DCs, promoting cross-presentation through an endosome escape-independent pathway [155, 156]. In addition to enhancing antigen uptake and cross-presentation, saponins activate the NLRP3 inflammasome and caspase-1 in lymph node-resident CD169+ macrophages, which recruit innate immune cells and promote DC maturation [157]. Furthermore, the aldehyde group of saponins can form Schiff base adducts with T-cell surface receptors (e.g., CD2), delivering costimulatory signals that increase Th1 cytokine production (IFN-γ and IL-2) [158].
The cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) synthesizes cyclic dinucleotides (CDNs) upon binding to cytosolic double-stranded DNA. CDNs subsequently activate STING (stimulator of interferon genes) on the endoplasmic reticulum, triggering IRF3- and NF-κB–dependent signaling pathways that drive robust production of type I IFN and other proinflammatory cytokines. The cGAS–STING axis orchestrates both innate and adaptive immune responses and is central to the antitumor effects elicited by radiotherapy and various chemotherapeutic agents [159]. Thus, STING agonists have been explored as adjuvants in cancer vaccination. In one preclinical model, the STING agonist ABM5 was covalently linked to tumor antigen peptides to facilitate delivery to the endoplasmic reticulum, thereby promoting antigen cross-presentation while simultaneously activating STING. Unlike TLR agonists, STING agonists must reach the cytosol to bind their receptor, necessitating specialized delivery systems for STING-based vaccines. The ABM5-peptide conjugate was encapsulated in lipid nanoparticles to ensure cytosolic release. This vaccine formulation was well tolerated in vivo and demonstrated significantly greater efficacy than conventional adjuvants, such as Poly-ICLC and ISCOMs [160]. In the clinic, certain patients harbor STING variants—for example, the R232H polymorphism—that exhibit reduced responsiveness to canonical CDN agonists [161]. Thus, several novel STING agonists have been developed to overcome these limitations. Li et al. reported a pH-sensitive polymer, PC7A, that activates STING by inducing STING–PC7A biomolecular condensates. PC7A binds a distinct, noncompetitive site on STING—separate from the cGAMP-binding pocket—enabling the activation of R232H STING variants [162]. When used as a vaccine adjuvant, PC7A induced potent T-cell responses [163]. Although STING agonist–based peptide vaccines have shown promise in preclinical studies, clinical exploration remains limited, and further research is needed to validate their efficacy in humans.
Current peptide-based cancer vaccines under clinical evaluation often consist of a simple admixture of TLR agonists, such as poly-ICLC, with antigenic peptides [164]. In some formulations, a water-in-oil emulsion adjuvant such as Montanide ISA 51 is included to enhance antigen retention at the injection site. However, owing to the low molecular weights of both peptides and adjuvants, these components rapidly diffuse into the systemic circulation. As a result, only a limited fraction of the antigen is internalized by local APCs. Moreover, potent adjuvants may induce systemic innate immune activation, leading to dose-limiting toxicity [165, 166]. To overcome these limitations, next-generation peptide vaccine strategies in preclinical development focus on engineering delivery platforms that codeliver both antigens and adjuvants directly to APCs, increase antigen uptake and cross-presentation, and stimulate APCs for T-cell priming while minimizing systemic exposure and toxicity.
Toward next-generation peptide vaccination
Various delivery platforms, including PLGA nanoparticles, hydrogels, inorganic nanomaterials, and liposomes, have been developed to increase the efficacy of peptide-based cancer vaccines. Among these, lipid-based carriers have received the most extensive investigation because of their favorable biocompatibility, structural versatility, and ease of functionalization. In the following section, we focus on lipid-based platforms and discuss the diverse strategies employed to optimize lipid carrier design for the efficient codelivery of antigens and adjuvants to APCs (Fig. 3).

Developmental trajectory of peptide vaccines. Upper panel: Early clinical peptide vaccine formulations combining minimal CD8 + T-cell epitopes with adjuvants such as incomplete Freund’s adjuvant (IFA), creating an antigen “depot” at the injection site. However, this leads to antigen presentation by nonprofessional APCs, causing sustained chronic inflammation at the injection site. Antigen-specific T cells accumulate at the injection site and undergo tolerance and dysfunction. Middle panel: Many current clinical trials use synthetic long peptides (SLPs) mixed with Toll-like receptor (TLR) agonists, such as polyICLC. SLPs promote antigen presentation by professional APCs and may contain CD4 + T-cell epitopes, which help recruit CD4 + T cells to assist in CD8 + T-cell priming. This improves T-cell priming and effector T-cell generation. However, owing to the small size of TLR agonists and SLPs, most of the injected reagents leak into the circulation, with limited antigen and adjuvant uptake by APCs. Additionally, some TLR agonists can induce systemic proinflammatory responses, resulting in toxicity. Lower panel: New-generation peptide vaccines use particulate delivery systems for antigens and adjuvants, which improve lymphoid tissue targeting. Antigens and adjuvants can be designed to self-assemble into nanometer-scale complexes or be incorporated into lipid-based nanoparticles, which are preferentially taken up by APCs. This design allows for the efficient codelivery of antigens and adjuvants, reducing systemic inflammatory responses and inducing robust T-cell responses. However, large-scale production and quality control of lipid nanoparticle-based vaccines remain significant challenges. Another strategy involves conjugating peptides or adjuvants to moieties that bind albumin, which increases APC uptake and enhances T-cell responses
Physicochemical properties influencing lymph node drainage and cellular uptake
The physicochemical properties of lipid nanoparticles—including size, surface charge, hydrophobicity, and flexibility—play critical roles in determining their lymphatic drainage and cellular uptake [167]. Particle size is a key determinant of biodistribution: nanoparticles smaller than 10 nm in diameter are prone to rapid diffuse into the bloodstream, whereas those larger than 100 nm are often retained at the injection site. Intermediate-sized particles are more likely to enter lymphatic vessels and reach draining lymph nodes [168]. Smaller particles are generally internalized by immune cells through multiple endocytic pathways, including clathrin- and caveolae-mediated endocytosis, which are efficient routes for antigen processing. In contrast, larger particles are primarily taken up by phagocytosis, a process typically restricted to professional phagocytes and associated with lower cross-presentation efficiency [169]. Surface charge and hydrophobicity also influence biodistribution and cellular uptake in opposing ways. Positively charged or hydrophobic particles are more likely to interact with and become entrapped in the negatively charged interstitial matrix, potentially limiting lymphatic drainage. However, such particles more readily associate with the negatively charged plasma membrane, facilitating endocytosis and enhancing endosomal escape. Conversely, hydrophilic or negatively charged particles exhibit improved lymphatic transport but may be less efficiently internalized by APCs [167, 170]. Flexibility represents an additional, independent factor influencing lymph node trafficking. In one study, lipid nanodiscs exhibited greater endothelial penetration and lymphatic drainage than liposomes did, which was attributed to their greater deformability [171]. However, the effect of particle elasticity on cellular uptake remains controversial. For example, Palomba et al. demonstrated that rigid nanoparticles were internalized five times more efficiently than their soft counterparts were internalized, whereas another study reported that liposomes with higher membrane fluidity were more readily taken up by APCs [50, 172]. These conflicting findings may reflect differences in nanoparticle composition and experimental context, suggesting that the role of particle elasticity in immune cell uptake is likely material- and system dependent, warranting further investigation.
Lipid component selection for enhanced delivery efficiency
The lipid composition of nanoparticles plays a pivotal role in determining their physicochemical properties, biodistribution, and immunological performance. The chemical structure of the lipid headgroup, in particular, modulates surface charge and immunomodulatory characteristics [173]. Cationic lipids confer a positive surface charge, which can enhance cellular uptake but may also induce undesired inflammatory responses through activation of the complement system and myeloid cells [174, 175]. Additionally, lipid headgroups can interact electrostatically or hydrophobically with serum proteins, leading to the formation of a protein corona that significantly influences nanoparticle fate in vivo, often enhancing clearance by the reticuloendothelial system [176, 177]. To mitigate nonspecific protein adsorption, polyethylene glycol (PEG)-conjugated lipids are frequently incorporated into lipid formulations. While PEGylation can reduce protein corona formation and prolong the circulation time, a high density of PEG chains may hinder their cellular uptake and impair endosomal escape [178]. Alternatively, tailoring the lipid headgroup to favor interactions with specific serum proteins can promote selective delivery to target tissues via receptor-mediated mechanisms [50, 179]. For example, precoating lipid particles with targeting moieties such as anti-CD40 antibodies has emerged as a strategy to direct particles toward antigen-presenting cells (APCs) and enhance their in vivo bioactivity [177]. The lipid tail also critically influences nanoparticle behavior. Unsaturated, short, or branched lipid tails increase membrane fluidity, facilitating faster antigen release and endosomal escape, but may compromise thermal stability [180,181,182]. In a systematic study evaluating the effects of both lipid headgroups and tails in a liposome-based peptide vaccine platform, phosphoethanolamine headgroups were found to preferentially bind serum complement protein C3, mediating spleen-targeted delivery in a C3-dependent manner. Additionally, lipids bearing 1,2-dioleoyl-sn-glycero tails demonstrated enhanced fluidity and a favorable phase transition temperature, leading to improved cellular uptake and robust T-cell activation [50]. In addition to the structural lipids that form the lipid bilayer, functional excipients such as PEGylated lipids and cholesterol are commonly incorporated to increase particle stability and circulation time [180]. Moreover, lipids can be chemically modified to allow covalent conjugation with key functional molecules such as antigenic peptides or immune-stimulatory adjuvants, further expanding their utility in rational vaccine design.
Incorporating antigens and adjuvants into lipid carriers
Antigenic peptides and adjuvants can be integrated into lipid-based delivery systems either by noncovalent or covalent attachment to the lipid surface or by encapsulation within the lipid core. Additionally, small hydrophobic adjuvants can be embedded within the lipid bilayers of liposomes [183]. Compared with noncovalent attachment or encapsulation, covalent conjugation allows for precise control over the surface density of antigens or adjuvants by modulating the chemical linkers incorporated into the lipid structure. Covalent linkages also increase the molecular loading efficiency and improve the consistency and stability of lipid particles [184]. Preclinical studies have demonstrated that lipid carriers covalently conjugated with peptides and adjuvants exhibit potent immunogenicity. For example, Kuai et al. successfully conjugated antigens and CpG oligonucleotides to high-density lipoprotein (HDL)-mimicking nanodiscs, markedly enhancing codelivery to lymphoid organs and sustaining antigen presentation by dendritic cells. This strategy resulted in a 47-fold increase in neoantigen-specific cytotoxic T lymphocytes (CTLs) compared with soluble vaccines and a 31-fold increase compared with the level of CpG in Montanide, one of the most potent adjuvants currently in clinical use [185]. Another study investigated the effects of different antigen–adjuvant conjugation strategies on the immunogenicity of spherical nucleic acid (SNA)-based liposome vaccines. In this study, CpG oligonucleotides were anchored on the liposome surface, while antigens were either (i) encapsulated in the liposome core (E structure), (ii) covalently conjugated to surface-anchored oligonucleotides (A structure), or (iii) hybridized to CpG through complementary oligonucleotides chemically linked to the antigen (H structure). Among these, the H structure enables the most efficient codelivery of antigen and adjuvant in vivo and best synchronized antigen presentation with costimulatory molecule expression, thereby eliciting superior T-cell responses [186]. Furthermore, the chemical linkers used for covalent conjugation can be engineered to allow either rapid or controlled release of antigens in response to the acidic and redox conditions of endosomes. In one study, antigens were conjugated via either noncleavable or redox-responsive cleavable linkers, and faster-releasing linkers led to stronger immunostimulatory effects [187]. Collectively, these findings highlight that the method of incorporating antigens and adjuvants into lipid particles, along with the design of chemical linkers and the structural configuration of the vaccine, plays a crucial role in determining vaccination efficacy.
Identifying optimal lipid particle formulations by screening
Due to the numerous technical variables influencing the efficacy of lipid particle–based peptide vaccines, rational and systematic large-scale screening may be necessary to identify optimal formulations. While many studies have focused on optimizing the lipid composition of lipid nanoparticles to increase mRNA delivery and translation, relatively few have systematically evaluated the key factors that govern the immunogenicity of peptide-loaded lipid particles [188, 189]. Notably, Gokay et al. employed high-throughput screening combined with machine learning to investigate the design of spherical nucleic acid (SNA)-based peptide vaccines. They evaluated 11 design parameters—including the core diameter, lipid composition, conjugation chemistry, and density of antigens and adjuvants—by generating hundreds of structurally distinct SNAs. A mass spectrometry–based assay was used to assess immune activation rapidly, and machine learning models were applied to quantitatively predict immunostimulatory effects. Through this approach, they identified and ranked critical factors influencing vaccine efficacy and developed a nonlinear model for predicting the immunogenic potential of SNAs [190]. Despite this progress, further efforts are needed to systematically define generalizable design principles for lipid particle–based peptide vaccines.
Barriers to the clinical application of lipid carrier-based peptide vaccination
Despite remarkable success in preclinical studies, significant challenges remain in synthesizing and scaling up clinical-grade complex formulations containing multiple components, hindering the application of diverse vaccine nanoparticles in clinical trials. Additionally, the instability of such formulations—both physically and chemically—limits long-term storage and clinical applicability [191, 192]. Current neoantigen-based vaccination strategies further complicate the clinical translation of lipid particle–based peptide vaccines. While preclinical studies often employ well-defined model antigens, such as the MHC-I epitope of ovalbumin, the HPV16 E7 protein, or the ADPGK neoantigen in MC38 cells, human neoantigen vaccination requires the synthesis of multiple, patient-specific peptides. These neoantigens often differ in their chemical properties, leading to inconsistent encapsulation efficiencies in lipid particles. Moreover, when conjugated to lipid particle surfaces, these peptides can significantly alter surface characteristics such as hydrophobicity and charge, resulting in final products with varying stability and physicochemical profiles. These factors pose substantial challenges for the clinical application of lipid particle–based neoantigen peptide vaccines.
Another strategy to improve lymph node trafficking and APC uptake is “albumin hitchhiking,” where adjuvants and peptides are linked to albumin-binding lipids, promoting lymphatic drainage and efficient APC targeting in vivo [193,194,195,196,197,198]. For example, Zhu et al. developed self-assembling albumin-vaccine nanocomplexes (AlbiVax) by conjugating molecular vaccines with Evans blue, a dye with a high affinity for albumin. Compared with the benchmark IFA adjuvant, AlbiVax achieved 100-fold more efficient codelivery of CpG and antigens to lymph nodes, eliciting ten-fold higher frequencies of peripheral antigen-specific CD8+ T cells with immune memory [193]. Another study utilized a similar albumin hitchhiking strategy by tagging peptides and adjuvants via a PEG linker to amphiphilic lipid tails that bind to albumin in vivo. This amphiphilic peptide vaccine also accumulated in lymph nodes and enhanced the generation of antigen-specific CD8 + T cells [194, 198]. These designs enhance vaccine delivery efficiency through simple molecular modifications, enabling stable, large-scale production suitable for clinical application.
In summary, next-generation peptide vaccines aim to codeliver antigens and adjuvants to the APCs of draining lymph tissues while minimizing peripheral toxicity via various delivery systems. While lipid–nanoparticle formulations have exhibited superior efficacy in preclinical studies, their clinical application is hindered by complex manufacturing procedures and challenges in large-scale, consistent production. Other strategies, such as albumin hitchhiking, may be clinically practicable; however, further investigations are needed on reliable delivery systems for clinical application.
Clinical trials
Peptide vaccines have demonstrated good safety profiles in clinical trials. However, several large phase III cancer vaccine studies in patients with advanced disease have reported limited survival benefits [121,122,123,124,125]. The failure of these early clinical trials can be attributed to several factors, including the use of tolerant TAA epitopes, ineffective adjuvants, insufficient antigen delivery, and a short half-life inside dLN [199].
Some ongoing clinical studies using neoantigen peptide vaccines have shown promising preliminary results (Table 1). One study tested a poly-ICLC adjuvant neoantigen peptide vaccine in glioblastoma patients following surgery and radiotherapy. In patients who do not receive dexamethasone, vaccination induces the generation of a memory immunity with polyfunctional neoantigen-specific CD4+ and CD8+ T cells in circulation. Single-cell TCR analysis revealed that TCRαβ clonotypes were shared among tumor-specific T cells from tumor tissue and peripheral blood, indicating that T cells migrated from the peripheral blood to the tumor. Intratumoral T cells upregulate multiple inhibitory molecules, providing a rationale for combining this therapy with ICB [148].
