
Abstract
Layered transition metal oxides (TMOs) have been utilized for centuries for their abundance and diverse applications in various fields. Developing efficient synthesis methods of layered TMOs is crucial for exploring novel properties and potential applications. This work introduces a direct microwave synthesis method for producing transition metal oxides and crystals. Using alpha molybdenum trioxide as a representative example, a microwave synthesis protocol is outlined for the rapid and scalable synthesis of layered transition metal oxides. Transition metal oxides have been synthesized using various state-of-the-art (SOTA) techniques, such as hydrothermal and sol-gel, among others. In contrast to these established methods, our direct microwave process is simple, requiring only a microwave and minimal handling, and it is highly scalable (1 g/h of α-MoO3 crystals). Additionally, it consumes just 0.5 kWh of energy per gram of α-MoO3, which is 8-140 times better than typical SOTA methods. It is environmentally friendly as it produces ~10-150 times less CO2 emissions (0.3 kg CO2 eq) compared to SOTA methods. It produces long crystals with a length of up to 8 mm, which is comparable to hazardous chemical and physical vapor deposition methods. We further demonstrate the advantages of these high-quality crystals by fabricating a MoO3-based Metal-Interlayer-Oxide-Semiconductor (MIOS) memristor incorporating an ultrathin Al2O3 interlayer. The device exhibits low SET (~ −2 V) and RESET (~2 V) voltages with stable endurance over multiple cycles, attributed to field-driven oxygen vacancy migration. These findings establish microwave synthesis as a transformative approach for producing high-performance metal oxides and underscore the significant potential of MoO3 for next-generation energy-efficient and reliable memory technologies.
Similar content being viewed by others


Introduction
Layered Transition metal oxides have garnered significant attention from researchers due to their remarkable electrical1, optical2, and mechanical properties3. Among these transition metal oxides (TMOs), molybdenum trioxide (MoO3) is the most studied, owing to its ability to adopt diverse morphologies such as nanorods4, nanotubes5, nanobelts6, nanoplatelets7, nanofibers8, nanoflowers9, nanospheres10, and nanoflakes11. These diverse morphologies arise from MoO3’s inherent layered structure and intriguing physical properties12. Functioning as an n-type semiconductor with a wide bandgap ranging from 2.8 to 3.6 eV13, MoO3 exhibits versatile applications, including photochromic14 and electrochromic devices15, photoconductive devices16, smart windows17, thermoelectric materials18, catalysis19, lithium-ion batteries20, supercapacitors21, solar cells22, and gas sensors23.
MoO3 exists in three distinct crystalline polymorphs (α, β, h-MoO3), each exhibiting unique structural characteristics and properties12. The first polymorph, α-MoO3, presents a dynamically stable phase featuring double layers of [MoO6] octahedral units24. These layers are arranged parallel to the (010) plane, offering open channels conducive to ion intercalation, thus enabling efficient charge storage25. In contrast, the β-MoO3 polymorph adopts a three-dimensional structure formed by corner-sharing [MoO6] octahedral units26, indicative of a metastable monoclinic phase27. Meanwhile, the h-MoO3 polymorph, also metastable, showcases a hexagonal crystalline structure characterized by zigzag chains of [MoO6] octahedra28, facilitating one-dimensional tunnel behavior29. Among the crystal structures of MoO3, α-MoO3 is particularly widely studied, the most studied phase due to its unique layered architecture30. A key characteristic of α-MoO3 is its layered structure, which can accommodate large quantities of positive ions in its tetrahedral and octahedral holes and extension channels31, enabling bandgap manipulation32. This layered configuration enhances charge transfer capabilities33, improving its electrochemical properties34. Various synthesis methods have been employed to prepare α-MoO3, each with advantages and limitations. State-of-the-art (SOTA) techniques, including this work, are compared in detail.
To the best of our knowledge, this is the first work to synthesize α-MoO3 from molybdenum disulfide (MoS2: a naturally occurring ore of Mo metal) as a starting material for large-scale production. Our in-depth analysis of SOTA methods demonstrates that microwave synthesis offers a compelling alternative for α-MoO3 production due to a combination of cost-effectiveness, speed, simplicity, scalability, crystal length, and energy efficiency. The crystal length achieved through microwave synthesis is approximately 7 mm, surpassing most common methods except for Chemical Vapor Deposition (CVD) and Physical Vapor Deposition (PVD), which produce crystals in a similar to higher size range. Microwave synthesis boasts a rapid production time of approximately 2 min to start forming MoO3 crystals, which is ~50–600 times faster than SOTA methods. The process is simple and scalable, requiring only a microwave, minimal handling, and only 1 h to produce 1 gram of α-MoO3, up to 600 times better than SOTA. Additionally, microwave synthesis is the most energy-efficient method, consuming just 0.5 kWh per gram of α-MoO3, up to 140 times better than SOTA methods. These features make microwave synthesis more practical and efficient for α-MoO3 production. In contrast to conventional methods in literature, this work pioneers the direct synthesis of MoO3 from naturally abundant MoS2 as the starting material, eliminating costly and energy-intensive intermediate purification steps and offering a cost-effective and scalable route for the large scale of MoO3 and TMOs production.
