
Abstract
Chronic liver disease represents a significant global health burden. Regardless of etiology, its pathogenesis is driven by persistent liver inflammation, which can lead to fibrosis, cirrhosis, and an increased risk of cancer development. Myeloid cells, including neutrophils, eosinophils, monocytes, macrophages, and dendritic cells, play diverse and critical roles in hepatic immunity and the maintenance of tissue homeostasis but are also involved in liver injury, disease progression, and resolution. With the emergence of high-resolution omics technologies and in vivo fate-mapping models, our understanding of myeloid cell ontogeny and functional heterogeneity has been significantly refined. In this review, we discuss current insights into the myeloid cell landscape in nonviral chronic liver inflammatory conditions and summarize the roles of myeloid cell subsets in disease pathogenesis.
Similar content being viewed by others
Introduction
Liver disease is a major global healthcare burden, with estimates suggesting that over 840 million people worldwide suffer from chronic liver disease (CLD), which is associated with a mortality rate of ~2 million deaths per year [1]. Recent data indicate a rising prevalence of CLD, mainly due to the increasing incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-associated liver disease (ALD) [2,3,4]. As such, there is increasing interest in identifying factors that contribute to disease development and progression.
CLD encompasses a range of etiologies, each highlighted by its unique pathogenesis but underlined by the premise that persistent liver inflammation begs fibrosis, progression to end-stage scarring termed cirrhosis, and increased risk of cancer development. MASLD, which is globally recognized as the fastest growing cause of CLD, particularly in the Western world (with a prevalence of 25–30%) [5, 6], is characterized by the development of steatosis (fat accumulation in the liver) in the presence of metabolic risk factors, such as obesity and type 2 diabetes. MASLD encompasses a spectrum of conditions, ranging from benign steatosis to a more aggressive, inflammatory form named metabolic dysfunction-associated steatohepatitis (MASH), which is associated with fibrosis and cirrhosis development [7]. ALD refers to liver disease in the context of sustained alcohol misuse. Similarly, this can be associated with benign steatosis, steatohepatitis and acutely, severe alcohol abuse and alcohol-associated hepatitis (AH) [3]. In ALD and AH, the inflammatory milieu is complex and is dictated by direct alcohol-induced hepatotoxicity, increased gut permeability leading to increased bacterial translocation, and innate immune dysregulation [8, 9].
Autoimmune hepatitis (AIH) is characterized by the presence of autoantibodies and histologic interface hepatitis, with a variable clinical presentation (asymptomatic, acute liver failure, or CLD) [10]. Liver diseases characterized by major bile duct alterations, or cholangiopathies, include primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), both of which are of idiopathic origin. PBC is a chronic inflammatory autoimmune disease characterized by cholestasis and progressive small bile duct cholangitis [11], whereas PSC encompasses an inflammatory cholangiopathy of largely unknown cause [12, 13], variably affecting both the small and large bile ducts depending on the subtype.
Chronic liver injury, regardless of etiology, leads to persistent activation of hepatic immune responses and subsequent inflammation. This in turn leads to aberrant activation of extracellular matrix (ECM)-producing cells such as hepatic stellate cells (HSCs) [14] and the deposition of fibrillar matrix proteins, including collagens, causing self-perpetuating liver inflammation and, as the disease progresses, cirrhosis. If left untreated or if the underlying causative factor is not removed, this may lead to the development of liver cancer, particularly hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (iCCA) [15, 16]. Undoubtedly, the immune system plays significant roles in CLD development and progression. In this review, we discuss the current understanding of the functional heterogeneity and roles of neutrophils, eosinophils, monocytes, macrophages, and dendritic cells (DCs) in various nonviral chronic inflammatory liver diseases.
The liver: a central immunological organ
The liver is the largest solid organ in the body. It is conventionally considered a nonimmunological organ that is primarily responsible for lipid metabolism, nutrient storage, and detoxification. To fulfill its physiological functions, the liver is primarily composed of hepatocytes, which are parenchymal cells that account for ~60–70% of the total cell count and 80% of the liver mass [17, 18]. The remaining cells are nonparenchymal cells, including endothelial cells, HSCs, Kupffer cells (KCs), and other immune cell populations [17, 18]. Anatomically, the liver is organized into repeating hexagonal lobules, each outlined by six “portal triads”, which consist of a bile duct, portal venule and hepatic arteriole (Fig. 1A). The liver has a dual blood supply: ~80% of the blood originates from the gut and flows via the portal vein, where it carries nutrients and gut-derived molecules, whereas the remaining 20% is oxygenated arterial blood delivered through the hepatic artery. These blood supplies mix within specialized capillaries known as liver sinusoids, which are lined with liver sinusoidal endothelial cells (LSECs) and drain into the central vein [19].

A (Left) Macroscopic view of the liver: it receives a dual blood supply, with nutrient-rich blood from the portal vein (PV) and oxygenated blood from the hepatic artery (HA); the blood exits through the hepatic vein (HV). (Right) Schematic view of the hepatic lobules: the repeated hexagonal anatomical units of the liver. Each lobule consists of hepatocytes (main parenchymal cells) arranged around a central vein (CV) and is demarcated by a portal triad, containing a PV, an HA, and a bile duct (BD), at each corner. B Schematic view of liver cell subsets under homeostatic conditions. Nonparenchymal cells include immune cells, liver sinusoidal endothelial cells (LSECs), and hepatic stellate cells (HSCs). The HA and PV converge at the portal triads, and the resulting mixed blood flows through the sinusoids toward the CV, generating gradients of oxygen and nutrients that drive the functional zonation of the parenchyma into three distinct zones: periportal, midlobular, and pericentral. Blood immune cells enter and mix in the sinusoids; some cells may circulate, temporarily patrol the sinusoids and parenchyma, or become tissue-resident. Kupffer cells (KCs), the most abundant immune cell subset involved in homeostasis, reside within the sinusoids in close contact with LSECs and extend protrusions that enable interactions with HSCs (residing in the space of Disse) and hepatocytes. This position enables KCs to recognize antigens and pathogens and maintain immune tolerance. Bone marrow monocyte-derived macrophage subsets can also be found around BDs and CVs. Dendritic cells are located primarily periportally. This figure was created with BioRender (biorender.com)
The unique structure of liver sinusoids, characterized by fenestrated endothelial cells lacking a basement membrane, facilitates low-pressure blood flow, enabling direct interactions between immune cells and hepatocytes, as well as the exchange of substances. Variations in the microenvironment, including Wnt signaling, oxygen levels, and nutrient gradients, give rise to functional liver zonation [20, 21], with the hepatic lobule divided into three zones: periportal (zone 1), mid-lobular (zone 2) and pericentral (zone 3) [17, 21, 22]. Functional zonation has been described in hepatocytes [23,24,25], LSECs [26], HSCs [27], and immune cells such as KCs [28,29,30].
The liver plays a critical role in immune surveillance and tissue homeostasis because of its strategic location at the confluence of the dual blood supply (Fig. 1A) [31, 32]. As the gut serves as a major entry point for pathogens, the liver must recognize, neutralize, and clear gut-derived pathogens before the blood re-enters the systemic circulation [32, 33]. This process requires a delicate balance between immune activation—targeting harmful pathogens—and tolerance—toward harmless molecules such as dietary antigens—to prevent inappropriate inflammation [33, 34]. This finely tuned homeostatic equilibrium is tightly regulated and essential for maintaining proper liver function.
Under homeostatic conditions, the liver is predominantly populated by innate immune cells, including macrophages, DCs, and natural killer (NK) cells [32, 34] (Fig. 1B). Among these, KCs, which are liver-resident macrophages, serve as key effector cells responsible for detecting microbial molecules, clearing pathogens, and producing cytokines and chemokines to mediate inflammatory responses [35, 36]. Numerous studies have demonstrated that KCs play a vital role in controlling and resolving infections caused by bacteria such as Staphylococcus aureus and Escherichia coli (E. coli) [28, 37,38,39,40,41,42,43,44,45,46,47,48].
The ability of the liver to recognize and neutralize danger signals relies on the broad expression of pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and NOD-like receptors [49, 50]. These receptors are expressed by KCs, hepatocytes, and professional antigen-presenting cells (APCs), allowing them to detect a variety of pathogen-associated molecular patterns (PAMPs) from microorganisms and damage-associated molecular patterns (DAMPs) from host tissue damage. Overall, the liver plays a key role as an immunological organ, particularly at the interface between the gut and systemic circulation, and is critical for host defense.
Myeloid cells in chronic liver inflammation
Myeloid cells constitute a significant proportion of leukocytes in the circulation and peripheral tissues, including the skin, lungs, gut, and liver [51]. They include granulocytes (neutrophils, eosinophils, and basophils), monocytes, macrophages, and DCs. These cells represent major cellular compartments of the innate immune system and play key roles in antimicrobial defense and inflammation, as well as in tissue development, homeostasis, and repair. With the advent of high-resolution single-cell omics technologies and in vivo fate-mapping models, the fields of myeloid cell ontogeny, heterogeneity, maturation, and specialization in homeostasis and inflammatory diseases [51,52,53,54,55,56], including cancer [57, 58], have been extensively revised. Recent studies suggest that, beyond their origin, a multitude of diverse microenvironmental cues can influence myeloid fate and function, with increasing evidence indicating that granulocytic subsets, such as neutrophils and eosinophils, are more heterogeneous than previously appreciated. This article provides a comprehensive overview of the phenotypical and functional properties of neutrophils [59], eosinophils [60], monocytes/macrophages [61,62,63,64,65,66,67], and DCs [68,69,70,71,72] in the context of liver health and disease, highlighting key discoveries.
Neutrophils
Origin and functions of neutrophils
Neutrophils, the predominant type of circulating leukocyte, are endowed with antipathogenic and inflammatory effector functions and are therefore critical for maintaining host immunity and tissue homeostasis [73, 74]. The multifaceted activities of these cells underscore their remarkable phenotypic and functional diversity [75] under both steady-state and stress conditions. As explored below, neutrophils were previously thought to be transcriptionally silent, terminally differentiated cells; however, this view has recently been challenged, and neutrophil plasticity is now widely recognized.
Neutrophils develop through granulopoiesis, an intricately regulated process by which immune cells in the bone marrow undergo progressive modifications to their transcriptomic, proteomic, structural, and consequently functional properties before being released into the circulation [52]. Under homeostatic and emergency conditions, granulopoiesis directs neutrophil differentiation and development from hematopoietic stem cells to common myeloid progenitors (CMPs), then to granulocyte‒macrophage progenitors (GMPs), and subsequently to lineage-specific maturation. Neutrophil commitment can be divided into two stages: (1) determination: a stage of differentiation to preneutrophils, after which only effector functions can be acquired, and (2) specification: whereby the neutrophil lineage is committed to, in the form of pro-neutrophils [52]. This process of specification, alongside the concurrent development of effector functions, is tightly guided by granulocytic transcription factors, primarily growth factor independence-1 (GFI-1) and CCAAT-enhancer binding protein-ε (C/EBPε) [52, 76]. Functional maturation is preestablished through pro-neutrophil chromatin remodeling, with specific effector roles peaking at different stages of subsequent maturation [74].
In the event of emergency granulopoiesis triggered by physiological stress, de novo neutrophil generation occurs [77]. Noncommitted precursor and immature committed low-density neutrophils (LDNs) expand and are prematurely released into the circulation, where they coexist with terminally differentiated subsets. In this context, these early neutrophil lineage subsets exhibit altered effector function capabilities compared with their mature counterparts [52, 74]. Heterogeneity under steady-state and stress conditions has thus been described, with phenotypic and functional alterations ultimately determined by physiological and pathologic contexts [74]. These contexts are highly dynamic and are shaped by combined influences such as homeostatic fluctuations (e.g., granulopoiesis state origin) [74], local bone marrow niche composition [78, 79], circadian rhythm [80, 81], stage of maturation postspecification, and the local microenvironment (circulating [82] or tissue-resident [83]). Further research is now needed to explore heterogeneity within individual circulating and tissue-resident neutrophil subsets.
Neutrophils are among the first innate immune cells to mobilize to sites of tissue injury, orchestrating the host immune response during inflammation while also coordinating critical antipathogen responses. They perform several antipathogen effector functions that, even in a state of health, vary depending on neutrophil density (determined by nuclear morphology and granule content), maturation stage, or activation state [74] as described below. Chemotaxis and adhesion are closely interlinked processes that enable neutrophils to respond effectively to sites of injury or infection. Chemotaxis involves their migration toward chemoattractants, such as chemokines, DAMPs and PAMPs [59] while adhesion is facilitated by proinflammatory agents [e.g., interleukin-1 (IL-1), IL-17, and tumor necrosis factor alpha (TNF-α)], which increase vascular permeability and stimulate the expression of adhesion molecules on the endothelium (selectins and integrins) and constitutively expressed receptors on neutrophils (P-selectin ligand 1 and L-selectin) [9, 84]. Together, these processes ensure that neutrophils can not only locate sites of inflammation but also firmly adhere to the endothelium.
Neutrophils capture and kill pathogens through several mechanisms. They interact directly with pathogens or via receptors such as Fc receptors (FccRIIIB/CD16 and FccRIIA/CD32), integrins (CD11b/CD18), and complement receptors (CR1/CD35 and CR3) to facilitate phagocytosis [8, 9]. Once internalized, pathogens are destroyed within phagolysosomes by intracellular granules and reactive oxygen species (ROS) [9, 52, 59]. Neutrophils generate ROS primarily through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which produces superoxide anions and hydrogen peroxide, and secondarily through free hydroxyl radicals formed by myeloperoxidase (MPO) [85, 86].
Pathogen interactions and inflammatory signals can also induce neutrophil degranulation, leading to the release of proteases, MPO, and defensins that contribute to pathogen destruction. In addition, neutrophils produce various chemokines [C-X-C motif chemokine ligand 1 (CXCL1), CXCL2, CXCL8 and CXCL12] and cytokines, including proinflammatory (IL-1α, IL-1β, IL-6, TNF-α), anti-inflammatory [IL-10, transforming growth factor beta (TGF-β)], and immunoregulatory (IL-22, IL-23) mediators [9, 52, 59]. Uncontrolled release of proinflammatory cytokines by aberrantly or persistently activated neutrophils can exacerbate local and systemic inflammation, promoting a “cytokine storm”. However, whether the degree to which neutrophil cytokine release coordinates systemic responses in a manner similar to that of other immune cells (e.g., lymphocytes) remains unclear.
Neutrophil extracellular trap (NET) formation, or NETosis, is another key function of these cells. NETs consist of extruded decondensed DNA, citrullinated histones, and a mixture of proteins of nuclear, cytosolic, and granular origin, including neutrophil elastase (NE) and MPO [9, 52, 59]. This process is ultimately driven by ROS production, wherein MPO activation leads to granule release into the cytoplasm and subsequently into the nucleus, promoting chromatin decondensation, whereas PAD4 activation induces histone citrullination [52, 87]. The decondensed DNA then mixes with the cytoplasmic granular content and is ultimately released into the surrounding environment following disintegration of the neutrophil plasma membrane. NETs are produced to capture, immobilize, and kill pathogens.
In homeostasis, the effector functions of neutrophils are well-regulated and harmoniously coordinated, leading to the resolution of inflammation, preservation of tissue integrity through injury healing, and efficient eradication of invading pathogens. However, in states of aberrant activation or sustained inflammation, neutrophil dysfunction can prevail, resulting in tissue damage and disease progression, depending on the underlying pathology.
Neutrophils in chronic liver inflammation
MASLD
In steatotic liver disease, lipid accumulation within hepatocytes induces metabolic stress. This triggers the activation of resident immune cells and the subsequent recruitment of circulating immune cells in response to the release of proinflammatory cytokines and chemokines. Hepatic neutrophil infiltration is a hallmark of MASH development [88]. This is not only evident in human liver biopsies [89, 90] but also observed in the early stages of high-fat diet (HFD) feeding in mice [91, 92], in which neutrophil depletion via Ly6G-neutralizing antibodies abrogates metabolic syndrome development (weight gain and increased blood glucose), initial liver inflammation and injury [91, 93].
Neutrophil-derived factors such as NE and human neutrophil peptide-1 (HNP-1) can activate KCs and recruited macrophages while also inducing the proliferation of HSCs [94, 95]. Together, these effects drive inflammation and fibrogenesis. In HFD-fed mouse models, CXCL1 [96], CXCR2 (induced via lipocalin-2) [97], and IL-8-mediated neutrophil infiltration have been shown to contribute to MASH pathogenesis through various mechanisms, including granule enzyme (e.g., MPO) and ROS release and increased activation of stress kinases [90, 98]. Notably, elevated in the serum of patients with MASH [99], MPO can increase oxidative stress, contributing to injury through hepatocyte death while activating HSCs [90]. Furthermore, NE disrupts lipid homeostasis through hepatic ceramide synthesis, resulting in insulin resistance [100]. Compared with NE knockout mice, wild-type (WT) mice fed a Western diet (WD) for 24 weeks presented significantly greater weight gain, biochemical derangements (high serum lipid/triglyceride levels and transaminitis), and histological fibrosis progression [100].
Aberrant neutrophil activation further leads to NETosis, with elevated serum NET levels observed in MASH patients [8]. In HFD-fed mouse models (with/without additional alcohol insult), NET formation results in increased inflammation and fibrosis [92, 101]. Treatment with deoxyribonuclease (DNase) attenuated monocyte-derived macrophage infiltration [101] and the resulting tissue injury, although it did not halt disease progression [102]. Overall, neutrophils appear to contribute to the early pathogenesis of MASH, wherein hepatocyte injury is potentiated by inflammatory mediators, ROS, and NETs. Although a potential restorative role for neutrophils, through the regulation of the macrophage phenotype, has been described in MASH/fibrosis (as further discussed below), their precise functions across the MASLD spectrum require further investigation.
ALD
In ALD, neutrophil recruitment depends on multiple variables, including neutrophil priming, circulating chemokines, and the activation of LSECs [103]. Neutrophil priming and activation may be prerequisites for their recruitment into the liver parenchyma, with enhanced activation of circulating neutrophils characterized by reduced L-selectin (CD62L) expression observed in AH patients [104]. Several chemokines released from hepatocytes, HSCs, KCs, and LSECs, including CXCL1, CXCL2, and CXCL8, promote neutrophil recruitment [105, 106]. Once recruited, neutrophils adhere to the endothelium and transmigrate into the hepatic microenvironment.
In AH, gastrointestinal dysbiosis and increased gut permeability lead to a greater bacterial load in the enterohepatic circulation. Recognition of PAMPs activates neutrophils and their arsenal of antimicrobial functions. Evidence suggests that neutrophils in AH are hyperresponsive, producing increased levels of inflammatory cytokines and chemokines [107] and demonstrate increased baseline oxidative burst capacity [108]. Compared with healthy controls, NET formation has been described as a driver of chronic inflammation in AH, with high serum NE and citrullinated histone H3 (H3Cit) levels and increased spontaneous NET formation capability [109]. Interestingly, these neutrophils release fewer NETs upon phorbol myristate acetate (PMA) stimulation [109], suggesting that they are primed but exhibit a defective antimicrobial response to subsequent challenge [103]. NETs further induce monocyte differentiation into CD14+CD16+ intermediate inflammatory phenotypes and reduce macrophage efferocytosis, thereby perpetuating chronic inflammation and tissue damage [109].
Recent single-cell RNA sequencing (scRNA-seq) of hepatic neutrophils in AH has revealed an expanded IL-8+ subset within an already elevated total neutrophil population compared with ALD patients and healthy controls [110]. Pathway analysis revealed that the top three differentially expressed hallmark pathways in this subset were related to TNF-α, interferon gamma (IFN-γ), and inflammatory responses [110]. Given that IL-8 is key for neutrophil activation, the accumulation of IL-8+ neutrophils in AH may contribute to a self-sustaining proinflammatory environment, thus representing a potential therapeutic target for disease management.
Overall, in the context of AH, neutrophils exhibit a proinflammatory, typically low-density phenotype that is aberrantly activated in response to dysbiosis. This activation drives the recruitment and differentiation of various innate immune cell subtypes. When unregulated, this process perpetuates inflammation, contributing to increased morbidity and mortality.