Another study combined a poly-ICLC adjuvant neoantigen peptide vaccine with nivolumab in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. This combination therapy induced durable neoantigen-specific T cells with cytotoxic potential, which migrated and infiltrated into tumor tissues. Importantly, vaccination also helps induce T-cell responses against nonvaccinated neoantigen epitopes, which may prevent epitope escape and tumor relapse. Notably, patients who experienced this epitope spreading response had longer progression-free survival (PFS) than those who did not [149]. Similar findings were reported in melanoma patients vaccinated with neoantigens, where memory T-cell formation and epitope spreading were observed [150].
For shared tumor antigens, novel vaccine formulations have yielded promising clinical results. For example, a synthetic TLR2 ligand was conjugated to SLPs derived from the HPV16 virus. This vaccination approach effectively induced robust HPV16-specific T-cell immunity without eliciting severe side effects in patients with HPV16-positive (pre)malignancies [200]. Another study conjugated mutant KRAS (mKRAS) peptides and a CpG adjuvant to albumin-binding lipids via a PEG linker. This vaccine was well tolerated in vivo, with mKRAS-specific T-cell responses observed in 21 of the 25 patients and tumor biomarker responses (longitudinal ctDNA or tumor antigen) noted in 21 of the 25 patients, including biomarker clearance in six of the patients. T-cell responses are correlated with PFS; patients with T-cell responses above the median exhibit delayed cancer recurrence compared with those with responses below the median [198].
These studies suggest that the clinical success of peptide vaccines may depend on several factors: the use of multiple neoantigen epitopes with potent adjuvants to overcome immune tolerance and epitope loss, the combination of vaccination with immune modulators such as immune checkpoint blocking antibodies to mitigate immune suppression, and the application of novel vaccine formulations that efficiently target antigens and adjuvants to APCs. Whether and how vaccines can be combined with first-line therapies, such as radiation and chemotherapy, remain to be explored.
mRNA vaccine
DNA-based vaccination was widely explored before the breakthrough of mRNA-based vaccination. However, complex electroporation procedures for delivering DNA into the nucleus for transcription and translation are needed, but show low efficiency and introduce the risk of genomic integration of transfected DNA molecules. The majority of DNA molecules are taken up by muscle cells but not APCs, leading to insufficient antigen presentation in the dLN [201]. The mRNA vaccine delivers mRNA molecules into the cytosol, allowing for high quantities of antigen production through translation and preventing the risk of genomic integration. It is relatively easy to include multiple antigen-encoding minigenes in the designed constructs. Thus, mRNA vaccination is currently more intensively explored [202].
The concept of mRNA-based therapeutics dates back to 1984, when the first biologically active mRNA was synthesized to express proteins in vitro [203]. By the early 1990s, researchers demonstrated that naked or liposome-encapsulated mRNAs could induce protein expression and activate antigen-specific cytotoxic T cells in vivo [204, 205]. These findings establish mRNA as a promising therapeutic strategy because of its low risk of genome integration, ease of design and modification, ability to encode multiple antigens, and rapid antigen production. In 1995, the first cancer mRNA vaccine encoding carcinoembryonic antigen (CEA) successfully induced anti-CEA antibodies in mice [206]. However, the development of mRNA-based cancer vaccines has been slower than that of other cancer vaccine platforms because of multiple challenges such as mRNA instability, inefficient delivery methods, and high intrinsic immunogenicity. Notably, the cornerstone discovery that nucleoside modifications reduce immunogenicity while enhancing protein production was instrumental in the successful application of mRNA vaccines against COVID-19 [207,208,209]. These findings have inspired the development of mRNA-based cancer vaccines. Current studies focus on optimizing the mRNA structure to improve stability and enhance antigen expression levels or duration, developing efficient delivery systems to increase transfection efficiency, targeting mRNA delivery to specific tissues or cells, modulating the adjuvant effects of mRNA vaccines to elicit stronger adaptive immune responses, and combining mRNA vaccines with other immunotherapies to achieve superior tumor control.
In vitro transcribed mRNA
The in vitro transcribed (IVT) mRNA comprises a 5ʹ cap, a 5ʹ untranslated region (UTR), an open reading frame (ORF) that encodes the antigen, a 3ʹ UTR, and a poly(A) tail. Optimizing these structural elements can increase mRNA stability and improve translation efficiency [210, 211]. High-throughput screening technologies, combined with machine learning algorithms, are employed to identify optimal 5ʹ and 3ʹ UTR sequences from UTR libraries [212,213,214,215]. Furthermore, coding sequence (CDS) optimization is achieved through codon optimization, GC content adjustment, and secondary structure improvement via computer algorithms [216,217,218,219]. Maintaining the length of the poly(A) tail within 120–150 nucleotides and incorporating short sequences or thiophosphates into poly(A) sequences have been shown to protect mRNAs from deacetylation and degradation [202, 211, 220,221,222,223].
Linear mRNA is rapidly degraded following translation, which limits the production of sufficient antigen for triggering immune responses. Various RNA types have been explored to increase the stability and duration of RNA. Commonly investigated RNA molecules include circular RNA (circRNA), self-amplifying RNA (saRNA), and trans-amplifying RNA (taRNA). CircRNAs exhibit increased stability due to the absence of free ends, which prevents recognition and degradation by nucleic acid exonucleases [224,225,226,227,228]. SaRNAs contain a sequence encoding RNA-dependent RNA polymerase (RDRP), which enables their self-amplification. This design extends protein expression while reducing the required mRNA vaccine dose [49, 229]. TaRNA is a modified form of saRNA generated by splitting saRNA into two distinct RNA molecules, one encoding the replicase sequence and the other encoding the protein of interest, which is amplified by the replicase to prolong antigen expression [202, 230]. Several of these RNA designs have been evaluated in preclinical cancer vaccine studies and have demonstrated potent antitumor immunity [49, 227, 228].
Current mRNA vaccination formulations in the clinic
Naked mRNA is rapidly degraded after administration because of the ubiquitous presence of ribonucleases (RNases). Additionally, its negative charge and hydrophilic nature limit its cellular uptake and result in inefficient endosomal escape, thereby restricting its therapeutic application [231]. Therefore, delivery systems that protect mRNAs from degradation, facilitate their uptake by APCs, and enable them to escape from endosomes for cytosolic protein translation are crucial for the effective delivery of mRNA vaccines (Fig. 4) [232].

Developmental trajectory of cancer mRNA vaccines. Early clinical trials of mRNA vaccines focused on the direct injection of unmodified, naked mRNA via intranodal administration. However, this approach faces many challenges, including the instability and high immunogenicity of mRNAs, low transfection efficiency into antigen-presenting cells (APCs), and low protein production. Therefore, some formulated vectors, such as cationic protamine and liposomes, have been developed to encapsulate unmodified mRNAs. While these formulations improve mRNA stability and transfection efficiency in vitro, their in vivo efficacy remains constrained by low transfection efficiency and protein production and the toxicity associated with cationic components. More recently, LNP- or LPX-encapsulated mRNA vaccines have been frequently utilized, along with modified (m1ψ) mRNAs, which exhibit reduced immunogenicity. Thus, the transfection efficiency and protein production are enhanced in vivo. These mRNA vaccines are generally well tolerated and possess inherent adjuvant effects that stimulate innate immune responses, triggering antigen-specific T-cell activation. Moreover, certain delivery systems, such as spleen-targeting mRNA-LPX, can deliver mRNAs more specifically to the spleen
mRNA‒lipid nanoparticles (LNPs)
LNPs are the preferred delivery system for mRNAs because of their proven success in COVID-19 mRNA vaccines, such as mRNA-1273 and BNT162b2 [233, 234]. LNPs typically consist of four lipid components: an ionizable lipid, cholesterol, a phospholipid, and a PEGylated lipid. LNPs encapsulate mRNA payloads via electrostatic interactions, forming uniform spheres with diameters ranging from 50 to 150 nm. The ionizable lipid, which carries a positively charged head group at low pH, facilitates the encapsulation of negatively charged nucleic acid payloads. The head group becomes uncharged at physiological pH, which reduces toxicity [235]. Following cellular uptake, the ionizable lipid is protonated within acidic endosomes, where it interacts with anionic endosomal phospholipids to disrupt the membrane, thereby enabling endosomal escape and cytosolic release of the payloads [236]. The clinically approved ionizable lipids include DLin-MC3-DMA (MC3), SM-102, and ALC-0315, which have been used in patisiran, mRNA-1273, and BNT162b2, respectively [237]. Cholesterol enhances the structural stability of LNPs and promotes membrane fusion. Phospholipids such as DOPE, DSPC, and DOPG act as helper lipids, enhancing structural stability and promoting endosomal escape [232]. PEGylated lipids, such as PEG-DMG and PEG-DSPE, are hydrophilic and assemble on the LNP surface. These lipids reduce nanoparticle aggregation and prolong their circulation time by reducing clearance by the mononuclear phagocytic system [238]. Additionally, functionalized PEGylated lipids can modify the LNP surface, allowing for conjugation with specific ligands or molecules for targeted delivery to specific cells or tissues. While PEGylation helps prolong circulation time, it may reduce LNP cellular uptake by limiting serum protein adsorption, membrane fusion, or endosome escape [178, 239, 240].
RNA-lipoplex (RNA-LPX)
RNA-LPX, developed by BioNTech, is a widely used lipoplex for delivering mRNA vaccines against cancer via intravenous administration. RNA-LPX is formed through the self-assembly of RNA with dedicated cationic liposomes, resulting in a topological transformation from liposomes into compact nanoparticles with diameters of 200–400 nm. Notably, RNA-LPX mainly targets RNA to the spleen because of the charge ratio between the cationic lipid and the RNA. The negative charge caused by excess RNA results in the accumulation of RNA-LPX in the spleen [241]. Following intravenous (i.v.) administration, splenic macrophages exhibit the highest uptake of RNA-LPX within one hour, whereas conventional dendritic cells (cDCs) demonstrate the greatest efficiency in protein translation [242]. The primary mechanism for RNA-LPX internalization by immature DCs is macropinocytosis rather than receptor-mediated endocytosis or phagocytosis [242].
Other delivery systems
Several other delivery platforms have been developed to deliver mRNA vaccines in vivo. Protamine–mRNA complexes utilize cationic, arginine-rich protamine peptides to condense and deliver mRNA. Lipopolyplex (LPP) consists of a polymeric polyplex mRNA core encapsulated within a phospholipid bilayer shell. Like RNA-LPX, LPP enters DCs through macropinocytosis [243]. Recently, “onion-like” multilamellar RNA lipid particle aggregates (LPAs) with a diameter of 200–500 nm have been developed. Through novel packaging strategies, LPAs enhance payload packaging and immunogenicity [244]. Following i.v. injection, LPAs are entrapped in reticuloendothelial organs such as the spleen, lymph nodes, liver, lungs, and kidneys. Unlike other platforms that deliver mRNA to APCs, LPAs transfect fibroblastic reticular cells (FRCs) and may provide antigens to APCs via locoregional release and direct contact.
In conclusion, the diameter, charge, and molecular composition of nanoparticles are critical factors in determining their tissue distribution and cellular targeting, although the underlying mechanisms remain unclear. Further surface modifications of nanoparticles with targeting moieties, such as antibodies or small-molecule ligands, can achieve active targeting to specific cell types [236]. However, as various lipid components and delivery carriers are used in RNA vaccination, their pharmacodynamic properties and in vivo metabolism need to be thoroughly evaluated. Clinical procedures for assessing the efficacy and safety of these carriers should also be established to guide the development of clinically viable RNA carriers.
Adjuvant effect
Adjuvants are key components of vaccines that increase the magnitude, breadth, and durability of the immune response [245]. Both lipid nanoparticles and mRNAs may function as effective adjuvants, stimulating innate immune responses that support the priming and maintenance of adaptive immunity. This built-in adjuvant effect elicits robust antitumor activity, obviating the need for additional adjuvants [246].
Adjuvant effect of mRNAs
Synthetic mRNAs can activate innate immune sensors, including Toll-like receptors (TLR3, TLR7, and TLR8), leading to the production of proinflammatory cytokines [207]. Additionally, double-stranded RNA (dsRNA), a byproduct generated during in vitro transcription, can stimulate various innate immune pathways via sensors such as TLR3, RIG-I, and MDA5 [245, 246]. Induced innate immune responses by mRNAs may lead to side effects, including the production of unwanted proinflammatory cytokines that cause toxicity, as well as mRNA degradation and protein translation inhibition by antiviral molecules such as RNA-dependent protein kinase (PKR) and RNase [209, 247, 248]. Researchers have improved purification strategies and explored nucleoside modifications to mitigate undesired immune activation and enhance translation efficiency. Clinically approved mRNA vaccines employ purification techniques, such as high-performance liquid chromatography (HPLC), cellulose adsorption, and other protocols, to remove dsRNA contaminants. These vaccines also incorporate a Cap1 structure (m⁷GpppNm) at the 5’ end of the mRNA, which reduces RIG-I recognition. Additionally, mRNAs containing pseudouridine exhibit enhanced translational efficiency and reduced immunogenicity, as evidenced by lower serum levels of IFN-α in murine models [207,208,209]. Importantly, N1-methylpseudouridine (m1ψ) modification has been successfully incorporated into two approved COVID-19 mRNA vaccines, namely, mRNA-1273 (Moderna) and BNT162b2 (Pfizer-BioNTech).
Base-modified mRNAs with reduced immunogenicity have been highly successful in prophylactic antiviral vaccination, which requires high protein production and robust antibody responses. However, owing to immune-suppressive mechanisms and suboptimal T-cell priming in cancer settings, cancer vaccination requires more robust Th1-prone inflammatory responses to induce potent CTL responses. In this context, nonimmunogenic base-modified mRNAs may not be ideal for stimulating antitumor T-cell responses. Some studies have suggested that unmodified mRNA or the deliberate addition of dsRNA mimics can induce comparable or superior antitumor responses in murine tumor models [49, 249]. Thus, there is no consensus on which type of mRNA offers better antitumor efficacy, and both base-modified and unmodified mRNAs are being tested in clinical trials for cancer vaccination.
In addition to the immunogenic properties of mRNA itself, the nanocarriers used for delivery can also act as adjuvants, contributing synergistically to innate immune activation. Therefore, mRNA-based vaccine platforms must carefully balance the combined immunostimulatory effects of mRNAs and nanocarriers to elicit the desired Th1-skewed immune response while minimizing excessive innate activation, which could result in systemic toxicity. In the following sections, we will examine the adjuvant effects of several mRNA cancer vaccine platforms currently under clinical evaluation, with a focus on the synergistic roles of mRNA molecules and their delivery vehicles in shaping innate immune responses.
Adjuvant effect of RNA-LNPs
Empty LNPs possess self-adjuvant activity. For example, when empty LNPs are mixed with antigen proteins, robust antigen-specific T follicular helper (Tfh) cell and germinal center (GC) B-cell responses are elicited following a single intramuscular (i.m.) immunization [250]. In this study, the adjuvant activity of empty LNPs relied on the induction of interleukin-6 (IL-6) and the use of the ionizable lipid DLinDMA. The adjuvant effect of empty LNPs did not rely on the MyD88 or MAVS signaling pathways, as Tfh and GC B-cell responses remained intact in Myd88–/– or Mavs–/– mice [250]. In another study, LNPs containing heterocyclic ionizable lipid head groups activated the STING signaling pathway, enhancing the immunostimulatory effects on APCs in LNs and improving antitumor efficacy upon subcutaneous (s.c.) administration [251]. Moreover, C1 lipid-like LNPs act as self-adjuvants by triggering TLR4 signaling, promoting DC activation, and inducing the expression of proinflammatory cytokines such as IL-6, IL-12, and IL-1β [252]. These findings highlight the dual functionality of LNPs as both delivery vehicles and self-adjuvants, wherein ionizable lipids play a pivotal role in activating distinct pattern recognition receptor pathways. However, repeated LNP administration can induce anti-PEG antibody production, leading to high systemic reactogenicity [253].