Mechanism growth of alpha molybdenum trioxide crystals
Various mechanisms (i.e., dielectric loss, Joule loss, and magnetic loss) are briefly discussed to understand the dominant heating mechanism during microwave irradiation comprehensively; however, the different interactions between materials and microwaves cannot be explained by a single theory. In general, microwave power absorption and penetration depth must be considered for the microwave processing of materials. The former, P (W/m3), is evaluated by Eq. (S1). The latter, Dp (m), represents a depth inside the material where the power of the wave reaches. Calculated according to Eq. (S2): Regarding Eqs. (S1) and (S2), materials’ electrical conductivity, permittivity, and permeability play vital roles during microwave heating. Three different mechanisms that would contribute to microwave heating are presented in Eq. (S1): Conduction (Joule) loss (first term, W/m3) is a dominant heating mechanism of materials with high electrical conductivity, such as metals. This mechanism results from eddy current loss generated in an alternating magnetic field and mainly depends on the material’s electrical conductivity. Dielectric loss (second term, W/m3) is due to either the polarization of materials and collision of dipoles or the electric charge’s collision under an alternating electric field. The dielectric polarization loss includes the polarization of either electron, dipolar, atoms, ions, or interfacial polarization. The contribution of magnetic loss (third term, W/m3) to the heating is caused by hysteresis, eddy currents, domain wall, and electron spin resonance in an alternating magnetic field.
Eq. (S2) explains the key difference between conventional and microwave heating methods. The shallow penetration depth of infrared waves during conventional heating results in the heating of the surface of materials followed by heat transfer from the surface toward the core of the sample, whereas a great penetration depth of microwaves during microwave heating results in the volumetric heating of the materials. In microwave heating, the core temperature is higher than the surface due to the heat loss from the surface, which results in the heat transfer from the core toward the sample’s surface.
Therefore, factors such as particle size (d), shape, and relative density, which directly/indirectly influence these equations, must be considered during microwave heating. For instance, electrical conductivity depends on a pressed sample’s particle size, shape, and compaction. Moreover, at a low particle size ratio to penetration depth (d/Dp), the particle is considered a transparent material and cannot absorb microwaves. By contrast, at a very high d/Dp ratio, a major portion of the microwaves are reflected and cannot heat the sample well, and heating is only limited to the sample’s surface. In the case of the thermal oxidation of MoS2 to MoO3 crystals, Rahman et al.35. investigated the impact of high-temperature oxidation on the mechanical performance of MoS2 using ReaxFF, proposing a detailed thermal oxidation mechanism. Initially, oxygen molecules (O₂) adsorb onto sulfur atoms on the basal plane of MoS₂, a process that is thermodynamically stable but kinetically hindered under ambient conditions. O₂ dissociates into atomic oxygen upon adsorption, forming covalent sulfur-oxygen (S-O) bonds. This oxidation leads to volatile sulfur oxides (e.g., SO2) that desorb from the surface, creating sulfur vacancies. These vacancies are subsequently filled by oxygen atoms, forming more stable molybdenum-oxygen (Mo-O) bonds. As the oxidation progresses, a molybdenum oxy-sulfide (MoS2-xOx) solid solution forms, which can further oxidize into MoO3 at higher temperatures. The process intensifies above 1500 K, with severe crystalline distortion of MoS2 and significant MoO₃ formation occurring around 1800 K. In 2023, Reidy et al.36. explained the atomic-scale mechanism of MoS2 oxidation to MoO3. The thermal oxidation of MoS2 begins with the oxidation of the MoS2 surface. This process occurs readily at elevated temperatures (e.g., 500 °C in air) and produces MoO3. Following the initial oxidation, the volatile species MoOx (x can vary) forms and sublimates from the surface. This sublimated species then undergoes vapor-phase mass transport. Then, the MoOx species redeposits and crystallizes into α-MoO3 on the MoS2 substrate or nearby surfaces. This results in the growth of crystalline α-MoO3, which is often textured and aligned with the underlying MoS2 substrate.
This oxidation is exothermic, releasing heat as the reaction progresses. The formation of MoO3 typically occurs as a gradual transformation of MoS2, resulting in MoO3 as a white crystalline material, as shown in Fig. 1. The reaction progresses by the oxidation of MoS2, where sulfur atoms are oxidized to form sulfur dioxide (SO2). Simultaneously, molybdenum atoms combine with oxygen atoms to form MoO3 according to Eq. 1:

Schematic illustration of the microwave synthesis setup for α-MoO3 crystal growth from MoS2 powder.
After completion of the oxidation reaction, the MoO3 crystals are allowed to cool down gradually to ambient temperature before they are collected. Digital images of the process are shown in Supplementary Figs 1 and 2, and α-MoO3 crystals are shown in Supplementary Figs. 5 and 6. This process could be used for more members of the TMO family, as shown in Supplementary Figs. 10,11, and 11.