AIH, PBC, and PSC
Dysregulation of immune responses to self-antigens is a hallmark of autoimmune disorders [52]. Neutrophils have been implicated in a variety of autoimmune conditions, including systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease [111]. In general, tissue-infiltrating neutrophils are involved in autoantigen presentation, the release of proinflammatory effector molecules/cytokines, and altered ROS production [111]. These multifaceted functions have also been demonstrated in immune-mediated CLD.
In AIH, the presence of an expanded population of circulating LDNs has been described [112]. The presence of NETosis, alongside dominant autoantibodies [e.g., antinuclear and anti-alpha smooth muscle actin (α-SMA) antibodies], suggests that NETosis contributes to abnormal autoimmune responses to self-antigens, thus driving disease progression and flares [112,113,114,115]. This hypothesis is further supported by the increased presence of MPO+ LDNs in AIH patients compared with healthy individuals [112, 115]. As such, the presence of LDNs may serve as a biomarker indicative of disease severity. However, the extent to which NETs contribute to AIH pathogenesis and whether circulating or liver-infiltrating neutrophils are more significantly implicated remain unclear.
In biliary injury, particularly in PBC, proinflammatory cytokines, including IL-6, IL-8, and TNF-α, are released [116], promoting the recruitment and activation of innate and adaptive immune cells, as well as mesenchymal cells, to initiate biliary repair [116, 117]. IL-8 has been observed in small bile ducts in PBC patients, particularly in patients with cirrhosis [117, 118]. IL-8 positivity in early disease is relatively infrequent [117], suggesting that its persistence contributes to disease progression but may not represent the initial driver. Clinically, the neutrophil‒lymphocyte ratio (NLR) is positively associated with nonresponse to ursodeoxycholic acid (UDCA) treatment [119].
In PSC, a mixed biliary tract inflammatory cell infiltrate, including lymphocytes, plasma cells, neutrophils, NK cells, and macrophages, has been described [116, 120]. Histologically, periductular inflammation in acinar Zone 1, a process associated with marked neutrophil infiltration, is observed [116, 121]. Neutrophil infiltration has been described in the context of biliary-resident T helper 1 and T helper 17 (Th1/Th17) cell activation; these cells display a CD103+ CD69 effector memory phenotype with a CXCL8-associated transcriptional profile [118] and are capable of producing IL-17 and IL-22 [122]. Additionally, the chemoattraction of neutrophils is promoted by C–C motif chemokine ligand 24 (CCL24), which is released from damaged cholangiocytes [123]. Although an increased proportion of MPO+ neutrophils are histologically detected, the absence of MPO in mouse models [Mdr2-deficient (Mdr2−/−)] did not confer protection from cholangiocyte injury [124]. The precise mechanisms by which neutrophils contribute to the PSC have yet to be fully elucidated.
Overall, neutrophils have not been widely explored in patients with liver autoimmune conditions, which may be challenging because of the significant histological overlap among AIH, PBC, and PSC. Further work is needed to comprehensively evaluate phenotypic variations in circulating and liver-infiltrating neutrophils to better understand their role in disease pathogenesis.
Fibrosis
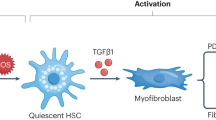
Although they are driven primarily by HSC activation and transdifferentiation, neutrophils play an active role in sustaining inflammation and thereby promoting liver fibrogenesis (Fig. 2A). As observed in all etiologies of CLD, neutrophils are recruited in the context of liver inflammation, contributing to fibrogenesis through the activation of KCs and the recruitment of other leukocytes, as shown in mouse models [125,126,127]. These processes are driven by a combination of their degranulation, ROS production, and NETosis. Neutrophil cytokine production is also implicated in fibrogenesis, with IL-17 released by neutrophils and Th17 cells playing a prominent role in the activation of TGF-β signaling pathways in HSCs [128]. IL-17 sensitizes HSCs to TGF-β by inducing the upregulation of TGF-β receptor II (TGFBR2) while also increasing the activation of downstream kinases [125]. In combination, HSC differentiation into myofibroblasts is induced, ultimately resulting in collagen deposition, thereby aggravating fibrosis.

A A schematic overview of the intricate interactions between neutrophils and other cells in the liver during fibrogenesis and fibrolysis across different chronic liver disease etiologies. B A schematic overview of the dysregulated effector functions of peripheral neutrophils in patients with cirrhosis. Abbreviations: HNP-1 human neutrophil peptide-1, HSC hepatic stellate cell, H3Cit citrullinated histone H3, KCs Kupffer cells, miR-223 microRNA-223, MMP matrix metalloproteinase, MPO myeloperoxidase, NE neutrophil elastase, NET neutrophil extracellular trap, ROS reactive oxygen species. This figure was created with BioRender (biorender.com)
Emerging evidence suggests that neutrophils also play a critical role in the resolution phase of injury. To successfully promote fibrolysis and degradation of the ECM, the underlying drivers of injury and inflammation must be resolved for HSCs to return to a quiescent phenotype [125]. To encourage this, neutrophils (with/without phagocytic stimuli) have been shown in mice to induce a functional switch in macrophages from a proinflammatory (Ly6Chi CX3CR1lo) type to a prorestorative (Ly6Clo CX3CR1hi) type [125, 129], which is capable of supporting tissue regeneration. Neutrophil depletion during the restorative phase is associated with ongoing fibrosis [130]. Apoptotic neutrophils can be phagocytosed by macrophages [131], suggesting that neutrophil apoptosis may serve as a cue for injury resolution and tissue repair [103].
Neutrophils also appear to directly promote fibrosis regression (Fig. 2A). In chronic carbon tetrachloride (CCl4)-induced injury mouse models, neutrophils have been suggested to contribute to fibrolysis and vascular regrowth [132] through the release of anti-inflammatory cytokines, hepatocyte growth factor (HGF), and anti-fibrotic matrix metalloproteinases (MMPs) 8 and 9 (MMP8 and MMP9). In MASH, extracellular vesicles released by neutrophils induce the downregulation of proinflammatory, profibrotic, and oncogenic hepatocyte genes through microRNA-223 (miR-223), the most abundant microRNA in neutrophils [133, 134]. In keeping with this functional role, miR-223 deletion in AH is associated with increased liver injury in the context of ROS production [135]. miR-223 regulates neutrophil infiltration in steatotic liver disease through the suppression of IL-6 and p47phox expression, indicating that their upregulation contributes to liver injury [130, 136]. Additionally, miR-223 has been suggested to induce the conversion of proinflammatory macrophages to an anti-inflammatory phenotype, thereby restoring liver homeostasis [130, 136].
Cirrhosis: Driving the development of multiorgan failure and death in patients with acutely decompensated cirrhosis (AD) and acute-to-chronic liver failure (ACLF) is increased susceptibility to bacterial infections [137, 138]. This is compounded by the development of high-grade systemic inflammation and immunoparesis, underpinned by defects in both innate and adaptive immune responses [139,140,141,142]. A summary of how neutrophil effector functions are impacted in cirrhosis is presented in Fig. 2B. It is postulated that persistent DAMP- and/or PAMP-driven immune cell stimulation in the AD and ACLF states drives systemic inflammation and ineffective responses to intercurrent infections [143, 144]. There is growing evidence that neutrophil antipathogen dysfunction in cirrhosis contributes to poor clinical outcomes, with the NLR shown to be an independent prognostic predictor of survival, irrespective of liver disease severity scores [145].
Neutrophil chemotaxis is reduced in cirrhosis patients because of a combination of intrinsic cellular defects and the impaired or inhibited chemoattractant ability of cirrhotic serum [146]. Neutrophils from cirrhotic patients demonstrate reduced migration toward healthy sera and IL-8 [147], decreased transendothelial migration in response to N-formyl-met-leu-phe (fMLF), increased adhesion to endothelial cells, and altered expression of adhesion receptors [59]. In AD and ACLF patients, CXCL1 and CXCL8 levels are highly elevated, even compared with those in patients with compensated cirrhosis [148, 149]; however, impaired neutrophil migration persists and is associated with adverse outcomes in these cohorts [150]. Overall, the degree of decreased chemotaxis in cirrhosis patients is pathway-dependent and multifactorial.
Impaired phagocytosis capacity has been widely described in patients with cirrhosis. Phagocytic capacity in the context of E. coli infection, a common cause of spontaneous bacterial peritonitis, in patients with advanced cirrhosis has been shown to be impaired in whole blood and isolated neutrophils [151]. The number of phagocytic neutrophils has also been shown to be reduced in patients with cirrhosis [152, 153]. In a similar manner, pathogen-killing capacity appears to be diminished in circulating neutrophils, irrespective of their phagocytic capacity [154]. Although impaired intracellular killing of bacteria and fungi (e.g., Candida albicans) [155] has been described in the literature, this has not always been replicated, with some studies reporting no change, albeit in the context of different cirrhosis etiologies.
Impaired neutrophil ROS production and degranulation have been described in cirrhosis [151, 156], with varying alterations in capability, production, and function reported [59]. There is an expanded pool of neutrophils with elevated “resting” oxidative burst potential in cirrhosis [151, 157, 158], although a consensus regarding basal ROS production — outside of the hyperinflammatory ACLF state, where it is increased [157, 159]—has not been reached. Cirrhotic neutrophils respond to low physiological stimuli such as fMLF [151, 156, 158], suggesting prepriming from persistent systemic inflammation. However, in patients with active infection, ROS production has been shown to be unaltered in response to fMLF or E. coli stimulation. The number of neutrophils that produce ROS in response to more potent stimulation, such as E. coli, has been reported to be unchanged [151, 153] or decreased in cirrhosis [158]. Importantly, however, the level of intracellular ROS is decreased, suggesting the possibility of immune exhaustion secondary to persistent inflammation [59].
The intracellular neutrophil enzyme content and its release from neutrophil granules upon stimulation and activation have been shown to be reduced in patients with cirrhosis. The number of cirrhotic neutrophils that produce MPO, as well as the total MPO produced, has been reported to be reduced [160]. However, other studies suggest that MPO’s extracellular release [154] or end function is impaired [161] rather than being the total initially produced. Currently, there are limited observations to support variation in the cytokine production capability of neutrophils in cirrhosis [159], particularly given the generally low levels of cytokines produced by neutrophils.
NET formation has been implicated in sustained liver injury and inflammation. Dysfunctional NETosis and impaired clearance of apoptotic neutrophils (efferocytosis) following binge drinking in mouse models of AH have been shown to exacerbate liver injury associated with sepsis [109]. NET formation in response to infection, fMLF, and PMA is elevated in patients with compensated cirrhosis and ACLF [159]. Elevated NET markers such as H3Cit-DNA and MPO-DNA have been found in cirrhosis and ACLF [162, 163], and plasma from patients with decompensated cirrhosis and ACLF has been shown to induce NET formation in healthy control neutrophils [164]. NETs remain a relatively novel area of investigation in cirrhosis pathogenesis. At present, there is a limited understanding of the role of neutrophil death in cirrhosis. Although some studies have suggested that the viability of neutrophils is reduced [165] and the rate of apoptosis is increased [166], this phenomenon has not been widely explored.
HCC
HCC is the most common primary liver cancer and typically develops in the context of underlying CLDs of various etiologies. Although liver carcinogenesis is a multifactorial process, with predisposing factors varying depending on the cancer subtype [15], chronic inflammation is believed to be central to tumor pathogenesis, driving development, progression, and metastasis [167]. As such, recognition of the complex immunological landscape underpinning HCC pathogenesis is critical for the development of targeted immunotherapies [168], particularly as disease progression limits the number of therapeutic options available for HCC treatment [169]. In the context of carcinogenesis, the immune-regulatory role of the liver in maintaining tolerance amid persistent inflammatory stimuli is critical for preventing tissue injury while sustaining systemic immune tolerance. This state is characterized by complex interactions between circulating leukocytes and liver-resident cells, as well as the fine regulation of pro- and anti-inflammatory cytokines. As such, the hepatic tumor microenvironment (TME) is unique, with preneoplastic lesions able to bypass surveillance, maintain evasion from cytotoxic lymphocytes, and ultimately develop into HCC tumors [83].
The HCC microenvironment is particularly immunosuppressive, with immune cell infiltrates being primarily composed of heterogeneous tumor-associated neutrophil (TAN) types. Depending on environmental signals, TANs can be polarized into antitumor (N1) or protumor (N2) phenotypes [83]. However, there is significant overlap in their morphology (typically mature with segmented nuclei), cell-surface expression markers (CD10hi CD11bhi CD16hi CXCR2+ CXCR4-), and functional characteristics [170, 171]. Although various phenotypic markers have been described as differentially expressed between these populations, functional assays are predominantly used to distinguish them. N1 neutrophils exhibit comparatively increased migratory, oxidative burst, and phagocytosis capacity [170, 171], enhancing their cytotoxic, antitumour functions.
Notably, polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) constitute an immature neutrophil population (CD33+ HLA-DR−/lo) that is functionally immunosuppressive [172]. Under the influence of tumor-released growth factors [e.g., granulocyte colony-stimulating factor (GM-CSF)] and cytokines (e.g., IL-6), aberrant granulopoiesis is stimulated, resulting in the expansion of MDSCs. These cells, in turn, can further facilitate disease progression, as they are recruited into the circulation and TME [172, 173]. Within the hepatic TME, PMN-MDSCs progressively increase their protumorigenic capacity—promoting tissue remodeling through MMP and TGF-β release [174] and supporting angiogenesis via vascular growth factor (VEGF) [175]—and exerting their immune-suppressive effects—through inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX2), prostaglandin E2 (PGE2), and IL-10 [83]. Although these cells appear to be distinct from the N2 phenotype, typically of low density, it remains unclear whether their differentiation or expansion reflects a specific state of neutrophil maturation or follows an entirely separate trajectory.
Neutrophil recruitment into the tumor and polarization to a pro-tumorigenic type are crucial for tumor development. HCC cells and stromal cells produce chemokines to drive neutrophil recruitment. Various CXC chemokine receptors are upregulated in HCC tumor cells [83], with CXCL2 and CXCL8 released by tumor-associated monocytes [176] and with CXCL12 released by cancer-associated fibroblasts (CAFs) and stromal cells [177, 178]. Neutrophils begin to accumulate in hypoxic regions in HCC via the activity of hypoxia-inducible factors [179]. In HCC, elevated circulating IL-6, GM-CSF, TGF-β, and PGE2 polarize neutrophils to a protumor phenotype. Moreover, circulating factors in the TME may promote further polarization. GM-CSF, in addition to promoting granulopoiesis, may bias the release of MDSCs [83]. Whether neutrophils first undergo polarization to a protumor phenotype in the circulation and are then recruited to the tumor, or vice versa, remains unclear [83]. Upon accumulation, TANs can promote tumorigenesis by creating an immunosuppressive environment, increasing tumor cell proliferation and survival, promoting extracellular tumor matrix remodeling, and stimulating angiogenesis.
Neutrophils suppress antitumoral immune responses [e.g., via programmed death-ligand 1 (PD-L1) expression], inhibit CD4+ and CD8+ T-cell proliferation, cytokine production, and cytotoxicity, and promote the expansion of regulatory (Treg) cells [180]. TANs also contribute to the recruitment of macrophages and Treg cells through the expression of CCL2 and CCL17 and via TLR4 signaling. This phenomenon has been shown to promote resistance to sorafenib, a tyrosine kinase inhibitor [181], with neutrophil depletion leading to improved efficacy in preclinical models. ROS production inhibits T-cell activation and promotes IL-10 and TGF-β expression by CD4+ T cells while suppressing CD8+ T cells [182]. Additionally, through the release of nitric oxide (NO) and arginase 1 (ARG1), neutrophils suppress T-cell proliferation and promote apoptosis [183] while impairing NK cell cytotoxicity [184]. Neutrophils may also be implicated in the induction of stem-like cancer cells, which are capable of rapid proliferation and show enhanced survival capacity [185]. These factors, in turn, seem to drive neutrophil recruitment through CXCL5 [185], leading to further tumor propagation.
The abundance of tumoral and circulating neutrophils has been reported to independently predict adverse outcomes in patients with HCC [186, 187]. The presence of peritumoral neutrophils correlates with metastasis, which is consistent with the expansion of circulating TANs, increasing the dissemination capacity of the tumor [188]. TANs directly enhance the metastatic capability of HCC cells through the release of growth factors (HGF, VEGF), oncostatin M (OSM), and MMP9 [175]. MMP9 stimulates ECM remodeling, HGF/c-MET upregulation, and VEGF angiogenesis, all of which facilitate tumor budding, survival, invasion, and metastasis. Compared with those in nonmetastatic HCC, neutrophils in HCC display increased NETosis capacity, with increased NETs observed in metastatic HCC [189]. HCC cells can internalize NETs, activating TLR signaling and leading to transformation into a more aggressive phenotype, with increased invasiveness and cell survival capability [83, 179]. In summary, neutrophils exhibit diverse functions and are implicated in HCC pathogenesis, from local tumor development and progression to metastasis (summarized in Fig. 3).

Schematic overview of the role of neutrophils in the development and progression of hepatocellular carcinoma (HCC). Abbreviations: CXCL C-X-C motif chemokine ligand, MMP matrix metalloproteinase, PMN-MDSCs polymorphonuclear myeloid-derived suppressor cells, ROS reactive oxygen species, TAN tumor-associated neutrophil, Treg T regulatory cell, VEGF vascular endothelial growth factor. This figure was created with BioRender (biorender.com)
Eosinophils
Origin and functions of eosinophils
Eosinophil development, termed eosinophilopoiesis, involves the differentiation of bone marrow hematopoietic stem cells into CMPs, followed by GMPs, before committing to the eosinophil lineage through eosinophil lineage-committed progenitors, which subsequently mature into eosinophils [60, 190]. Notably, heterogeneity in eosinophil lineage commitment has been shown, suggesting that the GMP stage may not be essential for human eosinophilopoiesis, unlike in mice [191, 192]. Eosinophil lineage commitment, differentiation, and maturation are regulated by a synergistic network of transcription factors, including GATA-binding proteins 1 and 2 (GATA-1 and GATA-2), CCAAT enhancer-binding protein alpha (c/EBPa), PU.1 (a member of the ETS family), Friend of GATA-1 (FOG1), and X-box binding protein 1 (XBP1) [190, 193,194,195,196,197,198,199]. In addition to transcriptional regulation, cytokines such as IL-3, IL-5, and GM-CSF play critical roles in eosinophilopoiesis [200], with IL-33 being implicated in facilitating eosinophil maturation via an IL-5/IL-5R-dependent mechanism [201].
Eosinophils are recruited via eotaxins such as CCL11 and CCL24, which are recognized by C–C motif chemokine receptor 3. They secrete lipid mediators and several granule proteins, including major basic protein 1 and 2 (MBP-1 and MBP-2), eosinophil cationic protein (ECP), eosinophil-derived neurotoxin, and eosinophil peroxidase [60, 202]. These granules also contain various cytokines and chemokines (e.g., IL-4, IL-6, IL-10, IL-12, IL-13, and TNF-α), contributing not only to the cytotoxic effects of eosinophils on host defense but also to potential tissue damage [60, 202]. Like NETosis, these cells produce eosinophil extracellular traps, which play a role in inflammatory reactions [203]. In addition to their cytotoxic functions, eosinophils are involved in both innate and adaptive immune regulation—for example, they express major histocompatibility complex class II (MHC-II) and costimulatory molecules—as well as in tissue repair. These diverse roles underscore their importance in maintaining homeostasis and contributing to the pathogenesis of various diseases [204, 205].