The innate immune responses triggered by mRNA vaccines are crucial for eliciting robust antigen-specific adaptive immune responses. The mechanisms underlying the innate and adaptive immunity of the COVID-19 vaccine BNT162b2, which consists of m1ψ nucleoside-modified mRNAs encapsulated in LNPs, are well characterized [254]. BNT162B2-induced antibody and T-cell responses do not depend on multiple canonical innate immune pathways, including TLR2/3/4/5/7 signaling, the cGAS-STING pathway, NLRP3-dependent inflammasome activation, nor on necroptosis and pyroptosis. Notably, MDA5-mediated type I IFN signaling plays a crucial role in activating innate immune cells and promoting the expansion and effector functions of antigen-specific CD8 + T cells [254]. The activation of MDA5 may be due to the spontaneous formation of dsRNA structures in the RNA-LNP vaccine.
Adjuvant effects of RNA-LPX and RNA-LPA
Unmodified RNA-LPXs are widely used in cancer vaccines. LPX-encapsulated unmodified mRNA but not LPX alone promoted IFNα production and enhanced the maturation and activation of splenic DCs [242]. However, m1Ψ-modified RNA-LPX is noninflammatory and can induce immune tolerance by expanding antigen-specific CD4 regulatory T cells [255]. These findings suggest the need to adjust the adjuvant effect of mRNA on the basis of the delivery system. Unmodified RNA-LPX induced TLR7-mediated biphasic IFN-α production by plasmacytoid dendritic cells (pDCs) and macrophages as early as 3 hours after i.v. injection [242]. In this context, the IFN-α response is essential for antigen-specific CD8+ T cells to acquire effector functions. The incorporation of unmodified RNA into RNA-LPAs also increased the induction of cytokines and chemokines, including type I/II IFNs, IL-12, CCL2, CCL4, and CXCL9. However, the potent innate stimulatory activity and antitumor ability of RNA-LPA depend on RIG-I but not TLRs or MDA5 [244].
To ensure safety, mRNA vaccines must achieve a balance between immunogenicity and reactogenicity, moderately stimulating the innate immune system to avoid excessive inflammation [246]. The toxic effects associated with RNA-LPX arise from its ability to stimulate IL-1β production, which subsequently triggers the release of a broad spectrum of proinflammatory cytokines [256]. The production of IL-1β may be dependent on TLR7/8 recognizing unmodified mRNAs to synthesize pro-IL-1β, as well as liposomes that activate the NLRP3 inflammasome to cleave pro-IL-1β. Humans exhibited greater sensitivity to RNA-LPX than laboratory mice because mice can upregulate anti-inflammatory IL-1ra, which competes with IL-1β for binding to IL-1R1. However, m1ψ-modified RNA-LPX failed to generate IL-1β and related cytokines in human PBMCs in vitro. This study also demonstrated that the toxicity of modified RNA-LNPs is influenced by the ionizable lipids used in their formulation. For example, modified RNA formulated in SM-102 LNPs induced robust IL-1β production and cytokine release, whereas formulation in MC3 LNPs resulted in weaker immune stimulation. Despite the toxicity associated with IL-1β, its immunogenic properties contribute to the expansion of antigen-specific T cells, as demonstrated in IL-1ra-deficient mice [256].
In summary, the components of nanoparticles, base modifications, and secondary structures of mRNA molecules can influence innate immune responses via various signaling pathways. The adjuvant activity of mRNA vaccines in clinical settings depends on both the chemical modification of the mRNA, which modulates recognition by innate immune sensors, and the nature of the lipid carrier used for delivery. In LNP formulations, mRNA is encapsulated within the core and remains inaccessible to membrane-bound innate immune sensors until it is released into the cytosol, where it is recognized primarily by cytosolic receptors such as RIG-I and MDA5. In contrast, in formulations such as LPX, mRNA is complexed on the surface of lipid particles, making it readily accessible to endosomal TLRs. However, how to modify the adjuvant effects of both RNA and nanoparticles to synergistically enhance DC activation and facilitate T-cell priming while minimizing side effects still needs to be further explored. This phenomenon is further complicated by the fact that several innate immune signaling pathways may lead to mRNA degradation and reduced antigen production.
Toward next-generation mRNA vaccines
Several synergistic approaches have been explored to improve next-generation mRNA vaccines: optimizing LNP formulations to increase delivery efficiency and immunomodulatory functions, engineering mRNA structural elements to maximize protein expression, and incorporating mRNAs encoding immune-stimulatory adjuvants to amplify adaptive immunity.
Novel LNP formulation
Novel LNP formulations have been designed for targeted mRNA delivery, which reduce nonspecific toxicity effects and improve mRNA expression. Conventional LNPs exhibit significant liver accumulation, which may lead to undesired antigen expression in the liver. Adjusting the LNP composition enables selective delivery to target tissues. By screening LNP components, an endogenous lymph node-targeting LNP named 113-O12B, which can transfect DCs and macrophages and elicit better CD8+ T-cell responses following subcutaneous injection, was explored [257]. Another study achieved lung-, spleen-, and liver-targeted delivery by adding different selective organ-targeting molecules (SORTs) to conventional LNPs [258]. In addition to nonspecific organ accumulation, current LNP formulations exhibit low endosomal escape efficiency and induce multiple undesired inflammatory responses [259,260,261]. Individual LNP components—including ionizable lipids, helper lipids, sterols, and PEG lipids—can be substituted with novel molecules to improve mRNA expression or modulate innate inflammatory pathways [262]. For example, poly(carboxybetaine) (PCB) lipids have been used in place of PEG lipids and result in increased transfection and mRNA expression due to enhanced membrane fusion and endosomal escape, resulting in a better safety profile with lower inflammatory cytokine induction and limited antipolymer antibody responses [263]. Ionizable lipids are the major component affecting endosomal escape efficacy and are largely responsible for LNP-associated toxicity. Researchers have reported that ionizable lipids, which promote increased mRNA expression and endosomal escape, typically cause more severe inflammatory responses by generating large, irreparable endosomal holes recognized by cytosolic galectins. These galectins bind inner-membrane sugars and trigger downstream inflammation. Strikingly, rapidly biodegradable ionizable lipids preferentially create smaller endosomal holes that are reparable by the ESCRT pathway, thereby reducing inflammatory responses while preserving high mRNA expression [264]. Another study screened an ionizable lipid library and identified a lipid with an unsaturated tail, a dihydroimidazole linker, and cyclic amine head groups that activate the STING pathway when formulated in LNPs, resulting in increased antitumor efficacy in the context of mRNA vaccination [251]. Other components, such as cholesterol and helper lipids, can also be replaced with analogs to increase mRNA expression [265, 266]. However, significant challenges remain in systematically evaluating the safety profiles of these novel LNP components in humans, and clinical standards for assessing their safety have yet to be established.
Designing novel mRNA molecules
Extensive research has focused on enhancing the translational efficacy of mRNA vaccines. Key strategies include nucleotide modification, machine learning-guided mRNA design, and the exploration of noncanonical structures, such as circular and self-amplifying mRNAs [49, 217, 227]. Novel chemical synthesis methodologies are also being leveraged to design next-generation mRNA vaccines. Chen et al. developed a chemically synthesized branched poly(A) tail. The branch termini were modified with phosphorothioate (PS) and 2’-methoxyethyl (2MOE) groups to confer resistance to exonuclease degradation. This modified mRNA exhibited enhanced binding to cytoplasmic poly(A)-binding proteins (PABPCs), significantly prolonging protein expression duration and increasing expression levels [267]. The same group also engineered chemical modifications within the 5ʹUTR and cap structure. They introduced locked nucleic acid (LNA) and 2ʹ-O-methylation (2ʹOMe) modifications and created a branched 5’ structure by chemically linking two LNAm⁷G caps. These modifications collectively confer resistance to the decapping enzyme hDcp2 and increase the valency of eIF4E binding, resulting in improved protein translation [268]. These chemical innovations, which target both the 5ʹ and 3ʹ mRNA regions, illuminate pathways for designing mRNAs with increased antigen production in vivo. Furthermore, basic research into the intracellular fate of exogenously delivered mRNAs offers crucial insights for future vaccine design. A recent study revealed that Moderna’s mRNA-1273 features a 100-nt poly(A) tail terminating in a mΨCΨAG sequence. This structure enables readenylation by the TENT5A poly(A) polymerase in macrophages, prolonging protein expression. This mechanism was absent in Pfizer-BioNTech’s BNT162b2, which possesses a composite poly(A) tail (30A-10 N-70A) [269]. These findings reveal a novel mechanism for mRNA persistence in vivo and inform the design of future mRNA constructs.
Addition of adjuvant mRNA molecules
Beyond structural modifications to increase protein expression, incorporating adjuvant mRNAs encoding immune modulatory molecules (e.g., cytokines and innate immune activators) is an emerging strategy to increase vaccination efficacy. Peng et al. screened and identified IL-12 as a potent T-cell-stimulating cytokine that synergizes with an mRNA cancer vaccine. To mitigate the peripheral toxicity of IL-12, they tethered IL-12 to the surface of APCs by linking to a transmembrane domain. This design reduced systemic toxicity while preserving antitumor efficacy. Specifically, they reported that a preeffector T-cell population was induced by membrane-bound IL-12 and differentiated into highly potent effector T cells for enhanced tumor control [270]. Similarly, another study corroborated that IL-12 enhances mRNA vaccine-induced T-cell responses by driving effector T-cell differentiation [271]. IL-27, which acts downstream of mRNA vaccine-induced type-I interferon signaling, was found to directly support CD8⁺ T-cell expansion [272]. IL-27 mRNA-LNPs enhanced the proliferation of antigen-specific CTLs without altering their differentiation state, highlighting their potential as direct adjuvants to increase cytolytic CD8⁺ T-cell responses. Zhivaki et al. reported an mRNA encoding a self-DNA-reactive cGAS variant (cGASAN) that produces cGAMP upon binding intramitochondrial DNA. When delivered to DCs, this adjuvant mRNA upregulated lymph node-homing receptors, enhanced antigen presentation and costimulatory signaling, and promoted proinflammatory cytokine secretion, collectively contributing to stronger T-cell responses [273]. These studies highlight the potential of incorporating mRNA vaccines with adjuvant mRNA sequences encoding immune-stimulating molecules.
Balancing the adjuvant effect and antigen translation
One of the major challenges in developing mRNA vaccines is to moderate the activation of the innate immune system, which can exert both beneficial and detrimental effects [274]. mRNA vaccination leads to the early production of type I IFNs, which can increase the maturation and activation of APCs to promote T-cell priming and activation [275] or directly modulate T-cell responses [276]. However, type I IFNs simultaneously upregulate RNA-dependent protein kinase (PKR) and oligoadenylate synthetase (OAS), which inhibit mRNA translation and mediate mRNA degradation. Studies have shown that type I IFN signaling impairs the ability of mRNA-LPX vaccines to elicit CTL responses following subcutaneous, intradermal, and intranodal administration [277]. Similarly, in self-amplifying RNA (saRNA) LNP vaccines, type I IFNs reduce antigen expression and dampen CTL responses [278]. However, other studies have demonstrated that although type I IFNs reduce antigen expression, they can support T-cell responses when mRNA-LNPs are administered intramuscularly [254, 279] or when negatively charged mRNA-LPXs are delivered intravenously [242, 280]. In these cases, knockout or blockade of the type I IFN receptor (IFNAR) results in decreased quantity and cytotoxic ability of antigen-specific T cells. One key factor contributing to the divergent effects of type I IFNs is the route of administration [276]. Following subcutaneous injection of mRNA-LPX, type I IFN signaling impairs the induction of antigen-specific T cells, whereas it enhances T-cell responses following intravenouse administration. This discrepant role of type I IFN in T-cell activation may be attributed to the relative timing of the activation of IFNAR and TCR signaling in T cells. Additionally, the type of mRNA may also determine the effect of type I IFNs. SaRNAs are engineered for self-replication, enabling prolonged antigen expression at low microgram doses. However, this also increases its susceptibility to degradation and sensitivity to type I IFNs [281]. Furthermore, self-replication generates dsRNA intermediates, which are potent inducers of type I IFNs. To counteract this, encoding virus-derived innate inhibitory proteins—such as E3L from vaccinia virus, NS1 from influenza A virus, or the leader protein from cardioviruses—into saRNA vaccines has been proposed to suppress interferon signaling and enhance mRNA translation [281,282,283,284,285]. Notably, most studies assessing the role of type I IFNs in mRNA vaccine efficacy have employed high-affinity model antigens, such as ovalbumin (OVA), which may be less dependent on antigen expression levels and cannot simulate clinical scenarios. Future research should investigate how innate immune responses affect the efficacy of cancer mRNA vaccines across various nanoparticle formulations, routes of administration, and RNA platforms, particularly in the context of low-affinity tumor antigens.
Owing to immune-suppressive mechanisms and suboptimal T-cell priming in cancer, effective vaccination requires robust innate immune activation to generate potent CTL responses. As previously noted, potent adjuvants such as STING agonists are commonly used in conjunction with mRNA vaccines. However, careful balancing of agonist dosage and the mRNA payload is essential to prevent excessive type I IFN responses, which can complicate clinical evaluation [286, 287]. Therefore, adjuvant strategies incorporating mRNA-encoded specific cytokines or costimulatory molecules, which enhance T-cell responses without suppressing antigen translation, represent a preferable approach. Further research should explore novel mRNA vaccines that evade innate immune-driven side effects and elucidate the mechanisms underlying mRNA vaccine-induced T-cell activation. This knowledge will facilitate the rational design of vaccines capable of optimally orchestrating innate and adaptive immunity for cancer treatment.
Clinical trials
In recent years, the development of cancer mRNA vaccines has been encouraging, with some clinical results advancing to phase III (Table 1) [288, 289]. In this section, we discuss the reasons for the failure of early clinical trials, highlight promising ongoing studies, and discuss the challenges faced by mRNA cancer vaccines.
Early clinical trials
One of the earliest clinical trials involved the direct injection of autologous amplified total tumor-derived naked mRNA combined with GM-CSF into stage III/IV metastatic melanoma patients [290, 291]. However, this phase I/II study did not demonstrate clinical efficacy, despite the observation of antitumor humoral immune responses in some patients. The lack of clinical effectiveness may be attributed to the heterogeneous amplification of total tumor mRNA in vitro, leading to the loss of lower-abundance mRNAs that encode tumor antigens [292]. Additionally, the insufficient expression and presentation of tumor antigens, likely due to competition with nonimmunogenic RNA-coding proteins, may have contributed to the failure. Therefore, subsequent clinical trials focused on selected TAAs or TSAs. Several early clinical trials utilizing unmodified naked tumor-antigen mRNA vaccines demonstrated their feasibility and safety, inducing antigen-specific CD4+ and CD8 + T-cell responses [293, 294]. However, naked mRNA vaccines are unstable, which encourages the development of formulated vectors. Early trials focused primarily on protamine–mRNA complexes. Four protamine-mRNA vaccines—CV9103 [291], CV9104 [295], CV9201 [296], and CV9202 [297]—were designed to encode several TAAs for the treatment of advanced castration-resistant prostate cancer or non-small cell lung cancer (NSCLC), with CV9202 being combined with local radiation therapy. The overall survival (OS) benefits of these protamine-mRNA vaccines have been limited. The lack of clinical responses may be attributed to several factors, including suboptimal mRNA modification and delivery, central immune tolerance to the selected TAAs, the highly immunosuppressive microenvironment in advanced-stage tumors, and insufficient induction of long-term memory responses required for durable clinical benefit. These challenges underscore the need for optimized tumor antigen selection, more efficient delivery systems, and potential combination therapies.
