
Abstract
PANoptosis, a newly defined inflammatory programmed cell death, plays key roles in tumor development and progression. This process involves the assembly of PANoptosome complexes under various stimuli, which activate multiple cell death pathways simultaneously. By integrating key sensors and effector molecules, PANoptosis enhances immunogenic cell death while counteracts immune evasion mechanisms. This review focuses on current research of PANoptosis in cancer. Clinically, PANoptosis-related signatures show clinical value for predicting patient survival, discerning tumor immune microenvironment (TIME) characteristics and evaluating the therapeutic response. Mechanistically, complex signaling networks regulate PANoptosis, which in turn influences tumor behavior through dynamic interactions with TIME components. Therapeutically, targeting PANoptosis-related pathways, including nanomedicine approaches, demonstrate encouraging preclinical results. Particularly, combining PANoptosis modulation with radiotherapy, chemotherapy, or immunotherapy enhances anti-tumor efficacy. These findings position PANoptosis as a promising therapeutic target for reshaping TIME, overcoming treatment resistance, and improving cancer outcomes. Future research will focus on elucidating context-dependent PANoptosome regulation and translating these insights into precision oncology strategies.
Similar content being viewed by others

Introduction
Dysregulated cell death, a fundamental hallmark of cancer, is closely associated with tumorigenesis and development [1]. The death modes, such as necrosis, apoptosis, and pyroptosis, can induce tumor cell death through different molecular mechanisms and signaling pathways to respond to exogenous environments and intracellular disorders [2, 3]. Inflammatory programmed cell death (PCD) refers to a regulated form of cell death that not only eliminates damaged, infected, or unnecessary cells but also actively triggers an inflammatory immune response, mainly including necroptosis, pyroptosis, and ferroptosis (an iron-dependent form of cellular demise instigated by lipid reactive oxygen species (ROS)) [4]. Besides, emerging evidence suggests that caspase-mediated apoptosis can also be inflammatory [4, 5]. Briefly, necroptosis, an lytic cell death pathway, is immunologically active, driven by receptor-interacting protein kinase 1/3 (RIPK1/3) and the effector mixed lineage kinase domain-like pseudokinase (MLKL) [6]. Pyroptosis is marked by inflammatory reactions that usually take place following the formation and stimulation of inflammasomes and executed by members of the gasdermin family [7, 8]. Apoptosis is a caspase-mediated pathway distinguished by the generation of apoptotic bodies, initiated by both internal and external cues [9]. Interestingly, cells carry out multiple PCD programs at the same time and exist extensive crosstalk under specific conditions, which is defined as PANoptosis.
PANoptosis, conceptualized by Kanneganti’s research team in 2019, refers to the phenomenon in which cells undergo pyroptosis, apoptosis and necroptosis simultaneously [10]. It acts as a unique inflammatory PCD pathway and a defensive response to external stimuli and pathogens, contributing to the maintenance of cellular homeostasis and overall stability, which was initially observed in macrophages responding to the influenza A virus [11]. It is worth noting that PANoptosis harbors the essential characteristics of above pathways, but cannot be explained by any one of them alone [12, 13]. Emerging evidence has established PANoptosis is governed by the assembly of PANoptosome complexes under various stimuli, which contains upstream receptors and molecular signals essential for activating multiple cell death pathways simultaneously, such as Z-DNA binding protein 1 (ZBP1)-PANoptosome and absent in melanoma 2 (AIM2)-PANoptosome.
The regulation of PANoptosis has been associated with various infections and inflammatory disorders [11, 14, 15]. Recently, investigations have revealed the significant role of PANoptosis in various malignancies [13, 16, 17]. PANoptosis-based molecular clustering and prognostic signature holds great potential in predicting patient survival and discerning tumor microenvironment (TME) traits [18, 19]. Targeting PANoptosis shows novel necessity in tumor treatment through multiple cell death pathways. For instance, in colorectal cancer, the increased cell death induced by cysteine desulfurase (NFS1) or Wilms tumor 1-associating protein (WTAP) depletion under oxaliplatin treatment cannot be completely reversed by any of the inhibitors of apoptosis, pyroptosis and necroptosis alone [13, 17]. PANoptosis triggers the release of a large number of inflammatory factors (such as IL-1β and IL-18), which significantly enhance anti-tumor immunity and can help reverse the immunosuppressive microenvironment typically induced by necroptosis alone [20, 21]. Wang et al. accurately localize tumor-specific PANoptosis activation using a “logic-gated” strategy, thereby avoiding the systemic inflammatory storm risk associated with targeted necroptosis induction [22]. Moreover, PANoptosis not only allows for the elimination of cancer cells by multiple cell death pathways, but also plays an important role in regulating the enrichment of effector or regulatory immune cells, thus participating in fine-tuning anti-tumor immunity within the TME and enhancing anti-tumor therapy effectiveness [23]. In addition, active investigation of the mechanisms and potential therapeutic agents, such as PANoptosis related agonists or inhibitors, that can regulate PANoptosis in cancer cells is likely to yield effective cancer treatments and improve patient outcomes [24]. Therefore, understanding the connection between PANoptosis and cancer may provide deeper insights into the occurrence and treatment of cancer, and may be critical in exploring new therapeutic strategies by interfering with the escape mechanism of PANoptosis in cancer cells. However, studies on these mechanisms are still in their infancy and require further exploration. This review discusses the regulatory mechanisms and prognostic role of PANoptosis in cancers. We focus primarily on the interplay of PANoptosis between cancer cells and the tumor immune microenvironment (TIME), and highlight the therapeutic implications of PANoptosis-targeting strategies in cancer therapy.
From independent cell death pathways to PANoptosis as a “death continuum”
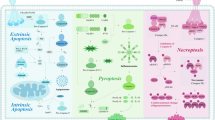
Each inflammatory PCD exhibit distinct morphological features, molecular mechanisms, and pathophysiological roles (Table 1). These processes are tightly regulated by upstream initiators and signaling cascades that assemble multimeric complexes to serve as activation platforms for downstream executioners. Given their critical role, these signaling molecules provide attractive target points for therapeutic intervention. Notably, emerging evidence highlights extensive crosstalk among these pathways under pathogenic or inflammatory stimuli, ultimately leading to PANoptosis [25]. The molecular machinery of PANoptosis revolves around the dynamic assembly of the PANoptosome, a multiprotein complex that integrates sensors, adapters, and effectors to synchronize pyroptosis, apoptosis, and necroptosis [26] (Table 2). The currently reported PANoptosomes include ZBP1-PANoptosome, AIM2-PANoptosome, NLRP3-PANoptosome, RIPK1-PANoptosome, NLRP12-PANoptosome, and NLRC5-PANoptosome (Fig. 1).

Mechanisms of PANoptosis activation. Under different extracellular or intracellular stimuli, such as pathogen infection, anti-tumor drugs treatment, cytokine storm and metabolic dysregulation, the ZBP1, AIM2, NLRP3, RIPK1, NLRP12 and NLRC5 sensor interact and recruit several adapter molecules to form PANoptosome, including ZBP1-PANoptosome (ZBP1, NLRP3, ASC, FADD, caspase-1/6/8 and RIPK1/3), AIM2-PANoptosome (AIM2, Pyrin, ZBP1, ASC, caspase-1/8, FADD and RIPK1/3), NLRP3-PANoptosome (NLRP3, ASC, RIPK3, caspase-1/8), RIPK1-PANoptosome (RIPK1/3, NLRP3, ASC, caspase-1/8), NLRP12-PANoptosome (NLRP12, NLRP3, ASC, caspase-1/8, and RIPK3), and NLRC5-PANoptosome (NLRC5, NLRP12, NLRP3, ASC, caspase-1/8 and RIPK3). These PANoptosomes further induce caspase-3/7 activation, GSDMB/C/D/E cleavage, and MLKL phosphorylation, resulting in apoptosis, pyroptosis, necroptosis
ZBP1-PANoptosome
ZBP1 contains two Zα domains that recognize Z-form nucleic acids (Z-DNA/Z-RNA) and a RHIM domain mediating interactions with RIPKs [27]. ZBP1-PANoptosome was first identified during influenza A virus (IAV) infection, which includes core sensors ZBP1 and NOD-like receptor family pyrin domain-containing 3 (NLRP 3); adapters apoptosis-associated speck-like protein containing a CARD (ASC), and Fas-associated protein with a death domain (FADD) along with effectors caspase-1/6/8 and RIPK1/3 [28]. Caspase-6 can enhance complex stability by reinforcing RIPK3-ZBP1 interactions [14, 28, 29]. The ZBP1-PANoptosome coordinates apoptosis via caspase-3/7, necroptosis via MLKL, and pyroptosis via gasdermin D (GSDMD)/GSDME to eliminate infected cells [11]. The ZBP1-induced inflammatory pathway interacts with the cyclic GMP-AMP synthase (cGAS)- stimulator of IFN genes (STING) pathway, amplifying antiviral immunity [30]. Notably, genetic ablation of ZBP1 abolishes PANoptosis, impairing viral clearance [31], while inflammatory injury in acute pancreatitis and the pathological process of spinal cord injury can be significantly improved by inhibiting ZBP1- PANoptosome-mediated PANoptosis [32, 33].
AIM2-PANoptosome
AIM2, an interferon-inducible cytosolic DNA sensor, plays a central role in antimicrobial defense. Its pyrin domain (PYD) domain binds ASC, while the HIN domain detects microbial dsDNA [14]. The AIM2-PANoptosome integrates signals from inflammasomes and recruits ZBP1, pyrin, ASC, FADD, caspase-1/8, and RIPK1/3 to drive pyroptosis, apoptosis, and necroptosis [14, 34, 35]. AIM2-knockout mice exhibit complete loss of PANoptosis during HSV-1 infection, leading to high mortality, whereas pyrin or ZBP1 deletion only partially reduces cell death, underscoring AIM2’s pivotal role [14]. AIM2-PANoptosome assembly depends on type I interferon (IFN-I) signaling, which upregulates ZBP1 and pyrin [14]. Single-cell RNA-seq data reveal that AIM2 deletion reduces ZBP1 expression in HSV-1-infected macrophages, indicating a feedback loop between AIM2 and ZBP1 [14]. Beyond infections, AIM2-PANoptosome contributes to autoimmune disorders. In systemic lupus erythematosus, AIM2 senses self-DNA released from neutrophil extracellular traps, promoting macrophage PANoptosis and type I interferon production [14]. This perpetuates autoantibody generation and organ damage.