Eosinophils in chronic liver inflammation
AIH, PBC, and PSC
Eosinophils are present in the bone marrow, blood, and peripheral organs but are infrequently detected in the liver during homeostasis [202]. To date, few studies have explored the role of eosinophils in noninfectious chronic liver inflammation. Despite some controversy, elevated intrahepatic levels of IL-2, IL-5, IFN-γ, and TGF-β have been reported to be more pronounced in PBC patients than in AIH patients [206, 207]. Increased numbers of eosinophils in the blood and liver of PBC patients, primarily in the vicinity of damaged bile ducts, have been observed and correlate positively with increased IL-5 expression [206,207,208]. Intriguingly, unlike neutrophils, eosinophilia is more prominent in the early stages of PBC, highlighting the potential of this subset as a diagnostic marker and a target for early therapeutic intervention [208]. Although eosinophil infiltration might contribute via degranulation to early tissue damage, its role in PBC disease progression remains unclear. The first-line therapeutic option for PBC, UDCA, has been shown not only to reduce eosinophil infiltration in the liver but also to inhibit degranulation [209]. Although eosinophilia has been described in AIH blood and PSC livers, minimal evidence exists regarding the role of these cells in their immunopathogenesis [210,211,212]. One study in children with AIH investigated single-nucleotide polymorphisms in cytokines associated with eosinophil maturation, such as IL-4, IL-5, and IL-13, and suggested a potential link between eosinophils and AIH pathogenesis in pediatric patients [213].
Fibrosis
Eosinophils have been implicated in combating parasitic infections such as Schistosoma mansoni infection [214, 215], which can lead to liver disease, during which a marked increase in both blood and tissue eosinophil recruitment, accompanied by increased IgE levels, has been observed. This response is mediated by IL-4 and IL-5, which increase eosinophil activation and facilitate parasite elimination [216]. The cytotoxic effects of eosinophils are attributed primarily to two mechanisms: intracellular killing—via phagocytosis and the production of ECP and MBP—and extracellular killing mediated by degranulation [60, 217]. However, these same mechanisms may also contribute to tissue damage. In IL-5 knockout mice infected with Schistosoma mansoni, eosinophil depletion and reduced levels of the profibrotic cytokine IL-13 have been observed, suggesting that IL-5 blockade may mitigate liver damage and fibrosis [218]. Hence, these cells play dual roles, both protective and tissue damaging. The significance of eosinophils in liver ischemia‒reperfusion and drug-induced liver injury has recently attracted interest [60]. Eosinophil recruitment to the liver was found in both patients and mice with acetaminophen-induced acute liver injury, where these cells appear to exert a protective effect [219]. Compared with those in WT mice, more severe injury was observed in eosinophil-deficient (DdblGata1) mice, as evidenced by elevated serum ALT levels and increased hepatocyte necrosis. The recruitment of eosinophils to the liver is thought to occur in a macrophage-dependent manner [219,220,221]. Finally, eosinophil accumulation has also been found to coincide with hepatocyte proliferation during liver regeneration [222].
HCC
The role of eosinophils in cancer development is controversial, as they have been demonstrated to play both pro- and antioncogenic roles across different cancer types [223, 224]. Eosinophils generally exhibit antitumoural characteristics in HCC. They directly mediate tumor cytotoxicity when cocultured with MH134 cells (an HCC cell line) in the presence of several mediators, such as IL-5 and CCL11 [225]. In IL-5/CCL11 knockout mice or eosinophil-deficient mice, tumor growth is significantly enhanced [226]. Additionally, the inhibition of dipeptidyl peptidase-4 (DPP4/CD26) promotes eosinophil infiltration into solid tumors by increasing CCL11 levels, leading to significant suppression of tumor growth [227]. Eosinophils may also exert indirect cytotoxic effects through the expression of NK cell surface markers, suggesting that NK cell-like activities are involved in antitumour immunity. Similarly, eosinophils are proposed to interact with various lymphocyte subsets, including NK cells and CD4+ and CD8+ T cells, under the regulation of cytokines and eotaxins, thereby orchestrating antitumour immune responses [228]. Moreover, clinical studies suggest a potential prognostic role for eosinophils in patients with HCC receiving sorafenib treatment. Low blood eosinophil counts prior to sorafenib administration are negatively associated with overall survival and progression-free survival, further highlighting their relevance in HCC prognosis [229].
Monocytes and macrophages
Origin and functions of monocytes/macrophages
Monocytes are phagocytes derived from hematopoietic stem cells that are typically found in peripheral blood and are able to differentiate into tissue macrophages or DCs during pathological inflammatory conditions [230]. Human peripheral monocytes are classified into three distinct subsets on the basis of the expression of the surface markers CD14 and CD16: classical monocytes (CD14+ CD16lo), nonclassical monocytes (CD14lo CD16hi), and intermediate monocytes (CD14+ CD16+) [231]. In mice, monocytes are generally described as Ly6C+ or Ly6C− cells, corresponding to their classical and nonclassical human counterparts, respectively. Tissue macrophages, such as skin Langerhans cells, lung alveolar macrophages, and hepatic KCs, are considered the first line of defense against pathogens and play significant roles in tissue development and maintenance of tolerance [53]. In mice, many resident tissue macrophage populations arise during embryogenesis, where erythro-myeloid progenitors (EMPs) give rise to premacrophages (p-Macs) and monocytes that seed embryonic tissues [232], subsequently acquiring resident cell phenotypes in response to niche-specific cues and transcriptional activators [233].
Under homeostatic conditions, the liver macrophage compartment (Fig. 4) is composed almost exclusively of resident Kupffer cells (Res-KCs), which originate primarily from yolk sac-derived EMPs. These progenitors migrate to the fetal liver on embryonic day (E8.5) and give rise to (p-Macs) and fetal liver monocytes. p-Macs, along with some fetal liver monocytes, eventually differentiate into KCs during organogenesis. This differentiation process is significantly regulated by the TGF-β-dependent transcription factor ID3 and the evolutionarily conserved ALK-1/BMP9 axis [29, 53, 61, 232, 234]. KCs are long-lived and have self-renewal capacity, with their identity maintained in part through interactions with LSECs [235], and are linked to the expression of Nr1h3 [gene encoding liver X receptor α], a transcription factor involved in cholesterol processing [236].

Macrophages in chronic liver inflammation. Schematic overview of the roles of hepatic macrophage subsets in homeostasis and different chronic liver diseases. Homeostasis: Embryonically derived resident Kupffer cells (Res-KCs) reside within sinusoids, possess self-renewal capacity, and perform various functions, including phagocytosis of pathogens, regulation of lipid and iron metabolism [e.g., removal of red blood cells (RBCs)], clearance of cellular debris, and maintenance of immune tolerance. The following additional liver macrophage subsets derived from bone marrow (BM) monocytes are present: central vein macrophages (CV-Macs) and bile duct lipid-associated macrophages (BD-LAMs). The composition of the hepatic macrophage pool is altered during chronic inflammation. Cholestasis: Bile duct damage [e.g., primary sclerosing cholangitis (PSC)] triggers the release of chemoattractants (e.g., CCL2 and IL-18), leading to increased infiltration and periportal accumulation of monocyte-derived macrophages (mo-Macs). In this context, monocyte-derived KCs (Mo-KCs) exhibit potential for communication with HSCs via factors such as Gas6 and increased phagocytosis capacity. The presence of LAM-like macrophages has also been described. Alcohol: In alcohol-related liver disease (ALD), increased gut permeability results in increased bacterial translocation and entry of pathogen-associated molecular patterns (PAMPs) into the liver, e.g., lipopolysaccharide (LPS); persistent exposure to these PAMPs activates liver macrophages, causing TNF-α and reactive oxygen species (ROS) production and perpetuating liver injury. Experimental depletion of KCs in mice led to aberrant hepatocyte proliferation, mimicking human disease. MASLD/MASH: In metabolic dysfunction-associated steatotic liver disease (MASLD) and metabolic dysfunction-associated steatohepatitis (MASH), a reduction in Res-KC numbers occurs due to impaired self-renewal and overactivation by insults, e.g., free fatty acids (FFAs), that may trigger apoptosis. Furthermore, KCs undergo ferroptosis after hepdicin-induced increased iron uptake, which is stimulated by macrophage-derived NCF1, whereas hypoxia-induced factors such as HIF-2α can promote lysosomal cell death. Mo-Macs are recruited in response to hepatocyte-derived damage-associated molecular patterns (DAMPs) and chemokines released from activated KCs (e.g., CCL2, IL-1β, and TNF-α) and cholangiocytes (CCL2, CCL5, and CXCL1). LAMs of both KC and BM origin emerge and expand, expressing the TREM2, SPP1, and GPNMB markers. Fibrosis: Liver inflammation is perpetuated by activated KCs, some of which express TREM1, as well as by CCL2/CCR2-recruited monocytes and mo-Macs. KCs facilitate immune cell recruitment through the production of various chemokines (e.g., CCL9, CXLC2, and CXCL3) and promote HSC activation and differentiation via the production of profibrotic factors (e.g., PDGF and TGF-β). LAM-like Macs are also observed, along with profibrogenic TREM2+ CD9+ SPP1+ scar-associated macrophages (SAMs) and FABP5-expressing macrophages around the fibrotic niche. Syncytial macrophage structures have been shown to play a role in KC in mouse fibrosis models. Cancer: Tumor-associated macrophages (TAMs), monocytic myeloid-derived suppressor cells (M-MDSCs), and Res-KCs promote tumorigenesis and dampen immune responses in hepatocellular carcinoma (HCC). CD68+ CD11b+ CD16- inflammatory macrophages (Inflammatory Macs) and CCR2+ S100A9+ TAMs accumulate periportally and around irregular blood vessels within tumors. In a hypoxic environment, the expression of matrix metalloproteinases (MMPs) and SPP1 denotes protumorigenic TAMs, which play roles in cancer stemness, epithelial‒mesenchymal transition (EMT) and neovascularization and dampen antitumor responses. PD-L1+ TAMs can have antitumour functions by facilitating the recruitment and activation of CXCR3+ effector memory T (TEM) cells. Abbreviations: CCR chemokine receptor, CCL C-C motif chemokine ligand, CXCL C-X-C motif chemokine ligand, TLR toll-like receptor, LSECs liver sinusoidal endothelial cells. This figure was created with BioRender (biorender.com)
KCs are located within the hepatic sinusoids, predominantly in the periportal regions. Their membrane protrusions extend toward the peri-sinusoidal space, enabling close contact with HSCs and hepatocytes. Interplay with these cells, as well as interactions with gut commensals and TLR4/MyD88-mediated LSEC microbial sensing, have been proven crucial for the maintenance of this spatial distribution, which, in turn, is of paramount importance for the preservation of tissue homeostasis [22, 28,29,30].
The main functions of KCs in homeostasis include the phagocytosis of pathogens, the maintenance of an immune-tolerant microenvironment [28, 37,38,39,40,41,42,43,44,45,46,47,48], the clearance of erythrocytes, aged platelets, and apoptotic cells [237,238,239], and the regulation of cholesterol homeostasis via the scavenger receptor CD36 [50, 240, 241]. Immune tolerance is of paramount importance in the liver to prevent tissue damage. Blood entering through the portal vein carries a diverse array of antigens, including food-derived components, commensal microorganisms, pathogens, and microbial byproducts. KCs, along with LSECs and DCs, localized within and around the sinusoids are in a privileged position to recognize these antigens and mount an appropriate, self-limiting immune response [49]. KCs contribute to local immune tolerance through multiple mechanisms: sequestration of PAMPs via scavenger receptors such as MARCO, secretion of anti-inflammatory cytokines [30], passenger T-cell arrest or promotion of local regulatory T-cell proliferation [242], and the expression of regulatory molecules, including immune checkpoints [243].
A multitude of KC markers have been described, reflecting their diverse physiological duties. Both mouse and human KCs express T-cell immunoglobulin and mucin domain containing 4 (TIMD4) and V-set and immunoglobulin domain-containing 4 (VSIG4), which play significant roles in liver-mediated iron homeostasis [244] and phagocytosis [245], respectively. Mouse KCs express the macrophage markers CD64 and F4/80, in addition to C-type lectin domain family 4 member F (CLEC4F) (not detected in human KCs); CLEC4F is expressed relatively late during KC maturation [246,247,248]. A full consensus has yet to be reached regarding a set of markers that outline a bona fide human KC population. However, the surface markers VSIG4 and folate receptor beta (FOLR2) are increasingly favored, along with other markers, such as CD163 and MARCO [29]. Under certain inflammatory conditions, in mice, bone marrow-derived monocytes are recruited to the liver and may differentiate into monocyte-derived KCs (mo-KCs) [29, 249, 250], but current knowledge does not allow for a clear demarcation between Res-KCs and mo-KCs in humans. Recently, CLEC2 has been proposed as a marker to discriminate between KCs and monocyte-derived macrophages (mo-Macs), as it emerges early during KC development [251].
In addition to KCs, three other macrophage subsets have been described in healthy livers. Liver capsule macrophages (LCMs) are monocyte-derived cells (expressing CX3CR1 and CD207 in mice) lacking KC markers such as VSIG4 [29, 252]. Another subset has been shown to be localized around bile ducts under homeostatic conditions [29] (Fig. 4); these cells express genes such as Gpnmb, Spp1, Trem2, and Cd9, resembling the transcriptomic profile of lipid-associated macrophages (LAMs) observed in the fatty liver [253]. Molecular cartography data have also highlighted the presence of a CD207+ macrophage subset distinct from LCMs, localized around the central vein, named central vein macrophages [29]. Notably, transcriptomic analyses have revealed the existence of two phenotypically and functionally distinct KC subsets, namely, CD206lo endothelial cell-selective adhesion molecule (ESAM)− KC1s and CD206hi ESAM+ KC2s [254, 255]; however, this remains a subject of ongoing debate within the field [29, 256]. As discussed below, the hepatic macrophage landscape becomes more complex in chronic liver inflammation.
Monocytes/macrophages in chronic liver inflammation
MASLD
In MASLD/MASH, the total number of hepatic macrophages increases due to the recruitment of circulating monocytes, which differentiate into macrophages in the liver (Fig. 4) [50]. KCs exhibit a dysregulated phenotype in MASLD/MASH, resulting in either a direct or a cellular interaction-mediated effect on the disease trajectory [66]. Steatosis-associated factors, such as free fatty acids, can activate KCs via TLR2 and TLR4 [257], and an inflammatory response can also be triggered through DAMP-mediated inflammasome activation. This leads to the release of proinflammatory cytokines such as IL-1β, TNF-α, and CCL2 [258, 259], which act as chemoattractants for peripheral monocytes [260]. CCR2+ monocytes are periportally distributed in the livers of patients with MASH, and their abundance is positively correlated with disease severity and the extent of fibrosis [261]. Rapid accumulation of monocytes has also been observed in mouse models of diet- and toxin-induced CLD [262]. Recently, osteopontin (SPP1) has been implicated in the recruitment of inflammatory Ly6Chi monocytes in MCD fibrosis and in two additional mouse models. Coombes et al. showed that SPP1 (which in humans is associated with portal inflammation, fibrosis, and the MASH stage) promotes cholangiocyte chemokine production (e.g., CCL2, CCL5, and CXCL1) via the modulation of the noncanonical NF-kB pathway and affects monocyte recruitment and fibrosis levels, highlighting the complexity of cellular interactions in fibrosis [263].
KC numbers are consistently decreased in mouse MASLD/MASH models, with liver-infiltrating monocytes differentiating into mo-KCs and occupying their place [251, 253, 264]. KC apoptotic death seems to affect total KC numbers, as evidenced by TUNEL staining in mouse MASH livers [264]. MASLD-related stimuli (e.g., free fatty acids, cholesterol, and hepatocyte-derived DAMPs) are contributing factors to increased KC death and influence their role in disease progression [265, 266]. Recently, oxidized phospholipids induced by macrophage-derived neutrophil cytosolic factor 1 were shown to promote hepcidin production by hepatocytes, leading to iron deposition in KCs and subsequent ferroptosis [267]. Moreover, the accumulation of free cholesterol in macrophages following the engulfment of dead hepatocytes triggers lysosomal stress and profibrotic activation in macrophages, particularly in the WD model [268]. Hypoxia has also been implicated in KC (over)activation and death in MASLD, with hypoxia-inducible factor 2 alpha having been shown to mediate increased KC death and impaired efferocytosis through transcription factor EB-mediated lysosomal stress [269]. However, the view that KC activation is a key mediator of MASLD pathology has been challenged by recent in vivo WD studies [253] and observations in obese patients [270], which do not support a traditional KC activation phenotype in MASLD.
In addition, impairment of the proliferative capacity of KCs has been shown to affect KC numbers. A diminished ability to self-renew, accompanied by the emergence of mo-KCs, has been reported in a model of L. monocytogenes infection in the context of MALSD [39], as well as in an MCD diet model of steatohepatitis [251], where embryonic (as opposed to monocyte-derived) KCs confer tissue protection through hepatic triglyceride storage. While the loss of KCs has been considered a key driver of monocyte recruitment, recent results from human patient samples and two mouse short-term disease models suggest that monocyte recruitment could be initiated long before the KC niche is vacated—at early stages of the disease—to support the sequestration of lipid-laden hepatocytes [271].
The existence of LAMs and their human analogs is generally considered ubiquitous in MASLD, with their niche-specific localization linked to disease progression. Increased proportions of LAMs in patient livers have been found to correlate with more advanced steatosis [29]. Spatial transcriptomics has revealed that, in the steady state, LAMs are primarily found periportally. However, in human steatotic and mouse WD-induced MASLD liver samples, their distribution shifts, with a higher abundance observed in pericentral regions and steatotic zones [29]. In mice, LAMs express high levels of osteopontin [253], whereas TREM2hi macrophages, termed MASH-associated macrophages, express genes associated with endocytosis, lysosomal degradation, MHC-II antigen presentation, and ECM remodeling [272]. The development of LAMs derived from infiltrating Ly6Chi monocytes has been shown to depend on Egr2, a transcription factor that is increased in mouse monocytes and macrophages in MASH. The long-chain fatty acid-Egr2 pathway promotes the differentiation of hepatic monocytes toward LAMs, whereas Egr2 deficiency drives a KC-like differentiation pathway, which is associated with reduced progression from benign steatosis to fibrosis [273].
There is a growing body of research on the role of TREM2+ macrophages in MASLD. In the WD model, mo-KCs, which are abundant during the early stages of disease, are progressively replaced by LAMs during regression and play a significant role in tissue restoration. Both subsets express Trem2, which is maintained in LAMs during regression and appears to be crucial in MASH. The absence of TREM2 impairs disease resolution by restricting the emergence of a less inflammatory, collagenolytic LAM population [274]. A similar protective effect has been proposed in the MCD mouse model. In this context, TREM2+ macrophages localize around areas of fibrosis, whereas TREM2-deficient macrophages display a decreased lipid-handling capacity, lower viability, and a profibrogenic phenotype [275]. Finally, TREM2 expression on macrophages has been suggested to drive the MASH-resolving effect of bariatric surgery in obese patients by preventing inflammation and augmenting efferocytosis [276]. In addition to TREM2, another receptor relevant to MASLD is macrophage scavenger receptor 1. In MASLD patients, MSR1 is expressed in both KCs and lipid-laden macrophages, and its expression is positively correlated with the degree of steatosis. In a mouse model, Msr1 deficiency mitigated proinflammatory macrophage activation by preventing lipid accumulation, suggesting an overall protective role [277].
ALD
One of the hallmarks of alcohol-related liver inflammation is increased gut permeability. As the disease progresses, elevated quantities of gut-derived bacteria, e.g., E. coli and bacterial products, pass through the portal circulation and are encountered by KCs [278]. Indeed, KCs from rats in a chronic ethanol feeding model have been reported to be activated, with LPS-stimulated KC-derived TNF-α and ROS being implicated in tissue damage [279]. Chronic exposure to increased concentrations of LPS, due to increased gut permeability, leads to TLR4-dependent NF-kB-mediated activation of macrophages and monocytes in chronic ALD patients [280]. Circulating monocytes also display a dysfunctional phenotype in AH. Monocytes from patients with severe AH presented impaired oxidative burst capacity, which was correlated with diminished NADPH oxidase expression. This defect was associated with an increased risk of infection within two weeks, as well as reduced survival rates at 28 days postadmission [281]. Mirroring the results observed in other CLDs, the infiltration of mo-Macs into the liver aggravates hepatic pathology. This has been reported in a mouse model of chronic alcohol feeding, where infiltration of Ly6Chi monocytes expressing TNF-α, IL-1β, and chemokines such as CCL2 was associated with increased tissue damage [282].