TAAs cancer mRNA vaccines
Nonformulated mRNAs are easily degraded by extracellular RNases. Consequently, formulated LPX and LNP-encapsulated mRNA vaccines are frequently utilized in ongoing clinical trials. FixVac (BNT111) is an intravenously administered RNA-LPX vaccine encoding four full-length melanoma TAAs—NY-ESO-1, MAGE-A3, TPTE, and tyrosinase (NCT02410733, NCT04526899) [37]. The administration of FixVac alone or in combination with anti-PD1 therapy induced durable objective responses in patients with stage IIIB-C and IV melanoma, with mild to moderate adverse events. In a phase I study, circulating antigen-specific T cells were expanded by several orders of magnitude following a repeat boost strategy, and these cells were fully functional, suggesting that selective TAAs may disrupt immune tolerance. More than 75% of the analyzed patients developed immune responses against at least one TAA, particularly NY-ESO-1 and MAGE-A3. Additionally, vaccine-induced T cells in some patients were sustained for over one year, which may contribute to long-term clinical benefits. FixVac combined with PD1 blockade was more effective than monotherapy (partial response rate: 12% vs. 35%). These findings support the potential of combining mRNA vaccines with ICB therapy for patients with low mutational burdens. A phase II trial of FixVac combined with cemiplimab is ongoing. RNA-LNP vaccines are also designed to target and eliminate immunosuppressive regulatory cells as well as cancer cells, thereby mitigating the immunosuppressive TME and enhancing T-cell effector functions. For example, mRNA-4359, which encodes both PD-L1 and indoleamine 2,3-dioxygenase 1 (IDO1) antigens, is being evaluated in patients with advanced solid tumors [298].
TSAs as cancer mRNA vaccines
It is very convenient to include multiple minigenes encoding mutated antigens in mRNA vaccines. BNT122 is a personalized RNA-LPX vaccine containing up to 20 neoepitopes presented on MHC class I and MHC class II molecules [299]. In a phase I study (NCT04161755), patients with surgically resected pancreatic ductal adenocarcinoma (PDAC) were sequentially treated with anti-PD-L1, BNT122, and a modified four-drug chemotherapy regimen (mFOLFIRINOX). The combination treatment is well tolerated and induces de novo, high-magnitude neoantigen-specific T-cell responses in 50% of patients (8/16). Half of these T-cell responses targeted more than one vaccine neoantigen. Moreover, over 80% of these specific T cells were detectable up to three years after administration in responders, with 75% of responders remaining disease free. These results suggest that neoantigen-based mRNA vaccines can generate potent T-cell responses and long-lived T cells for tumor control [300]. A phase II trial of BNT122 is ongoing (NCT04161755). mRNA-4157 (V940) is another individualized, nucleoside-modified RNA-LNP vaccine encoding up to 34 melanoma neoantigens that is administered via intramuscular injection [301]. In a phase 2 trial, mRNA-4157, with or without pembrolizumab (anti-PD-1), was used to treat patients with completely resected melanoma (stage IIIB-IV). The 2.5-year overall survival (OS) and recurrence-free survival (RFS) rates for the combination treatment were 96% and 74.8%, respectively, whereas they were 90.2% and 55.6%, respectively, for pembrolizumab alone. Ongoing phase III clinical trials are evaluating mRNA-4157 in combination with pembrolizumab as adjuvant therapy for patients with resected high-risk melanoma (NCT05933577) and non-small cell lung cancer (NSCLC) (NCT06077760), making mRNA-4157 the first cancer mRNA vaccine to reach phase III. However, despite the potential of neoantigens to generate tumor-reactive T cells with high affinity due to a lack of thymic negative selection, not all neoantigen-based mRNA vaccines tested in clinical trials have demonstrated clinical efficacy. For example, mRNA-4650, which encodes up to 20 neoantigens expressed by autologous tumors, failed to elicit objective clinical responses in patients with gastrointestinal cancer, potentially because of the lower responsiveness to vaccination caused by lymphodepletion during the TIL therapeutic regimen [302], which suggests the importance of appropriate schedules and combination therapies.
Self-amplifying mRNA cancer vaccines
In addition to nonreplicating mRNAs, self-amplifying mRNAs (saRNAs) have been utilized to increase protein expression levels. In a phase I study (NCT03639714), a heterologous prime/boost vaccination strategy was evaluated [303]. Specifically, heterologous chimpanzee adenovirus (ChAd68) for priming followed by neoantigen saRNA-LNP boosts, in combination with nivolumab and ipilimumab, were administered to patients with advanced metastatic solid tumors. This regimen was well tolerated and induced functional neoantigen-specific effector memory CD8+ T-cell responses in all patients. A phase II/III study has been initiated to further investigate this strategy in patients with metastatic colorectal cancer [304]. Moreover, an “off-the-shelf” vaccine utilizing this heterologous prime/boost regimen in combination with ICB therapy was assessed in a phase I trial (NCT03953235) [305]. This vaccine encodes 20 shared oncogenic mutations within a single vector, such as KRAS mutations (G12D, G12C, G12V, and G13D) and TP53 mutations, and is intended to circumvent the need for individualized neoantigen prediction. Despite the generation of antigen-specific T-cell responses in most patients, the overall response rate was 0%, with median recurrence-free survival (RFS) and overall survival (OS) rates of 1.9 and 7.9 months, respectively. Interestingly, T-cell responses were predominantly directed toward tumor-unrelated TP53 epitopes rather than the KRAS mutations present in the tumors, indicating the existence of a potential hierarchy of immunodominance in the processing and presentation of multiple epitopes within the same APCs. To address this issue, researchers have split mRNAs into separate vectors to ensure delivery to distinct APCs, and encoding repeated copies of each epitope could optimize KRAS presentation in mice [305]. An optimized shared vaccine encoding only four highly prevalent KRAS-derived mutations (each of which is repeated four times) is currently being evaluated in a phase II study (NCT03953235).
Combination therapy with CAR-T cells and RNA vaccine
Chimeric antigen receptor (CAR)-T cells have shown impressive efficacy in patients with B-cell malignancies, where CAR-T cells are easily expanded and reactivated by circulating B cells. However, transferred CAR-T cells targeting solid tumors show limited persistence and reactivation due to the lack of efficient in vivo stimulation. CAR-T-cell-enhancing mRNA vaccines have been designed to deliver CAR antigens for the expansion and reactivation of CAR-T cells in a proper environment inside lymphoid tissues. The CARVac is an RNA-LPX vaccine designed to deliver and induce the expression of CLDN6 on the surface of APCs within lymph tissues. The combination therapy of CLDN6-specific CAR-T cells with CARVac is currently undergoing phase I/II trials (NCT04503278) [306, 307]. CARVac significantly expanded the transferred CAR-T cells in secondary lymphoid tissues and had a superior antitumor effect on a mouse model. In a phase I study, the ORR was 38%, and the DCR was 69%, with CAR-T cells observed to persist beyond 90 days in some patients [308].
Although substantial progress has been made in the development of cancer mRNA vaccines, several challenges remain unresolved. First, balancing innate and adaptive immunity to achieve rapid and durable clinical effects remains a challenge. Unlike prophylactic vaccines, therapeutic cancer vaccines must induce a strong inflammatory environment to overcome peripheral and intratumoral immunosuppression for efficient antigen-specific T-cell priming. In this context, optimizing the adjuvant effect of cancer mRNA vaccines, which can induce a proinflammatory milieu without impairing mRNA translation, might be necessary. Compared with modified mRNAs, unmodified mRNAs may elicit equal or even more robust antitumor responses in some contexts [49, 309]. Systemic administration may elicit more rapid systemic innate immune responses; however, potential toxicity must be carefully considered. Second, most tumor antigens, particularly TAAs, are poorly immunogenic and often subject to immune tolerance [310]. Strategies to overcome immune tolerance and generate a large pool of antigen-specific effector T cells during the priming phase are essential. Third, while many clinical cancer mRNA vaccines encode both CD8+ and CD4+ T-cell epitopes, the observed immune responses are predominantly mediated by CD4+ T cells rather than CD8+ T cells [291, 294, 297, 302]. mRNA vaccines may preferentially elicit antigen-specific CD4+ T cells, potentially due to the broader clonal heterogeneity of CD4+ T cells. Questions remain regarding whether both CD8+ and CD4+ epitopes are necessary [220] and whether vaccine-induced CD4+ T-cell responses can promote CD8 + T-cell priming and differentiation for improved tumor control [311,312,313]. Both the adjuvanticity and the antigen hierarchy of mRNA vaccines require further investigation to optimize T-cell responses.
Administration route
The route of administration significantly influences the innate and adaptive immune responses of different formulations of cancer vaccines. Therefore, selecting the appropriate administration route is critical and should be carefully evaluated to ensure successful vaccination outcomes.
Peptide vaccines
The injection route can significantly influence both the immunogenicity and toxicity of peptide vaccines. Common injection routes include s.c., i.m., and i.v. injections. I.m. injections are commonly used because of their ability to administer larger vaccine volumes and their favorable safety profile. S.c. injections, while facilitating more efficient vaccine drainage into lymph nodes through a dense network of lymphatic vessels, are limited by the smaller volume that can be injected. I.v. injections allow for systemic exposure of vaccines to lymphoid organs but may also induce systemic side effects [314, 315].
Hussein et al. compared T-cell responses associated with different injection routes using TriVax (which contains a palmitoylated peptide, anti-CD40 monoclonal antibody, and poly-ICLC) and reported that i.v. injection generated substantially greater CTL responses than i.m. and s.c. injections did [315]. I.v. injections may promote systemic antigen uptake by a larger number of APCs, which allows for more efficient recruitment and activation of antigen-specific CTL precursors. However, systematic exposure to adjuvants may increase toxicity and limit clinical applicability. Another study assessed a self-assembling peptide conjugated to a TLR7/8 agonist (SNP-7/8a) and reported that the same vaccination dose generated fewer antigen-specific T cells via the i.v. route (SNP-IV) than via the s.c. route (SNP-SC). Interestingly, the antigen-specific T cells generated after SNP-IV vaccination retained more Tcf1+ stem-like cells. When the vaccination dose for SNP-IV was increased to induce a comparable number of T cells to SNP-SC, the SNP-IV-generated T cells exhibited enhanced synergy with checkpoint blockade therapy and resulted in better tumor control. This study revealed that the duration of antigen presentation by DCs was critical in shaping the quantity and quality of CD8+ T-cell responses, with SNP-IV leading to transient antigen presentation by DCs, fostering the generation of stem-like T cells [316].
Intranodal (i.n.) injection is less commonly employed because of the complexity of the injection process and volume limitations. Nonetheless, studies have demonstrated that, compared with traditional vaccination routes, a dose of 106-fold reduction in antigen and 100-fold reduction in adjuvant achieves enhanced protection and fewer side effects in i.n. injections [317, 318]. Mechanistic studies on i.n. injection might inspire new designs of vaccines to mimic this microenvironment.
mRNA vaccines
The route of administration significantly influences the organ distribution of nanoparticles, the types of immune cells that take up mRNA, the kinetics of antigen expression, and the resulting local or systemic immune responses, all of which influence the therapeutic efficacy of cancer mRNA vaccines. Common administration routes include i.m., intradermal (i.d.), s.c., and i.v. injections. The choice of the administration route is typically determined by the characteristics of the delivery nanoparticles and the desired immune response, whether localized or systemic.
Local administration
Skin and muscle tissues contain abundant resident APCs, and innate immune cells, which can take up RNA-LNPs locally and then migrate to dLNs after s.c. or i.m. vaccination. Furthermore, lymphatic vessels in these tissues facilitate the direct flow of RNA-LNPs into dLNs, where they transfect resident APCs [319, 320]. Intramuscular injection has been widely employed during the administration of COVID-19 mRNA vaccines. In murine and nonhuman primate (NHP) models, following i.m. injection, RNA-LNPs are highly taken up by stromal cells at the injection site, including endothelial cells, pericytes, and fibroblasts, as well as by myeloid cells such as macrophages, neutrophils, mast cells, and DCs [279, 319, 321]. Fibroblasts express IFNβ and chemokines in response to RNA-LNPs, which recruit inflammatory immune cells to the injection site [279]. The mRNA-encoded protein is expressed primarily in macrophages, fibroblasts, and adipocytes at the injection site, whereas it is highly expressed in macrophages, DCs, and monocytes in dLNs [254, 279, 319, 321]. Although RNA-LNPs are located primarily in the injection site and dLNs, mRNA concentrations have also been detected in the spleen, liver, and blood [319, 322]. Consistently, protein expression was detected as early as 4 hours postadministration at both the injection site and dLNs in nonhuman primates (NHPs). The skin harbors many epidermal Langerhans cells, dermal DCs, and macrophages, which suggests that i.d. delivery may elicit stronger immune responses and prolong protein expression [321]. However, most studies have shown comparable immune responses following i.d., i.m., and s.c. vaccinations [323]. Additionally, owing to technical difficulties and increased adverse effects, i.d. and s.c. injections are used less frequently than i.m. injections [324].
Systemic administration
Systemic injection of RNA vaccines allows circulation to the spleen, where abundant antigen-presenting cells, particularly cDC1s, are present. Compared with s.c. injection, intravenous injection of spleen-targeting RNA-LPX elicits a stronger immune response and induces a greater proportion of antigen-specific CD8 + T cells [242]. In contrast, RNA-LNP-based cancer vaccines are less commonly administered via i.v. injection because of liver accumulation, which may promote antigen presentation in an immune-tolerant microenvironment [322, 325]. However, modified LNP formulations designed for spleen-targeted delivery of mRNAs, such as SORT LNPs [258] and antibody/peptide-modified LNPs [326, 327], have been developed, although their clinical exploration for cancer vaccines remains limited.
The kinetics of induced proinflammatory cytokine release could be influenced by the RNA vaccine injection route, which affects the induction of the antigen-specific T-cell response. Studies suggest that different administration routes may induce opposing effects of type-I IFN signaling on T-cell responses. The induction of type-I IFN promoted antigen-specific T-cell responses when mRNA-LPX was i.v. injected but suppressed T-cell responses when the vaccine was delivered s.c. or i.d [276, 277]. A potential explanation for these opposing effects is the relative timing of the activation of IFNAR and TCR signaling in T cells. When IFNAR signaling coincides with TCR stimulation, type I IFNs promote effector T-cell responses. In contrast, IFNAR signaling preceding TCR engagement may lead to the formation of different STAT complexes, contributing to an inhibitory effect [328, 329]. Thus, the route of administration likely influences the kinetics of T-cell exposure to type I IFN and TCR engagement, ultimately impacting vaccination outcomes. However, a comprehensive understanding of the immune responses elicited by different administration routes and a direct comparison of their antitumor effects remain insufficiently explored.
Concluding and future perspectives
Early cancer vaccines against TAAs have shown limited therapeutic efficacy because of antigen immune tolerance and the lack of a potent adjuvant and delivery system. Recent therapeutic cancer vaccines have undergone a remarkable transformation, driven by advances in antigen identification, delivery platforms, and immune modulation strategies. Advancements in next-generation sequencing technology, computational capabilities, and predictive algorithms informed by the accumulation of wet-laboratory data have facilitated the identification of nontolerant neoantigen candidates for cancer vaccination. Furthermore, novel delivery platforms, including peptide-based vaccines and RNA vaccines, have demonstrated the ability to prime potent CD4+ and CD8 + T-cell responses, not only against neoantigens but also against TAAs. Despite these achievements, several challenges may impede the clinical efficacy of cancer vaccines.
Identifying shared neoantigens
Current neoantigen-based cancer vaccines require highly personalized antigen identification and vaccine formulation, resulting in formidable medical costs that are inaccessible to many cancer patients. Although some clinical trials have demonstrated the efficacy of TAA-based vaccination with novel platforms, the risk of autoimmune diseases poses a significant barrier, particularly for cancer types in which autoimmunity could have fatal consequences. Identifying shared tumor-specific antigens may address these limitations. A substantial proportion of neoantigens have been found to originate from noncoding and “dark matter” regions of the genome, as well as from noncanonical translation processes, which need further exploration for shared epitopes. Further advancements in NGS technology, mass spectrometry-based MHC ligandome analysis, and novel computational algorithms could unveil those antigens that can truly activate patients’ T cells, enabling their evaluation in clinical studies.