NLRP3-PANoptosome
NLRP3, the most versatile inflammasome sensor, is selectively expressed in myeloid cells and comprises three core domains: N-terminal PYD, a central NACHT domain with ATPase activity, and C-terminal leucine-rich repeats (LRRs) [36, 37]. Upon the activation by diverse stimuli (such as ATP, crystals, K⁺/Ca2+/Na+ influx or mitochondrial ROS), oligomeric complexes termed “specks” are formed in the cytoplasm, composed predominantly of ASC [38,39,40]. These specks recruit ASC, RIPK3, and caspase-1/8 complexes to form PANoptosome [41]. Beyond contributing to specific PANoptosomes, NLRP3 is integral to ZBP1-/RIPK1-/NLRP12-/NLRC5- PANoptosomes [42]. Several NLRP3 inhibitors are now in clinical trials for neurological and cardiovascular diseases, while some reported inhibitors exhibit poly-pharmacology, potentially affecting other cellular targets [36].
RIPK1-PANoptosome
RIPK1 functions as a dual-function regulator, with its kinase activity initiating necroptosis and its scaffolding role facilitating apoptosis [43]. Under transforming growth factor beta-activated kinase 1 (TAK1) deficiency or TNF-α stimulation, RIPK1 orchestrates PANoptosome formation by recruiting NLRP3, ASC, RIPK3, and caspase-1/8 complexes [44]. TAK1 critically regulates this process, as both genetic ablation and pharmacological inhibition of TAK1 promote RIPK1-containing PANoptosome assembly [10]. However, subsequent investigations reveal that TAK1 ablation also activates RIPK1-independent PANoptosis through a distinct RIPK3-MLKL signaling axis, highlighting the existence of parallel regulatory mechanisms in inflammatory cell death programming [45]. Notably, RIPK1 deletion abolishes Yersinia-triggered pyroptosis and apoptosis but enhances ZBP1-mediated necroptosis, reflecting functional redundancy [44]. RIPK1 inhibitors are promising for treating inflammatory bowel disease, where RIPK1-PANoptosome activation drives epithelial cell death and barrier dysfunction [43, 46, 47]. In contrast, RIPK1 kinase-dead knock-in mice exhibit resistance to TNF-induced shock but increased susceptibility to bacterial infections, highlighting the delicate balance required for therapeutic intervention [48, 49].
NLRP12-PANoptosome
NLRP12, a sensor with a leucine-rich repeat (LRR) domain, detects bacterial LPS, fungal components, and heme [50, 51]. It forms the NLRP12-PANoptosome with NLRP3, ASC, caspase-1/8, and RIPK3, driving PANoptosis during infections [50, 52]. TLR2/4 and IRF1 pathways regulate this complex, influencing intestinal immunity [50, 53]. NLRP12-PANoptosome activation correlates with acute kidney injury, and its knockout alleviates pathology [50]. In sickle cell disease, heme overload activates NLRP12-PANoptosome in renal tubular cells, exacerbating vaso-occlusive crises [50].
NLRC5-PANoptosome
Recent studies have also revealed that NLRP12 can interact with NOD-like receptor family CARD domain-containing 5 (NLRC5), a critical innate immune sensor, inducing inflammatory cell death under hemolytic and inflammatory conditions [28]. The interaction between NLRC5 and NLRP12 facilitates the recruitment of NLRP3, ASC, RIPK3, caspase-1/8 to assemble the NLRC5-PANoptosome [54]. NLRC5 and NLRP12 exhibit distinct functional roles. A key distinction lies in NLRP12’s regulation of caspase-1 activation and inflammasome-dependent cytokine release, whereas NLRC5 does not influence these processes. Notably, the NLRP3-caspase-1 axis operates independently of NLRC5 [50, 54]). These findings highlight the subtle features of NLR family sensors within PANoptosome-mediated inflammatory cell death pathways.
PANoptosis involves extensive crosstalk regulated by PANoptosome complexes. For example, the activation of inflammasomes within the body, can activate the caspase protein family (such as caspase-1/4/5/11) or granzymes (such as granzyme A/B) to cleave and activate gasdermin proteins [8, 55]. As verified in apoptosis, caspase-3 can cleave the GSDME to potentially block pyroptosis via a different site from the inflammatory caspases that inactivate the protein [56, 57]. Caspase-8 is not only a key component of the exogenous apoptosis pathway, but also serves as a common molecular switch for apoptosis, necroptosis, and pyroptosis, functioning as a signaling hub for the overall mechanism [58]. Notably, RIPK1 deficiency abolishes pyroptosis and apoptosis induced by Yersinia but enhances ZBP1-mediated necroptosis [44]. Moreover, the same triggers can simultaneously activate multiple PANoptosomes, which is verified by the activated ZBP1- and AIM2-PANoptosome induced by viral infection (Table 2). In addition, activated ZBP1 promotes IFN-I secretion through the TANK binding kinase 1 (TBK1)–interferon regulatory factor 3 (IRF3) axis, thereby enhancing the formation of ZBP1- and AIM2-PANoptosomes and ultimately inducing PANoptosis [59]. Cytoplasmic mtDNA accumulation activates the AIM2 inflammasome, facilitating ZBP1-RIPK3 PANoptosome formation and inducing PANoptosis [60]. These highlight the crosstalk between the different cell death pathways. However, there is still much to learn regarding the regulators upstream of PANoptosome assembly and execution, and more PANoptosome complex may be discovered in the future.
PANoptosis in cancer prognosis and TIME interplay
Cancer progression and treatment outcomes are the dynamic result of interaction and co-evolution between malignant cells and their surrounding TME, particularly within the TIME, whose main components include tumor cells, immune populations, various cytokines and inflammatory mediators [61, 62]. Recent research demonstrates the clinical relevance of PANoptosis patterns and its association with TIME in several types of cancers.
The clinical relevance of PANoptosis-related signatures
PANoptosis exhibits key genetic and molecular features that regulate tumor progression [63]. Studies have reported that developing PANoptosis-related signatures (PANRS) through systematic analysis of PANoptosis-related genes (PRGs) or PANoptosis-related noncoding RNAs provides valuable clinical insights. These signatures enable effective prognostic stratification and precise assessment of therapeutic responses to anti-tumor therapy, such as chemotherapy or immunotherapy, highlighting the potential of PANRS to be used as an independent prognostic indicator.
Prognostic models and molecular subtyping systems based on PANRS demonstrate tumor-specific survival patterns across multiple malignancies. In prostate adenocarcinoma, comprehensive pathway analysis reveals interactions between PANoptosis-related genetic alterations (mutations, transcriptional dysregulation, methylation changes) and clinical features. Patients with elevated PANRS scores show improved survival, enhanced immunotherapy response, and elevated mutation burden [20]. Contrasting patterns emerge in hepatobiliary and gastrointestinal malignancies (biliary tract cancer, colorectal carcinoma, hepatocellular carcinoma), where high-risk PANRS groups correlate with poor prognosis, while low-risk patients demonstrate increased chemosensitivity and potential immunotherapy benefits [19, 64, 65]. Similarly, in breast cancer, He et al. download breast cancer data and GSE176078 single-cell sequencing dataset, construct prognostic models, COX and LASSO regression to establish PANoptosis-based molecular classification, and show low-risk clusters exhibit favorable prognosis and higher degree of microsatellite instability (MSI), suggesting immunotherapy responsiveness [66]. Transcriptomic analyses from 32 different cancer types obtaining from TCGA also reveal tissue-specific prognostic associations of PANoptosis effectors. Adverse outcomes correlate with ZBP1/ADAR/caspase-2,3,4,8/GSDMD overexpression in low grade gliomas and renal carcinomas, while AIM2/caspase-3,4/TNFRSF10 upregulation associates with favorable prognosis in skin cutaneous melanomas [67]. In breast cancer, PANoptosis model based on multi-center cohorts indicates that high-risk patients show higher recurrence and mortality risk and worse prognosis, which was confirmed by immunohistochemistry outcomes of 30 patients [68]. In addition, studies further reveal the predictive role of PANoptosis-related noncoding RNA signatures in cancers. Risk stratification using PANoptosis-related microRNAs (clear cell renal cell carcinoma) or long non-coding RNAs (hepatocellular/thyroid carcinomas) identifies high-risk groups with advanced tumor stages and reduced survival, yet increased responsiveness from immunotherapy and chemotherapy [69,70,71]. Similar studies have explored this approach in other tumors, including head and neck carcinoma [72, 73], ovarian cancer [74], melanoma [75] lung adenocarcinoma [76, 77]. These studies reveal PANRS’s prognostic and therapeutic implications across cancers.