More recently, the role of macrophage CRIg (VSIG4) expression has been highlighted in the context of AH. Compared with WT animals, CRIg-deficient mice developed more severe liver injury after chronic plus binge ethanol feeding, exhibiting significantly greater steatosis and liver inflammation. Additionally, these mice showed a diminished ability to clear gut-translocated E. faecalis, creating a setting that could contribute to exacerbated liver disease or systemic infection in patients [283]. Finally, through scRNA analysis, a recent study using a model of concurrent Western diet and alcohol feeding identified the replacement of KCs by monocyte-derived cells, while KC ablation led to loss of liver function and increased hepatocyte proliferation, findings that are consistent with observations in human AH [284].
PSC and PBC
Compared with those in control tissues, increased numbers of periportal/peribiliary and fibrotic region-adjacent CD68+ macrophages have been observed in human PSC livers (Fig. 4) [285, 286]. The macrophages localized to these areas in PSCs were CCR2+, and gene expression analysis revealed that a proinflammatory phenotype predominated in peribiliary mo-Macs. Cholangiocyte insults were identified as the driving factor for the recruitment of peripheral monocytes through the secretion of CCL2 and IL-18, as demonstrated by in vitro experiments in which activated and senescent cholangiocytes were used. Treatment with cenicriviroc (a dual CCR2/CCR5 inhibitor) in this model reduced fibrosis and associated markers such as Tgfb and Acta2 [285]. IBA1 expression has been described as a marker shared by all macrophage subpopulations (CD16+, CD163+, and CD68+) found in PSC tissue samples, indicating that an activated macrophage phenotype is positively correlated with disease progression. In mice, the accumulation of IBA1+ CLEC4F− mo-Macs and a decrease in IBA1+ CLEC4F+ KCs were associated with disease progression in the Mdr2−/− model of PSC. Interestingly, IBA1-expressing cells were found near CK19+ ductular cells in both Mdr2-/- livers and human MASLD/MASH and PSC tissues. Similar histological findings were also observed in liver samples from patients with AH. In mice, IBA1+ Macs located near ductular cells presented lower expression levels of IBA1, CLEC4F, CD206, and MERTK [286].
Biliary epithelial dysregulation has been shown to drive the development of scar tissue in a mouse model of congenital hepatic fibrosis, primarily in a macrophage-dependent manner. In this model, homozygous deletion of exon 4 of the Pkhd1 gene led to the secretion of chemokines, including CXCL1, CXCL10, and CXCL12, by cholangiocytes, resulting in the recruitment of bone marrow-derived macrophages. The recruited macrophages secreted cytokines such as TNF-α, which induced the upregulation of ανβ6 integrin, a local activator of latent TGFβ1, thereby promoting collagen deposition. Notably, integrin ανβ6 levels are correlated with both peribiliary CD45+ cell accumulation and portal fibrosis [287].
Analysis of KCs in experimental cholestatic liver injury induced by common bile duct ligation (CBDL) [288] revealed that compared with resident KCs, mo-KCs display increased in vivo and ex vivo proliferation, along with antiapoptotic properties. Additionally, mo-KCs exhibited more pronounced phagocytic and reparative potential. The latter was indicated by increased expression of genes such as Trem2, Cd36, Igf1, and Lpl, as well as increased bioparticle phagocytosis ex vivo. In addition, mo-KCs also exhibited greater potential for HSC communication and ECM remodeling through the expression of genes such as Gas6 and Mmp12. Depletion of resident KCs highlighted the protective role of mo-KCs in the CBDL model, as evidenced by reduced necrosis, tissue damage, and fibrosis [288].
In a study using two models of experimental PSCs (0.1% DDC-containing diet or CBDL), monocyte infiltration was prominent, while both CLEC4F+ TIM4+ Res-KCs and CLEC4F+ TIM4− mo-KCs were found to be activated in the early stages, exhibiting increased proliferation after 2 days and upregulation of Tnfα and Ccl2 after 4 days of injury. In both models, liver-resident KC numbers decreased, with apparent KC death demonstrated by positive TUNEL staining of CLEC4F+ cells. Depletion of CLEC4F+ KCs at the onset of PSC did not affect disease progression or myeloid cell infiltration at the end stage [289]. In a subsequent study, the same team reported that in CBDL mice, mo-Macs upregulated Spp1, Trem2, and Gpnmb, resulting in an LAM-like phenotype. Compared with TIM4+ KCs, mo-Macs expressed higher levels of Spp1, Il1b, Tnfa, and Tgfb 4 weeks after CBDL surgery. Immunofluorescence data indicated that Spp1+ mo-Macs accumulated near fibrotic regions in both human PSC samples and experimental models (CBDL or Mdr2−/− animals). The functional relevance of Spp1 was highlighted in the CBDL model, where genetic knockout led to the development of a proinflammatory milieu [290]. Additionally, in a recent bulk RNA-seq study of patient liver tissue, a PSC-specific profibrogenic gene signature was identified, implicating SPP1 along with pathways such as the Wnt and PI3K/AKT signaling pathways in the development of biliary fibrosis [291]. Finally, a significantly increased abundance of immune-suppressive MARCO+ cells was recently described in human PSC liver samples compared with healthy controls, suggesting a possible correlation with disease progression [30].
Fibrosis
Macrophage-derived triggers can contribute to HSC differentiation toward a myofibroblast-like phenotype [292]. Profibrotic cytokines such as KC-derived TGF-β and platelet-derived growth factor (PDGF) [293, 294], along with proinflammatory cytokines such as IL-1β and TNF-α, and chemokines such as the CCL2/CCR2 and CCL1/CCR8 axes [295], as well as inflammatory mediators (ROS and iNOS) [296] and microRNAs (e.g., miRNA-342-3p) [297], can directly promote HSC differentiation, proliferation, and ECM production. There is a growing body of work aimed at elucidating monocyte and macrophage heterogeneity in liver fibrosis and cirrhosis. Monocyte infiltration of the liver is prominent during fibrosis, with chemokine signaling (CCL2/CCR2) playing a significant role [50, 261]. In addition to the bone marrow, the spleen can act as a reservoir of immune cells for a rapid response to inflammatory conditions. In CCl4-induced fibrosis in mice, splenic Ly6Chi CX3CR1lo monocytes have been shown to migrate specifically to the fibrotic liver in large numbers and localize around the central vein within the hepatic lobules, thereby exacerbating fibrosis and increasing hepatic proinflammatory cytokine levels. Interestingly, this also led to the mobilization of endogenous hepatic CX3CR1+ cells, which displayed greater somatic motility and membrane indices of cellular crosstalk. In support of these findings, splenectomy mitigated these effects [298].
Monocyte-derived, CD9+ TREM2+ SPP1+ scar-associated macrophages (SAMs), which can promote collagen production by primary HSCs in vitro, are expanded in both human and mouse fibrotic livers, particularly around fibrotic regions [299]. In the same study, a reduction in a TIMD4- MARCO+ KC subset was observed, although no significant difference was noted in the total number of KCs between healthy and cirrhotic livers [299]. These results were corroborated by another study, which reported that tissue monocytes were abundant around the portal triad during the early stages of human liver fibrosis, whereas KCs, SAMs, and tissue monocytes populated the portal area at later stages [300]. Additionally, a monocyte-derived SPP1+ GPNMB+ FABP5+ cell subset within the CD9+ TREM2+ population has been found to accumulate around fibrotic lesions in both human samples and mouse CCl4 or HFD-induced liver fibrosis models. Blockade of TGF-β, IL-17A, or GM-CSF in the CCl4 model reduced the accumulation of this subset and was associated with decreased fibrosis. On the basis of these findings and supporting transcriptomic data, the authors propose a profibrotic function for this macrophage population [301].
In addition to infiltrating mo-Macs (Fig. 4), KCs have been shown to actively promote hepatic inflammation and fibrogenesis in experimental CCl4 fibrosis. TREM1 expression on CD11b- F4/80+ cells was found to be upregulated early and maintained throughout the disease course and was associated with increased tissue damage [302]. Interestingly, TREM1+ macrophages have also been identified in liver samples from patients with severe fibrosis. In this context, TREM1+ KCs release proinflammatory cytokines, facilitate the recruitment of inflammatory cells via CCL9, CXCL2, and CXCL3, and promote fibrogenesis through TGF-β-mediated KC-HSC crosstalk [302]. Remodeling of the KC niche during fibrosis, such as sinusoidal strictures and the formation of collateral vessels, can lead to a loss of KC identity [40]. In the CCl4 model, it was recently proposed that KCs transdifferentiate, losing expression of TIM4 and VSIG4, and consequently display impaired functions, including phagocytosis and bacterial killing. Infiltrating monocytes have been shown to form aggregates that acquire KC-like functions, a process driven by the intestinal microbiota. CD44 upregulation on intrahepatic vessels promoted monocyte adhesion and CD36-dependent formation of syncytial structures capable of capturing bacteria within larger vessels. These macrophage syncytia have also been reported in human liver disease [40], although these findings have not yet been independently replicated, and their functional significance in CLD remains unclear.
Hepatic macrophages have also been implicated in the regression of fibrosis, notably through the production of ECM-degrading MMPs, for example, via KC-derived MMP9 [303], and the secretion of Treg-inducing anti-inflammatory cytokines, such as IL-10 and IL-12, which are key in modulating resolution [304]. During peak fibrosis regression in the CCl4 model, flow cytometric analysis revealed that the recruited Ly6Clo subset constituted the dominant liver macrophage subpopulation. These cells exhibited a highly phagocytic and restorative phenotype while also expressing genes involved in ECM degradation (Mmp9 and Mmp12). Interestingly, this subset was shown to be derived from Ly6Chi monocytes. Depletion of Ly6Clo cells resulted in diminished scar remodeling capacity, whereas experimental induction of a phagocytic phenotype in vivo augmented fibrolytic activity [305].
A recent study corroborated the previously established importance [274] of TREM2 in macrophages for fibrosis resolution. Specifically, the recruitment of monocyte-derived LAMs occurs in both HFD- and CCl4-induced fibrosis, and a subset of both resident and monocyte-derived KCs adopts an LAM-like phenotype localized near injured tissue. This LAM phenotype appears to be driven by efferocytosis, as indicated by ex vivo and in vitro experiments. TREM2 was found to regulate efferocytic capacity, and TREM2 deficiency impaired tissue repair [250]. In addition, ferroptotic death of recruited “inflammatory” macrophages has been observed in CCl4 and CBDL models [306], whereas necroptotic death of mo-KCs in a MASLD model [307] was similarly associated with fibrosis mitigation. These findings underscore the pivotal role of recruited macrophages in directing the disease trajectory toward either progression or regression.
Cirrhosis: As discussed above, a significant proportion of patients with cirrhosis may progress to ACLF, which is characterized by peripheral monocyte dysfunction and an increased risk of bacterial infections [137, 138]. MDSCs contribute to dampening systemic immune responses, and their relevance to liver disease has been reported in several studies [308, 309]. In human ACLF, a significant expansion of CD14+ CD15− CD11b+ HLA-DR− monocytic MDSCs (M-MDSCs) was observed in the peripheral blood of patients with cirrhosis, which was correlated with disease prognosis. M-MDSCs cocultured with T cells resulted in reduced T-cell proliferation. M-MDSCs exhibited a diminished capacity for the production of proinflammatory cytokines (TNF-α and IL-6) after LPS stimulation as well as reduced bacterial phagocytosis. Interestingly, ex vivo TLR3 stimulation led to the upregulation of HLA-DR and partially restored M-MDSC function [310].
The expression of the receptor protein tyrosine kinases AXL and MERTK, as well as checkpoint receptors on monocytes and macrophages, is indicative of a tolerogenic or prorestorative type [311]. AXL+ highly efferocytic mature monocytes are expanded in the peripheral blood of patients with cirrhosis, which is correlated with complications and poor outcomes. These cells exert a T-cell-suppressive function and exhibit diminished TNF-α and IL-6 production [312]. AXL is broadly expressed on liver macrophages during homeostasis; however, in cirrhosis, AXL expression is downregulated in response to HSC-derived growth arrest-specific 6. This reduction was highlighted in both the CCl4 model and in human cirrhotic liver samples, paralleling disease progression [313]. Monocytes from patients with cirrhosis also express MERTK in a manner that reflects disease severity. Furthermore, MERTKhi CD163hi macrophages are significantly expanded in the livers of ACLF patients, particularly around hepatic sinusoids [314, 315].
Decreased HLA-DR expression, upregulation of IL-10, and an anti-inflammatory transcriptomic profile are associated with a defective antibacterial response (phagocytosis, oxidative burst) in ACLF monocytes ex vivo [316]. Given the involvement of checkpoint molecules in CLD, the programmed cell death protein 1 (PD-1) axis has been shown to be crucial for the function of monocytes/macrophages in this context. Compared with those from controls, human liver macrophages, as well as peripheral monocytes from patients with cirrhosis, overexpress PD-L1, while monocyte PD-L1 is correlated with disease severity and infection incidence [317]. Mouse and human liver macrophages displayed diminished in vivo phagocytosis of S. aureus particles and 99mTc-phytate colloids, respectively. In a DDC mouse model, PD-L1 blockade improved macrophage function, reducing the expression of immunosuppressive markers (MERTK, MARCO, and PDGF-β) and enhancing bacterial phagocytic capacity [317].
Recent transcriptomic analyses have provided new insights into monocyte heterogeneity in cirrhosis. Circulating monocytes from patients with compensated and not acutely decompensated (NAD) cirrhosis were partitioned into seven clusters on the basis of scRNA-seq data. Interestingly, a shift toward a phagocytic phenotype in compensated cirrhosis and the emergence of M-MDSCs in NAD were observed [318]. In another study focusing on ACLF, two distinct functional and metabolic profiles of circulating monocytes, termed recovery (R) and nonrecovery (NR) monocytes, were described on the basis of patient outcome. ACLF-R monocytes highly expressed resistin and S100A8/9/11 proinflammatory alarmins and were characterized by a tolerant state with reduced phagocytic capacity. On the other hand, ACLF-NR monocytes appeared activated, overexpressing regulators of inflammasome activation (VIM and SGK1) and genes involved in antigen presentation and antibacterial responses (HLA-A/B, CD74, B2M, LYZ). Metabolic states were also found to be significantly dissimilar between the two monocyte groups, with ACLF-NR showing lower levels of metabolites such as valine, glycine, α-ketoglutarate, and adenosine, indicating reduced activity in the pentose phosphate and leukotriene synthesis pathways [319].
HCC and iCCA
The predominant hypothesis is that tumor-associated macrophages (TAMs) and MDSCs act in a protumorigenic manner in HCC [167, 320]. While the traditional dichotomy of the peri- and intratumoural areas remains relevant, imaging mass cytometry and single-cell analysis have provided a more granular dissection of the TME. Three distinct niches have been described within HCC TMEs—normal, fibrotic, and cancerous regions—each of which is differentially infiltrated by macrophage subtypes. Normal regions display an abundance of resident KCs, along with a periportal accumulation of CD68+ CD11b+ CD16− “inflammatory macrophages”. These cells, along with M-MDSCs, are expanded in fibrotic areas (characterized by increased a-SMA and collagen I levels), whereas within Ki67+ cancerous regions, inflammatory macrophages surround irregular blood vessels. Notably, ex vivo work has shown that KCs dampen CD8+ T-cell activation. Furthermore, in vivo depletion of KCs in an experimental c-Myc/NRAS HCC model increased intratumoral T-cell infiltration and reduced the number of PD-1+ CD8+ cells [321].
PD-L1 expression is generally associated with HCC immune suppression [322]. Interestingly, in an analysis of HCC patient liver tissue, PD-L1 expression on tumor cells was indeed found to be negatively associated with overall and relapse-free survival, but notably, PD-L1 expression on CD68+ macrophages positively correlated with overall survival [323]. In patient samples, PD-L1+ ICOS+ MDSCs and TAMs were found to infiltrate MASH-HCC tumors, colocalizing both peritoumorally and intratumorally with PD-1+ CD8+ T cells. CD4+ and CD8+ T cells from MASH-HCC exhibited diminished cytolytic capacity, a phenotype not observed in viral HCC, suggesting an etiology-dependent mechanism of TAM/MDSC-mediated suppression [324]. Furthermore, CSF1R+ PD-L1+ TAMs were shown to colocalize with MAIT cells in a human HCC cohort, appearing to induce a dysfunctional phenotype marked by impaired tumor infiltration and cytotoxicity. Coculture experiments revealed a cell contact-mediated effect, and PD-1/PD-L1 axis blockade in vitro and in two tumor-bearing mouse models restored MAIT cell IFN-γ production and enhanced MAIT cell tumor infiltration, respectively [325]. Recently, scRNA-seq analysis revealed a subset of CXCL10+ PD-L1+ TAMs that was positively associated with the response to immunotherapy. These macrophages expressed genes involved in T-cell recruitment (e.g., CXCL9 and CXCL10) and IFN-γ signaling (e.g., STAT1, indoleamine-2,3-dioxygenase 1 (IDO1), and GBP1), and CellChat analysis predicted their involvement in CXCR3+ effector-memory T-cell recruitment and activation in the TME [326]. Another checkpoint implicated in the role of macrophages in HCC is TIM-3. KCs in HBV-related HCC have been shown to express galectin-9, a ligand for TIM-3. Galectin-9+ KCs colocalized with TIM-3+ T cells, which exhibited diminished proliferative capacity and a senescent phenotype. In vitro blockade of this interaction enhances T-cell proliferation and increases IL-2 and IFN-γ production [327].
In humans, CCR2+ S100A9-expressing TAMs have been observed at the HCC border and colocalize with newly formed CD31+ blood vessels (Fig. 4). In contrast, TAMs located at the center of tumors appear to be immunosuppressive, as indicated by CD163 expression. In a fibrosis-HCC model (diethylnitrosamine and 16 weeks of CCl4 injections), three tumor-associated myeloid populations were isolated and characterized by cell sorting and mRNA profiling. Gr1lo MHC-IIhi CCR2+ TAMs (TAM1) are highly inflammatory (Il-1β, S100a9) and angiogenic (Vegfa, Mmp9). Gr1lo MHC-IIlo (TAM2) expressed Ccl6, Arg1, Mrc1, and NfkBia, displaying an anti-inflammatory phenotype. This population overlapped slightly with the Gr1hi MHC-IIlo/int (MI) subset, which also expressed Ccl1 and Ccr2. Inhibition of CCL2 led to reduced vascularization and hepatic blood volume, a trend toward decreased tumor volume, and the presence of nodules with central necrosis. These changes corresponded with a reduction in the Gr1lo MHCIIhi CCR2+ subpopulation [328].
Comparative transcriptomic analysis of human HCC, normal liver tissue, and fetal liver revealed the presence of distinct macrophage populations in tumor tissue, initially subdivided by high (TAM1s) or low (TAM2/3 s) expression of CD163. Further analysis revealed FOLR2+ TAM1s, SPP1+ TAM2s, and metallothionein 1G (MT1G)-enriched TAM3s, which were supported by flow cytometric and RNA-FISH data. Notably, TAM1s and TAM2s exhibited distinct spatial localizations within the tumor tissue. Interestingly, TAM1s presented increased expression of hairy and enhancer of split-1 and displayed similarities with fetal liver macrophages, suggesting the acquisition of a fetal-like phenotype with a possible role of Notch signaling in this transition [329].
On the basis of single-cell transcriptomic data, macrophages expressing TREM2 accumulate within HCC tumors. TREM2+ macrophages display M2-like polarization [330] and are associated with an anti-inflammatory function but exhibit the capacity to differentiate into protumorigenic MMP9+ macrophages [331]. SPP1 and MMP expression have recently been described to define protumorigenic TAM subsets. SPP1+ MMP9+ macrophages sorted from primary HCC tumors were found to promote HCC cell migration ex vivo, as well as tube formation by human umbilical vein endothelial cells, indicating the promotion of angiogenesis. PPAR-γ was found to be crucial for both the differentiation and protumorigenic effects of these macrophages [331]. HCC patients with increased numbers of cancer stem cells also displayed an accumulation of a HIF-1α-driven SPP1+ macrophage subset characterized by the expression of MMPs (MMP9, MMP12, and MMP17), which may enhance cancer epithelial‒mesenchymal transition. These cells are correlated with poor patient prognosis and an inadequate response to immunotherapy, as evidenced by CD8+ T-cell exclusion from areas of SPP1+ macrophage clustering, as well as suppression of immune-related pathways such as antigen processing and presentation, as indicated by scRNA analysis [332].