Opportunities in other vaccine platforms
In addition to peptide- and mRNA-based cancer vaccines, several other vaccine platforms have been developed for cancer immunotherapy, including DC vaccines, in situ vaccines, viral vector-based vaccines, and bacteria-based vaccines. DC vaccines have been extensively explored in the clinic for cancer vaccination and exhibit good tolerance and elicit antigen-specific T-cell responses. Several excellent reviews have provided valuable insight into this vaccination platform [330,331,332,333]. In situ vaccines aim to increase antigen release from tumor cells at the tumor site to elicit adaptive immune responses without the need for prior identification of tumor antigens [334]. Key determinants of in situ vaccine efficacy include the adequate release of tumor antigens from cancer cells and the induction of robust antigen processing by APCs through the activation of immunogenic cell death (ICD) and innate immune responses [335]. Thus, various strategies have been employed to enhance the ICD and antigen processing. These include intratumoral injection of chemotherapeutic hydrogels for local drug retention and sustained release [336, 337]; the design of nanosensitizers for radiotherapy, photodynamic therapy, or sonodynamic therapy [338,339,340,341,342,343]; and the codelivery of pattern recognition receptor (PRR) agonists [338, 344] or ICD inducers [345]. Additionally, the intratumoral administration of oncolytic viruses has emerged as a promising in situ vaccination approach, offering advantages such as selective replication within tumor cells, direct oncolysis, and the activation of innate immune-sensing pathways [346, 347]. Current research is focused on genetically engineering oncolytic viruses to express tumor-irrelevant bystander epitopes [348], tumor-associated antigens, cytokines, or immune checkpoint inhibitors, thereby increasing their antitumor efficacy [349]. Viral vector-based vaccines are typically engineered by deleting pathogenic genes and incorporating genes encoding tumor antigens [350]. Through genetic modification, viral vectors can target APCs with high transduction efficiency while minimizing pathogenicity. Commonly used viral vectors in clinical studies include adenovirus (Ad), adeno-associated virus (AAV), herpes simplex virus (HSV), and retrovirus (RV) [351, 352]. Bacteria-based vaccines exploit the unique properties of bacteria, such as their intrinsic tumor-targeting capabilities, preferential proliferation in hypoxic and immunosuppressive TMEs, and strong stimulation of the innate immune system, which contributes to TME remodeling [353,354,355]. Engineered bacterial strains, such as Listeria monocytogenes [356, 357] and the probiotic Escherichia coli [358], have been developed to express and release high levels of tumor antigens following systemic administration. The integration of synthetic biology and other immunotherapeutic strategies with these vaccine platforms holds promise for broader clinical application.
Combination therapy for optimized tumor control
The immunosuppressive TME poses a significant barrier to the infiltration, activation, proliferation, and effector functions of vaccine-induced T cells in tumors [359, 360]. Insufficient T-cell infiltration, driven by the lack of attractant chemokines and abnormal tumor vasculature, significantly limits the effectiveness of vaccine-induced T-cell trafficking into the TME. Moreover, infiltrated T cells often encounter immunosuppressive signals from myeloid cells, such as tumor-associated macrophages, which provide suboptimal TCR and costimulatory signals while expressing inhibitory surface markers and soluble factors that drive T-cell exhaustion [361, 362]. The scarcity of professional APCs within the TME and the lack of T-cell-supporting cytokines exacerbate these challenges, leading to limited T-cell proliferation, survival, and effector function. Combining cancer vaccines with other immunomodulatory therapies may help overcome these barriers and achieve optimal tumor control. Angiogenesis inhibitors can normalize the tumor vasculature, enhancing T-cell infiltration. Radiotherapy and in situ vaccination agents, such as STING agonists, TLR agonists, and oncolytic viruses, can create an inflammatory TME that supports T-cell infiltration while providing additional tumor antigens to enhance T-cell priming in a feed-forward loop [6]. Immune checkpoint inhibitors and tumor-targeting cytokines can further enhance T-cell survival and effector differentiation, as demonstrated in early-phase clinical trials. Owing to the heterogeneity of cancer, antigen-negative cell clones may be selected after vaccination, leading to tumor relapse. Combination therapies may also induce epitope spreading in T-cell responses, which broadens the T-cell repertoire and triggers additional tumor antigen-directed immune responses [149, 150]. Further investigation of the mechanisms of epitope spreading will help identify a more effective combination therapy strategy to completely control tumors.
Identifying predictive biomarkers to predict patients’ response to vaccines
Identifying predictive biomarkers for selecting patients who will respond to cancer vaccine therapy is also essential. Early-phase clinical trials have shown that a subset of patients who receive neoantigen-based vaccination exhibit a response characterized by robust T-cell activation and prolonged survival [299]. However, it remains uncertain whether robust T-cell activation represents a direct survival benefit conferred by vaccination or is a biomarker reflecting preexisting antitumor immunity and patients’ responsiveness to vaccination. Clinical evidence suggests that while cancer vaccines targeting unselected patient populations may fail to produce statistically significant survival benefits, they may offer substantial advantages for subsets of patients with specific biomarkers or favorable physiological conditions [363].
In conclusion, while therapeutic cancer vaccines have yet to achieve their full clinical potential, the integration of novel antigen discovery pipelines, cutting-edge delivery technologies, and rational combinatory strategies could transform them into a cornerstone of cancer immunotherapy. The accumulated knowledge from preclinical and clinical research will help better understand the immune mechanism of cancer vaccines and design novel vaccine formulations for durable and effective tumor control.
References
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol: Mechanisms Dis. 2021;16:223–49.
Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet. 2021;398:1002–14.
Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol. 2022;40:45–74.
Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. 2024;42:1825–63.
Sellars MC, Wu CJ, Fritsch EF. Cancer vaccines: building a bridge over troubled waters. Cell. 2022;185:2770–88.
Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. 2022;3:911–26.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discov. 2022;21:261–82.
Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125:3401–12.
Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med. 2011;135:55–62.
Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov. 2023;22:101–26.
Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Invest. 2001;107:477–84.
Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27.
Bakker AB, Schreurs MW, de Boer AJ, Kawakami Y, Rosenberg SA, Adema GJ, et al. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med. 1994;179:1005–9.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8 + T cells. J Exp Med. 2003;198:569–80.
Cunha AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens: comparative analysis of transcript levels in prostate and nonprostatic tissues. Cancer Lett. 2006;236:229–38.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.
Noguchi M, Fujimoto K, Arai G, Uemura H, Hashine K, Matsumoto H, et al. A randomized phase III trial of personalized peptide vaccination for castration‑resistant prostate cancer progressing after docetaxel. Oncol Rep. 2021;45:159–68.
Hardwick NR, Frankel P, Ruel C, Kilpatrick J, Tsai W, Kos F, et al. p53-Reactive T Cells Are Associated with Clinical Benefit in Patients with Platinum-Resistant Epithelial Ovarian Cancer After Treatment with a p53 Vaccine and Gemcitabine Chemotherapy. Clin Cancer Res. 2018;24:1315–25.
Chung V, Kos FJ, Hardwick N, Yuan Y, Chao J, Li D, et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin Transl Oncol. 2019;21:363–72.
Schoen RE, Boardman LA, Cruz-Correa M, Bansal A, Kastenberg D, Hur C, et al. Randomized, double-blind, placebo-controlled trial of MUC1 peptide vaccine for prevention of recurrent colorectal adenoma. Clin Cancer Res. 2023;29:1678–88.
Kjeldsen JW, Lorentzen CL, Martinenaite E, Ellebaek E, Donia M, Holmstroem RB, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. 2021;27:2212–23.
Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65:1339–52.
Sugiyama H. WT1 (Wilms’ Tumor Gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40:377–87.
Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37.
Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018;2:224–34.
Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, et al. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol. 2018;9:947.
Bethune MT, Li XH, Yu J, McLaughlin J, Cheng D, Mathis C, et al. Isolation and characterization of NY-ESO-1–specific T-cell receptors restricted on various MHC molecules. Proc Natl Acad Sci. 2018;115:E10702–E10711.
Zeng G, Wang X, Robbins PF, Rosenberg SA, Wang RF. CD4( + ) T-cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci USA. 2001;98:3964–9.
Odunsi K, Qian F, Matsuzaki J, Mhawech-Fauceglia P, Andrews C, Hoffman EW, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T-cell responses in ovarian cancer. Proc Natl Acad Sci USA. 2007;104:12837–42.
Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18:6497–508.
Gasser O, Sharples KJ, Barrow C, Williams GM, Bauer E, Wood CE, et al. A phase I vaccination study with dendritic cells loaded with NY-ESO-1 and α-galactosylceramide: induction of polyfunctional T cells in high-risk melanoma patients. Cancer Immunol Immunother. 2018;67:285–98.
Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–27.
Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. 2020;585:107–12.
Esfandiary A, Ghafouri-Fard S. MAGE-A3: an immunogenic target used in clinical practice. Immunotherapy. 2015;7:683–704.
Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small cell lung cancer: phase II randomized study results. J Clin Oncol. 2013;31:2396–403.
Dreno B, Thompson JF, Smithers BM, Santinami M, Jouary T, Gutzmer R, et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): a double-blind, randomized, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19:916–29.
Slingluff CL, Lewis KD, Andtbacka R, Hyngstrom J, Milhem M, Markovic SN, et al. Multicenter, double-blind, placebo-controlled trial of seviprotimut-L polyvalent melanoma vaccine in patients with post-resection melanoma at high risk of recurrence. J Immunother Cancer. 2021;9:e003272.
Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumor virology. Nat Rev Cancer. 2010;10:878–89.
Chen YP, Zhang WN, Chen L, Tang LL, Mao YP, Li WF, et al. Effect of latent membrane protein 1 expression on overall survival in Epstein‒Barr virus-associated cancers: a literature-based meta-analysis. Oncotarget. 2015;6:29311–23.
Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein‒Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808.
Zhao GX, Bu GL, Liu GF, Kong XW, Sun C, Li ZQ, et al. mRNA-based vaccines targeting the T-cell epitope-rich domain of Epstein Barr virus latent proteins elicit robust anti-tumor immunity in mice. Adv Science. 2023;10:2302116.
Taylor GS, Jia H, Harrington K, Lee LW, Turner J, Ladell K, et al. A recombinant modified vaccinia ankara vaccine encoding Epstein‒Barr Virus (EBV) target antigens: a phase I trial in UK patients with EBV-positive cancer. Clin Cancer Res. 2014;20:5009–22.
Pal A, Kundu R. Human papillomavirus E6 and E7: the cervical cancer hallmarks and targets for therapy. Front Microbiol. 2020;10:3116.
Lechner M, Liu J, Masterson L, Fenton TR. HPV-associated oropharyngeal cancer: epidemiology, molecular biology and clinical management. Nature Reviews Clin Oncol. 2022;19:306–27.
Ramos da Silva J, Bitencourt Rodrigues K, Formoso Pelegrin G, Silva Sales N, Muramatsu H, de Oliveira Silva M, et al. Single immunizations of self-amplifying or nonreplicating mRNA-LNP vaccines control HPV-associated tumors in mice. Sci Transl Med. 2023;15:eabn3464.
He J, Wang C, Fang X, Li J, Shen X, Zhang J, et al. Tuning the fluidity and protein corona of ultrasound-responsive liposomal nanovaccines to program T-cell immunity in mice. Nat Commun. 2024;15:8121.
Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16–related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5:67–73.
Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomized, double-blind, placebo-controlled phase 2b trial. Lancet. 2015;386:2078–88.
Harper DM, Nieminen P, Donders G, Einstein MH, Garcia F, Huh WK, et al. The efficacy and safety of Tipapkinogen Sovacivec therapeutic HPV vaccine in cervical intraepithelial neoplasia grades 2 and 3: Randomized controlled phase II trial with 2.5 years of follow-up. Gynecol Oncol. 2019;153:521–9.
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–29.
Hu Z, Ott PA, Wu CJ. Toward personalized, tumor specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18:168–82.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Puig-Saus C, Sennino B, Peng S, Wang CL, Pan Z, Yuen B, et al. Neoantigen-targeted CD8 + T-cell responses with PD-1 blockade therapy. Nature. 2023;615:697–704.
Lowery FJ, Krishna S, Yossef R, Parikh NB, Chatani PD, Zacharakis N, et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science. 2022;375:877–84.
Zheng C, Fass JN, Shih YP, Gunderson AJ, Sanjuan Silva N, Huang H, et al. Transcriptomic profiles of neoantigen-reactive T cells in human gastrointestinal cancers. Cancer Cell. 2022;40:410–423.e7.
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4 + T cells in human melanoma. Nat Med. 2015;21:81–85.
Zhao M, Schoenfeld JD, Egloff AM, Hanna GJ, Haddad RI, Adkins DR, et al. T-cell dynamics with neoadjuvant immunotherapy in head and neck cancer. Nat Rev Clin Oncol. 2024;22:83–94.
Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative tumor-specific antigens. Nat Rev Cancer. 2019;19:465–78.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25:389–402.
Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:386 p.
Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nature Reviews. Clin Oncol. 2021;18:645–61.
Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–90.
Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumor-specific neoantigens and the immunogenic phenotype: a pancancer analysis. Lancet Oncol. 2017;18:1009–21.
Roudko V, Bozkus CC, Orfanelli T, McClain CB, Carr C, O'Donnell T, et al. Shared immunogenic poly-epitope frameshift mutations in microsatellite unstable tumors. Cell. 2020;183:1634–1649.e17.
Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227–238.e3.
Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukemia. Nature. 1985;315:550–4.
Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, et al. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol. 2018;36:1056–8.
Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–224.e6.
Ehx G, Larouche JD, Durette C, Laverdure JP, Hesnard L, Vincent K, et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity. 2021;54:737–752.e10.
Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10:eaau5516.
Chong C, Coukos G, Bassani-Sternberg M. Identification of tumor antigens with immunopeptidomics. Nat Biotechnol. 2022;40:175–88.
Kacen A, Javitt A, Kramer MP, Morgenstern D, Tsaban T, Shmueli MD, et al. Posttranslational modifications reshape the antigenic landscape of the MHC I immunopeptidome in tumors. Nat Biotechnol. 2023;41:239–51.
Attermann AS, Bjerregaard AM, Saini SK, Grønbæk K, Hadrup SR. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann Oncol. 2018;29:2183–91.
Apavaloaei A, Zhao Q, Hesnard L, Cahuzac M, Durette C, Larouche J-D, et al. Tumor antigens preferentially derive from unmutated genomic sequences in melanoma and non-small cell lung cancer. Nat Cancer. 2025. https://doi.org/10.1038/s43018-025-00979-2.
Weber D, Ibn-Salem J, Sorn P, Suchan M, Holtsträter C, Lahrmann U, et al. Accurate detection of tumor-specific gene fusions reveals strongly immunogenic personal neo-antigens. Nat Biotechnol. 2022;40:1276–84.
Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019;9:1022–35.
Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413–21.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Raine KM, Hinton J, Butler AP, Teague JW, Davies H, Tarpey P, et al. cgpPindel: identifying somatically acquired insertion and deletion events from paired end sequencing. Curr Protoc Bioinforma. 2015;52:15.7.1–15.7.12.
Moncunill V, Gonzalez S, Beà S, Andrieux LO, Salaverria I, Royo C, et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotechnol. 2014;32:1106–12.
Huang D, Zhu X, Ye S, Zhang J, Liao J, Zhang N, et al. Tumor circular RNAs elicit antitumor immunity by encoding cryptic peptides. Nature. 2024;625:593–602.
Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–26.
Huber F, Arnaud M, Stevenson BJ, Michaux J, Benedetti F, Thevenet J, et al. A comprehensive proteogenomic pipeline for neoantigen discovery to advance personalized cancer immunotherapy. Nat Biotechnol. 2024. https://doi.org/10.1038/s41587-024-02420-y.
Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, Gannon PO, et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput Biol. 2017;13:e1005725.
Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol. 2019;37:1283–6.
Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199:3360–8.
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406.
Gfeller D, Guillaume P, Michaux J, Pak HS, Daniel RT, Racle J, et al. The length distribution and multiple specificity of naturally, presented HLA-I ligands. J Immunol. 2018;201:3705–16.
Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteom. 2015;14:658–73.
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung. Cancer Cancer Discov. 2017;7:264–76.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Richman LP, Vonderheide RH, Rech AJ. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019;9:375–382.e4.
Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–6.
Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumor response to checkpoint blockade immunotherapy. Nature. 2017;551:517–20.
Poudel R, Dai D, Li F, Yin X, Kou Y, Gomes ALC. Abstract B003: Cross-reacting antigens between the intestinal microbiome and tumor neoantigen are associated with immune checkpoint inhibitor response in a gastrointestinal tumor cohort. Cancer Res. 2022;82:B003–B003.