PANRS not only predict survival and response to anti-tumor therapy in tumor patients, but also identify suitable populations for different therapeutic regimens. In hepatocellular carcinoma, elevated HPAN-index scores (derived from PANoptosis-associated differentially expressed genes) correlate with poorer prognosis but high response to immunotherapy, whereas low-scoring patients show enhanced sensitivity to targeted therapies like sorafenib [78]. Comprehensive analyses in prostate cancer reveal PANRS-associated risk models can predict response to 56 chemotherapeutic agents, with high-risk groups demonstrating lower IC50 values for paclitaxel, sepantronium, and tozasertib, while low-risk groups exhibiting increased sensitivity to oxaliplatin, sorafenib, and irinotecan [79]. Moreover, cutaneous melanoma patients show reversed patterns with low-risk individuals responding better to paclitaxel, dasatinib, imatinib, and lapatinib, while high-risk patients benefiting more from sorafenib [80]. Breast cancer stratification reveals high-risk patients have worse outcomes but respond to chemotherapies like BI-2536 and ispinesib, whereas low-risk patients benefit more from immunotherapy [68]. Gastric cancer analyses demonstrate low PANoptosis scores associate with favorable prognosis and heightened sensitivity to immunotherapy plus multiple chemotherapeutics (paclitaxel, rapamycin, cisplatin), contrasting with high-risk group responsiveness to EGFR/HER2/AKT inhibitors [81], which is further validated by multiple independent cohorts [82]. In addition, through consensus clustering from GSE89749 cohorts and validation with biliary tract cancer cohort in TCGA, PRGs score system classifies patients into cluster A and cluster B. Cluster A remains high level of PANoptosis, better prognosis and more sensitive to docetaxel, while cluster B remains low level of PANoptosis, shorter overall survival and more beneficial from immunotherapy [65]. Pancreatic cancer stratification via PRG expression identifies two subgroups: PANcluster A with poorer prognosis and more sensitivity to erlotinib, selumetinib and trametinib, whereas patients in PANcluster B are highly sensitive to irinotecan, oxaliplatin and sorafenib [83]. These findings collectively establish PANRS as critical biomarkers for guiding personalized therapeutic strategies, enabling optimized selection of immunotherapy, chemotherapy, and targeted agents based on individual molecular profiles.
The interplay of PANoptosis between cancer cells and TIME
TIME play a crucial role in immune surveillance and immune clearance by inducing tumor cells death, but tumor cells develop immune tolerance mechanisms to evade these surveillance systems [84]. The distinct roles of apoptosis, pyroptosis, and necroptosis in modulating TIME and influencing therapeutic outcomes in cancer have been highlighted (Table 3). PANoptosis, a specific form of inflammatory cell death, is considered as an immunogenic cell death accompanied by the release of damage-associated molecular patterns (DAMPs), such as calreticulin (CRT), high mobility group protein B1 (HMGB1), ATP and so on [23]. Thus PANoptosis not only induces cancer cell death directly, but also orchestrate immune activation by recruiting antigen-presenting cells, stimulating cytokine storm, and reprogramming immunosuppressive cellular networks within the TIME (Fig. 2). Mechanistic studies demonstrate the immune stimulatory potential of PANoptosis. Wang et al. reveal that oncolytic virus (OV)-based therapies trigger PANoptosis, which amplifies inflammatory responses through immune activation associated dendritic cell (DCs) maturation, CD8+ T cell infiltration, and M2-to-M1 macrophage repolarization, while markedly depleting regulatory T cells (Tregs) [85]. Parallel investigations in hepatocellular carcinoma models show DNASE1L3-mediated PANoptosis enhances anti-tumor immunity. High DNASE1L3 expression correlates with elevated inflammatory mediators and increased CD4+/CD8+ T cells, natural killer (NK) cells, and macrophages compared with low-expression group [86]. Furthermore, PANoptosis induction in breast cancer and glioblastoma models consistently reduces immunosuppressive tumor-associated macrophages and Tregs while expands mature DCs, memory T cells, and tumor-infiltrating CD8+ T cells [87, 88]. These findings collectively demonstrate that PANoptosis is a critical driver of anti-tumor immunity by coordinating immune activation and immunosuppressive cell depletion.

The interplay of PANoptosis between cancer cells and TIME. On the one hand, PANoptotic tumor cells release inflammatory factors or DAMPs, which enhance DCs maturation, M2-to-M1 macrophage repolarization, CD4+/CD8+ T cell infiltration and activate NK cells, while deplete Tregs to orchestrate immune activation and subsequently suppress tumor development. On the other hand, macrophages are induced to PANoptosis via TNF-α + IFN-γ—JAK/STAT1—IRF1—RIPK1/caspase-8 axis, IFNs+NEIs—ADAR1—ZBP1 axis, or baicalin—mitochondrial injury—ZBP1 axis to enhance inflammatory responses and immune activation (CBL0137 is the agonist of ZBP1), while neutrophils are induced to PANoptosis via diABZI—STING—AIM2/NLRP3 axis and the interaction of GATA2—HMGB1—TIM-3 axis with DCs to inhibit immune activation
Studies also highlight the critical roles of PANRS in delineating TME traits across various cancers. In thyroid cancer, pathway enrichment analysis demonstrates that PRGs predominantly associate with immunomodulatory processes and TNF signaling pathway, suggesting their potential involvement in macrophage-mediated immune dysregulation and lymphocyte activation [89]. Distinct immune landscape patterns emerge when analyzing PRG-based risk stratification in colorectal and hepatocellular carcinomas. High-risk scores are positively correlated with pro-tumor immune components (M0/M2 macrophages, activated mast cells, and neutrophils), while negatively associated with anti-tumor immune elements (activated DCs, M1 macrophages, resting mast cells, and memory CD4+/CD8+ T cells) [18, 19]. Notably, microenvironmental polarization shows tumor-type specificity. Aggressive glioma, clear cell renal cell carcinoma, prostate adenocarcinoma and hepatocellular carcinoma display enhanced anti-tumor immunity in high-risk groups, evidenced by elevated immune cell infiltration, upregulated checkpoint molecules, and low tumor immune dysfunction and low exclusion scores [20, 64, 71, 87]. Conversely, immunosuppressive microenvironments dominate high-risk kidney renal clear cell carcinoma and breast cancer cases, characterized by reduced cytotoxic lymphocyte infiltration (CD4+ T/NK cells), diminished checkpoint expression, and increased M2 macrophages/Treg populations [66, 90]. In addition, pan-cancer pathway analysis further establishes significant associations between PANoptosis signatures and key immune regulatory mechanisms, including inflammatory response pathways, immune cell infiltration patterns, and immune-related gene networks [82, 91]. These different immune regulatory profiles may explain the different immunotherapy response among patients with distinct PANRS.
Immune cells not only express PANoptosis-related genes but also undergo PANoptosis, significantly influencing tumor immune dynamics (Fig. 2). Experimental models reveal distinct activation mechanisms in macrophages and neutrophils. In macrophage, adenosine deaminase acting on RNA 1 (ADAR1), an RNA editor to maintain homeostasis of post-transcriptional RNA base modification, suppresses PANoptosis by interacting with the Zα2 domain of ZBP1 to limit ZBP1 and RIPK3 interactions, while combining interferons (IFNs) and nuclear export inhibitors (NEIs: KPT-330 or LMB) treatment induces PANoptosis in ZBP1-dependent manner [92]. TNF-α/IFN-γ co-stimulation also trigger caspase-8/FADD-mediated PANoptosis through JAK/STAT1/IRF1 axis activation and nitric oxide production [15]. Pharmacological modulation studies highlight therapeutic potential. Baicalin (a scutellaria baicalensis flavonoid) suppresses PANoptosis, whereas curaxin CBL0137 promotes ZBP1-PANoptosome assembly in macrophages, respectively inhibiting or inducing inflammatory responses, which may activate the TIME[93, 94]. In addition, The DNA damage adapter stimulator of interferon genes (STING) agonist diamidobenzimidazole (diABZI) induces neutrophilic inflammation and PANoptosis via Toll-like receptor 9 (TLR9) upregulation coupled with NLRP3/AIM2 inflammasome activation [95]. Tumor-associated neutrophils (TANs) display elevated PANoptosis gene expression and play pro-tumor activity with DCs via GATA-binding protein 2 (GATA2)-HMGB1-TIM-3 pathway, thereby hindering anti-tumor immunity and facilitating immune evasion of non-small cell lung cancer cells [96]. Relevant sequencing analyses also reveal cell-type specific PANoptosis patterns. Yi et al. find CD8+ T cells, B cells, and DCs show higher PANoptosis signature scores compared with other immune populations in prostate adenocarcinoma, indicating that these cells have more severe PANoptosis [20]. Similarly, Dai et al. show that high PANscore clustered relatively in B cells, and low PANscore in CD16+ and CD14+ monocytes and megakaryocyte progenitors based on single-cell sequencing data [97]. Hepatocellular carcinoma studies further demonstrate differential PRG expression across immune cells, such as upregulated CRADD in tumor-infiltrating Tregs versus elevated TNF in cytotoxic FGFBP2+ T cells [27,28,29]. These studies provided critical insights to understand the mechanism of PANoptosis in cancers and tumor microenvironment. Investigating the distinct types of immune cells and their ability to mediate anti-tumor or pro-tumor upon cellular context is crucial, particularly their interaction with PANoptosis, which inspired us the therapeutic intervention to regulate the interaction between PANoptosis and TIME.
Therapeutic implications for targeting PANoptosis in cancer
Targeting PCD regulators through genetic or pharmacological interventions has gained significant progress in oncology research. Various compounds or inhibitors targeting the sensor or execution proteins of PCD, such as caspases, NLRP3 or RIPK1/3, have been proposed to regulate cell death [98, 99]. Notably, recent advances demonstrate that multiple anticancer agents and nanoparticle-based systems can effectively trigger PANoptosis signaling pathways to offer novel therapeutic strategies for tumor treatment [88, 100]. Targeting PANoptosis not only synergistically activates multiple cell death pathways, overcome therapy resistance, but also promotes anti-tumor immune responses by recruiting antigen-presenting cells, stimulating cytokine storm, and reprogramming immunosuppressive cellular networks within the TIME [85, 101]. PANoptosis related sensors, PANoptosome components, or upstream regulatory networks present promising candidates for developing combination therapies to overcome therapeutic challenges in cancer management.