In accordance with most data from HCC, TAM infiltration in iCCA has been correlated with immune evasion, worse patient prognosis, and a diminished antitumor response [333, 334], with PD-L1 expression on CD68+ macrophages predicting an unfavorable prognosis [335]. The presence of CD86+ CD206+ TAMs has been shown to serve as a prognostic marker for iCCA [336], whereas the presence of CD163+ macrophages was correlated with worse overall survival in a retrospective study of human tumor samples [337]. In another iCCA cohort, neovascularization, regulatory T-cell infiltration, and poor disease-free survival were correlated with CD163+ macrophages [338]. A recent multiomics analysis of iCCA tissue revealed three novel molecular subtypes (chromatin remodeling, metabolism, chronic inflammation), each with a distinct prognosis [339]. TAMs, and more specifically APOE+ C1QB+ macrophages, were significantly overrepresented in the chronic inflammation subtype, whereas a high APOE+ C1QB+ TAM signature score was correlated with worse overall survival. In a mouse model in which CSF1R blockade was used to deplete TAMs, reduced TNF-α production by T cells was observed, indicating a dampened T-cell-mediated inflammatory response [339].
Dendritic cells
Origin and functions of dendritic cells
DCs are professional APCs with predominant MHC-II expression that sense pathogens through PRRs and orchestrate immune responses by connecting innate and adaptive immunity [340]. They are classified as conventional DCs (cDCs) or plasmacytoid DCs (pDCs) [340]. The former can be further subdivided into CD103+/CD8a+ type 1 cDCs (cDC1s) and CD11b+ type 2 cDCs (cDC2s). DCs are derived from bone marrow hematopoietic stem cells, which give rise to CMPs, which subsequently differentiate into monocyte‒dendritic cell progenitors and common dendritic cell progenitors (CDPs) [340, 341]. Development from CDPs, the precursors of cDCs (precDCs), an intermediate stage between CDPs and cDCs, migrate from the bone marrow to lymphoid and nonlymphoid organs via the blood and eventually differentiate into cDCs [340, 342]. On the other hand, pDCs complete their development and maturation in the bone marrow, diverging from precDCs and developing directly from CDPs [343]. However, a recent study using clonal tracing of hematopoietic stem cells and Cx3cr1+ progenitors suggested that cDC1s are more closely related to pDCs than to cDC2s [343, 344]. While Flt3 ligand (Flt3L) is an essential cytokine that mediates DC development through its receptor Flt3, transcription factors, including interferon regulatory factor 8, basic leucine zipper ATF-like 3 transcription factor (BATF3), and nuclear factor interleukin-3-regulated protein, are also involved in this process [342, 345,346,347,348].
Studies using scRNA-seq and high-dimensional protein profiling techniques have revealed markers for DC subpopulation identification and mapped the DC lineage, revealing the complex DC lineage and heterogeneity [346,347,348,349]. cDC1s specialize in antigen cross-presentation on MHC-I to activate CD8+ T cells in response to viral infections, whereas cDC2s express a wide spectrum of TLRs and are more prone to present antigens to CD4+ T cells [341, 350]. Clec9a, which is expressed on cDC1s, contributes to their superior ability to take up dying cells and present antigens to T cells; the deficiency of cross-presentation in mice has been shown to result in failure in priming antiviral or antitumour immunity [350, 351]. Compared with cDC1s, cDC2s are characterized by their expression of various cytokines and their ability to stimulate Th1 and Th17 responses in CD4+ T cells [352]. In contrast, pDCs express lower levels of MHC-II and a narrower array of PRRs [345]. Nonetheless, these cells are characterized by their ability to recognize pathogen-derived nucleic acids through high expression of TLR7 and TLR9 and to rapidly produce a large amount of type I IFN in response to viruses [353]. Interestingly, cDC1s express high levels of type III IFN when encountering poly I:C; thus, they are likely to interact with pDCs to promote type I IFN production [350, 354]. Upon stimulation, cDCs migrate to the T-cell zone or lymph nodes, which is primarily mediated by the CCR7‒CCL21 axis, for antigen presentation and T-cell activation [349, 355].
Dendritic cells in chronic liver inflammation
Hepatic DCs, along with resident KCs, contribute to immune tolerance within the tissue, partly because of their low phagocytic capacity and limited ability to stimulate T-cell responses, as highlighted by increased IL-10 production [356, 357]. All three DC subpopulations have been identified in both healthy or chronically inflamed human and mouse livers [29, 250, 253, 358, 359]. Recently, spatial transcriptomics analysis has placed cDC1s (Xcr1, Clec9a) and cDC2s (Cd209a, Mgl2, Clec10a) predominantly in the periportal regions [29] of the homeostatic liver, with the former located mainly around the lymphatics and the latter surrounding the biliary tree [360].
MASLD
The extent to which DCs contribute to MASLD has yet to be delineated, with data largely indicating contradictory conclusions [67, 72, 361]. Early research suggested that the presence of lipids and their engulfment by DCs in human and mouse livers can affect their immunogenic potential, shifting them toward a proinflammatory state [362]. Both cDC1s and cDC2s, along with pDCs, have been shown to be numerically expanded in various dietary models [WD, choline deficient high fat diet (CDHFD), methionine choline deficient diet (MCDD)] [253, 363,364,365]. In the MASH model, the expansion of total cDCs coincided with a shift in the ratio between the two subsets due to a relative increase in cDC2s [366] and decrease in cDC1s. Furthermore, in WD-fed mice, hepatic cDC2s exhibited heterogeneous expression patterns of Tbet, Mgl1, and CCR2 [253], whereas the total cDC population was shown to express lower levels of the antimicrobial peptide Slpi [359]. In both the two-week MCD diet and 6-month CDHFD models, flow cytometry and scRNA-seq data revealed an expansion of XCR1+ cDC1s, which was attributed to increased bone marrow and blood progenitor cell proliferation and was correlated with liver pathology. [364]. DC maturation, activation, and a shift toward a more immunogenic phenotype have been observed in MASH models, with the upregulation of markers such as CD80 and CD86, as well as increased TNF-α, IL-6, MCP-1, and IL-10 production [362, 363, 367]. Chu et al. reported that CCL20, an immature DC chemoattractant, is upregulated in the fibrotic MASLD liver. LX-2 cells (a human HSC line) exposed to fatty acids upregulated CCL20 expression in vitro, suggesting that HSCs might be responsible for the influx of immature DCs in the MASLD liver [368].
Recently, a mechanism through which DCs can influence the progression of HFD-induced steatosis in mice has been proposed; the activation of liver DCs is accompanied by the phosphorylation of liver kinase B1 (LKB1), a kinase that plays a regulatory role in DCs. DC-specific deletion of LKB1 led to increased insulin resistance; increased proportions of hepatic regulatory T cells (Tregs) and Th17 cells; increased expression of Cd36 and fibrotic markers; worsened steatosis; and elevated triglyceride and cholesterol levels. This effect was mitigated by the blockade of IL-17A, while ex vivo, cDC2s from LKB1 knockout mice exhibited increased production of the Th17-promoting cytokines Il6 and Il1β, collectively suggesting that DCs impact MASH pathology through the regulation of hepatic Th17 responses [367]. Finally, a Ly6Chi monocyte-derived F4/80+ CD11chi MHCII+ DC subset expressing high levels of CX3CR1 emerged during MCD-induced MASH and produced TNF-α, which correlated with hepatic injury in advanced stages of the model [369].
DCs have also been implicated in MASH regression. In MCD-fed mice, CD11c diphtheria toxin receptor (DTR)-mediated depletion of DCs led to exacerbation of inflammation and fibrosis, as well as delayed resolution of pathology [363]. Notably, the CD11c-DTR system is unlikely to be specific to DCs or particular DC subsets. In addition to recent data describing cDC1s as drivers of pathology, a protective role of CD103+ cDC1s against MASH pathology has also been proposed. In Batf3−/− mice, which lack cDC1s, feeding a high sucrose diet (HSD) for two weeks caused increased recruitment of inflammatory monocytes, the development of a proinflammatory milieu (characterized by elevated IL-1ra, CCL2, CCL-5, and TNF-α), progression toward steatohepatitis, and alterations in lipid metabolism. However, no effect on collagen deposition was observed in either MCDD-fed or HSD-fed Batf3−/− mice [365]. Human data regarding DCs in MASLD/MASH remain limited. Deep immunophenotyping of blood samples from patients with MASH before and after lifestyle intervention revealed a positive correlation of cDC2s (HLA-DR+ CD123- CD11c+ CD141−) with MASH activity, particularly hepatocyte ballooning and lobular inflammation. In contrast, cDC1s (HLA-DR+ CD123− CD11c+ CD141+) and pDCs (HLA-DR+ CD123+) were inversely correlated with MASH and glucose levels [366].
Fibrosis
Research on the role of DCs in humans and experimental fibrosis outside the context of MASLD remains limited [71, 370]. Both cDC subsets, differentiated from peripheral blood monocytes, have been reported in proximity to the fibrotic niche in human cirrhosis [299]. The expansion and activation of DCs have been observed in a mouse thioacetamide model, where depletion of CD11c+ cells led to decreased hepatic TNF-α levels [371, 372]. Additionally, ex vivo coculture of CD11c+ cells with LSECs enhanced T-cell activation and proliferation [371, 372]. However, these studies relied solely on CD11c as a marker to identify DCs, raising the possibility of contamination by non-DC CD11c⁺ cells. A study by Pradere et al. revealed that cDCs and pDCs play a nonsignificant role in promoting fibrosis [373]. In contrast, an antifibrotic effect has been reported, wherein therapeutic Flt3L-mediated expansion of DCs accelerated fibrosis regression in a CCl4 model, partly through upregulation of MMP9 [374]. In support of this, recent in vitro data have shown that DC-derived IL-10 can induce apoptosis, reduce proliferation, and downregulate α-SMA RNA, TGF-β1, and SMAD3 in LX-2 cells, further suggesting a potential antifibrotic role of immature DCs in HSCs [375]. In patients with cirrhosis of various etiologies, analysis of peripheral blood revealed a reduction in DC numbers and an increased pDC/cDC ratio compared with those in healthy controls [376].
HCC and iCCA
Mounting a sufficient immune response against tumors is highly dependent on DC-mediated recruitment and activation of tumoricidal lymphocytes [71, 377, 378]. Hepatic DCs are intrinsically programmed to maintain tolerance and have been shown, in vitro, to induce IL-10-dependent hyporesponsiveness in T cells, as well as to promote the generation of CD4+ CD25+ FoxP3+ Tregs [379]. This immunosuppressive effect of hepatic DCs can be further enhanced by the TME, with both tumor-associated cells and proteins influencing DC function. Hepatic CAFs are particularly capable of modulating the TME. One study demonstrated that IDO-producing regulatory DCs could be induced by CAFs via IL-6 secretion in vitro [380]. Additionally, HCC tumor-derived alpha-fetoprotein (AFP) has been shown to disrupt DC metabolism by inhibiting mTORC1 signaling, resulting in defective oxidative phosphorylation, impaired mitochondrial integrity, and a diminished capacity to stimulate T cells in vitro [381]. The expression of immune checkpoint molecules by DCs has also been described in human HCC samples, where tumor-infiltrating DCs express PD-L1 and galectin-9 (ligands for PD-1 and TIM3, respectively), among others [382]. Furthermore, PD-1 was found to be overexpressed on peripheral blood mDCs and pDCs from HCC patients [383]. In the periphery, a CD14+ CTLA4+ DC subset present in the peripheral blood of HCC patients was able to suppress T-cell responses via IDO in addition to IL-10, inducing systemic immunosuppression [384].
Studies on the effects of pDCs in liver cancer are limited [385], but existing data show a correlation with poor prognosis. The infiltration of tumor tissue by pDCs has been associated with worse overall survival, a shorter time to relapse, and increased tumor invasion in both HCC [386, 387] and iCCA [388], as well as with ICOS-ICOSL-mediated activation of a CD4+ FoxP3− IL-13− IL-10+ Tr1 regulatory cell subset that can suppress the antitumor response [389]. Vaccines targeting DC function in HCC are currently in various stages of clinical and preclinical development; however, this topic is beyond the scope of this article and has been recently reviewed elsewhere [390].
Future perspectives
On the basis of the latest insights in the field, a substantial amount of information is still lacking regarding the functional and phenotypic heterogeneity of myeloid cells in the liver, particularly in humans. Two key examples that highlight efforts to bridge this knowledge gap involve recent observations of macrophages and neutrophils. Under both steady-state and inflammatory conditions, hepatic macrophages are highly heterogeneous, comprising various niche- and context-instructed subsets (resident and recruited) with distinct transcriptional profiles and activation states. Future studies employing targeted approaches to modulate specific subsets and/or genetically manipulate expressed factors are needed to elucidate their precise functions and contributions to CLD pathophysiology and identify targets for the development of macrophage-tailored therapies. Regarding neutrophils—cells once considered largely transcriptionally inactive—advances in sequencing technologies adapted to their volatile nature have revealed a new level of complexity, including a dynamic transcriptional program that remains to be fully explored in different CLD settings.
The acquisition of this wealth of new information can be attributed, in part, to advancements in novel multiomics approaches. The integration of high-resolution microscopy with proteomic and single-cell or single-nucleus transcriptomic analyses continues to shed light on the functional and phenotypic variations of myeloid subpopulations and to facilitate the discovery of unrecognized subsets through the identification of novel markers. Hence, researchers are now equipped to study cell subsets that have previously remained elusive and to implement a more granular approach in analyzing the involvement of specific pathways or molecules in various forms of CLD. These new approaches may also help overcome challenges associated with studying rare cell populations by enabling more precise molecular and functional distinctions from phenotypically similar cells.
The discovery of novel cell subset markers will be particularly crucial for advancing DC research. The broad use of CD11c-targeted modifications in vivo (e.g., CD11c-cre, CD11c-DTR) does not allow for a more detailed study of the intricacies of pDC or cDC subset biology and their disease relevance. Moreover, DC subset-specific transgenic models (e.g., XCR1-cre) have yet to be employed in the context of CLD. Although DC-targeted therapeutic approaches are already being explored, particularly in oncology with DC vaccines and TME modulation, targeting T-cell/DC or LSEC/DC interactions may also represent promising avenues for antifibrotic therapies.
Finally, spatially resolved omics will further contextualize the role of cell type localization in CLD progression and regression. The spatial distribution of immune cells during homeostasis is vital for supporting their function and often reflects it. As discussed above, the niche-specific accumulation of immune subsets has been linked to various types and stages of CLD, with transcriptomic data providing an additional layer of insight into their function and potential for therapeutic targeting. In the coming years, the broader application of spatial omics to human liver tissue holds great promise for advancing our understanding of liver myeloid cell biology and identifying novel therapeutic strategies.
References
Marcellin P, Kutala BK. Liver diseases: a major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018;38:2–6.
Younossi ZM, Wong G, Anstee QM, Henry L. The global burden of liver disease. Clin Gastroenterol Hepatol. 2023;21:1978–91.
Mackowiak B, Fu Y, Maccioni L, Gao B. Alcohol-associated liver disease. J Clin Investig. 2024;134. https://doi.org/10.1172/JCI176345.
Åberg F, Byrne CD, Pirola CJ, Männistö V, Sookoian S. Alcohol consumption and metabolic syndrome: clinical and epidemiological impact on liver disease. J Hepatol. 2023;78:191–206.
Lan Y, Wang H, Weng H, Xu X, Yu X, Tu H, et al. The burden of liver cirrhosis and underlying etiologies: results from the Global Burden of Disease Study 2019. Hepatol Commun. 2023;7:e0026.
Paik JM, Golabi P, Younossi Y, Mishra A, Younossi ZM. Changes in the global burden of chronic liver diseases from 2012 to 2017: the growing impact of NAFLD. Hepatology. 2020;72. https://doi.org/10.1002/hep.31173/suppinfo.
Tacke F, Horn P, Wai-Sun Wong V, Ratziu V, Bugianesi E, Francque S, et al. EASL–EASD–EASO clinical practice guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024;81:492–542.
Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol. 2021;18:38–44. https://doi.org/10.1038/s41423-020-00560-0.
Bernsmeier C, van der Merwe S, Périanin A. Innate immune cells in cirrhosis. J Hepatol. 2020;73. https://doi.org/10.1016/j.jhep.2020.03.027.
Hahn JW, Yang HR, Moon JS, Chang JY, Lee K, Kim GA et al. Global incidence and prevalence of autoimmune hepatitis, 1970–2022: a systematic review and meta-analysis. EClinicalMed. 2023;65. https://doi.org/10.1016/j.eclinm.2023.102280.
European Association for the Study of the Liver. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–72.
Chazouilleres O, Beuers U, Bergquist A, Karlsen TH, Levy C, Samyn M, et al. EASL clinical practice guidelines on sclerosing cholangitis. J Hepatol. 2022;77:761–806.
Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis—a comprehensive review. J Hepatol. 2017;67:1298–323.
Schwabe RF, Brenner DA. Hepatic stellate cells: balancing homeostasis, hepatoprotection and fibrogenesis in health and disease. Nat Rev Gastroenterol Hepatol. 2025. https://doi.org/10.1038/s41575-025-01068-6.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7:6.
Brindley PJ, Bachini M, Ilyas SI, Khan SA, Loukas A, Sirica AE et al. Cholangiocarcinoma. Nat Rev Dis Primers. 2021;7. https://doi.org/10.1038/s41572-021-00300-2.
Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol. 2019;16:395–410.
Gao B, Jeong W-I, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–36.
Nemeth E, Baird AW, O’Farrelly C. Microanatomy of the liver immune system. Semin Immunopathol. 2009;31:333–43.
Cheng X, Kim SY, Okamoto H, Xin Y, Yancopoulos GD, Murphy AJ, et al. Glucagon contributes to liver zonation. Proc Natl Acad Sci USA. 2018;115:E4111–19.
Paris J, Henderson NC. Liver zonation, revisited. Hepatology. 2022;76:1219–30.
Kawashima K, Andreata F, Beccaria CG, Iannacone M. Priming and maintenance of adaptive immunity in the liver. Annu Rev Immunol. 2024;42:375–99.
Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, et al. Single-cell spatial reconstruction reveals global division of labor in the mammalian liver. Nature. 2017;542:352–6.
MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9. https://doi.org/10.1038/s41467-018-06318-7.
Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204.
Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, Massasa EE, et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol. 2018;36:962–70.
Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–47.e8.
Gola A, Dorrington MG, Speranza E, Sala C, Shih RM, Radtke AJ, et al. Commensal-driven immune zonation of the liver promotes host defense. Nature. 2021;589:131–6.
Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379–96.e38.
Miyamoto Y, Kikuta J, Matsui T, Hasegawa T, Fujii K, Okuzaki D, et al. Periportal macrophages protect against commensal-driven liver inflammation. Nature. 2024;629:901–9.
Strnad P, Tacke F, Koch A, Trautwein C. Liver-guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14:55–66.
Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. 2018;36:247–77.
Stamataki Z, Swadling L. The liver as an immunological barrier redefined by single-cell analysis. Immunology. 2020;160:157–70.
Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006.
Campana L, Esser H, Huch M, Forbes S. Liver regeneration and inflammation: from fundamental science to clinical applications. Nat Rev Mol Cell Biol. 2021;22:608–24.
Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13:267–76.
Sun D, Sun P, Li H, Zhang M, Liu G, Strickland AB et al. Fungal dissemination is limited by liver macrophage filtration of the blood. Nat Commun. 2019;10. https://doi.org/10.1038/s41467-019-12381-5.
Zhao D, Yang F, Wang Y, Li S, Li Y, Hou F et al. ALK1 signaling is required for the homeostasis of Kupffer cells and prevention of bacterial infection. J Clin Investig. 2022;132. https://doi.org/10.1172/JCI150489.
Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 2015;42:145–58.