Zitvogel L, Kroemer G. Cross-reactivity between microbial and tumor antigens. Curr Opin Immunol. 2022;75:102171.
Roerden M, Castro AB, Cui Y, Harake N, Kim B, Dye J, et al. Neoantigen architectures define immunogenicity and drive immune evasion of tumors with heterogenous neoantigen expression. J Immunother Cancer. 2024;12:e010249.
Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–101.
Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T-cell-based immunotherapy of solid tumors. Cancer Cell. 2020;38:454–72.
Kina E, Larouche JD, Thibault P, Perreault C. The cryptic immunopeptidome in health and disease. Trends Genet. 2024;41:162–9.
Starck SR, Shastri N. Nowhere to hide: unconventional translation yields cryptic peptides for immune surveillance. Immunol Rev. 2016;272:8–16.
Ely ZA, Kulstad ZJ, Gunaydin G, Addepalli S, Verzani EK, Casarrubios M, et al. Pancreatic cancer–restricted cryptic antigens are targets for T-cell recognition. Science. 2025;388:eadk3487.
Dopkins N, Nixon DF. Activation of human endogenous retroviruses and its physiological consequences. Nat Rev Mol Cell Biol. 2024;25:212–22.
Cherkasova E, Weisman Q, Childs RW. Endogenous retroviruses as targets for antitumor immunity in renal cell cancer and other tumors. Front Oncol. 2013;3:243.
Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018;128:4804–20.
Saini SK, Ørskov AD, Bjerregaard AM, Unnikrishnan A, Holmberg-Thydén S, Borch A, et al. Human endogenous retroviruses form a reservoir of T-cell targets in hematological cancers. Nat Commun. 2020;11:5660.
Jiang Q, Braun DA, Clauser KR, Ramesh V, Shirole NH, Duke-Cohan JE, et al. HIF regulates multiple translated endogenous retroviruses: implications for cancer immunotherapy. Cell. 2025;188:1807–1827.e34.
Ng KW, Boumelha J, Enfield K, Almagro J, Cha H, Pich O, et al. Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature. 2023;616:563–73.
Kristensen LS, Jakobsen T, Hager H, Kjems J. The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol. 2022;19:188–206.
Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15:461–70.
Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6:404–14.
Kalita P, Tripathi T. Methodological advances in the design of peptide-based vaccines. Drug Discov Today. 2022;27:1367–80.
Reed SG, Tomai M, Gale MJ Jr. New horizons in adjuvants for vaccine development. Curr Opin Immunol. 2020;65:97–101.
Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–96.
Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small cell lung cancer (MAGRIT): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:822–35.
Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumor cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51:2321–9.
Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small cell lung cancer (START): a randomized, double-blind, phase 3 trial. Lancet Oncol. 2014;15:59–68.
Rini BI, Stenzl A, Zdrojowy R, Kogan M, Shkolnik M, Oudard S, et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicenter, open-label, randomized, controlled, phase 3 trial. Lancet Oncol. 2016;17:1599–611.
Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomized, phase 3 trial. Lancet Oncol. 2014;15:829–40.
Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275–84.
Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8( + ) T-cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–72.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol. 2008;38:1033–42.
Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–60.
Rabu C, Rangan L, Florenceau L, Fortun A, Charpentier M, Dupré E, et al. Cancer vaccines: designing artificial synthetic long peptides to improve presentation of class I and class II T-cell epitopes by dendritic cells. Oncoimmunology. 2019;8:e1560919.
Guo M, Liu MYR, Brooks DG. Regulation and impact of tumor-specific CD4( + ) T cells in cancer and immunotherapy. Trends Immunol. 2024;45:303–13.
Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol. 2013;43:2554–65.
Zhang H, Hong H, Li D, Ma S, Di Y, Stoten A, et al. Comparing pooled peptides with intact protein for accessing cross-presentation pathways for protective CD8+ and CD4 + T cells. J Biol Chem. 2009;284:9184–91.
Livingston BD, Newman M, Crimi C, McKinney D, Chesnut R, Sette A. Optimization of epitope processing enhances immunogenicity of multiepitope DNA vaccines. Vaccine. 2001;19:4652–60.
Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, et al. Heteroclitic immunization induces tumor immunity. J Exp Med. 1998;188:1553–61.
Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, et al. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13:529–38.
Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T-cell epitopes. J Immunol. 1994;153:5586–92.
Cole DK, Edwards ES, Wynn KK, Clement M, Miles JJ, Ladell K, et al. Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T-cell recognition. J Immunol. 2010;185:2600–10.
Cavalluzzo B, Ragone C, Mauriello A, Petrizzo A, Manolio C, Caporale A, et al. Identification and characterization of heteroclitic peptides in TCR-binding positions with improved HLA-binding efficacy. J Transl Med. 2021;19:89.
Tagliamonte M, Mauriello A, Cavalluzzo B, Ragone C, Manolio C, Luciano A, et al. MHC-Optimized Peptide Scaffold for Improved Antigen Presentation and Anti-Tumor Response. Front Immunol. 2021;12:769799.
Ciesielski MJ, Ahluwalia MS, Munich SA, Orton M, Barone T, Chanan-Khan A, et al. Antitumor cytotoxic T-cell response induced by a survivin peptide mimic. Cancer Immunol Immunother. 2010;59:1211–21.
Gupta RK. Aluminum compounds as vaccine adjuvants. Adv Drug Deliv Rev. 1998;32:155–72.
Hogenesch H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front Immunol. 2012;3:406.
Zhao T, Cai Y, Jiang Y, He X, Wei Y, Yu Y, et al. Vaccine adjuvants: mechanisms and platforms. Signal Transduct Target Ther. 2023;8:283.
Gurung P. Innate immune sensors in health and disease. Immunol Rev. 2025;330:e70008 p.
Cao LL, Kagan JC. Targeting innate immune pathways for cancer immunotherapy. Immunity. 2023;56:2206–17.
Yin Q, Fu TM, Li J, Wu H. Structural biology of innate immunity. Annu Rev Immunol. 2015;33:393–416.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T-cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–9.
Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell. Lung Cancer, or Bladder Cancer Cell. 2020;183:347–362.e24.
Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T-cell responses and epitope spreading in patients with melanoma. Nat Med. 2021;27:515–25.
Sultan H, Salazar AM, Celis E. Poly-ICLC, a multifunctional immune modulator for treating cancer. Semin Immunol. 2020;49:101414.
Sultan H, Wu J, Kumai T, Salazar AM, Celis E. Role of MDA5 and interferon-I in dendritic cells for T-cell expansion by anti-tumor peptide vaccines in mice. Cancer Immunol Immunother. 2018;67:1091–103.
Wang P. Natural and Synthetic Saponins as Vaccine Adjuvants. Vaccines (Basel). 2021;9:222.
Lacaille-Dubois MA. Updated insights into the mechanism of action and clinical profile of the immunoadjuvant QS-21: A review. Phytomedicine. 2019;60:152905.
den Brok MH, Büll C, Wassink M, de Graaf AM, Wagenaars JA, Minderman M, et al. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat Commun. 2016;7:13324.
Huis In 't Veld LG, Cornelissen LA, van den Bogaard L, Ansems M, Ho NI, Adema GJ. Saponin-based adjuvant uptake and induction of antigen cross-presentation by CD11b+ dendritic cells and macrophages. npj Vaccines. 2025;10:15.
Detienne S, Welsby I, Collignon C, Wouters S, Coccia M, Delhaye S, et al. Central role of CD169(+) lymph node resident macrophages in the adjuvanticity of the QS-21 component of AS01. Sci Rep. 2016;6:39475.
Marciani DJ. Elucidating the mechanisms of action of saponin-derived adjuvants. Trends Pharm Sci. 2018;39:573–85.
Samson N, Ablasser A. The cGAS–STING pathway and cancer. Nature Cancer. 2022;3:1452–63.
Wang X, Huang Z, Xing L, Shang L, Jiang J, Deng C, et al. STING agonist-based ER-targeting molecules boost antigen cross-presentation. Nature. 2025;641:202–10.
Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8:e77846.
Li S, Luo M, Wang Z, Feng Q, Wilhelm J, Wang X, et al. Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng. 2021;5:455–66.
Wilhelm J, Quiñones-Pérez M, Wang J, Wang X, Basava VS, Gao J. Antigen folding improves loading efficiency and antitumor efficacy of PC7A nanoparticle vaccine. J Control Release. 2021;329:353–60.
Niemi JVL, Sokolov AV, Schiöth HB. Neoantigen Vaccines; Clinical Trials, Classes, Indications, Adjuvants and Combinatorial Treatments. Cancers (Basel). 2022;14:5163.
Black M, Trent A, Tirrell M, Olive C. Advances in the design and delivery of peptide subunit vaccines with a focus on toll-like receptor agonists. Expert Rev Vaccines. 2010;9:157–73.
Ilyinskii PO, Roy CJ, O'Neil CP, Browning EA, Pittet LA, Altreuter DH, et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014;32:2882–95.
Wang Y, Wang J, Zhu D, Wang Y, Qing G, Zhang Y, et al. Effect of physicochemical properties on in vivo fate of nanoparticle-based cancer immunotherapies. Acta Pharmaceutica Sin B. 2021;11:886–902.
Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–64.
Kuhn DA, Vanhecke D, Michen B, Blank F, Gehr P, Petri-Fink A, et al. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J Nanotechnol. 2014;5:1625–36.
Yue Z-G, Wei W, Lv PP, Yue H, Wang LY, Su ZG, et al. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules. 2011;12:2440–6.
Dane EL, Belessiotis-Richards A, Backlund C, Wang J, Hidaka K, Milling LE, et al. STING agonist delivery by tumor-penetrating PEG-lipid nanodiscs primes robust anticancer immunity. Nat Mater. 2022;21:710–20.
Palomba R, Palange AL, Rizzuti IF, Ferreira M, Cervadoro A, Barbato MG, et al. Modulating phagocytic cell sequestration by tailoring nanoconstruct softness. ACS Nano. 2018;12:1433–44.
Li J, Wang X, Zhang T, Wang C, Huang Z, Luo X, et al. A review on phospholipids and their main applications in drug delivery systems. Asian J Pharm Sci. 2015;10:81–98.
Gao J, Ochyl LJ, Yang E, Moon JJ. Cationic liposomes promote antigen cross-presentation in dendritic cells by alkalizing the lysosomal pH and limiting the degradation of antigens. Int J Nanomed. 2017;12:1251–64.
Heuts J, Jiskoot W, Ossendorp F, van der Maaden K. Cationic Nanoparticle-Based Cancer Vaccines. Pharmaceutics. 2021;13:596.
Gao Y, Huang Y, Ren C, Chou P, Wu C, Pan X, et al. Looking back, moving forward: protein corona of lipid nanoparticles. J Mater Chem B. 2024;12:5573–88.
Kim W, Ly NK, He Y, Li Y, Yuan Z, Yeo Y. Protein corona: Friend or foe? Co-opting serum proteins for nanoparticle delivery. Adv Drug Deliv Rev. 2023;192:114635.
Zalba S, Ten Hagen T, Burgui C, Garrido MJ. Stealth nanoparticles in oncology: facing the PEG dilemma. J Control Release. 2022;351:22–36.
Dilliard SA, Cheng Q, Siegwart DJ. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc Natl Acad Sci USA. 2021;118:52.
Nsairat H, Khater D, Sayed U, Odeh F, Al Bawab A, Alshaer W. Liposomes: structure, composition, types, and clinical applications. Heliyon. 2022;8:e09394.
Han X, Zhang H, Butowska K, Swingle KL, Alameh MG, Weissman D, et al. An ionizable lipid toolbox for RNA delivery. Nat Commun. 2021;12:7233.
Abumanhal-Masarweh H, da Silva D, Poley M, Zinger A, Goldman E, Krinsky N, et al. Tailoring the lipid composition of nanoparticles modulates their cellular uptake and affects the viability of triple negative breast cancer cells. J Controlled Release. 2019;307:331–41.
Huang Z, Callmann CE, Wang S, Vasher MK, Evangelopoulos M, Petrosko SH, et al. Rational vaccinology: harnessing nanoscale chemical design for cancer immunotherapy. ACS Cent Sci. 2022;8:692–704.
Meckes B, Banga RJ, Nguyen ST, Mirkin CA. Enhancing the Stability and Immunomodulatory Activity of Liposomal Spherical Nucleic Acids through Lipid-Tail DNA Modifications. Small. 2018;14:1702909.
Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16:489–96.
Wang S, Qin L, Yamankurt G, Skakuj K, Huang Z, Chen PC, et al. Rational vaccinology with spherical nucleic acids. Proc Natl Acad Sci. 2019;116:10473–81.
Skakuj K, Teplensky MH, Wang S, Dittmar JW, Mirkin CA. Chemically tuning the antigen release kinetics from spherical nucleic acids maximizes immune stimulation. ACS Central Science. 2021;7:1838–46.
Rhym LH, Manan RS, Koller A, Stephanie G, Anderson DG. Peptide-encoding mRNA barcodes for the high-throughput in vivo screening of libraries of lipid nanoparticles for mRNA delivery. Nat Biomed Eng. 2023;7:901–10.
Khirallah J, Xu Q. A high-throughput approach of screening nanoparticles for targeted delivery of mRNA. Cell Rep Methods. 2023;3:100572.
Yamankurt G, Berns EJ, Xue A, Lee A, Bagheri N, Mrksich M, et al. Exploration of the nanomedicine-design space with high-throughput screening and machine learning. Nat Biomed Eng. 2019;3:318–27.
Liu D, Liu L, Li X, Wang S, Wu G, Che X. Advancements and challenges in peptide-based cancer vaccination: a multidisciplinary perspective. Vaccines (Basel). 2024;12:950.
Ezike TC, Okpala US, Onoja UL, Nwike CP, Ezeako EC, Okpara OJ, et al. Advances in drug delivery systems, challenges and future directions. Heliyon. 2023;9:e17488.
Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat Commun. 2017;8:1954.
Moynihan KD, Holden RL, Mehta NK, Wang C, Karver MR, Dinter J, et al. Enhancement of peptide vaccine immunogenicity by increasing lymphatic drainage and boosting serum stability. Cancer Immunol Res. 2018;6:1025–38.
Martin JT, Hartwell BL, Kumarapperuma SC, Melo MB, Carnathan DG, Cossette BJ, et al. Combined PET and whole-tissue imaging of lymphatic-targeting vaccines in nonhuman primates. Biomaterials. 2021;275:120868.
Ma L, Hostetler A, Morgan DM, Maiorino L, Sulkaj I, Whittaker CA, et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. Cell. 2023;186:3148–3165.e20.
Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–22.
Pant S, Wainberg ZA, Weekes CD, Furqan M, Kasi PM, Devoe CE, et al. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: the phase 1 AMPLIFY-201 trial. Nat Med. 2024;30:531–42.
Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–24.
Zom GG, Welters MJ, Loof NM, Goedemans R, Lougheed S, Valentijn RR, et al. TLR2 ligand-synthetic long peptide conjugates effectively stimulate tumor-draining lymph node T cells of cervical cancer patients. Oncotarget 2016;7:67087–100.
Gary EN, Weiner DB. DNA vaccines: prime time is now. Curr Opin Immunol. 2020;65:21–27.
Liu C, Shi Q, Huang X, Koo S, Kong N, Tao W. mRNA-based cancer therapeutics. Nat Rev Cancer. 2023;23:526–43.
Melton DA, Krieg PA, Rebagliati MR, Maniatis T, Zinn K, Green MR. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984;12:7035–56.
Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;2471:1465–8.
Martinon F, Krishnan S, Lenzen G, Magné R, Gomard E, Guillet JG, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993;23:1719–22.
Conry RM, LoBuglio AF, Wright M, Sumerel L, Pike MJ, Johanning F, et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995;55:1397–400.
Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–75.
Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16:1833–40.
Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38:5884–92.
Jin L, Zhou Y, Zhang S, Chen SJ. mRNA vaccine sequence and structure design and optimization: advances and challenges. J Biol Chem. 2024;301:108015.
Yao R, Xie C, Xia X. Recent progress in mRNA cancer vaccines. Hum Vaccin Immunother. 2024;20:2307187.
Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, et al. Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol. 2019;37:803–9.
Castillo-Hair SM, Seelig G. Machine learning for designing next-generation mRNA therapeutics. Acc Chem Res. 2022;55:24–34.