Directly modulating PANoptosome components
ZBP1-mediated PANoptosis plays an important role in the anti-tumor effects in various tumors. Small-molecule inhibitors, such as necrosulfonamide, targeting ZBP1’s RHIM domain have shown efficacy in reducing intestinal damage in murine models [102]. Depletion of ZBP1 markedly decrease PANoptotic cell death in adrenocortical carcinoma, breast cancer and head and neck squamous cell carcinoma cells, and results in a significant tumorigenesis in colorectal cancer, melanoma and hemophagocytic lymphohistiocytosis mice models [85, 92, 93, 103]. Conversely, ZBP1 agonists, such as CBL0137, are under investigation to enhance tumor cell death in ZBP1-deficient cancers [24]. AIM2 PANoptosome also acts as a tumor suppressor by eliminating transformed cells through PANoptosis in colorectal cancer [104], and the downregulation of AIM2 led to accelerated proliferation of melanoma cells [105]. However, chronic inflammation in the TME often silences AIM2 via promoter hypermethylation, enabling immune evasion. Therapeutic strategies to demethylate AIM2 or deliver AIM2-encoding vectors are being explored to restore its tumor-suppressive function [106]. In addition, the pan-caspase inhibitor emricasan can inhibit the PANoptosis induced by co-treatment of IFN-γ and TNF-α and reduce cell death in DNA mismatch repair deficiency (dMMR) tumor [107]. However, due to its complex mechanism, the role of PANoptosis mediated by caspase-5, caspase-8 and NLRP12 in cancer remains controversial. Yang et al. show that reducing caspase-5 level suppresses the growth, migration, and invasion of clear cell renal cell carcinoma cells [108]. For caspase-8, high expression of caspase-8 in the nucleus of tumor cells promotes tumor progression, while inhibits tumor growth by acting as a downstream of granzymes, specifically inducing GSDME cleavage [109]. For NLRP12, it is shown to suppress colitis-associated colorectal cancer by limiting IL-1β production, while exist pro-tumor effects via NF-κB activation in prostate cancer [110, 111]. These findings highlight the conditional therapeutic value of PANoptosome targeting, emphasizing the need for context-specific modulation strategies in cancer treatment.
Targeting the regulatory network of PANoptosis
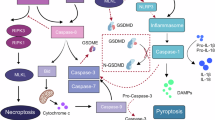
PANoptosis is regulated by multiple upstream signaling pathways under the stimulation of the intracellular and extracellular stimuli (Fig. 3). Targeting these pathways can induce PANoptosis to provide therapeutic strategies for tumor treatment. Metabolic reprogramming, a crucial hallmark of malignant cells, plays a crucial role in regulating PANoptosis [1]. Central to this regulation is ROS accumulation from oxidative stress imbalance, which amplifies PANoptosis through effector protein activation. Antioxidants like N-acetyl-l-cysteine (NAC) effectively neutralize ROS to block this process [13, 17]. Mechanistic studies identify multiple ROS regulatory nodes. Depletion of NFS1 disrupts iron-sulfur (Fe-S) cluster biogenesis, inhibition of m6A methyltransferase WTAP reduces the stability of NRF2 mRNA in an m6A-dependent manner, and loss of RIPK4 dephosphorylate methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) to inhibit NADPH production, all these result in the increase of intracellular levels of ROS, which subsequently significantly triggers PANoptosis to attenuate CRC cell growth [13, 17, 112]. Consistently, phenyllactic acid enhances prostate cancer cell survival by promoting ATP generation, enhancing NFS1 expression, reducing TNF-α levels, thereby inhibiting PANoptosis, suggesting phenyllactic acid as a novel therapeutic target [113]. Moreover, DNA damage response pathways also show significant effect on PANoptosis. In diffuse large B-cell lymphoma, sterile alpha motif and HD domain-containing protein 1 (SAMHD1) deficiency induces PANoptosis and inhibits tumor growth by inducing DNA damage and promoting the expression of STING, which promotes the formation of caspase-8/RIPK3/ASC complex [114]. Deoxyribonuclease 1 like 3 (DNASE1L3) facilitates double-strand deoxyribonucleic acid (dsDNA) breaks generation and activates AIM2 pathway during sorafenib treatment to trigger PANoptosis and decrease the hepatocellular carcinoma cell survival [86]. In addition, genetic interventions targeting PPM1B/USP10-Y-box binding protein 1 (YBX1) axis or lysophosphatidylcholine acyltransferase 1 (LPCAT1) enhance PANoptosis-related protein activation, effectively suppressing gastric and hepatocellular carcinoma progression [16, 64].

The regulatory network of PANoptosis in cancer cells. In cancer cells, PANoptosis is induced by ROS accumulation, which can be revised by NFS1—Fe-S clusters axis, WTAP—NRF2—NADPH axis, RIPK4—MTHFD1—NADPH axis, sulconazole—HK—glycolysis/mitochondrial function axis, and fusobacterium nucleatum—SLC7A5—mTORC1—mitochondrial function axis (rapamycin is the inhibitor of mTORC1, chrysoeriol and AM-A are natural drug that inhibit the mitochondrial function). Besides, the SAMHD1/DNASE1L3—DNA damage—STING axis and TNF-α + IFN-γ activate AIM2-PNAoptosome, and oHSVs—IFN—ISGs—Z-RNA axis, mebendazole—TUBA1A—cell cycle axis, and TNF-α + IFN-γ activate ZBP1-PNAoptosome to trigger PANoptosis. Moreover, the PANoptosis is inhibited by CurE—CDK1 axis, IFN-γ + NEIs—ADAR1 axis, PM2.5—piR-27222—eIF4B—WTAP axis, and PPM1B/USP10—YBX1 axis
Pharmacological agents exhibit PANoptosis-modulating capacities through distinct pathways. The antifungal agent sulconazole promotes PANoptosis and inhibits the proliferation and migration of esophageal cancer by triggering mitochondrial oxidative stress and inhibiting glycolysis via downregulation of hexokinases (HKs) [115]. Chrysoeriol, a natural flavonoid, shows significant suppressive effects on melanoma progression via inducing PANoptosis by altering mitochondrial membrane potential, increasing ROS production and up-regulating molecules linked to PANoptosis [105]. Atramacronoid A (AM-A), a unique natural sesquiterpene lactone, reduces the proliferation of breast cancer by activating caspase-3/PARP-GSDMD-MLKL cascades through mitochondrial dysfunction [116]. Moreover, the CDK1 inhibitor CurE induces ZBP1-dependent PANoptosis and suppresses the proliferation of adrenocortical carcinoma cells, and adenosine deaminase acting on RNA 1 (ADAR1) depletion exerts similar effects under the treatment of NEIs plus IFN-γ in colorectal cancer and melanoma models [92, 103]. In addition, mebendazole (MBD), an FDA-approved cost-effective anthelmintic drug, inhibits tubulin α1A (TUBA1A), blocks cell cycle and promotes ZBP-1 mediated PANoptosis, simultaneously inhibits tumorigenesis and invasion of acute myeloid leukemia cells [117]. Notably, microbial interactions further modulate PANoptosis dynamics. Fusobacterium nucleatum infection attenuates radiation-induced PANoptosis in nasopharyngeal carcinoma cells via the SLC7A5/leucine-mTORC1 axis to maintain energy metabolism and ROS reduction. mTORC1 inhibitor rapamycin significantly reverses the effects mediated by Fusobacterium nucleatum [118]. Given the emerging critical roles of microbiota in tumorigenesis and progression, the interplay between other microbial species and PANoptosis-related signaling pathways needs systematic investigation to elucidate their potential impact on tumor intervention.
Cytokine combinations, particularly TNF-α and IFN-γ, demonstrate broad PANoptosis-inducing capacity across 13 human cancer models, where MLH1 deficiency activates the AIM2-ZBP-ASC-RIPK1-RIPK3-caspase8 complex, accompanied by hyperactivation of PANoptosis-related effector proteins [107]. The intratumoral administration of TNF-α and IFN-γ exhibits pan-cancer therapeutic efficacy [119]. Notably,IRF1 orchestrates PANoptosis in both myeloid and epithelial cells to induce cell death, suppressing colorectal tumorigenesis through dual-compartment regulation [26]. The interferon response, induced by HSV-1-based OVs (oHSVs), can facilitate the accumulation of endogenous Z-RNA and subsequent activation of ZBP1-mediated PANoptosis to achieve an anti-tumor effect [85]. Nuclear export inhibitors synergize with IFN-γ to enhance melanoma cell death via ZBP1 upregulation and PANoptotic marker activation [67]. In addition, Environmental carcinogenesis studies reveal PM2.5 pollution impairs PANoptosis and promotes lung cancer through piR-27222-mediated eIF4B/WTAP/m6A axis. This effects notably reversed by piR-27222 knockdown [120]. Collectively, these findings demonstrate that targeting the regulatory network of PANoptosis represents a promising therapeutic strategy across multiple cancer types.
Targeting PANoptosis via nanomedicine
In nanomedicine research, engineered nanoparticles including extracellular vesicles, liposome carriers, dendrimer structures, and polymer matrices serve as versatile platforms for precise delivery of chemotherapeutic agents, genetic materials, and bioactive compounds [121, 122]. The integration of nanotechnology in cancer treatments has emerged as a transformative approach that effectively addresses key challenges in conventional drug delivery systems. These nanoscale innovations significantly enhance tumor-targeting precision, improve water solubility of therapeutic reagents and optimize bioavailability, while minimize systemic toxicity [123,124,125]. Notably, recent advances demonstrate their capacity to induce PANoptosis and promote the therapeutic effect of cancers.