Peiseler M, Araujo David B, Zindel J, Surewaard BGJ, Lee W-Y, Heymann F, et al. Kupffer cell-like syncytia replenish resident macrophage function in the fibrotic liver. Science. 2023;381:eabq5202.
Triantafyllou E, Gudd CL, Mawhin M-A, Husbyn HC, Trovato FM, Siggins MK et al. PD-1 blockade improves kupffer cell bacterial clearance in acute liver injury. J Clin Investig. 2021;131. https://doi.org/10.1172/JCI140196.
Balmer ML, Slack E, de Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. 2014;6:237ra66.
David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. 2016;151:1176–91.
Zeng Z, Surewaard BGJ, Wong CHY, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe. 2016;20:99–106.
Nakagaki BN, Mafra K, de Carvalho É, Lopes ME, Carvalho-Gontijo R, de Castro-Oliveira HM, et al. Immune and metabolic shifts during neonatal development reprogram liver identity and function. J Hepatol. 2018;69:1294–307.
Zeng Z, Surewaard BGJ, Wong CHY, Guettler C, Petri B, Burkhard R, et al. Sex-hormone-driven innate antibodies protect females and infants against EPEC infection. Nat Immunol. 2018;19:1100–11.
Wang J, An H, Ding M, Liu Y, Wang S, Jin Q, et al. Liver macrophages and sinusoidal endothelial cells execute vaccine-elicited capture of invasive bacteria. Sci Transl Med. 2023;15:eade0054.
Araujo David B, Atif J, Vargas e Silva Castanheira F, Yasmin T, Guillot A, Ait Ahmed Y, Peiseler M, et al. Kupffer cell reverse migration into the liver sinusoids mitigates neonatal sepsis and meningitis. Sci Immunol. 2024;9:eadq9704.
Heymann F, Tacke F. Immunology in the liver-from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Ng LG, Liu Z, Kwok I, Ginhoux F. Origin and heterogeneity of tissue myeloid cells: a focus on GMP-derived monocytes and neutrophils. Annu Rev Immunol. 2023;41:375–404.
Aroca-Crevillén A, Vicanolo T, Ovadia S, Hidalgo A. Neutrophils in physiology and pathology. Annu Rev Pathol. 2024;19. https://doi.org/10.1146/annurev-pathmechdis-051222-015009.
Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. 2023;23:563–79.
Herro R, Grimes HL. The diverse roles of neutrophils from protection to pathogenesis. Nat Immunol. 2024;25:2209–19.
Zhao J, Andreev I, Silva HM. Resident tissue macrophages: key coordinators of tissue homeostasis beyond immunity. Sci Immunol. 2024;9:eadd1967.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485–98.
Barry ST, Gabrilovich DI, Sansom OJ, Campbell AD, Morton JP. Therapeutic targeting of tumor myeloid cells. Nat Rev Cancer. 2023;23:216–37.
van Vlerken-Ysla L, Tyurina YY, Kagan VE, Gabrilovich DI. Functional states of myeloid cells in cancer. Cancer Cell. 2023;41:490–504.
Balazs I, Stadlbauer V. Circulating neutrophil anti-pathogen dysfunction in cirrhosis. JHEP Rep. 2023;5. https://doi.org/10.1016/j.jhepr.2023.100871.
Xie L, Zhang H, Xu L. The role of eosinophils in liver disease. Cell Mol Gastroenterol Hepatol. 2025;19:101413.
Kulle A, Thanabalasuriar A, Cohen TS, Szydlowska M. Resident macrophages of the lung and liver: the guardians of our tissues. Front Immunol. 2022;13:1029085.
De Ponti FF, Liu Z, Scott CL. Understanding the complex macrophage landscape in MASLD. JHEP Rep. 2024;6:101196.
Barreby E, Chen P, Aouadi M. Macrophage functional diversity in NAFLD-more than inflammation. Nat Rev Endocrinol. 2022;18:461–72.
Guilliams M, Scott CL. Liver macrophages in health and disease. Immunity. 2022;55:1515–29.
Bhattacharya M, Ramachandran P. Immunology of human fibrosis. Nat Immunol. 2023;24:1423–33.
Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of nonalcoholic fatty liver disease. JHEP Rep. 2022;4:100524.
Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77:1136–60.
Wang B, Yang L, Yuan X, Zhang Y. Roles and therapeutic targeting of dendritic cells in liver fibrosis. J Drug Target. 2024;32:647–54.
Li W, Chen G, Peng H, Zhang Q, Nie D, Guo T et al. Research progress on dendritic cells in hepatocellular carcinoma immune microenvironments. Biomolecules. 2024;14. https://doi.org/10.3390/biom14091161.
Méndez-Sánchez N, Córdova-Gallardo J, Barranco-Fragoso B, Eslam M. Hepatic dendritic cells in the development and progression of metabolic steatohepatitis. Front Immunol. 2021;12:641240.
Lurje I, Hammerich L, Tacke F. Dendritic cell and T-cell crosstalk in liver fibrogenesis and hepatocarcinogenesis: implications for prevention and therapy of liver cancer. Int J Mol Sci. 2020;21:7378.
Pinto AT, Lukacs-Kornek V. The role of dendritic cells in MASH: friends or foes? Front Immunol. 2024;15:1379225.
Lehman HK, Segal BH. The role of neutrophils in host defense and disease. J Allergy Clin Immunol. 2020;145. https://doi.org/10.1016/j.jaci.2020.02.038.
Montaldo E, Lusito E, Bianchessi V, Caronni N, Scala S, Basso-Ricci L et al. Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nat Immunol. 2022;23. https://doi.org/10.1038/s41590-022-01311-1.
Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19. https://doi.org/10.1038/s41577-019-0141-8.
Herro R, Grimes HL. The diverse role of neutrophils from protectin to pathogenesis. Nat Immunol. 2024;25:2209–19.
Kwok AJ, Allcock A, Ferreira RC, Cano-Gamez E, Smee M, Burnham KL et al. Neutrophils and emergency granulopoiesis drive immune suppression and an extreme response endotype during sepsis. Nat Immunol. 2023;24. https://doi.org/10.1038/s41590-023-01490-5.
Zhang J, Wu Q, Johnson CB, Pham G, Kinder JM, Olsson A et al. In situ mapping identifies distinct vascular niches for myelopoiesis. Nature. 2021;590. https://doi.org/10.1038/s41586-021-03201-2.
Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. 2018;21:1209–17. https://doi.org/10.1038/s41593-018-0213-2.
Wang C, Kay Lutes L, Barnoud C, Scheiermann C. The circadian immune system. Sci Immunol. 2022;7. https://doi.org/10.1126/sciimmunol.abm2465.
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153:1025–35. https://doi.org/10.1016/j.cell.2013.04.040.
Wigerblad G, Cao Q, Brooks S, Naz F, Gadkari M, Jiang K, et al. Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils. J Immunol. 2022;209:772–82. https://doi.org/10.4049/jimmunol.2200154.
Geh D, Leslie J, Rumney R, Reeves HL, Bird TG, Mann DA. Neutrophils as potential therapeutic targets in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2022;19:257–73. https://doi.org/10.1038/s41575-021-00568-5.
Mitroulis I, Alexaki VI, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T. Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharm Ther. 2015;147:123–35. https://doi.org/10.1016/j.pharmthera.2014.11.008.
Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5–23. https://doi.org/10.1038/cmi.2014.89.
Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem. 2016;85:765–92. https://doi.org/10.1146/annurev-biochem-060815-014442.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47. https://doi.org/10.1038/nri.2017.105.
Huby T, Gautier EL. Immune cell-mediated features of nonalcoholic steatohepatitis. Nat Rev Immunol. 2022;22:429–43. https://doi.org/10.1038/s41577-021-00639-3.
Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–405. https://doi.org/10.1002/hep.26937.
Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175:1473–82. https://doi.org/10.2353/ajpath.2009.080999.
Zang S, Wang L, Ma X, Zhu G, Zhuang Z, Xun Y, et al. Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem Biophys. 2015;73:479–87. https://doi.org/10.1007/s12013-015-0682-9.
Zhao X, Yang L, Chang N, Hou L, Zhou X, Yang L, et al. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. 2020;11:379. https://doi.org/10.1038/s41419-020-2582-1.
Ou R, Liu J, Lv M, Wang J, Wang J, Zhu L, et al. Neutrophil depletion improves diet-induced nonalcoholic fatty liver disease in mice. Endocrine. 2017;57:72–82.
Shrestha S, Jeon J-H, Hong C-W. Neutrophils in MASLD and MASH. BMB Rep. 2025;58:116–23.
Ibusuki R, Uto H, Arima S, Mawatari S, Setoguchi Y, Iwashita Y, et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. 2013;33:1549–56.
Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology. 2020;72:412–29. https://doi.org/10.1002/hep.31031.
Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 mediates nonalcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol. 2016;65:988–97. https://doi.org/10.1016/j.jhep.2016.05.041.
Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE. 2012;7:e52411 https://doi.org/10.1371/journal.pone.0052411.
Zang S, Ma X, Zhuang Z, Liu J, Bian D, Xun Y, et al. Increased ratio of neutrophil elastase to α1-antitrypsin is closely associated with liver inflammation in patients with nonalcoholic steatohepatitis. Clin Exp Pharm Physiol. 2016;43:13–21. https://doi.org/10.1111/1440-1681.12499.
Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z, et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun. 2019;518:691–7.
Babuta M, Morel C, de Carvalho Ribeiro M, Calenda C, Ortega-Ribera M, Thevkar Nagesh P, et al. Neutrophil extracellular traps activate hepatic stellate cells and monocytes via NLRP3 sensing in alcohol-induced acceleration of MASH fibrosis. Gut. 2024;73:1854–69.
van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68:1347–60. https://doi.org/10.1002/hep.29914.
Khan RS, Lalor PF, Thursz M, Newsome PN. The role of neutrophils in alcohol-related hepatitis. J Hepatol. 2023;79:1037–48. https://doi.org/10.1016/j.jhep.2023.05.017.
Taïeb J, Mathurin P, Elbim C, Cluzel P, Arce-Vicioso M, Bernard B, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol. 2000;32:579–86. https://doi.org/10.1016/S0168-8278(00)80219-6.
Liu M, Cao S, He L, Gao J, Arab JP, Cui H, et al. Super enhancer regulation of cytokine-induced chemokine production in alcoholic hepatitis. Nat Commun. 2021;12:4560. https://doi.org/10.1038/s41467-021-24843-w.
Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57. https://doi.org/10.1002/hep.22680.
Taı̈eb J, Delarche C, Paradis V, Mathurin P, Grenier A, Crestani B, et al. Polymorphonuclear neutrophils are a source of hepatocyte growth factor in patients with severe alcoholic hepatitis. J Hepatol. 2002;36:342–8. https://doi.org/10.1016/S0168-8278(01)00276-8.
Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372:1619–28. https://doi.org/10.1056/nejmoa1412278.
Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78:28–44. https://doi.org/10.1016/j.jhep.2022.08.029.
Guan Y, Peiffer B, Feng D, Parra MA, Wang Y, Fu Yet al. IL-8+ neutrophils drive inexorable inflammation in severe alcohol-associated hepatitis. J Clin Investig. 2024;134. https://doi.org/10.1172/JCI178616.
Herrero-Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. 2022;19:177–91. https://doi.org/10.1038/s41423-021-00832-3.
Domerecka W, Homa-Mlak I, Mlak R, Michalak A, Wilińska A, Kowalska-Kępczyńska A, et al. Indicator of inflammation and NETosis—low-density granulocytes as a biomarker of autoimmune hepatitis. J Clin Med. 2022;11:2174 https://doi.org/10.3390/jcm11082174.
Lee KH, Kronbichler A, Park DDY, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev. 2017;16:1160–73. https://doi.org/10.1016/j.autrev.2017.09.012.
Domerecka W, Kowalska-Kępczyńska A, Michalak A, Homa-Mlak I, Mlak R, Cichoż-Lach H, et al. Etiopathogenesis and diagnostic strategies in autoimmune hepatitis. Diagnostics. 2021;11:1418 https://doi.org/10.3390/DIAGNOSTICS11081418.
Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. 2018;15:206–21. https://doi.org/10.1038/nrgastro.2017.183.
Liaskou E, Hirschfield GM, Gershwin ME. Mechanisms of tissue injury in autoimmune liver diseases. Semin Immunopathol. 2014;36:553–68. https://doi.org/10.1007/s00281-014-0439-3.
Isse K, Harada K, Nakanuma Y. IL-8 expression by biliary epithelial cells is associated with neutrophilic infiltration and reactive bile ductules. Liver Int. 2007;27:672–80. https://doi.org/10.1111/j.1478-3231.2007.01465.x.
Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C, et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One. 2011;6:e21381 https://doi.org/10.1371/journal.pone.0021381.
Zhu H, Zheng M, He H, Lei H, Tai W, Yang J. High neutrophil-lymphocyte ratio indicates a worse response to ursodeoxycholic acid in primary biliary cholangitis: a retrospective cohort study. BMC Gastroenterol. 2023;23:400 https://doi.org/10.1186/s12876-023-03031-8.
Hashimoto E, Lindor KD, Homburger HA, Dickson ER, Czaja AJ, WIESNER RH, et al. Immunohistochemical characterization of hepatic lymphocytes in primary biliary cirrhosis in comparison with primary sclerosing cholangitis and autoimmune chronic active hepatitis. Mayo Clin Proc. 1993;68:1049–55. https://doi.org/10.1016/S0025-6196(12)60897-0.
Desmet VJ. Histopathology of cholestasis. Verh Dtsch Ges Pathol. 1995;79:233–40.
Zimmer CL, von Seth E, Buggert M, Strauss O, Hertwig L, Nguyen S, et al. A biliary immune landscape map of primary sclerosing cholangitis reveals a dominant network of neutrophils and tissue-resident T cells. Sci Transl Med. 2021;13:abb3107 https://doi.org/10.1126/scitranslmed.abb3107.
Greenman R, Snir T, Katav A, Aricha R, Mishalian I, Hay O, et al. The role of CCL24 in primary sclerosing cholangitis: bridging patient serum proteomics to preclinical data. Cells. 2024;13:209 https://doi.org/10.3390/cells13030209.
Hintermann E, Tondello C, Fuchs S, Bayer M, Pfeilschifter JM, Taubert R et al. Blockade of neutrophil extracellular trap components ameliorates cholestatic liver disease in Mdr2 (Abcb4) knockout mice. J Autoimmun. 2024;146:103229.
Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023;20:633–46. https://doi.org/10.1038/s41575-023-00807-x.
Liang W, Lindeman JH, Menke AL, Koonen DP, Morrison M, Havekes LM, et al. Metabolically induced liver inflammation leads to NASH and differs from LPS- or IL-1β-induced chronic inflammation. Lab Invest. 2014;94:491–502.
Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60:782–91.
Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. 2014;193:3925–33. https://doi.org/10.4049/jimmunol.1400861.
Kaufmann B, Reca A, Kim AD, Feldstein AE. Novel mechanisms for resolution of liver inflammation: therapeutic implications. Semin Liver Dis. 2021;41:150–62. https://doi.org/10.1055/s-0041-1723031.
Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest. 2019;129:4091–109.
Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut. 2018;67:333–47. https://doi.org/10.1136/gutjnl-2016-313615.
Saijou E, Enomoto Y, Matsuda M, Yuet-Yin Kok C, Akira S, Tanaka M, et al. Neutrophils alleviate fibrosis in the CCl4-induced mouse chronic liver injury model. Hepatol Commun. 2018;2:703–17. https://doi.org/10.1002/hep4.1178.
He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. 2019;70:1150–67. https://doi.org/10.1002/hep.30645.
He Y, Rodrigues RM, Wang X, Seo W, Ma J, Hwang S et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J Clin Investig. 2021;131. https://doi.org/10.1172/JCI141513.
Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47phox-oxidative stress pathway in neutrophils. Gut. 2017;66:705–15. https://doi.org/10.1136/gutjnl-2016-311861.
Kim Y, Park Y, Rho H, Yao T, Gao B, Hwang S. Inflammation in MASLD progression and cancer. JHEP Rep. 2025;7:101414.
Ekpanyapong S, Reddy KR. Infections in cirrhosis. Curr Treat Options Gastroenterol. 2019;17:254–70. https://doi.org/10.1007/s11938-019-00229-2.
Morrison MA, Artru F, Trovato FM, Triantafyllou E, McPhail MJ. Potential therapies for acute-on-chronic liver failure. Liver Int. 2025;45:e15545.
Singanayagam A, Triantafyllou E. Macrophages in chronic liver failure: diversity, plasticity and therapeutic targeting. Front Immunol. 2021;12:661182.
Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front Immunol. 2018;9:2948.
Geng A, Flint E, Bernsmeier C. Plasticity of monocytes and macrophages in cirrhosis of the liver. Front Netw Physiol. 2022;2:937739.
Yang AY-P, Wistuba-Hamprecht K, Greten TF, Ruf B. Innate-like T cells in liver disease. Trends Immunol. 2024;45:535–48.
Albillos A, Martin-Mateos R, Van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol. 2022;19:112–34.
Van der Merwe S, Chokshi S, Bernsmeier C, Albillos A. The multifactorial mechanisms of bacterial infection in decompensated cirrhosis. J Hepatol. 2021;75:S82–S100.
Biyik M, Ucar R, Solak Y, Gungor G, Polat I, Gaipov A, et al. Blood neutrophil-to-lymphocyte ratio independently predicts survival in patients with liver cirrhosis. Eur J Gastroenterol Hepatol. 2013;25:435–41.
Fiuza C, Salcedo M, Clemente G, Tellado JM. Granulocyte colony-stimulating factor improves deficient in vitro neutrophil transendothelial migration in patients with advanced liver disease. Clin Diagn Lab Immunol. 2002;9:433–9. https://doi.org/10.1128/CDLI.9.2.433-439.2002.
Clària J, Titos E, Jiménez W, Ros J, Ginès P, Arroyo V, et al. Altered biosynthesis of leukotrienes and lipoxins and host defense disorders in patients with cirrhosis and ascites. Gastroenterology. 1998;115:147–56. https://doi.org/10.1016/S0016-5085(98)70376-2.
Satsangi S, Duseja A, Sachdeva M, Tomer S, Arora SK, Taneja S, et al. Monocyte human leukocyte antigen–antigen D related, neutrophil oxidative burst and cytokine analysis in patients of decompensated cirrhosis with and without acute-on chronic liver failure. PLoS ONE. 2018;13:e0200644. https://doi.org/10.1371/journal.pone.0200644.
Xiao L, Tang S, Zhang L, Ma S, Zhao Y, Zhang F et al. Serum CXCL1 is a prognostic factor for patients with hepatitis B virus–related acute-on-chronic liver failure. Front Med. 2021;8. https://doi.org/10.3389/fmed.2021.657076.
Langer MM, Sichelschmidt S, Bauschen A, Bornemann L, Guckenbiehl S, Gunzer M, et al. Pathological neutrophil migration predicts adverse outcomes in hospitalized patients with liver cirrhosis. Liver Int. 2023;43:896–905. https://doi.org/10.1111/liv.15486.
Mookerjee RP, Stadlbauer V, Lidder S, Wright GAK, Hodges SJ, Davies NA, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology. 2007;46:831–40. https://doi.org/10.1002/hep.21737.
Taylor NJ, Manakkat Vijay GK, Abeles RD, Auzinger G, Bernal W, Ma Y, et al. The severity of circulating neutrophil dysfunction in patients with cirrhosis is associated with 90-day and 1-year mortality. Aliment Pharm Ther. 2014;40:705–15. https://doi.org/10.1111/apt.12886.
Tritto G, Bechlis Z, Stadlbauer V, Davies N, Francés R, Shah N, et al. Evidence of neutrophil functional defect despite inflammation in stable cirrhosis. J Hepatol. 2011;55:574–81. https://doi.org/10.1016/j.jhep.2010.11.034.
Boussif A, Rolas L, Weiss E, Bouriche H, Moreau R, Périanin A. Impaired intracellular signaling, myeloperoxidase release and bactericidal activity of neutrophils from patients with alcoholic cirrhosis. J Hepatol. 2016;64:1041–8. https://doi.org/10.1016/j.jhep.2015.12.005.