Li T, Liu G, Bu G, Xu Y, He C, Zhao G. Optimizing mRNA translation efficiency through rational 5′UTR and 3′UTR combinatorial design. Gene. 2025;942:149254.
Ma Q, Zhang X, Yang J, Li H, Hao Y, Feng X. Optimization of the 5’ untranslated region of mRNA vaccines. Sci Rep. 2024;14:19845.
Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, et al. Codon optimality is a major determinant of mRNA stability. Cell. 2015;160:1111–24.
Zhang H, Zhang L, Lin A, Xu C, Li Z, Liu K, et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature. 2023;621:396–403.
Wayment-Steele HK, Kim DS, Choe CA, Nicol JJ, Wellington-Oguri R, Watkins AM, et al. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucleic Acids Res. 2021;49:10604–17.
Leppek K, Byeon GW, Kladwang W, Wayment-Steele HK, Kerr CH, Xu AF, et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat Commun. 2022;13:1536.
Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumor immunity and response to immunotherapy. Nature. 2019;574:696–701.
Trepotec Z, Geiger J, Plank C, Aneja MK, Rudolph C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. Rna. 2019;25:507–18.
Xia X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines (Basel). 2021;9:734.
Li CY, Liang Z, Hu Y, Zhang H, Setiasabda KD, Li J, et al. Cytidine-containing tails robustly enhance and prolong protein production of synthetic mRNA in cell and in vivo. Mol Ther Nucleic Acids. 2022;30:300–10.
Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12:381–8.
Liu X, Zhang Y, Zhou S, Dain L, Mei L, Zhu G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J Control Release. 2022;348:84–94.
Obi P, Chen YG. The design and synthesis of circular RNAs. Methods. 2021;196:85–103.
Li H, Peng K, Yang K, Ma W, Qi S, Yu X, et al. Circular RNA cancer vaccines drive immunity in hard-to-treat malignancies. Theranostics. 2022;12:6422–36.
Amaya L, Grigoryan L, Li Z, Lee A, Wender PA, Pulendran B, et al. Circular RNA vaccine induces potent T-cell responses. Proc Natl Acad Sci USA. 2023;120:e2302191120.
Lundstrom K. Self-Amplifying RNA Viruses as RNA Vaccines. Int J Mol Sci. 2020;21:5130.
Beissert T, Perkovic M, Vogel A, Erbar S, Walzer KC, Hempel T, et al. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol Ther. 2020;28:119–28.
Weng Y, Li C, Yang T, Hu B, Zhang M, Guo S, et al. The challenge and prospect of mRNA therapeutics landscape. Biotechnol Adv. 2020;40:107534.
Zong Y, Lin Y, Wei T, Cheng Q. Lipid nanoparticle (LNP) enables mRNA delivery for cancer therapy. Adv Mater. 2023;35:e2303261.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384:403–16.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383:2603–15.
Kulkarni JA, Witzigmann D, Leung J, van der Meel R, Zaifman J, Darjuan MM, et al. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale. 2019;11:9023–31.
Kon E, Ad-El N, Hazan-Halevy I, Stotsky-Oterin L, Peer D. Targeting cancer with mRNA-lipid nanoparticles: key considerations and future prospects. Nat Rev Clin Oncol. 2023;20:739–54.
Samaridou E, Heyes J, Lutwyche P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv Drug Deliv Rev. 2020;154-155:37–63. p.
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51.
Mui BL, Tam YK, Jayaraman M, Ansell SM, Du X, Tam YY, et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol Ther Nucleic Acids. 2013;2:e139.
Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36:892–9.
Grabbe S, Haas H, Diken M, Kranz LM, Langguth P, Sahin U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: case study with RNA-lipoplexes for the treatment of melanoma. Nanomed (Lond). 2016;11:2723–34.
Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defense for cancer immunotherapy. Nature. 2016;534:396–401.
Persano S, Guevara ML, Li Z, Mai J, Ferrari M, Pompa PP, et al. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials. 2017;125:81–89.
Mendez-Gomez HR, DeVries A, Castillo P, von Roemeling C, Qdaisat S, Stover BD, et al. RNA aggregates harness the danger response for potent cancer immunotherapy. Cell. 2024;187:2521–2535 e21.
Pulendran B. S.A. P, and D.T. O’Hagan, Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov. 2021;20:454–75.
Verbeke R, Hogan MJ, Loré K, Pardi N. Innate immune mechanisms of mRNA vaccines. Immunity. 2022;55:1993–2005.
García MA, Meurs EF, Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811.
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–5.
Tockary TA, Abbasi S, Matsui-Masai M, Hayashi A, Yoshinaga N, Boonstra E, et al. Comb-structured mRNA vaccine tethered with short double-stranded RNA adjuvants maximizes cellular immunity for cancer treatment. Proc Natl Acad Sci USA. 2023;120:e2214320120.
Alameh MG, Tombácz I, Bettini E, Lederer K, Sittplangkoon C, Wilmore JR, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity. 2021;54:2877–2892 e7.
Miao L, Li L, Huang Y, Delcassian D, Chahal J, Han J, et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol. 2019;37:1174–85.
Zhang H, You X, Wang X, Cui L, Wang Z, Xu F, et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc Natl Acad Sci USA 2021;118:e2005191118.
Ju Y, Lee WS, Pilkington EH, Kelly HG, Li S, Selva KJ, et al. Anti-PEG antibodies boosted in humans by SARS-CoV-2 lipid nanoparticle mRNA vaccine. ACS Nano. 2022;16:11769–80.
Li C, Lee A, Grigoryan L, Arunachalam PS, Scott M, Trisal M, et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat Immunol. 2022;23:543–55.
Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science. 2021;371:145–53.
Tahtinen S, Tong AJ, Himmels P, Oh J, Paler-Martinez A, Kim L, et al. IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat Immunol. 2022;23:532–42.
Chen J, Ye Z, Huang C, Qiu M, Song D, Li Y, et al. Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8( + ) T-cell response. Proc Natl Acad Sci USA. 2022;119:e2207841119.
Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR‒Cas gene editing. Nat Nanotechnol. 2020;15:313–20.
Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6:1078–94.
Bitounis D, Jacquinet E, Rogers MA, Amiji MM. Strategies to reduce the risks of mRNA drug and vaccine toxicity. Nat Rev Drug Discov. 2024;23:281–300.
Cullis PR, Felgner PL. The 60-year evolution of lipid nanoparticles for nucleic acid delivery. Nat Rev Drug Discov. 2024;23:709–22.
Mehta M, Bui TA, Yang X, Aksoy Y, Goldys EM, Deng W. Lipid-based nanoparticles for drug/gene delivery: an overview of the production techniques and difficulties encountered in their industrial development. ACS Mater Au. 2023;3:600–19.
Luozhong S, Liu P, Li R, Yuan Z, Debley E, Chen Y, et al. Poly(carboxybetaine) lipids enhance mRNA therapeutics efficacy and reduce their immunogenicity. Nat Mater. 2025. https://doi.org/10.1038/s41563-025-02240-8.
Omo-Lamai S, Wang Y, Patel MN, Essien EO, Shen M, Majumdar A, et al. Lipid nanoparticle-associated inflammation is triggered by sensing of endosomal damage: engineering endosomal escape without side effects. bioRxiv, 2024 2024.04.16.589801.
Barbieri BD, Peeler DJ, Samnuan K, Day S, Hu K, Sallah HJ, et al. The role of helper lipids in optimizing nanoparticle formulations of self-amplifying RNA. J Controlled Release. 2024;374:280–92.
Patel S, Ashwanikumar N, Robinson E, Xia Y, Mihai C, Griffith JP, et al. Naturally occurring cholesterol analogs in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun. 2020;11:983.
Chen H, Liu D, Guo J, Aditham A, Zhou Y, Tian J, et al. Branched chemically modified poly(A) tails enhance the translation capacity of mRNA. Nat Biotechnol. 2025;43:194–203.
Chen H, Liu D, Aditham A, Guo J, Huang J, Kostas F, et al. Chemical and topological design of multicapped mRNA and capped circular RNA to augment translation. Nat Biotechnol. 2024. https://doi.org/10.1038/s41587-024-02393-y.
Krawczyk PS, Mazur M, Orzeł W, Gewartowska O, Jeleń S, Antczak W, et al. Readenylation by TENT5A enhances efficacy of SARS-CoV-2 mRNA vaccines. Nature. 2025;641:984–92.
Peng K, Zhao X, Li H, Fu YX, Liang Y. Membrane-IL12 adjuvant mRNA vaccine polarizes pre-effector T cells for optimized tumor control. J Exp Med. 2025;222:e20241454.
Aunins EA, Phan AT, Alameh MG, Dwivedi G, Cruz-Morales E, Christian DA, et al. An Il12 mRNA-LNP adjuvant enhances mRNA vaccine-induced CD8 T-cell responses. Sci Immunol. 2025;10:eads1328.
Phan AT, Aunins E, Cruz-Morales E, Dwivedi G, Bunkofske M, Eberhard JN, et al. The type I IFN-IL-27 axis promotes mRNA vaccine-induced CD8 ( + ) T-cell responses. bioRxiv. 2025.
Zhivaki D, Gosselin EA, Sengupta D, Concepcion H, Arinze C, Chow J, et al. mRNAs encoding self-DNA reactive cGAS enhance the immunogenicity of lipid nanoparticle vaccines. mBio. 2023;14:e02506–23.
Linares-Fernández S, Lacroix C, Exposito JY, Verrier B. Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol Med. 2020;26:311–23.
Mochida Y, Uchida S. mRNA vaccine designs for optimal adjuvanticity and delivery. RNA Biol. 2024;21:1–27.
Van Hoecke L, Roose K, Ballegeer M, Zhong Z, Sanders NN, De Koker S, et al. The Opposing effect of type I IFN on the T-cell response by nonmodified mRNA-lipoplex vaccines is determined by the route of administration. Mol Ther Nucleic Acids. 2020;22:373–81.
De Beuckelaer A, Pollard C, Van Lint S, Roose K, Van Hoecke L, Naessens T, et al. Type I interferons interfere with the capacity of mRNA lipoplex vaccines to elicit cytolytic T-cell responses. Mol Ther. 2016;24:2012–20.
Pepini T, Pulichino AM, Carsillo T, Carlson AL, Sari-Sarraf F, Ramsauer K, et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J Immunol. 2017;198:4012–24.
Kim S, Jeon JH, Kim M, Lee Y, Hwang YH, Park M, et al. Innate immune responses against mRNA vaccine promote cellular immunity through IFN-beta at the injection site. Nat Commun. 2024;15:7226.
Broos K, Van der Jeught K, Puttemans J, Goyvaerts C, Heirman C, Dewitte H, et al. Particle-mediated intravenous delivery of antigen mrna results in strong antigen-specific T-cell responses despite the induction of type I interferon. Mol Ther Nucleic Acids. 2016;5:e326.
Wojcechowskyj JA, Jong RM, Mäger I, Flach B, Munson PV, Mukherjee PP, et al. Controlling reactogenicity while preserving immunogenicity from a self-amplifying RNA vaccine by modulating nucleocytoplasmic transport. NPJ Vaccines. 2025;10:85.
Liu Y, Chin JM, Choo EL, Phua K. Messenger RNA translation enhancement by immune evasion proteins: a comparative study between EKB (vaccinia virus) and NS1 (influenza A virus). Sci Rep. 2019;9:11972.
Beissert T, Koste L, Perkovic M, Walzer KC, Erbar S, Selmi A, et al. Improvement of in vivo expression of genes delivered by self-amplifying RNA using vaccinia virus immune evasion proteins. Hum Gene Ther. 2017;28:1138–46.
Devoldere J, Dewitte H, De Smedt SC, Remaut K. Evading innate immunity in nonviral mRNA delivery: do not shoot the messenger. Drug Discov Today. 2016;21:11–25.
Blakney AK, McKay PF, Bouton CR, Hu K, Samnuan K, Shattock RJ. Innate inhibiting proteins enhance expression and immunogenicity of self-amplifying RNA. Mol Ther. 2021;29:1174–85.
Tse SW, McKinney K, Walker W, Nguyen M, Iacovelli J, Small C, et al. mRNA-encoded, constitutively active STING(V155M) is a potent genetic adjuvant of antigen-specific CD8( + ) T cell response. Mol Ther. 2021;29:2227–38.
Fan N, Chen K, Zhu R, Zhang Z, Huang H, Qin S, et al. Manganese-coordinated mRNA vaccines with enhanced mRNA expression and immunogenicity induce robust immune responses against SARS-CoV-2 variants. Sci Adv. 2022;8:eabq3500.
Weber JS, Luke JJ, Carlino MS, Khattak MA, Meehan RS, Brown M, et al. INTerpath-001: Pembrolizumab with V940 (mRNA-4157) versus pembrolizumab with placebo for adjuvant treatment of high-risk stage II-IV melanoma. J Clin Oncol. 2024;42:TPS9616–TPS9616.
Lee JM, Spicer J, Nair S, Khattak A, Brown M, Meehan RS, et al. The phase 3 INTerpath-002 study design: Individualized neoantigen therapy (INT) V940 (mRNA-4157) plus pembrolizumab vs placebo plus pembrolizumab for resected early-stage non–small-cell lung cancer (NSCLC). J Clin Oncol. 2024;42:TPS8116–TPS8116.
Carralot JP, Weide B, Schoor O, Probst J, Scheel B, Teufel R, et al. Production and characterization of amplified tumor-derived cRNA libraries to be used as vaccines against metastatic melanomas. Genet Vaccines Ther. 2005;3:6.
Kübler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, Vom Dorp F, et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J Immunother Cancer. 2015;3:26.
Carralot JP, Probst J, Hoerr I, Scheel B, Teufel R, Jung G, et al. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol Life Sci. 2004;61:2418–24.
Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther. 2011;19:990–9.
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–6.
Stenzl A, Feyerabend S, Syndikus I, Sarosiek T, Kübler H, Heidenreich A, et al. Results of the randomized, placebo-controlled phase I/IIB trial of CV9104, an mRNA based cancer immunotherapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol. 2017;28:v408–v409.
Sebastian M, Schröder A, Scheel B, Hong HS, Muth A, von Boehmer L, et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother. 2019;68:799–812.
Papachristofilou A, Hipp MM, Klinkhardt U, Früh M, Sebastian M, Weiss C, et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J Immunother Cancer. 2019;7:38.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase 1/2 study of mRNA-4359 administered alone and in combination with immune checkpoint blockade in adult participants with advanced solid tumors. J Clin Oncol. 2023;41:TPS2676–TPS2676.
Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618:144–50.
Sethna Z, Guasp P, Reiche C, Milighetti M, Ceglia N, Patterson E, et al. RNA neoantigen vaccines prime long-lived CD8( + ) T cells in pancreatic cancer. Nature. 2025;639:1042–51.
Weber JS, Carlino MS, Khattak A, Meniawy T, Ansstas G, Taylor MH, et al. Individualized neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomized, phase 2b study. Lancet. 2024;403:632–44.
Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, et al. mRNA vaccine-induced neoantigen-specific T-cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130:5976–88.
Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong SJ, et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med. 2022;28:1619–29.
Hecht JR, Shergill A, Goldstein MG, Fang B, Cho MT, Lenz HJ, et al. Phase 2/3, randomized, open-label study of an individualized neoantigen vaccine (self-amplifying mRNA and adenoviral vectors) plus immune checkpoint blockade as maintenance for patients with newly diagnosed metastatic colorectal cancer (GRANITE). J Clin Oncol. 2022;40:TPS3635–TPS3635.
Rappaport AR, Kyi C, Lane M, Hart MG, Johnson ML, Henick BS, et al. A shared neoantigen vaccine combined with immune checkpoint blockade for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med. 2024;30:1013–22.
Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–53.
Mackensen A, Haanen J, Koenecke C, Alsdorf W, Wagner-Drouet E, Borchmann P, et al. CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the phase 1 BNT211-01 trial. Nat Med. 2023;29:2844–53.
Haanen JBAG, Mackensen A, Schultze-Florey C, Alsdorf W, Wagner-Drouet E, Heudobler D, et al. 61°Updated results from BNT211-01 (NCT04503278), an ongoing, first-in-human, phase I study evaluating safety and efficacy of CLDN6 CAR T cells and a CLDN6-encoding mRNA vaccine in patients with relapsed/refractory CLDN6+ solid tumors. Ann Oncol. 2024;35:S489–S490.