Innovative delivery platforms induce PANoptosis for immune activation and anti-tumor therapy. Zhou et al. develop nano/genetically engineered extracellular vesicles that induce tumor highly immunogenic PANoptosis, characterized by DAMPs release (CRT exposure, HMGB1/ATP secretion) promoting DC maturation and macrophage polarization. This initiates cGAS-STING-mediated T cell responses, effectively overcoming tumor immunosuppression [126]. Similarly, mesoporous piezoelectric SrTiO3 nanoparticles (MeST NPs), combined with bioactive glasses scaffolds, generate ROS to disrupt cellular integrity, inducing PANoptosis to drive immune activation through DAMP release and enhanced T cell infiltration, which significantly suppress osteosarcoma growth [22]. Targeted co-delivery systems further demonstrate synergistic potential. pH-sensitive alginate-cholesterol-folic acid nanoparticles (FCA NPs) co-deliver a combination of metformin (MET) and doxorubicin (DOX), thus induce PANoptosis and block progression of melanoma cell through concurrent pyroptotic, apoptotic, and necroptotic pathways [100]. Moreover, sonodynamic therapy (SDT) utilizes sonosensitizers and oxygen to generate ROS and induce oxidative damage of tumor cells, but the efficiency of sonosensitizers in generating reactive ROS is often limited by rapid electron-hole recombination [127, 128]. Bi-based nanocomposites, such as BiF3@BiOI@Pt-PVP (BBP) and Bi@Bi2O3-Pt-PEG (BBOP), address ROS generation limitations to enhance SDT efficacy by prolonging electron-hole separation, amplifying mitochondrial damage and glutathione depletion to induce PANoptosis, thereby synergistically inhibit breast tumor growth and lung metastasis [129, 130]. In addition, emerging nanoplatforms combine multiple therapeutic modalities, such as covalent organic frameworks (COFs) that are porous crystals of high potential in biomedical application [131]. The COF-based PorSe-CuPt@CBL, preloaded with ZBP1 agonist CBL0137 under radiotherapy treatment, generates substantial amounts of ROS, which effectively trigger ZBP1-mediated PANoptosis and potentiate breast cancer treatment [88]. These observations indicate that targeting PANoptosis cell death via nanoengineering may offer unique opportunities to boost anti-tumor therapy.
Combination therapies involving PANoptosis modulation
Although the existing therapeutic regimens, including radiotherapy, chemotherapy, targeted agents and immunotherapy, have obtained curative effects for cancers, the associated side effects and hyposensitivity are the main factors contributing to treatment failure for refractory patients [132]. The studies on the role and mechanism of PANoptosis provide a promising strategy to enhance the efficacy of existing cancer therapies (Table 4).
Enhancing the efficacy of tumor chemoradiotherapy
Emerging evidence highlights PANoptosis activation as a strategic enhancer of chemoradiotherapy efficacy. Cytotoxic drugs are conventional first-line chemotherapy drugs for various cancers. In gastrointestinal cancers, NFS1, WTAP or YBX1 suppression significantly improves oxaliplatin efficacy by inducing PANoptosis, thereby overcoming chemoresistance in colorectal and gastric cancer models [13, 16, 17]. Similarly, FADD knockdown in head and neck squamous cell carcinoma not only inhibits tumor proliferation but also improves 5-fluorouracil responsiveness [72]. Mitotane is the only first-line treatment for adrenocortical carcinoma. The therapeutic effect of a combination of CDK1 inhibitor CurE and mitotane in adrenocortical carcinoma cells achieves superior therapeutic outcomes through PANoptosis activation compared to monotherapies [103]. Radiotherapy optimization studies reveal microenvironment-dependent mechanisms. Sulconazole is effective in inhibiting the growth of esophageal cancer cells, and the combination of low dose sulconazole and radiotherapy promotes PANoptosis through oxidative stress activation and glycolytic pathway inhibition to increase radiosensitivity [115]. In radioresistant nasopharyngeal carcinomas, microbiota modulation through mitochondrial-targeted antibiotics or leucine-restricted diets restores radiation sensitivity by reducing Fusobacterium nucleatum-mediated PANoptosis suppression [118]. In addition, nanotechnology applications show particular promise, with PorSe-CuPt@CBL nanoparticles effectively triggering PANoptosis to boost radiotherapy effectiveness in breast cancer [88]. Notably, ZBP1-mediated PANoptosis contributes to the toxic side effects in healthy tissues of colorectal cancer patients under DNA-damage therapies, suggesting ZBP1 can be a promising therapeutic target to alleviate chemotherapy-related side effects [133]. Therefore, combining PANoptosis inducers with radiotherapy or chemotherapy demonstrates synergistic anti-tumor effects, highlighting PANoptosis as a pivotal modulator of treatment response and a candidate for precision oncology.
Enhancing the efficacy of immunotherapy
Anti-tumor immunotherapies have arisen as fresh therapeutic pillars within oncology by countering tumor-induced immunosuppression, but their clinical effectiveness remains limited by acquired resistance and immune evasion mechanisms. Emerging evidence positions PANoptosis induction as a strategic enhancer of immunotherapy efficacy through TME reprogramming [86, 114]. Mechanistic studies demonstrate DNASE1L3-induced PANoptosis augment the activation of anti-tumor immunity within the TME, thereby enhancing the efficacy of sorafenib/programmed death-1 (PD-1) mAb combination therapy in hepatocellular carcinoma [86]. Similarly, STING pathway activation via genetic overexpression or agonist DMXAA promote PANoptosis to enhance programmed death-ligand 1 (PD-L1) blockade efficacy and suppress diffuse large B-cell lymphoma tumor growth [114]. Bioactive compounds like cinobufagin synergize with PD-1 inhibitors in glioblastoma models, achieving survival prolongation via PANoptosis-driven immune activation [87]. In addition, oncolytic viruses (OVs) are increasingly recognized as promising tools for cancer therapy, as they selectively infect and destroy tumor cells while leave healthy cells unharmed [134]. However, the potential of oncolytic virus (OV) for cancer therapy is limited by the efficiency of induced immune response [135, 136]. It shows particular promise when combining with PANoptosis induction. Herpes simplex virus-1 (HSV-1)-based OVs (oHSVs) or SMAC/DIABLO gene-inserted vesicular stomatitis virus (VSV)-based OVSs (VSVs) potentiate anti-tumor immunity by inducing PANoptosis, promoting T cell tumor infiltration and enhancing their cytotoxic capacity. oHSVs or VSV-S in combination with PD-1 blockade, produces a more potent anti-tumor effect than either therapy alone [137]. Innovative combinations using Fusobacterium nucleatum outer membrane vesicles (Fn-OMV) with oHSVs convert immunologically cold tumors to hot phenotypes, amplifying PANoptosis-mediated inflammation to improve PD-L1/CTLA-4 therapy [85]. In addition, nanotechnological advances further expand therapeutic possibilities, with nanocomposites (BBP/BBOP) demonstrating PANoptosis-triggering capacity and immunotherapy enhancement in breast cancer models [129, 130]. These strategies underscore PANoptosis as a key modulator of immune evasion, enabling precision oncology approaches. However, optimizing combinatorial regimens and addressing tumor heterogeneity remain critical for clinical translation.
Conclusive remarks and future perspectives
The evolving landscape of PANoptosis has established its central role in shaping tumorigenesis, immune evasion, and therapeutic response. By orchestrating apoptosis, pyroptosis and necroptosis through PANoptosome-mediated signaling, this multifaceted cell death pathway not only amplifies immunogenicity but also interacts with the TME to influence cancer progression. In clinical and preclinical models, integrating multi-omics data and machine learning, derived from the expression pattern of PANoptosis-related signatures reveal the great potential in predicting patient prognosis, discerning tumor microenvironment traits, and predicting response to existing anti-tumor therapy. However, due to the complex mechanism and the context heterogeneity of PANoptosis activation in cancers, its molecular drivers and downstream effects exhibit tumor-type specificity. For instance, the high-score group of PANoptosis signature correlate with better survival outcomes and better immunotherapeutic responses in prostate cancer, while exhibit poor prognosis, lower response to chemotherapeutic agents and immunotherapy in biliary tract cancer, colorectal carcinoma and hepatocellular carcinoma [19, 20, 64, 65]. This heterogeneity may arise from tissue-specific signaling contexts or differences in microbiota composition. This underscores the need for systematic single-cell and spatial multi-omics studies to dissect PANoptosis signatures across tumor subtypes and microenvironmental niches. In addition, patients with distinct PANRS profiles exhibit differential therapeutic sensitivities to various treatments, including chemotherapy, immunotherapy, or specific chemotherapeutic agents. The development of specific biomarkers is required to guide personalized therapeutic strategies for different tumor patients. However, most PANRS models are rarely validated by human cohort data, their predictive power must be strictly tested in large, multi-cohort studies, and need further validation by preclinical and clinical studies.
Studies have provided critical insights to understand the mechanism of PANoptosis in bridging direct cancer cell elimination with TIME, in which PANoptotic tumor cells can enhance anti-tumor immunity by promoting DC maturation, T-cell infiltration, or M1 macrophage polarization [86,87,88]. However, there exhibits contrasting effects of PANoptosis across different tumor types, which may be related to the activation differences of the TIME, tumor-tissue heterogeneity or complex molecular mechanisms. PANoptosis signatures correlate with immune activation in glioma or hepatocellular carcinoma but associate with immunosuppression in breast and kidney cancer [64, 66, 71, 90]. Moreover, immune cells themselves exhibit PANoptosis plasticity. For instance, tumor-associated neutrophils upregulate PANoptosis to promote immune evasion in non-small cell lung cancer, while ZBP1-dependent PANoptosis in macrophages amplifies anti-tumor responses [93, 96]. Additionally, RIPK1/RIPK3 mediated necroptosis upregulates the expression of CXCL1, increasing infiltration of immunosuppressive macrophages, resulting in the pro-tumoral role [138], while ZBP1-MLKL necroptotic cascade induces cytoplasmic DNA accumulation, activates cGAS—STING signaling and increases infiltration of IFN-γ+ TNF-α+ CD8+ T cells, mediating the antitumor immunity [139]. These findings underscore the dual role of PANoptosis in immune TME regulation and even tumor treatment, highlighting the necessity of context-specific therapeutic strategies. Moreover, the dynamic interplay between PANoptosis and other immune cells or tumor stromal cells in TME, such as cancer-associated fibroblasts, myeloid progenitors, needs detailed characterization. Moreover, continuous studies tracking PANoptosis-induced DAMP release and its systemic immune consequences will be essential to avoid immune-related adverse events. Overall, further studies of the relationship between TME and PANoptosis could help to gain a deeper understanding of the tumor immune escape mechanism and provide new ideas for the development of tumor immunotherapy strategies.