Knooihuizen SAI, Alexander NJ, Hopke A, Barros N, Viens A, Scherer A, et al. Loss of coordinated neutrophil responses to the human fungal pathogen, Candida albicans, in patients with cirrhosis. Hepatol Commun. 2021;5:502–15.
Stadlbauer V, Mookerjee RP, Hodges S, Wright GAK, Davies NA, Jalan R. Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis. J Hepatol. 2008;48:945–51. https://doi.org/10.1016/j.jhep.2008.02.015.
Makkar K, Tomer S, Verma N, Rathi S, Arora SK, Taneja S, et al. Neutrophil dysfunction predicts 90-day survival in patients with acute on chronic liver failure: a longitudinal case–control study. JGH Open. 2020;4:595–602. https://doi.org/10.1002/jgh3.12344.
Balazs I, Horvath A, Leber B, Feldbacher N, Sattler W, Rainer F et al. Serum bile acids in liver cirrhosis promote neutrophil dysfunction. Clin Transl Med. 2022;12. https://doi.org/10.1002/ctm2.735.
Wu W, Sun S, Wang Y, Zhao R, Ren H, Li Z et al. Circulating neutrophil dysfunction in HBV-related acute-on-chronic liver failure. Front Immunol. 2021;12. https://doi.org/10.3389/fimmu.2021.620365.
Rajkovic IA, Williams R. Abnormalities of neutrophil phagocytosis, intracellular killing and metabolic activity in alcoholic cirrhosis and hepatitis. Hepatology. 1986;6:252–62. https://doi.org/10.1002/hep.1840060217.
Garfia C, Garcı́a-Ruiz I, Solı́s-Herruzo JA. Deficient phospholipase C activity in blood polimorphonuclear neutrophils from patients with liver cirrhosis. J Hepatol. 2004;40:749–56. https://doi.org/10.1016/j.jhep.2004.01.004.
Blasi A, Patel VC, Adelmeijer J, Azarian S, Aziz F, Fernández J, et al. Plasma levels of circulating DNA are associated with outcome, but not with activation of coagulation in decompensated cirrhosis and ACLF. JHEP Rep. 2019;1:179–87. https://doi.org/10.1016/j.jhepr.2019.06.002.
Zenlander R, Havervall S, Magnusson M, Engstrand J, Ågren A, Thålin C, et al. Neutrophil extracellular traps in patients with liver cirrhosis and hepatocellular carcinoma. Sci Rep. 2021;11:18025. https://doi.org/10.1038/s41598-021-97233-3.
Agraz-Cibrián JM, Delgado-Rizo V, Segura-Ortega JE, Maldonado-Gómez HA, Zambrano-Zaragoza JF, Durán-Avelar MJ Vibanco-Perez N et al. Impaired neutrophil extracellular traps and inflammatory responses in the peritoneal fluid of patients with liver cirrhosis. Scand J Immunol. 2018;88. https://doi.org/10.1111/sji.12714.
KUSBAA N, Kumashiro R, Ogata H, Sata M, Tanikawa K. In Vitro study of neutrophil apoptosis in liver cirrhosis. Intern Med. 1998;37:11–17. https://doi.org/10.2169/internalmedicine.37.11.
Ramı́rez J, Titos E, Clària J, Navasa M, Fernández J, Rodés J. Increased apoptosis dependent on caspase-3 activity in polymorphonuclear leukocytes from patients with cirrhosis and ascites. J Hepatol. 2004;41:44–48. https://doi.org/10.1016/j.jhep.2004.03.011.
Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19:222–32.
Llovet JM, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer. 2022;3:386–401. https://doi.org/10.1038/s43018-022-00357-2.
Suddle A, Reeves H, Hubner R, Marshall A, Rowe I, Tiniakos D, et al. British Society of Gastroenterology guidelines for the management of hepatocellular carcinoma in adults. Gut. 2024;73:1235–68.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: ‘N1’ versus ‘N2’ TAN. Cancer Cell. 2009;16:183–94. https://doi.org/10.1016/j.ccr.2009.06.017.
Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10:562–73. https://doi.org/10.1016/j.celrep.2014.12.039.
Raskov H, Orhan A, Gaggar S, Gögenur I. Neutrophils and polymorphonuclear myeloid-derived suppressor cells: an emerging battleground in cancer therapy. Oncogenesis. 2022;11:22. https://doi.org/10.1038/s41389-022-00398-3.
Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22:173–87. https://doi.org/10.1038/s41577-021-00571-6.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. https://doi.org/10.1038/nri2506.
Xu R, Huang H, Zhang Z, Wang F-S. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. 2014;11:224–31.
Peng ZP, Jiang ZZ, Guo HF, Zhou MM, Huang YF, Ning WR, et al. Glycolytic activation of monocytes regulates the accumulation and function of neutrophils in human hepatocellular carcinoma. J Hepatol. 2020;73:906–17. https://doi.org/10.1016/j.jhep.2020.05.004.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422. https://doi.org/10.1038/s41419-018-0458-4.
Zhang H, He G, Kong Y, Chen Y, Wang B, Sun X, et al. Tumor-activated liver stromal cells regulate myeloid-derived suppressor cells accumulation in the liver. Clin Exp Immunol. 2017;188:96–108. https://doi.org/10.1111/cei.12917.
Chiu DKC, Xu IMJ, Lai RKH, Tse APW, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. 2016;64:797–813. https://doi.org/10.1002/hep.28655.
Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229:114–25. https://doi.org/10.1111/j.1600-065X.2009.00767.x.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to Sorafenib. Gastroenterology. 2016;150:1646–58.e17. https://doi.org/10.1053/j.gastro.2016.02.040.
Chang CJ, Yang YH, Chiu CJ, Lu LC, Liao CC, Liang CW, et al. Targeting tumor-infiltrating Ly6G+ myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int J Cancer. 2018;142:1878–89. https://doi.org/10.1002/ijc.31216.
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807. https://doi.org/10.1002/hep.23054.
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit t-cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–95. https://doi.org/10.4049/jimmunol.168.2.689.
Zhou SL, Yin D, Hu ZQ, Bin Luo C, Zhou ZJ, Xin HY, et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology. 2019;70:1214–30. https://doi.org/10.1002/hep.30630.
Li X, Xing YF, Lei AH, Xiao Q, Lin ZH, Hong YF, et al. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value of for hepatocellular carcinoma patients. Oncotarget. 2017;8:24380–8. https://doi.org/10.18632/oncotarget.15456.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45. https://doi.org/10.1038/nm.3909.
Wang Y, Yao R, Zhang D, Chen R, Ren Z, Zhang L. Circulating neutrophils predict poor survival for HCC and promote HCC progression through p53 and STAT3 signaling pathway. J Cancer. 2020;11:3736–44. https://doi.org/10.7150/jca.42953.
Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma by provoking tumorous inflammatory response. J Hematol Oncol. 2020;13:3 https://doi.org/10.1186/s13045-019-0836-0.
Blanchard C, Rothenberg ME. Biology of the eosinophil. Adv Immunol. 2009;101:81–121.
Iwasaki H, Mizuno S, Mayfield R, Shigematsu H, Arinobu Y, Seed B, et al. Identification of eosinophil lineage-committed progenitors in the murine bone marrow. J Exp Med. 2005;201:1891–7.
Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A, et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med. 2009;206:183–93.
Fulkerson PC, Rothenberg ME. Eosinophil development, disease involvement, and therapeutic suppression. Adv Immunol. 2018;138:1–34.
Fulkerson PC. Transcription factors in eosinophil development and as therapeutic targets. Front Med. 2017;4:115.
Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, Kato Y, et al. Essential and instructive roles of GATA factors in eosinophil development. J Exp Med. 2002;195:1379–86.
Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20:3010–21.
Walsh JC, DeKoter RP, Lee HJ, Smith ED, Lancki DW, Gurish MF, et al. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17:665–76.
Bettigole SE, Lis R, Adoro S, Lee A-H, Spencer LA, Weller PF, et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. 2015;16:829–37.
McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. J Exp Med. 2002;195:F43–7.
Fulkerson PC, Rothenberg ME. Targeting eosinophils in allergy, inflammation and beyond. Nat Rev Drug Discov. 2013;12:117–29.
Johnston LK, Hsu C-L, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol. 2016;197:3445–53.
Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol. 2017;17:746–60.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53.
Gigon L, Fettrelet T, Yousefi S, Simon D, Simon H-U. Eosinophils from A to Z. Allergy. 2023;78:1810–46.
Masterson JC, Menard-Katcher C, Larsen LD, Furuta GT, Spencer LA. Heterogeneity of intestinal tissue eosinophils: potential considerations for next-generation eosinophil-targeting strategies. Cells. 2021;10:426 https://doi.org/10.3390/cells10020426.
Martinez OM, Villanueva JC, Gershwin EM, Krams SM. Cytokine patterns and cytotoxic mediators in primary biliary cirrhosis. Hepatology. 1995;21:113–9.
Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–7.
Terasaki S, Nakanuma Y, Yamazaki M, Unoura M. Eosinophilic infiltration of the liver in primary biliary cirrhosis: a morphological study. Hepatology. 1993;17:206–12.
Yamazaki K, Suzuki K, Nakamura A, Sato S, Lindor KD, Batts KP, et al. Ursodeoxycholic acid inhibits eosinophil degranulation in patients with primary biliary cirrhosis. Hepatology. 1999;30:71–8.
Watanabe H, Ohira H, Kuroda M, Takagi T, Ishikawa H, Nishimaki T, et al. Primary sclerosing cholangitis with marked eosinophilic infiltration in the liver. J Gastroenterol. 1995;30:524–8.
Garrido I, Lopes S, Fonseca E, Carneiro F, Macedo G. Autoimmune hepatitis and eosinophilia: a rare case report. World J Hepatol. 2023;15:311–7.
Chowdry S, Rubin E, Sass DA. Acute autoimmune hepatitis presenting with peripheral blood eosinophilia. Ann Hepatol. 2012;11:559–63.
De Oliveira LC, Goldberg AC, Marin MLC, Schneidwind KR, Frade AF, Kalil J, et al. Autoimmune hepatitis in brazilian children: ige and genetic polymorphisms in associated genes. J Immunol Res. 2015;2015:1–9. https://doi.org/10.1155/2015/679813.
Ye Z, Huang S, Zhang Y, Mei X, Zheng H, Li M, et al. Galectins, eosinophiles, and macrophages may contribute to schistosoma japonicum egg-induced immunopathology in a mouse model. Front Immunol. 2020;11:146.
Malta KK, Palazzi C, Neves VH, Aguiar Y, Silva TP, Melo RCN. Schistosomiasis mansoni-recruited eosinophils: an overview in the granuloma context. Microorganisms. 2022;10:2022 https://doi.org/10.3390/microorganisms10102022.
Klion AD, Nutman TB. The role of eosinophils in host defense against helminth parasites. J Allergy Clin Immunol. 2004;113:30–7.
Ramirez GA, Yacoub M-R, Ripa M, Mannina D, Cariddi A, Saporiti N, et al. Eosinophils from physiology to disease: a comprehensive review. Biomed Res Int. 2018;2018:1–28.
Reiman RM, Thompson RW, Feng CG, Hari D, Knight R, Cheever AW, et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74:1471–9.
Xu L, Yang Y, Wen Y, Jeong JM, Emontzpohl C, Atkins CL, et al. Hepatic recruitment of eosinophils and their protective function during acute liver injury. J Hepatol. 2022;77:344–52.
Gouon-Evans V, Lin EY, Pollard JW. Requirement of macrophages and eosinophils and their cytokines/chemokines for mammary gland development. Breast Cancer Res. 2002;4:155–64.
Voehringer D, van Rooijen N, Locksley RM. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J Leukoc Biol. 2007;81:1434–44.
Yang Y, Xu L, Atkins C, Kuhlman L, Zhao J, Jeong JM, et al. Novel IL-4/HB-EGF-dependent crosstalk between eosinophils and macrophages controls liver regeneration after ischemia and reperfusion injury. Gut. 2024;73:1543–53. https://doi.org/10.1136/gutjnl-2024-332033.
O'Sullivan JA, Bochner BS. Eosinophils and eosinophil-associated diseases: an update. J Allergy Clin Immunol. 2018;141:505–17.
Grisaru-Tal S, Itan M, Klion AD, Munitz A. A new dawn for eosinophils in the tumor microenvironment. Nat Rev Cancer. 2020;20:594–607.
Kataoka S, Konishi Y, Nishio Y, Fujikawa-Adachi K, Tominaga A. Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol. 2004;23:549–60.
Simson L, Ellyard JI, Dent LA, Matthaei KI, Rothenberg ME, Foster PS, et al. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J Immunol. 2007;178:4222–9.
Hollande C, Boussier J, Ziai J, Nozawa T, Bondet V, Phung W, et al. Inhibition of the dipeptidyl peptidase DPP4 (CD26) reveals IL-33-dependent eosinophil-mediated control of tumor growth. Nat Immunol. 2019;20:257–64.
Grisaru-Tal S, Rothenberg ME, Munitz A. Eosinophil–lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat Immunol. 2022;23:1309–16.
Orsi G, Tovoli F, Dadduzio V, Vivaldi C, Brunetti O, Ielasi L, et al. Prognostic role of blood eosinophil count in patients with sorafenib-treated hepatocellular carcinoma. Target Oncol. 2020;15:773–85.
Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The myeloid cell compartment—cell by cell. Annu Rev Immunol. 2019;37:269–93.
Ziegler-Heitbrock L. Blood monocytes and their subsets: established features and open questions. Front Immunol. 2015;6:423.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–51.
Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353. https://doi.org/10.1126/science.aaf4238.
Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–49.
Gibert-Ramos A, Andrés-Rozas M, Pastó R, Alfaro-Retamero P, Guixé-Muntet S, Gracia-Sancho J. Sinusoidal communication in chronic liver disease. Clin Mol Hepatol. 2025;31:32–55.
Scott CL, T’Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity. 2018;49:312–25.e5.
Willekens FLA, Werre JM, Kruijt JK, Roerdinkholder-Stoelwinder B, Groenen-Döpp YAM, Van Den Bos AG, et al. Liver Kupffer cells rapidly remove red blood cell-derived vesicles from the circulation by scavenger receptors. Blood. 2005;105:2141–5.
Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020;217. https://doi.org/10.1084/jem.20190723.
Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18:45–56.
Fima R, Dussaud S, Benbida C, Blanchet M, Lanthiez F, Poupel L, et al. Loss of embryonically derived Kupffer cells during hypercholesterolemia accelerates atherosclerosis development. Nat Commun. 2024;15:8341.
Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42.
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62:279–91.
Triantafyllou E, Gudd CLC, Possamai LA. Immune-mediated liver injury from checkpoint inhibitors: mechanisms, clinical characteristics and management. Nat Rev Gastroenterol Hepatol. 2024;22:112–26. https://doi.org/10.1038/s41575-024-01019-7.
Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. 2016;22:945–51.
Liu B, Cheng L, Gao H, Zhang J, Dong Y, Gao W, et al. The biology of VSIG4: implications for the treatment of immune-mediated inflammatory diseases and cancer. Cancer Lett. 2023;553:215996. https://doi.org/10.1016/j.canlet.2022.215996.
Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol. 2016;65:758–68.
Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity. 2019;51:655–70.e8.
Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321. https://doi.org/10.1038/ncomms10321.
Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. 2021;34:108626. https://doi.org/10.1016/j.celrep.2020.108626.
De Ponti FF, Bujko A, Liu Z, Collins PJ, Schuermans S, Maueröder C, et al. Spatially restricted and ontogenically distinct hepatic macrophages are required for tissue repair. Immunity. 2025;58:1867–8. https://doi.org/10.1016/j.immuni.2025.01.002.
Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during nonalcoholic steatohepatitis. Immunity. 2020;53:627–40.
Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity. 2017;47:374–88.e6.
Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from Kupffer cells in the fatty liver. Immunity. 2020;53:641–57.e14.
Blériot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, Ficht X, et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity. 2021;54:2101–16.e6.
De Simone G, Andreata F, Bleriot C, Fumagalli V, Laura C, Garcia-Manteiga JM, et al. Identification of a Kupffer cell subset capable of reverting the T-cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089–2100.e8.
Hume DA, Offermanns S, Bonnavion R. Contamination of isolated mouse Kupffer cells with liver sinusoidal endothelial cells. Immunity. 2022;55:1139–40.
Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates the NLRP3 inflammasome in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 2018;332:111–20.
Tosello-Trampont A-C, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem. 2012;287:40161–72.
Song K, Kwon H, Han C, Chen W, Zhang J, Ma W, et al. Yes-associated protein in Kupffer cells enhances the production of proinflammatory cytokines and promotes the development of nonalcoholic steatohepatitis. Hepatology. 2020;72:72–87.
Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–26.
Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67:1270–83.
Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1310–G1321. https://doi.org/10.1152/ajpgi.00365.2011.
Coombes JD, Manka PP, Swiderska-Syn M, Vannan DT, Riva A, Claridge LC et al. Osteopontin promotes cholangiocyte secretion of chemokines to support macrophage recruitment and fibrosis in MASH. Liver Int. 2024;45. https://doi.org/10.1111/liv.16131.
Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057–74.e7.
Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16:145–59.
Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res. 2017;58:1067–79.
Zhang J, Wang Y, Fan M, Guan Y, Zhang W, Huang F, et al. Reactive oxygen species regulation by NCF1 governs ferroptosis susceptibility of Kupffer cells to MASH. Cell Metab. 2024;36:1745–1763.e6.
Itoh M, Tamura A, Kanai S, Tanaka M, Kanamori Y, Shirakawa I et al. Lysosomal cholesterol overload in macrophages promotes liver fibrosis in a mouse model of NASH. J Exp Med. 2023;220. https://doi.org/10.1084/jem.20220681.
Jeelani I, Moon J-S, da Cunha FF, Nasamran CA, Jeon S, Zhang X, et al. HIF-2α drives hepatic Kupffer cell death and proinflammatory recruited macrophage activation in nonalcoholic steatohepatitis. Sci Transl Med. 2024;16:eadi0284.
Morgantini C, Jager J, Li X, Levi L, Azzimato V, Sulen A, et al. Liver macrophages regulate systemic metabolism through noninflammatory factors. Nat Metab. 2019;1:445–59.
Chan MM, Daemen S, Beals JW, Terekhova M, Yang BQ, Fu CF, et al. Steatosis drives monocyte-derived macrophage accumulation in human metabolic dysfunction-associated fatty liver disease. JHEP Rep. 2023;5:100877.
Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. 2019;75:644–660.e5.
Iwata A, Maruyama J, Natsuki S, Nishiyama A, Tamura T, Tanaka M, et al. Egr2 drives the differentiation of Ly6Chi monocytes into fibrosis-promoting macrophages in metabolic dysfunction-associated steatohepatitis in mice. Commun Biol. 2024;7:681 https://doi.org/10.1038/s42003-024-06357-5.
Ganguly S, Rosenthal SB, Ishizuka K, Troutman TD, Rohm TV, Khader N et al. Lipid-associated macrophages’ promotion of fibrosis resolution during MASH regression requires TREM2. Proc Natl Acad Sci USA. 2024;121. https://doi.org/10.1073/pnas.2405746121.
Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Miličević N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2+ macrophages that localize to fibrotic areas and limit NASH. J Hepatol. 2022;77:1373–85.
Fredrickson G, Florczak K, Barrow F, Mahmud S, Dietsche K, Wang H, et al. TREM2 macrophages mediate the beneficial effects of bariatric surgery against MASH. Hepatology. 2024;81:1776–91. https://doi.org/10.1097/HEP.0000000000001098.
Govaere O, Petersen SK, Martinez-Lopez N, Wouters J, Van Haele M, Mancina RM, et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in nonalcoholic fatty liver disease. J Hepatol. 2022;76:1001–12.
Ait Ahmed Y, Lafdil F, Tacke F. Ambiguous Pathogenic Roles of Macrophages in Alcohol-Associated Liver Diseases. Hepat Med. 2023;15:113–27.
Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79:1348–56.
Mandrekar P, Szabo G. Signaling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–66.
Vergis N, Khamri W, Beale K, Sadiq F, Aletrari MO, Moore C, et al. Defective monocyte oxidative burst predicts infection in alcoholic hepatitis and is associated with reduced expression of NADPH oxidase. Gut. 2017;66:519–29.
Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. 2014;96:657–65.
Duan Y, Chu H, Brandl K, Jiang L, Zeng S, Meshgin N, et al. CRIg on liver macrophages clears pathobionts and protects against alcoholic liver disease. Nat Commun. 2021;12:7172. https://doi.org/10.1038/s41467-021-27385-3.
Sasaki K, Rooge S, Gunewardena S, Hintz JA, Ghosh P, Pulido Ruiz IA, et al. Kupffer cell diversity maintains liver function in alcohol-associated liver disease. Hepatology. 2024;81:870–87. https://doi.org/10.1097/hep.0000000000000918.
Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O'Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol. 2018;69:676–86.
Guillot A, Winkler M, Silva Afonso M, Aggarwal A, Lopez D, Berger H, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023;78:150–66.
Locatelli L, Cadamuro M, Spirlì C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63:965–82.
Li W, Yang Y, Yang L, Chang N, Li L. Monocyte-derived Kupffer cells dominate in the Kupffer cell pool during liver injury. Cell Rep. 2023;42:113164 https://doi.org/10.1016/j.celrep.2023.113164.
De Muynck K, Vanderborght B, De Ponti FF, Gijbels E, Van Welden S, Guilliams M, et al. Kupffer cells contested as early drivers in the pathogenesis of primary sclerosing cholangitis. Am J Pathol. 2023;193:366–79.
De Muynck K, Heyerick L, De Ponti FF, Vanderborght B, Meese T, Van Campenhout S, et al. Osteopontin characterizes bile duct-associated macrophages and correlates with liver fibrosis severity in primary sclerosing cholangitis. Hepatology. 2024;79:269–88.
Laschtowitz A, Lindberg EL, Liebhoff AM, Liebig LA, Casar C, Steinmann S, et al. Liver transcriptome analysis reveals PSC-attributed gene set associated with fibrosis progression. JHEP Rep. 2025;7:101267 https://doi.org/10.1016/j.jhepr.2024.101267.
Ran J, Yin S, Issa R, Zhao Q, Zhu G, Zhang H et al. Key role of macrophages in the progression of hepatic fibrosis. Hepatol Commun. 2024;9. https://doi.org/10.1097/HC9.0000000000000602.
Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3:785–97.
Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X, et al. CXCL6-EGFR-induced kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J Cell Mol Med. 2018;22:5050–61.
Diao S, Li L, Zhang J, Ji M, Sun L, Shen W, et al. Macrophage-derived CCL1 targets CCR8 receptor in hepatic stellate cells to promote liver fibrosis through JAk/STAT pathway. Biochem Pharm. 2025;237:116884 https://doi.org/10.1016/j.bcp.2025.116884.
Matsuda M, Seki E. Hepatic stellate cell-macrophage crosstalk in liver fibrosis and carcinogenesis. Semin Liver Dis. 2020;40:307–20.
Li W, Chen L, Zhou Q, Huang T, Zheng W, Luo F, et al. Liver macrophage-derived exosomal miRNA-342-3p promotes liver fibrosis by inhibiting HPCAL1 in stellate cells. Hum Genom. 2025;19:9. https://doi.org/10.1186/s40246-025-00722-z.
Han C, Zhai Y, Wang Y, Peng X, Zhang X, Dai B, et al. Intravital imaging of splenic classical monocytes modifying the hepatic CX3CR1+ cells motility to exacerbate liver fibrosis via spleen-liver axis. Theranostics. 2024;14:2210–31.
Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–8.
Lee J, Kim CM, Cha JH, Park JY, Yu YS, Wang HJ, et al. Multiplexed digital spatial protein profiling reveals distinct phenotypes of mononuclear phagocytes in livers with advanced fibrosis. Cells. 2022;11:3387 https://doi.org/10.3390/cells11213387.
Fabre T, Barron AMS, Christensen SM, Asano S, Bound K, Lech MP, et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol. 2023;8:eadd8945.
Nguyen-Lefebvre AT, Ajith A, Portik-Dobos V, Horuzsko DD, Arbab AS, Dzutsev A, et al. The innate immune receptor TREM-1 promotes liver injury and fibrosis. J Clin Invest. 2018;128:4870–83.
Feng M, Ding J, Wang M, Zhang J, Zhu X, Guan W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int J Biol Sci. 2018;14:1033–40.
Sun R, Xiang Z, Wu B. T cells and liver fibrosis. Portal Hypertens Cirrhosis. 2022;1:125–32.
Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA. 2012;109. https://doi.org/10.1073/pnas.1119964109.
Pang Q, Zhou S, Wang Y, Pan H, Wang Z, Qin X, et al. GAMG alleviates liver fibrosis through inducing ferroptosis in inflammatory macrophages via the IRF1/SLC7A11 signaling pathway. Redox Biol. 2025;80:103509 https://doi.org/10.1016/j.redox.2025.103509.
Park JS, Ma YQ, Wang F, Ma H, Sui G, Rustamov N, et al. A3AR antagonism mitigates metabolic dysfunction-associated steatotic liver disease by exploiting monocyte-derived Kupffer cell necroptosis and inflammation resolution. Metabolism. 2025;164:156114 https://doi.org/10.1016/j.metabol.2024.156114.
Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother. 2013;62:1421–30.
Hammerich L, Tacke F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J Gastrointest Pathophysiol. 2015;6:43.
Bernsmeier C, Triantafyllou E, Brenig R, Lebosse FJ, Singanayagam A, Patel VC, et al. CD14+CD15−HLA-DR− myeloid-derived suppressor cells impair antimicrobial responses in patients with acute-on-chronic liver failure. Gut. 2018;67:1155–67.
Flint E, Triantafyllou E, Bernsmeier C. TAM Receptors in the pathophysiology of liver disease. Livers. 2022;2:15–29.
Brenig R, Pop OT, Triantafyllou E, Geng A, Singanayagam A, Perez-Shibayama C, et al. Expression of AXL receptor tyrosine kinase relates to monocyte dysfunction and severity of cirrhosis. Life Sci Alliance. 2020;3:e201900465.
Pop OT, Geng A, Flint E, Singanayagam A, Ercan C, Possamai L et al. AXL expression on homeostatic resident liver macrophages is reduced in cirrhosis following GAS6 production by hepatic stellate cells. CMGH. 2023;16. https://doi.org/10.1016/j.jcmgh.2023.03.007.
Bernsmeier C, Pop OT, Singanayagam A, Triantafyllou E, Patel VC, Weston CJ, et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology. 2015;148:603–615.e14. https://doi.org/10.1053/j.gastro.2014.11.045.
Trovato FM, Zia R, Napoli S, Wolfer K, Huang X, Morgan PE, et al. Dysregulation of the lysophosphatidylcholine/autotaxin/lysophosphatidic acid axis in acute-on-chronic liver failure is associated with mortality and systemic inflammation by lysophosphatidic acid–dependent monocyte activation. Hepatology. 2021;74:907–25. https://doi.org/10.1002/hep.31738.
Korf H, Du Plessis J, van Pelt J, De Groote S, Cassiman D, Verbeke L, et al. Inhibition of glutamine synthetase in monocytes from patients with acute-on-chronic liver failure resuscitates their antibacterial and inflammatory capacity. Gut. 2019;68:1872–83.
Pose E, Coll M, Martínez-Sánchez C, Zeng Z, Surewaard BGJ, Català C, et al. Programmed death ligand 1 is overexpressed in liver macrophages in chronic liver diseases, and its blockade improves the antibacterial activity against infections. Hepatology. 2021;74:296–311.
Geng A, Brenig RG, Roux J, Lütge M, Cheng H-W, Flint EE, et al. Circulating monocytes upregulate CD52 and sustain innate immune function in cirrhosis unless acute decompensation emerges. J Hepatol. 2025;83:146–60. https://doi.org/10.1016/j.jhep.2024.12.031.
Feio-Azevedo R, Boesch M, Radenkovic S, van Melkebeke L, Smets L, Wallays M, et al. Distinct immunometabolic signatures in circulating immune cells define disease outcome in acute-on-chronic liver failure. Hepatology. 2025;81:509–22.
Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J Hepatol. 2020;72:167–82.
Sheng J, Zhang J, Wang L, Tano V, Tang J, Wang X, et al. Topological analysis of hepatocellular carcinoma tumor microenvironment based on imaging mass cytometry reveals cellular neighborhood regulated reversely by macrophages with different ontogeny. Gut. 2022;71:1176–91.
Kuang D-M, Zhao Q, Peng C, Xu J, Zhang J-P, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37.
Liu CQ, Xu J, Zhou ZG, Jin LL, Yu XJ, Xiao G, et al. Expression patterns of programmed death ligand 1 correlate with different microenvironments and patient prognosis in hepatocellular carcinoma. Br J Cancer. 2018;119:80–88.
Li M, Wang L, Cong L, Wong CC, Zhang X, Chen H, et al. Spatial proteomics of immune microenvironment in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Hepatology. 2024;79:560–74.
Ruf B, Bruhns M, Babaei S, Kedei N, Ma L, Revsine M, et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell. 2023;186:3686–3705.e32.
Cappuyns S, Philips G, Vandecaveye V, Boeckx B, Schepers R, Van Brussel T, et al. PD-1- CD45RA++ effector-memory CD8 T cells and CXCL10+ macrophages are associated with response to atezolizumab plus bevacizumab in advanced hepatocellular carcinoma. Nat Commun. 2023;14:7825. https://doi.org/10.1038/s41467-023-43381-1.
Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2012;56:1342–51.
Bartneck M, Schrammen PL, Möckel D, Govaere O, Liepelt A, Krenkel O, et al. The CCR2 + macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. CMGH. 2019;7:371–90.
Sharma A, Seow JJW, Dutertre CA, Pai R, Blériot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377–394.e21.
Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, Dunsmore G, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883–1900.e5.
Lu Y, Yang A, Quan C, Pan Y, Zhang H, Li Y, et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun. 2022;13:4594. https://doi.org/10.1038/s41467-022-32283-3.
Fan G, Xie T, Li L, Tang L, Han X, Shi Y. Single-cell and spatial analyses revealed the colocation of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. 2024;8:75. https://doi.org/10.1038/s41698-024-00564-3.
Tomlinson JL, Valle JW, Ilyas SI. Immunobiology of cholangiocarcinoma. J Hepatol. 2023;79:867–75.
Greten TF, Schwabe R, Bardeesy N, Ma L, Goyal L, Kelley RK, et al. Immunology and immunotherapy of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2023;20:349–65.
Tian L, Ma J, Ma L, Zheng B, Liu L, Song D, et al. PD-1/PD-L1 expression profiles within intrahepatic cholangiocarcinoma predict clinical outcome. World J Surg Oncol. 2020;18:303 https://doi.org/10.1186/s12957-020-02082-5.
Sun D, Luo T, Dong P, Zhang N, Chen J, Zhang S, et al. CD86 + /CD206+ tumor-associated macrophages predict prognosis of patients with intrahepatic cholangiocarcinoma. PeerJ. 2020;8:e8458 https://doi.org/10.7717/peerj.8458.
Kunk PR, Dougherty SC, Lynch K, Whitehair R, Meneveau M, Obeid JM, et al. Myeloid cell infiltration correlates with prognosis in cholangiocarcinoma and varies based on tumor location. J Immunother. 2021;44:254–63.
Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101:1913–9.
Bao X, Li Q, Chen J, Chen D, Ye C, Dai X, et al. Molecular subgroups of intrahepatic cholangiocarcinoma discovered by single-cell RNA sequencing-assisted multiomics analysis. Cancer Immunol Res. 2022;10:811–28.
Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis e Sousa C. Dendritic cells revisited. Annu Rev Immunol. 2021;39:131–66.
Yáñez A, Bono C, Goodridge HS. Heterogeneity and origins of myeloid cells. Curr Opin Hematol. 2022;29:201–8.
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571–8.
Adams NM, Das A, Yun TJ, Reizis B. Ontogeny and function of plasmacytoid dendritic cells. Annu Rev Immunol. 2024;42:347–73.
Feng J, Pucella JN, Jang G, Alcántara-Hernández M, Upadhaya S, Adams NM, et al. Clonal lineage tracing reveals shared origin of conventional and plasmacytoid dendritic cells. Immunity. 2022;55:405–422.e11.
Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604.
Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356:eaah4573 https://doi.org/10.1126/science.aah4573.
See P, Dutertre CA, Chen J, Günther P, McGovern N, Irac SE, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. 2017;356:aag3009 https://doi.org/10.1126/science.aag3009.
Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity. 2019;51:573–589.e8.
Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. 2014;40:642–56.
Balan S, Saxena M, Bhardwaj N. Dendritic cell subsets and locations. Int Rev Cell Mol Biol. 2019;348:1–68.
Theisen DJ, Davidson JT, Briseño CG, Gargaro M, Lauron EJ, Wang Q, et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science. 2018;362:694–9.
Leal Rojas IM, Mok W-H, Pearson FE, Minoda Y, Kenna TJ, Barnard RT, et al. Human blood CD1c+ dendritic cells promote Th1 and Th17 effector function in memory CD4+T cells. Front Immunol. 2017;8:971.
Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. 2019;50:37–50.
Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med. 2010;207:2703–17.
Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48.
Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10:753–66.
Lukacs-Kornek V, Schuppan D. Dendritic cells in liver injury and fibrosis: shortcomings and promises. J Hepatol. 2013;59:1124–6.
Matchett KP, Wilson-Kanamori JR, Portman JR, Kapourani CA, Fercoq F, May S, et al. Multimodal decoding of human liver regeneration. Nature. 2024;630:158–65.
Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut. 2020;69:551–63.
English K, Tan SY, Kwan R, Holz LE, Sierro F, McGuffog C, et al. The liver contains distinct interconnected networks of CX3CR1+ macrophages, XCR1+ type 1 and CD301a+ type 2 conventional dendritic cells embedded within portal tracts. Immunol Cell Biol. 2022;100:394–408.
Sawada K, Chung H, Softic S, Moreno-Fernandez ME, Divanovic S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metab. 2023;35:1852–71.
Ibrahim J, Nguyen AH, Rehman A, Ochi A, Jamal M, Graffeo CS, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. 2012;143:1061–72.
Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology. 2013;58:589–602.
Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. XCR1+ type 1 conventional dendritic cells drive liver pathology in nonalcoholic steatohepatitis. Nat Med. 2021;27:1043–54.
Heier E-C, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine CD103+ dendritic cells protect against steatosis progression toward steatohepatitis. J Hepatol. 2017;66:1241–50.
Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, Fleury S, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab. 2019;1:604–14.
van der Zande HJP, Brombacher EC, Lambooij JM, Pelgrom LR, Zawistowska-Deniziak A, Patente TA et al. Dendritic cell-intrinsic LKB1-AMPK/SIK signaling controls metabolic homeostasis by limiting the hepatic Th17 response during obesity. JCI Insight. 2023;8. https://doi.org/10.1172/jci.insight.157948.
Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, et al. CCL20 is upregulated in nonalcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J Transl Med. 2018;16:108. https://doi.org/10.1186/s12967-018-1490-y.
Sutti S, Locatelli I, Bruzzì S, Jindal A, Vacchiano M, Bozzola C, et al. CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin Sci. 2015;129:797–808.
Rahman AH, Aloman C. Dendritic cells and liver fibrosis. Biochim Biophys Acta Mol Basis Dis. 2013;1832:998–1004.
Dai K. Leptin administration exacerbates thioacetamide-induced liver fibrosis in mice. World J Gastroenterol. 2005;11:4822–6.
Connolly MK, Bedrosian AS, Mallen-St. clair J, Mitchell AP, Ibrahim J, Stroud A, Pachter HL, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-α. J Clin Investig. 2009;119:3213–25.
Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73.
Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi–Nejad Z, Ginhoux F, et al. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology. 2012;55:244–55.
Xu Y, Liu F, He D, Han L, Zheng X, Hu M, et al. Monocyte-derived immature dendritic cells negatively regulate hepatic stellate cells in vitro by secreting IL-10. Immunobiology. 2023;228:152315 https://doi.org/10.1016/j.imbio.2022.152315.
Cardoso CC, Matiollo C, Pereira CHJ, Fonseca JS, Alves HEL, da Silva OM, et al. Patterns of dendritic cell and monocyte subsets are associated with disease severity and mortality in liver cirrhosis patients. Sci Rep. 2021;11:5923. https://doi.org/10.1038/s41598-021-85148-y.
Cazzetta V, Franzese S, Carenza C, Della Bella S, Mikulak J, Mavilio D. Natural killer–dendritic cell interactions in liver cancer: Implications for immunotherapy. Cancers. 2021;13:2184 https://doi.org/10.3390/cancers13092184.
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015;194:2985–91.
Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T-cell hyporesponsiveness. J Immunol. 2009;182:1901–11.
Cheng J, Deng Y, Yi H, Wang G, Fu B, Chen W, et al. Hepatic carcinoma-associated fibroblasts induce ido-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198. https://doi.org/10.1038/oncsis.2016.7.
Santos PM, Menk AV, Shi J, Tsung A, Delgoffe GM, Butterfield LH. Tumor-derived α-fetoprotein suppresses fatty acid metabolism and oxidative phosphorylation in dendritic cells. Cancer Immunol Res. 2019;7:1001–12.
Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153:1107–1119.e10.
Lim TS, Chew V, Sieow JL, Goh S, Yeong JPS, Soon AL, et al. PD-1 expression on dendritic cells suppresses CD8++T-cell function and antitumor immunity. Oncoimmunology. 2016;5:e1085146 https://doi.org/10.1080/2162402X.2015.1085146.
Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, et al. Human CD14+CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59:567–79.
Hansen FJ, David P, Weber GF. The multifaceted functionality of plasmacytoid dendritic cells in gastrointestinal cancers: a potential therapeutic target? Cancers. 2024;16:2216 https://doi.org/10.3390/cancers16122216.
Zhou ZJ, Xin HY, Li J, Hu ZQ. Luo C Bin, Zhou SL. Intratumoral plasmacytoid dendritic cells as a poor prognostic factor for hepatocellular carcinoma following curative resection. Cancer Immunol, Immunother. 2019;68:1223–33.
Pang L, Ng KTP, Liu J, Yeung WHO, Zhu J, Chiu TLS, et al. Plasmacytoid dendritic cells recruited by HIF-1α/eADO/ADORA1 signaling induce immunosuppression in hepatocellular carcinoma. Cancer Lett. 2021;522:80–92.
Hu ZQ, Zhou ZJ, Luo CB, Xin HY, Xin J, Li SY, et al. Peritumoral plasmacytoid dendritic cells predict a poor prognosis for intrahepatic cholangiocarcinoma after curative resection. Cancer Cell Int. 2020;20:582. https://doi.org/10.1186/s12935-020-01676-z.
Pedroza-Gonzalez A, Zhou G, Vargas-Mendez E, Boor PP, Mancham S, Verhoef C, et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology. 2015;4:e1008355. https://doi.org/10.1080/2162402X.2015.1008355.
Jeng LB, Liao LY, Shih FY, Teng CF. Dendritic-cell-vaccine-based immunotherapy for hepatocellular carcinoma: clinical trials and recent preclinical studies. Cancers. 2022;14:4380. https://doi.org/10.3390/cancers14184380.
Acknowledgements
Figures were created with BioRender.com.
Funding
PR is supported by a UKRI Medical Research Council Senior Clinical Fellowship (MR/W015919/1), and ET is supported by a UKRI Medical Research Council New Investigator Research Grant (MR/X009904/1).
Author information
Authors and Affiliations
Contributions
Conceptualization: ET. Supervision: PR, ET. Visualization: DP, AEM, ZL, ET. Writing—original draft preparation: DP, AEM, ZL, ET. Writing—review and editing: PR, ET.
Corresponding author
Ethics declarations
Competing interests
PR has served as a consultant for Merck and Macomics and has received research grant funding from Genentech and Intercept.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patseas, D., El-Masry, A., Liu, Z. et al. Myeloid cells in chronic liver inflammation. Cell Mol Immunol 22, 1237–1261 (2025). https://doi.org/10.1038/s41423-025-01324-4
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01324-4