Sittplangkoon C, Alameh MG, Weissman D, Lin P, Tam YK, Prompetchara E, et al. mRNA vaccine with unmodified uridine induces robust type I interferon-dependent anti-tumor immunity in a melanoma model. Front Immunol. 2022;13:983000.
Kappler JW, Roehm N, Marrack P. T-cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–80.
de Graaf JF, Pesic T, Spitzer FS, Oosterhuis K, Camps M, Zoutendijk I, et al. Neoantigen-specific T cell help outperforms nonspecific help in multiantigen DNA vaccination against cancer. Mol Ther Oncol. 2024;32:200835.
Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–3.
Ninmer EK, Zhu H, Chianese-Bullock KA, von Mehren M, Haas NB, Ross MI, et al. Multipeptide vaccines for melanoma in the adjuvant setting: long-term survival outcomes and post hoc analysis of a randomized phase II trial. Nat Commun. 2024;15:2570.
Bouazzaoui A, Abdellatif AAH. Vaccine delivery systems and administration routes: Advanced biotechnological techniques to improve the immunization efficacy. Vaccine: X. 2024;19:100500.
Sultan H, Kumai T, Nagato T, Wu J, Salazar AM, Celis E. The route of administration dictates the immunogenicity of peptide-based cancer vaccines in mice. Cancer Immunol Immunother. 2019;68:455–66.
Baharom F, Ramirez-Valdez RA, Tobin K, Yamane H, Dutertre CA, Khalilnezhad A, et al. Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8( + ) T cells. Nat Immunol. 2021;22:41–52.
Johansen P, Häffner AC, Koch F, Zepter K, Erdmann I, Maloy K, et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur J Immunol. 2005;35:568–74.
von Beust BR, Johansen P, Smith KA, Bot A, Storni T, Kündig TM. Improving the therapeutic index of CpG oligodeoxynucleotides by intralymphatic administration. Eur J Immunol. 2005;35:1869–76.
Hassett KJ, Rajlic IL, Bahl K, White R, Cowens K, Jacquinet E, et al. mRNA vaccine trafficking and resulting protein expression after intramuscular administration. Mol Ther Nucleic Acids. 2024;35:102083.
Kim J, Eygeris Y, Gupta M, Sahay G. Self-assembled mRNA vaccines. Adv Drug Deliv Rev. 2021;170:83–112.
Liang F, Lindgren G, Lin A, Thompson EA, Ols S, Röhss J, et al. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol Ther. 2017;25:2635–47
Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. 2015;217:345–51.
Schnyder JL, Garcia Garrido HM, De Pijper CA, Daams JG, Stijnis C, Goorhuis A, et al. Comparison of equivalent fractional vaccine doses delivered by intradermal and intramuscular or subcutaneous routes: a systematic review. Travel Med Infect Dis. 2021;41:102007.
Lee Y, Jeong M, Park J, Jung H, Lee H. Immunogenicity of lipid nanoparticles and its impact on the efficacy of mRNA vaccines and therapeutics. Exp Mol Med. 2023;55:2085–96.
Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front Immunol. 2019;10:2525.
Tombácz I, Laczkó D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A, et al. Highly efficient CD4 + T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol Ther. 2021;29:3293–304.
Su FY, Zhao QH, Dahotre SN, Gamboa L, Bawage SS, Silva Trenkle AD, et al. In vivo mRNA delivery to virus-specific T cells by light-induced ligand exchange of MHC class I antigen-presenting nanoparticles. Sci Adv. 2022;8:eabm7950.
Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T-cell responses by type I interferons. Nat Rev Immunol. 2015;15:231–42.
van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–72.
Sayour EJ, Boczkowski D, Mitchell DA, Nair SK. Cancer mRNA vaccines: clinical advances and future opportunities. Nat Rev Clin Oncol. 2024;21:489–500.
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95.
Saxena M, Bhardwaj N. Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer. 2018;4:119–37.
Borges F, Laureano RS, Vanmeerbeek I, Sprooten J, Demeulenaere O, Govaerts J, et al. Trial watch: anticancer vaccination with dendritic cells. Oncoimmunology. 2024;13:2412876.
Liu N, Wang X, Wang Z, Kan Y, Fang Y, Gao J, et al. Nanomaterials-driven in situ vaccination: a novel frontier in tumor immunotherapy. J Hematol Oncol. 2025;18:45.
Gong N, Alameh MG, El-Mayta R, Xue L, Weissman D, Mitchell MJ. Enhancing in situ cancer vaccines using delivery technologies. Nat Rev Drug Discov. 2024;23:607–25.
Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z, et al. Localized cocktail chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. Sci Adv. 2020;6:eaaz4204.
Liu Q, Xu R, Shen J, Tao Y, Shao J, Ke Y, et al. In situ chemoimmunotherapy hydrogel elicits immunogenic cell death and evokes efficient antitumor immune response. J Transl Med. 2024;22:341.
Xu P, Ma J, Zhou Y, Gu Y, Cheng X, Wang Y, et al. RadIotherapy-triggered In Situ Tumor Vaccination Boosts Checkpoint Blockaded Immune Response Via Antigen-capturing Nanoadjuvants. ACS Nano. 2024;18:1022–40.
Liang G, Jin X, Zhang S, Xing D. RGD peptide-modified fluorescent gold nanoclusters as highly efficient tumor-targeted radiotherapy sensitizers. Biomaterials. 2017;144:95–104.
Li Y, Luo Y, Hou L, Huang Z, Wang Y, Zhou S. Antigen-capturing dendritic-cell-targeting nanoparticles for enhanced tumor immunotherapy based on photothermal-therapy-induced in situ vaccination. Adv Healthc Mater. 2023;12:e2202871.
Xiao G, Zhao Y, Wang X, Zeng C, Luo F, Jing J. Photothermally sensitive gold nanocage augments the antitumor efficiency of immune checkpoint blockade in immune “cold” tumors. Front Immunol. 2023;14:1279221.
Wang Z, You T, Su Q, Deng W, Li J, Hu S, et al. Laser-activatable in situ vaccine enhances cancer-immunity cycle. Adv Mater. 2023;35:e2307193.
Yue W, Chen L, Yu L, Zhou B, Yin H, Ren W, et al. Checkpoint blockade and nanosonosensitizer-augmented noninvasive sonodynamic therapy combination reduces tumor growth and metastases in mice. Nat Commun. 2019;10:2025.
Chen, J, Qiu M., Ye Z., Nyalile T., Li Y., Glass Z., et al., In situ cancer vaccination using lipidoid nanoparticles. Sci Adv. 2021. 7.
Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, et al. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829.
Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20:160–77.
Fu R, Qi R, Xiong H, Lei X, Jiang Y, He J, et al. Combination therapy with oncolytic virus and T cells or mRNA vaccine amplifies antitumor effects. Signal Transduct Target Ther. 2024;9:118.
Chen X, Zhao J, Yue S, Li Z, Duan X, Lin Y, et al. An oncolytic virus delivering tumor-irrelevant bystander T-cell epitopes induces anti-tumor immunity and potentiates cancer immunotherapy. Nat Cancer. 2024;5:1063–81.
Melcher A, Harrington K, Vile R. Oncolytic virotherapy as immunotherapy. Science. 2021;374:1325–6.
Wang S, Liang B, Wang W, Li L, Feng N, Zhao Y, et al. Viral vectored vaccines: design, development, preventive and therapeutic applications in human diseases. Signal Transduct Target Ther. 2023;8:149.
D'alise AM, Leoni G, Cotugno G, Siani L, Vitale R, Ruzza V, et al. Phase I trial of viral vector-based personalized vaccination elicits robust neoantigen-specific antitumor T-cell responses. Clin Cancer Res. 2024;30:2412–23.
Le Tourneau C, Delord JP, Lantz O, Dochy E, Ceppi M, Spring-Giusti C, et al. Randomized phase I trial of adjuvant personalized cancer vaccine TG4050 in resected locally advanced (LA) head and neck squamous cell carcinoma (HNSCC) patients (pts). J Clin Oncol. 2025;43:6016–6016.
Kwon SY, Thi-Thu Ngo H, Son J, Hong Y, Min JJ. Exploiting bacteria for cancer immunotherapy. Nat Rev Clin Oncol. 2024;21:569–89.
Gurbatri CR, Arpaia N, Danino T. Engineering bacteria as interactive cancer therapies. Science. 2022;378:858–64.
Nguyen DH, Chong A, Hong Y, Min JJ. Bioengineering of bacteria for cancer immunotherapy. Nat Commun. 2023;14:3553.
Gerstner GJ, Ramalingam SS, Lisberg AE, Farber CM, Morganstein N, Sanborn RE, et al. A phase 2 study of an off-the-shelf, multineoantigen vector (ADXS-503) in patients with metastatic non–small cell lung cancer either progressing on prior pembrolizumab or in the first-line setting. J Clin Oncol. 2022;40:9038–9038.
Hecht JR, Goldman JW, Hayes S, Balli D, Princiotta MF, Dennie JG, et al. Abstract CT007: Safety and immunogenicity of a personalized neoantigen-Listeria vaccine in cancer patients. Cancer Res. 2019;79:CT007–CT007.
Redenti A, Im J, Redenti B, Li F, Rouanne M, Sheng Z, et al. Probiotic neoantigen delivery vectors for precision cancer immunotherapy. Nature. 2024;635:453–61.
Saxena M, van der Burg SH, Melief C, Bhardwaj N. Therapeutic cancer vaccines. Nat Rev Cancer. 2021;21:360–78.
Baharom F, Hermans D, Delamarre L, Seder RA. Vax-Innate: improving therapeutic cancer vaccines by modulating T cells and the tumor microenvironment. Nat Rev Immunol. 2024;25:195–211.
Kersten K, Hu KH, Combes AJ, Samad B, Harwin T, Ray A, et al. Spatiotemporal codependency between macrophages and exhausted CD8( + ) T cells in cancer. Cancer Cell. 2022;40:624–638 e9.
Waibl Polania J, Hoyt-Miggelbrink A, Tomaszewski WH, Wachsmuth LP, Lorrey SJ, Wilkinson DS, et al. Antigen presentation by tumor-associated macrophages drives T cells from a progenitor exhaustion state to terminal exhaustion. Immunity. 2025;58:232–246.e6.
Jo JH, Kim YT, Choi HS, Kim HG, Lee HS, Choi YW, et al. Efficacy of GV1001 with gemcitabine/capecitabine in previously untreated patients with advanced pancreatic ductal adenocarcinoma having high serum eotaxin levels (KG4/2015): an open-label, randomized, phase 3 trial. Br J Cancer. 2024;130:43–52.
Ibrahim NK, Murray JL, Zhou D, Mittendorf EA, Sample D, Tautchin M, et al. Survival advantage in patients with metastatic breast cancer receiving endocrine therapy plus sialyl Tn-KLH vaccine: post hoc analysis of a large randomized trial. J Cancer. 2013;4:577–84.
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373–85.
Middleton GW, Valle JW, Wadsley J, Propper D, Coxon FY, Ross PJ, et al. A phase III randomized trial of chemoimmunotherapy comprising gemcitabine and capecitabine with or without telomerase vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer. J Clin Oncol. 2013;31:LBA4004–LBA4004.
Makino T, Miyata H, Yasuda T, Kitagawa Y, Muro K, Park JH, et al. A phase 3, randomized, double-blind, multicenter, placebo-controlled study of S-588410, a five-peptide cancer vaccine as an adjuvant therapy after curative resection in patients with esophageal squamous cell carcinoma. Esophagus. 2024;21:447–55.
Patel S, Thompson J, Patel M, Daugherty FJ, Rimawi MF. Phase III study to evaluate the efficacy and safety of GLSI-100 (GP2 + GM-CSF) in breast cancer patients with residual disease or high-risk PCR after both neoadjuvant and postoperative adjuvant anti-HER2 therapy: Flamingo-01. J Clin Oncol. 2023;41:TPS617–TPS617.
Melief CJM, Welters MJP, Vergote I, Kroep JR, Kenter GG, Ottevanger PB, et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci Transl Med. 2020;12:eaaz8235.
van Poelgeest MI, Welters MJ, Vermeij R, Stynenbosch LF, Loof NM, Berends-van der Meer DM, et al. Vaccination against oncoproteins of HPV16 for noninvasive vulvar/vaginal lesions: lesion clearance is related to the strength of the T-cell response. Clin Cancer Res. 2016;22:2342–50.
Speetjens FM, Welters MJP, Slingerland M, van Poelgeest MIE, de Vos van Steenwijk PJ, Roozen I, et al. Intradermal vaccination of HPV-16 E6 synthetic peptides conjugated to an optimized Toll-like receptor 2 ligand shows safety and potent T cell immunogenicity in patients with HPV-16 positive (pre-)malignant lesions. J Immunother Cancer. 2022;10:e005016.
Lou Y, Chumsri S, Asmann YW, Shi Q, Reynolds J, Necela B, et al. A phase I pilot study of personalized neoantigen peptide-based vaccine in combination with pembrolizumab in advanced solid tumors (PNeoVCA). J Clin Oncol. 2024;42:TPS2700–TPS2700.
Gillison ML, Awad MM, Twardowski P, Sukari A, Johnson ML, Stein MN, et al. Long term results from a phase 1 trial of GEN-009, a personalized neoantigen vaccine, combined with PD-1 inhibition in advanced solid tumors. J Clin Oncol. 2021;39:2613–2613.
Elez Fernandez ME, Maurel J, Morris V, Kopetz S, Galligan B, Ali S, et al. 29 P Characterization of T-cell responses induced by the individualized mRNA neoantigen vaccine autogene cevumeran in adjuvant stage II (high risk)/stage III colorectal cancer (CRC) patients (pts) from the biomarker cohort of the phase II BNT122-01 trial. Ann Oncol. 2024;35:S14–S15.
Saba NF, Klinghammer K, Castelluci E, Colevas AD, Rutkowski T, Thurner D, et al. 877P Exploratory efficacy and translational results from the safety run in of AHEAD-MERIT, a phase II trial of first line pembrolizumab plus the fixed-antigen cancer vaccine BNT113 in advanced HPV16 + HNSCC. Ann Oncol. 2024;35:S627.
Khattak MA, Sullivan RJ, Gutierrez M, Jimeno A, Thomas SS, Long GV, et al. 663P First-in-human, phase I/II, monotherapy, dose-escalation study of mRNA-4359, an mRNA-encoded PD-L1/IDO1 antigen-specific therapy, in advanced/refractory solid tumors. Ann Oncol. 2024;35:S521–S522.
Tabatabai G, Freres P, Glas M, Hau P, von Baumgarten L, van den Bent MJ, et al. 44°First in human study of the mRNA-based cancer vaccine CVGBM in patients (pts) with newly diagnosed and surgically resected MGMT-unmethylated glioblastoma (GBM): First results from the dose escalation phase. Ann Oncol. 2024;35:S406.
Arance Fernandez AM, Baurain JF, Vulsteke C, Rutten A, Soria A, Carrasco J, et al. A phase I study (E011-MEL) of a TriMix-based mRNA immunotherapy (ECI-006) in resected melanoma patients: analysis of safety and immunogenicity. J Clin Oncol. 2019;37:2641–2641.
Acknowledgements
This study was supported by funding from the National Science and Technology Major Project of the Ministry of Science and Technology of China (2023YFC2508505) and grants from the Natural Science Foundation of China (82250710684). Figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflicts of interest. Dr. Yang-Xin Fu is an editorial board member of Cellular & Molecular Immunology, but he has not been involved in peer review or decision-making related to the article.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, K., Zhao, X., Fu, YX. et al. Eliciting antitumor immunity via therapeutic cancer vaccines. Cell Mol Immunol 22, 840–868 (2025). https://doi.org/10.1038/s41423-025-01316-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01316-4
Keywords
This article is cited by
-
Immune checkpoint blockade in cancer: current insights and future horizons
Discover Oncology (2026)
-
Mapping the frontiers: a bibliometric perspective on t cell-based immunotherapy in pancreatic cancer since the twenty-first century
Journal of Cancer Research and Clinical Oncology (2025)