PANoptosis emerges as a therapeutic vulnerability in cancers and targeting PANoptosis-related pathways offers unique opportunities to boost anti-tumor efficacy. Many activators or inhibitors associated with PANoptosis have been discovered and verified to enhance tumor cell death, such as ZBP1 agonists CBL0137 [24]. However, the role of PANoptosis has mainly been assessed in vitro and in murine models. Due to the limitations in uniformly controlling the TME, the significant heterogeneity in the activation degree of immune cells among different tumor types and variability in patient responses with monotherapy, their safety profiles and more optimized combinatorial regimens remain uncertain, which may inadvertently trigger compensatory cell death to raise toxicity. Further investigation and clinical trials are essential to validate the safety and efficacy of PANoptosis targeted treatments. Clinical trials must adopt adaptive designs to accommodate the complexity of PANoptosis biology, including patient stratification based on genetic, epigenetic, and immunophenotypic markers. In addition, other forms of cell death include cuproptosis, disulfidptosis and alkaliptosis have also been documented as emerging PCD pathways [140,141,142]. The interplay and regulatory mechanism between PANoptosis and other emerging PCD pathways remain poorly understood. With the discovery of novel PANoptosome and mechanisms, the interaction between PANoptosis and ferroptosis or other PCD will be further analyzed. Unraveling these crosstalk mechanisms will be critical to avoiding unexpected consequences of PANoptosis modulation. Equally pressing is the development of delivery systems to selectively target PANoptosis components in the TME. Nanotechnology-based platforms, such as nanoparticles co-loaded with PANoptosis inducers and immune stimulants, hold translational potential but require optimization for scalability and applicability.
In conclusion, PANoptosis represents a paradigm-shifting concept. The therapeutic implications of PANoptosis in cancer are still emerging, and there is potential for the development of new treatments that target this pathway and improve conventional therapies. Integrating PANoptosis-targeted therapy with existing modalities is likely to pave the way for personalized and precise approaches to cancer treatment in the future.
References
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7:286.
Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187:235–56.
Christgen S, Tweedell RE, Kanneganti T-D. Programming inflammatory cell death for therapy. Pharmacol Ther. 2022;232:108010.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022;7:196.
Tong X, Tang R, Xiao M, Xu J, Wang W, Zhang B, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15:174.
Wei X, Xie F, Zhou X, Wu Y, Yan H, Liu T, et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol. 2022;19:971–92.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417.
Malireddi RKS, Kesavardhana S, Kanneganti T-D. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406.
Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045.
Pandian N, Kanneganti T-D. PANoptosis: a unique innate immune inflammatory cell death modality. J Immunol. 2022;209:1625–33.
Lin J-F, Hu P-S, Wang Y-Y, Tan Y-T, Yu K, Liao K, et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct Target Ther. 2022;7:54.
Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti T-D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597:415–9.
Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149–68.
Lin C, Lin P, Yao H, Liu S, Lin X, He R, et al. Modulation of YBX1-mediated PANoptosis inhibition by PPM1B and USP10 confers chemoresistance to oxaliplatin in gastric cancer. Cancer Lett. 2024;587:216712.
Tan Y-T, Li T, Wang R-B, Liu Z-K, Ma M-Y, Huang R-Z, et al. WTAP weakens oxaliplatin chemosensitivity of colorectal cancer by preventing PANoptosis. Cancer Lett. 2024;604:217254.
Zhang Z, Zhang F, Pang P, Li Y, Chen X, Sun S, et al. Identification of PANoptosis-relevant subgroups to evaluate the prognosis and immune landscape of patients with liver hepatocellular carcinoma. Front Cell Dev Biol. 2023;11:1210456.
Wang X, Sun R, Chan S, Meng L, Xu Y, Zuo X, et al. PANoptosis-based molecular clustering and prognostic signature predicts patient survival and immune landscape in colon cancer. Front Genet. 2022;13:955355.
Yi X, Li J, Zheng X, Xu H, Liao D, Zhang T, et al. Construction of PANoptosis signature: novel target discovery for prostate cancer immunotherapy. Mol Ther Nucleic Acids. 2023;33:376–90.
Hänggi K, Li J, Gangadharan A, Liu X, Celias DP, Osunmakinde O, et al. Interleukin-1α release during necrotic-like cell death generates myeloid-driven immunosuppression that restricts anti-tumor immunity. Cancer Cell. 2024;42:2015–31.
Wang X, Guo X, Ren H, Song X, Chen L, Yu L, et al. An “Outer Piezoelectric and Inner Epigenetic” logic-gated PANoptosis for osteosarcoma sono-immunotherapy and bone regeneration. Adv Mater. 2025;37:e2415814.
Oh S, Lee J, Oh J, Yu G, Ryu H, Kim D, et al. Integrated NLRP3, AIM2, NLRC4, Pyrin inflammasome activation and assembly drive PANoptosis. Cell Mol Immunol. 2023;20:1513–26.
Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature. 2022;606:594–602.
Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:237.
Karki R, Sharma BR, Lee E, Banoth B, Malireddi RKS, Samir P, et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight. 2020;5:e136720.
Kuriakose T, Kanneganti T-D. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 2018;39:123–34.
Liu K, Wang M, Li D, Duc Duong NT, Liu Y, Ma J, et al. PANoptosis in autoimmune diseases interplay between apoptosis, necrosis, and pyroptosis. Front Immunol. 2024;15:1502855.
Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J Biol Chem. 2020;295:8325–30.
Zhan J, Wang J, Liang Y, Wang L, Huang L, Liu S, et al. Apoptosis dysfunction: unravelling the interplay between ZBP1 activation and viral invasion in innate immune responses. Cell Commun Signal. 2024;22:149.
Karki R, Lee S, Mall R, Pandian N, Wang Y, Sharma BR, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. 2022;7:eabo6294.
Lou J, Mao Y, Jiang W, Shen H, Fan Y, Yu Q, et al. TRIM56 modulates YBX1 degradation to ameliorate ZBP1-mediated neuronal PANoptosis in spinal cord injury. Adv Sci. 2024;11:e2407132.
Li J, Jia Y-C, Lu J, Zhang H, Wang Z, Xie X, et al. Inhibition of Zbp1-PANoptosome-mediated PANoptosis effectively attenuates acute pancreatitis. Cell Death Discov. 2025;11:180.
Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–72.
Ma C, Li S, Hu Y, Ma Y, Wu Y, Wu C, et al. AIM2 controls microglial inflammation to prevent experimental autoimmune encephalomyelitis. J Exp Med. 2021;218:e20201796.
Coll RC, Schroder K. Inflammasome components as new therapeutic targets in inflammatory disease. Nat Rev Immunol. 2025;25:22–41.
Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. 2009;21:194–8.
Liu Y, Zhai H, Alemayehu H, Boulanger J, Hopkins LJ, Borgeaud AC, et al. Cryo-electron tomography of NLRP3-activated ASC complexes reveals organelle co-localization. Nat Commun. 2023;14:7246.
Paik S, Kim JK, Shin HJ, Park E-J, Kim IS, Jo E-K. Updated insights into the molecular networks for NLRP3 inflammasome activation. Cell Mol Immunol. 2025;22:563–96.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91.
Sharma BR, Choudhury SM, Abdelaal HM, Wang Y, Kanneganti T-D. Innate immune sensor NLRP3 drives PANoptosome formation and PANoptosis. J Immunol. 2025;214:1236–46.
Man SM, Kanneganti T-D. Innate immune sensing of cell death in disease and therapeutics. Nat Cell Biol. 2024;26:1420–33.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21.
Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti T-D. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons. 2020;4:789–96.
Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217:jem.20191644.
Zu R, Yu Z, Zhao J, Lu X, Liang W, Sun L, et al. Quantitative analysis of phosphoproteome in necroptosis reveals a role of TRIM28 phosphorylation in promoting necroptosis-induced cytokine production. Cell Death Dis. 2021;12:994.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193:1539–43.
Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. 2020;19:553–71.
Sundaram B, Pandian N, Mall R, Wang Y, Sarkar R, Kim HJ, et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell. 2023;186:2783–2801.
Zhang Y, Okamoto CT. Nucleotide binding domain and leucine-rich repeat pyrin domain-containing protein 12: characterization of its binding to hematopoietic cell kinase. Int J Biol Sci. 2020;16:1507–25.
Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107.
Chen L, Wilson JE, Koenigsknecht MJ, Chou W-C, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. 2017;18:541–51.
Sundaram B, Pandian N, Kim HJ, Abdelaal HM, Mall R, Indari O, et al. NLRC5 senses NAD+ depletion, forming a PANoptosome and driving PANoptosis and inflammation. Cell. 2024;187:4061–77.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Yu X, He S. GSDME as an executioner of chemotherapy-induced cell death. Sci China Life Sci. 2017;60:1291–4.
Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683–7.
Song Q, Fan Y, Zhang H, Wang N. Z-DNA binding protein 1 orchestrates innate immunity and inflammatory cell death. Cytokine Growth Factor Rev. 2024;77:15–29.
Guo J, Meng S, Zhang J, Wang N, Guo F. Zn2+ regulates mitochondrial DNA efflux to inhibit AIM2-mediated ZBP1-PANoptosome pathway and alleviate septic myocardial injury. Chem Biol Interact. 2025;417:111525.
Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51:27–41.
de Visser KE, Joyce JA. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374–403.
Nguyen LN, Kanneganti T-D. PANoptosis in viral infection: the missing puzzle piece in the cell death field. J Mol Biol. 2022;434:167249.
Zhu J, Huang Q, Peng X, Luo C, Liu Z, Liu D, et al. Identification of molecular subtypes based on PANoptosis-related genes and construction of a signature for predicting the prognosis and response to immunotherapy response in hepatocellular carcinoma. Front Immunol. 2023;14:1218661.
Liu D, Chen W, Han Z, Wang Y, Liu W, Ling A, et al. Identification of PANoptosis-relevant subgroups and predicting signature to evaluate the prognosis and immune landscape of patients with biliary tract cancer. Hepatol Int. 2024;18:1792–803.
He P, Ma Y, Wu Y, Zhou Q, Du H. Exploring PANoptosis in breast cancer based on scRNA-seq and bulk-seq. Front Endocrinol. 2023;14:1164930.
Mall R, Bynigeri RR, Karki R, Malireddi RKS, Sharma BR, Kanneganti T-D. Pancancer transcriptomic profiling identifies key PANoptosis markers as therapeutic targets for oncology. NAR Cancer. 2022;4:zcac033.
Wang S, Li Z, Hou J, Li X, Ni Q, Wang T. Integrating PANoptosis insights to enhance breast cancer prognosis and therapeutic decision-making. Front Immunol. 2024;15:1359204.
Li R, Zhao M, Sun M, Miao C, Lu J. Construction and validation of a PANoptosis-related lncRNA signature for predicting prognosis and targeted drug response in thyroid cancer. PeerJ. 2023;11:e15884.
Wang L, Wan P, Xu Z. A novel PANoptosis-related long non-coding RNA index to predict prognosis, immune microenvironment and personalised treatment in hepatocellular carcinoma. Aging. 2024;16:2410–37.
Wang Y, Zhou J, Zhang N, Zhu Y, Zhong Y, Wang Z, et al. A novel defined PANoptosis-related miRNA signature for predicting the prognosis and immune characteristics in clear cell renal cell carcinoma: a miRNA signature for the prognosis of ccRCC. Int J Mol Sci. 2023;24:9392.
Yang P, Huang G, Li Y, Yu L, Yin Z, Li Q. Identification of PANoptosis-related biomarkers and analysis of prognostic values in head and neck squamous cell carcinoma. Sci Rep. 2024;14:9824.
Wang L, Lin B, Wang F, Dai Z, Xie G, Zhang J. Exploring PANoptosis in head and neck cancer: a novel approach to cancer therapy. Ecotoxicol Environ Saf. 2025;289:117678.
Chen Y, Deng Z, Chen J, Lin J, Zou J, Li S, et al. PANoptosis-relevant subgroups predicts prognosis and characterizes the tumour microenvironment in ovarian cancer. J Inflamm Res. 2024;17:9773–93.
Xie J, Zhang P, Xu X, Zhou X, Zhao S, Zhang M, et al. PANoptosis-related signature in melanoma: transcriptomic mapping and clinical prognostication. Environ Toxicol. 2024;39:2545–59.
Han C, Danzeng Q, Li L, Bai S, Zheng C. Machine learning reveals PANoptosis as a potential reporter and prognostic revealer of tumour microenvironment in lung adenocarcinoma. J Gene Med. 2024;26:e3599.
Zhang C, Xia J, Liu X, Li Z, Gao T, Zhou T, et al. Identifying prognostic genes related PANoptosis in lung adenocarcinoma and developing prediction model based on bioinformatics analysis. Sci Rep. 2023;13:17956.
Song F, Wang C-G, Mao J-Z, Wang T-L, Liang X-L, Hu C-W, et al. PANoptosis-based molecular subtyping and HPAN-index predicts therapeutic response and survival in hepatocellular carcinoma. Front Immunol. 2023;14:1197152.
Ouyang G, Li Q, Wei Y, Dai W, Deng H, Liu Y, et al. Identification of PANoptosis-related subtypes, construction of a prognosis signature, and tumor microenvironment landscape of hepatocellular carcinoma using bioinformatic analysis and experimental verification. Front Immunol. 2024;15:1323199.
Wang W, Zhou Q, Lan L, Xu X. PANoptosis-related prognostic signature predicts overall survival of cutaneous melanoma and provides insights into immune infiltration landscape. Sci Rep. 2023;13:8449.
Liu Z, Sun L, Peng X, Zhu J, Wu C, Zhu W, et al. PANoptosis subtypes predict prognosis and immune efficacy in gastric cancer. Apoptosis. 2024;29:799–815.
Pan H, Pan J, Li P, Gao J. Characterization of PANoptosis patterns predicts survival and immunotherapy response in gastric cancer. Clin Immunol. 2022;238:109019.
Zhang B, Huang B, Zhang X, Li S, Zhu J, Chen X, et al. PANoptosis-related molecular subtype and prognostic model associated with the immune microenvironment and individualized therapy in pancreatic cancer. Front Oncol. 2023;13:1217654.
Zou W. Immune regulation in the tumor microenvironment and its relevance in cancer therapy. Cell Mol Immunol. 2022;19:1–2.
Wang S, Song A, Xie J, Wang Y-Y, Wang W-D, Zhang M-J, et al. Fn-OMV potentiates ZBP1-mediated PANoptosis triggered by oncolytic HSV-1 to fuel antitumor immunity. Nat Commun. 2024;15:3669.
Wang J, Chen Y, Xu Y, Zhang J, Yang S, Zhou Y, et al. DNASE1L3-mediated PANoptosis enhances the efficacy of combination therapy for advanced hepatocellular carcinoma. Theranostics. 2024;14:6798–817.
Cai Y, Xiao H, Xue S, Li P, Zhan Z, Lin J, et al. Integrative analysis of immunogenic PANoptosis and experimental validation of cinobufagin-induced activation to enhance glioma immunotherapy. J Exp Clin Cancer Res. 2025;44:35.
Zhang L, Wang S, Yang Q-C, Song A, Wang Y-Y, Wang W-D, et al. Radioactive diselenide bonded covalent organic framework. Adv Mater. 2025. https://doi.org/10.1002/adma.202413002.
Li Y, Wu D. Identification of signature genes and immune infiltration analysis in thyroid cancer based on PANoptosis related genes. Front Endocrinol. 2024;15:1397794.
Jiang Z, Wang J, Dao C, Zhu M, Li Y, Liu F, et al. Utilizing a novel model of PANoptosis-related genes for enhanced prognosis and immune status prediction in kidney renal clear cell carcinoma. Apoptosis. 2024;29:681–92.
Zhuang L, Sun Q, Huang S, Hu L, Chen Q. A comprehensive analysis of PANoptosome to prognosis and immunotherapy response in pan-cancer. Sci Rep. 2023;13:3877.
Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021;37:109858.
You Y-P, Yan L, Ke H-Y, Li Y-P, Shi Z-J, Zhou Z-Y, et al. Baicalin inhibits PANoptosis by blocking mitochondrial Z-DNA formation and ZBP1-PANoptosome assembly in macrophages. Acta Pharm Sin. 2025;46:430–47.
Li Y-P, Zhou Z-Y, Yan L, You Y-P, Ke H-Y, Yuan T, et al. Inflammatory cell death PANoptosis is induced by the anti-cancer curaxin CBL0137 via eliciting the assembly of ZBP1-associated PANoptosome. Inflamm Res. 2024;73:597–617.
Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 2022;13:269.
Hu Q, Wang R, Zhang J, Xue Q, Ding B. Tumor-associated neutrophils upregulate PANoptosis to foster an immunosuppressive microenvironment of non-small cell lung cancer. Cancer Immunol Immunother. 2023;72:4293–308.
Dai W, Zheng P, Wu J, Chen S, Deng M, Tong X, et al. Integrated analysis of single-cell RNA-seq and chipset data unravels PANoptosis-related genes in sepsis. Front Immunol. 2023;14:1247131.
Wang L, Zhu Y, Zhang L, Guo L, Wang X, Pan Z, et al. Mechanisms of PANoptosis and relevant small-molecule compounds for fighting diseases. Cell Death Dis. 2023;14:851.
Gao J, Xiong A, Liu J, Li X, Wang J, Zhang L, et al. PANoptosis: bridging apoptosis, pyroptosis, and necroptosis in cancer progression and treatment. Cancer Gene Ther. 2024;31:970–83.
Song M, Xia W, Tao Z, Zhu B, Zhang W, Liu C, et al. Self-assembled polymeric nanocarrier-mediated co-delivery of metformin and doxorubicin for melanoma therapy. Drug Deliv. 2021;28:594–606.
Gao L, Shay C, Teng Y. Cell death shapes cancer immunity: spotlighting PANoptosis. J Exp Clin Cancer Res. 2024;43:168.
Yang W, Tao K, Wang Y, Huang Y, Duan C, Wang T, et al. Necrosulfonamide ameliorates intestinal inflammation via inhibiting GSDMD-medicated pyroptosis and MLKL-mediated necroptosis. Biochem Pharm. 2022;206:115338.
Ren L, Yang Y, Li W, Zheng X, Liu J, Li S, et al. CDK1 serves as a therapeutic target of adrenocortical carcinoma via regulating epithelial-mesenchymal transition, G2/M phase transition, and PANoptosis. J Transl Med. 2022;20:444.
Zhang Z, Li X, Zhang Y, Zhu H, Qiao Z, Lu Y, et al. Absent in melanoma 2 attenuates proliferation and migration and promotes apoptosis of human colorectal cancer cells by activating P38MAPK signaling pathway. Oncol Res. 2023;32:353–60.
Liu Y, Wang L, Huang T, Li Y, Zhang H. Integrative gut microbiota and metabolomic analyses reveal the PANoptosis- and ferroptosis-related mechanisms of chrysoeriol in inhibiting melanoma. J Agric Food Chem. 2024;72:25173–85.
Woerner SM, Kloor M, Schwitalle Y, Youmans H, Doeberitz MvK, Gebert J, et al. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer. 2007;46:1080–9.
Li H, Ni H, Li Y, Zhou A, Qin X, Li Y, et al. Tumors cells with mismatch repair deficiency induce hyperactivation of pyroptosis resistant to cell membrane damage but are more sensitive to co-treatment of IFN-γ and TNF-α to PANoptosis. Cell Death Discov. 2024;10:227.
Yang K, Wang Y, Jian Y, Wang B, Du H, Xia Y, et al. CASP5 associated with PANoptosis promotes tumorigenesis and progression of clear cell renal cell carcinoma. Cancer Cell Int. 2025;25:8.
Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: a key protein of cross-talk signal way in “PANoptosis” in cancer. Int J Cancer. 2021;149:1408–20.
Karan D, Tawfik O, Dubey S. Expression analysis of inflammasome sensors and implication of NLRP12 inflammasome in prostate cancer. Sci Rep. 2017;7:4378.
Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. 2012;36:742–54.
Yu L, Zhou S, Xu Y-B, Zhang Z-J, Cheng X-M, Zhou C, et al. RIPK4 driven by TP53 mutations promotes resistance to redox stress of CRC by phosphorylating MTHFD1. bioRxiv 2024. https://doi.org/10.1101/2024.08.06.606759.
Hao Y, Xie F, He J, Gu C, Zhao Y, Luo W, et al. PLA inhibits TNF-α-induced PANoptosis of prostate cancer cells through metabolic reprogramming. Int J Biochem Cell Biol. 2024;169:106554.
Cai Y, Chen X, Lu T, Fang X, Ding M, Yu Z, et al. Activation of STING by SAMHD1 deficiency promotes PANoptosis and enhances efficacy of PD-L1 blockade in diffuse large B-cell lymphoma. Int J Biol Sci. 2023;19:4627–43.
Liu L-X, Heng J-H, Deng D-X, Zhao H, Zheng Z-Y, Liao L-D, et al. Sulconazole induces PANoptosis by triggering oxidative stress and inhibiting glycolysis to increase radiosensitivity in esophageal cancer. Mol Cell Proteom. 2023;22:100551.
Li J-R, Li L-Y, Zhang H-X, Zhong M-Q, Zou Z-M. Atramacronoid A induces the PANoptosis-like cell death of human breast cancer cells through the CASP-3/PARP-GSDMD-MLKL pathways. J Asian Nat Prod Res. 2024;26:1475–88.
Yang W, Xu Y, Liu S, Gao L, Li S, Xie X, et al. Mebendazole induces ZBP-1 mediated PANoptosis of acute myeloid leukemia cells by targeting TUBA1A and exerts antileukemia effect. J Adv Res. 2025. https://doi.org/10.1016/j.jare.2025.02.013.
Guo S, Xing S, Wu Z, Chen F, Pan X, Li Q, et al. Leucine restriction ameliorates Fusobacterium nucleatum-driven malignant progression and radioresistance in nasopharyngeal carcinoma. Cell Rep. Med. 2024;5:101753.
Malireddi RKS, Karki R, Sundaram B, Kancharana B, Lee S, Samir P, et al. Inflammatory cell death, PANoptosis, mediated by cytokines in diverse cancer lineages inhibits tumor growth. Immunohorizons. 2021;5:568–80.
Ma W, Xu L, Wang Y, Chen S, Li D, Huo X, et al. piR-27222 mediates PM2.5-induced lung cancer by resisting cell PANoptosis through the WTAP/m6A axis. Environ Int. 2024;190:108928.
Xia W, Tao Z, Zhu B, Zhang W, Liu C, Chen S, et al. Targeted delivery of drugs and genes using polymer nanocarriers for cancer therapy. Int J Mol Sci. 2021;22:9118.
Ding N, Xiao H, Zhen L, Li H, Zhang Z, Ge J, et al. Systemic cytokines inhibition with Imp7 siRNA nanoparticle ameliorates gut injury in a mouse model of ventilator-induced lung injury. Biomed Pharmacother. 2023;165:115237.
Rogalla S, Flisikowski K, Gorpas D, Mayer AT, Flisikowska T, Mandella MJ, et al. Biodegradable fluorescent nanoparticles for endoscopic detection of colorectal carcinogenesis. Adv Funct Mater. 2019;29:1904992.
Hasani-Sadrabadi MM, Majedi FS, Miller ML, Thauland TJ, Bouchard L-S, Li S, et al. Augmenting T-cell responses to tumors by in situ nanomanufacturing. Mater Horiz. 2020;7:3028–33.
Ling X, Jiang X, Li Y, Han W, Rodriguez M, Xu Z, et al. Sequential treatment of bioresponsive nanoparticles elicits antiangiogenesis and apoptosis and synergizes with a CD40 agonist for antitumor immunity. ACS Nano. 2021;15:765–80.
Zhou L, Lyu J, Liu F, Su Y, Feng L, Zhang X. Immunogenic PANoptosis-initiated cancer sono-immune reediting nanotherapy by iteratively boosting cancer immunity cycle. Adv Mater. 2024;36:e2305361.
Chen P, Zhang P, Shah NH, Cui Y, Wang Y. A comprehensive review of inorganic sonosensitizers for sonodynamic therapy. Int J Mol Sci. 2023;24:12001.
Song K, Du J, Wang X, Zheng L, Ouyang R, Li Y, et al. Biodegradable bismuth-based nano-heterojunction for enhanced sonodynamic oncotherapy through charge separation engineering. Adv Health Mater. 2022;11:e2102503.
He Z, Wang Q, Du J, Wu S, Miao Q, Li Y, et al. Overcoming tumor hypoxic bismuth-based ternary heterojunctions enable defect modulation-augmented tumor sonocatalytic immunotherapy. Biomaterials. 2025;315:122962.
Wu S, Wang Q, Du J, Zhu L, Yang F, Lu J, et al. Bi-Pt heterojunction cascade reaction platform for sono-immunotherapy of tumors via PANoptosis and ferroptosis. Adv Health Mater. 2024;13:e2401697.
Fang Q, Wang J, Gu S, Kaspar RB, Zhuang Z, Zheng J, et al. 3D porous crystalline polyimide covalent organic frameworks for drug delivery. J Am Chem Soc. 2015;137:8352–5.
Lin J-F, Liu Z-X, Chen D-L, Huang R-Z, Cao F, Yu K, et al. Nucleus-translocated GCLM promotes chemoresistance in colorectal cancer through a moonlighting function. Nat Commun. 2025;16:263.
Wang F, Li K, Wang W, Hui J, He J, Cai J, et al. Sensing of endogenous retroviruses-derived RNA by ZBP1 triggers PANoptosis in DNA damage and contributes to toxic side effects of chemotherapy. Cell Death Dis. 2024;15:779.
Shalhout SZ, Miller DM, Emerick KS, Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023;20:160–77.
Su W, Qiu W, Li S-J, Wang S, Xie J, Yang Q-C, et al. A dual-responsive STAT3 inhibitor nanoprodrug combined with oncolytic virus elicits synergistic antitumor immune responses by igniting pyroptosis. Adv Mater. 2023;35:e2209379.
Tian Y, Xie D, Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct Target Ther. 2022;7:117.
Chen F, Lang L, Yang J, Yang F, Tang S, Fu Z, et al. SMAC-armed oncolytic virotherapy enhances the anticancer activity of PD1 blockade by modulating PANoptosis. Biomark Res. 2025;13:8.
Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–9.
Yang Y, Wu M, Cao D, Yang C, Jin J, Wu L, et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci Adv. 2021;7:eabf6290.
Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D, et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology. 2018;154:1480–93.
Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25:404–14.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Ai Y, Meng Y, Yan B, Zhou Q, Wang X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol Cell. 2024;84:170–9.
Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25:379–95.
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20.
Galluzzi L, Kepp O, Chan FK-M, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–30.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Zhang Y-Y, Han Y, Li W-N, Xu R-H, Ju H-Q. Tumor iron homeostasis and immune regulation. Trends Pharm Sci. 2024;45:145–56.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Ocansey DKW, Qian F, Cai P, Ocansey S, Amoah S, Qian Y, et al. Current evidence and therapeutic implication of PANoptosis in cancer. Theranostics. 2024;14:640–61.
Gong T, Wang Q-D, Loughran PA, Li Y-H, Scott MJ, Billiar TR, et al. Mechanism of lactic acidemia-promoted pulmonary endothelial cells death in sepsis: role for CIRP-ZBP1-PANoptosis pathway. Mil Med Res. 2024;11:71.
Karki R, Kanneganti T-D. PANoptosome signaling and therapeutic implications in infection: central role for ZBP1 to activate the inflammasome and PANoptosis. Curr Opin Immunol. 2023;83:102348.
Bai Q, Wang C, Ding N, Wang Z, Liu R, Li L, et al. Eupalinolide B targets DEK and PANoptosis through E3 ubiquitin ligases RNF149 and RNF170 to negatively regulate asthma. Phytomedicine. 2025;141:156657.
Shi C, Wang Y, Guo J, Zhang D, Zhang Y, Gong Z. Deacetylated MDH1 and IDH1 aggravates PANoptosis in acute liver failure through endoplasmic reticulum stress signaling. Cell Death Discov. 2024;10:275.
Jadhav PS, Mahajan S, Man SM. NLRC5 PANoptosome: aquaman of the dead sea. Cell Res. 2025;35:9–10.
Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol. 2017;19:1116–29.
Zhao L, Liu P, Mao M, Zhang S, Bigenwald C, Dutertre C-A, et al. BCL2 inhibition reveals a dendritic cell-specific immune checkpoint that controls tumor immunosurveillance. Cancer Discov. 2023;13:2448–69.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–79.
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016;15:274–87.
Schmidt SV, Seibert S, Walch-Rückheim B, Vicinus B, Kamionka E-M, Pahne-Zeppenfeld J, et al. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget. 2015;6:8635–47.
Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol. 2019;4:eaaw2004.
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–40.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–6.
Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10:254–69.
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2022YFA1105300), Guangdong Basic and Applied Basic Research Foundation (2023B1515040030) and Young Talents Program of Sun Yat-sen University Cancer Center (YTP-SYSUCC-0019).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision: Ju HQ, Chen DL; Writing and editing: Lin JF, Wang TT, Huang ZR and Tan YT. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, JF., Wang, TT., Huang, RZ. et al. PANoptosis in cancer: bridging molecular mechanisms to therapeutic innovations. Cell Mol Immunol 22, 996–1011 (2025). https://doi.org/10.1038/s41423-025-01329-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41423-025-01329-z
Keywords
This article is cited by
-
Comprehensive analysis of PANoptosis-related molecular subtypes and prognostic model development of clear cell renal cell carcinoma
Discover Oncology (2026)
-
Targeting PANoptosis in atherosclerosis: bridging cell death mechanisms and therapy
Apoptosis (2026)
-
Therapeutic targeting of cell death-immune crosstalk in cancer to rewire the tumor immune microenvironment
Molecular Cancer (2025)
-
Characterization of PANoptosis-related expression pattern, prognosis and tumor microenvironment in head and neck squamous cell carcinoma
Discover Oncology (2025)