Characterization of alpha molybdenum trioxide crystals
The crystal structure of the samples was analyzed using X-ray diffraction (XRD). As shown in Fig. 2a, the distinctive characteristic diffraction peaks at 14.76°, 33.08°, 40.00°, and 50.19°, corresponding to (002), (100), (103), and (105) crystal planes of hexagonal phase MoS2 are consistent well with standard card of MoS2 (JCPDS card No. 77-1716)37, respectively. Figure 2b reveals peaks at 13.22°, 26.09°, and 39.36°, which correspond to (020), (040), and (060), respectively, of α-MoO3, which possesses an orthorhombic crystal system (JCPDS card No. 05-0508)38. The XRD peaks were analyzed employing a modified Scherrer equation to calculate the nano crystallite size, as detailed in Table S2. The (020), (040), and (060) phases exhibit respective particle sizes (Dp) of 38.02, 41.22, and 40.38, along with approximate d-spacing values of 6.7, 3.4, and 2.3, respectively. Notably, the strong specific (020), (040), and (060) peaks confirm the layered crystal structure of α-MoO3 and are consistent with highly anisotropic growth along the c-axis39. Furthermore, the absence of peaks from other phases, such as MoS2, suggests the high purity of the sample. Raman spectrum (Fig. 2c) of MoS2 powder shows characteristic peaks at 384 and 408 cm−1, corresponding to E12g (in-plane optical vibration of the Mo−S bond in opposite directions)40 and A1g (out-of-plane optical vibration of S atoms)41 active modes, respectively. The 24 cm−1 separation between these two peaks indicates the presence of multilayer MoS242, as the MoS2 used was purchased commercially and remained unaltered. Raman spectroscopy for the synthesized confirms the α-MoO3 phase (Fig. 2d), with peaks at 997 cm−1 and 822 cm−1 corresponding to asymmetrical and symmetrical stretching vibrations of terminal Mo=O bonds43. In contrast, the peak at 669 cm−1 corresponds to asymmetrical stretching vibrations of O–Mo–O bonds along with peaks from 200 to 450 cm−1 44,45. Additionally, the peaks between 50 and 200 cm−1 represent the skeletal modes of the chains of MoO4 tetrahedra, as reported previously46,47. A Raman repeatability study was conducted to test the validity of the material over a number of batches (Supporting Fig. 16).

a XRD of MoS2, (b) XRD of MoO3, (c) Raman of MoS2, (d) Raman of MoO3, (e) XPS spectrum of Mo 3 d, (f) XPS spectrum of O 1 s.
The XPS detailed spectral analysis (Fig. 2e, f, and Supplementary Fig. 9d) confirms the presence and oxidation states of molybdenum (Mo) and the chemical environment of oxygen (O) in the sample. High-resolution Mo 3 d spectrum (Fig. 2e), which can be deconvoluted into two 3 d doublets using Gaussian fitting, reveals major peaks at 232.8 eV and 236.0 eV characteristic of Mo6+ oxidation state, 3d5/2 and 3d3/2, respectively, without minor peaks. The O 1 s spectrum (Fig. 2f), with a peak around 530.1 eV, corresponds to oxygen in the MoO3 lattice.
The SEM images of MoS2 in Fig. 3a exhibit an uneven structure with agglomerated particles. revealing defined, multi-layered particles with a flake-like structure and particle size of a few micrometers. Additionally, EDX analysis confirms the purity of the MoS2 samples, showing the presence of only molybdenum and sulfur without any other elemental impurities (Supplementary Fig. 8a). The closer magnifications in Fig. 3b, c shows the layered belt-shaped structure of α-MoO3, confirming the successful transformation of MoS2 to the pure structure of α-MoO3 and their structural uniformity. EDX analysis of the MoO3 samples (Supplementary Fig. 8c) also confirms their purity, with only molybdenum and oxygen detected, verifying the absence of other elemental contaminants. In Fig. 3d, e, Optical microscope images of the MoO3 show well-defined, elongated belt-like crystals with lengths between 3 to 7 mm. Crystals were long and stable enough to be handled and gathered to form KU letters (Supplementary Fig. 6). Notably, the crystal lengths observed are among the highest recorded in the literature, up to 7 mm, surpassing all the synthesis methods, with CVD and PVD recording higher lengths in millimeters48. Figure 3f–h shows an optical microscope image of the MoO3 crystals at different magnifications. Atomic Force Microscopy (AFM) and Conductive Atomic Force Microscopy (C-AFM) were employed to evaluate the structural and electrical properties of the synthesized crystals. Figure 3i presents the topographical AFM image of a representative crystal, revealing a thickness of approximately 5 nm (additional topographical images are provided in Supplementary Fig. 3). The corresponding C-AFM measurement, depicted in Fig. 3j, indicates a current of approximately 30 nA along the crystal. To further explore the correlation between crystal thickness and electrical conductivity, we examined crystals of varying thicknesses. As shown in Supplementary Fig. 4, crystals with thicknesses of 70 nm, 213 nm, and 330 nm exhibited measured currents of 15 nA, 8 nA, and 10 nA, respectively. These observations suggest a potential dependence of charge transport properties on the crystal thickness. Transmission electron microscopy (TEM) images in BF-TEM mode provided compelling evidence for the thin rectangular morphology of the synthesized α-MoO3 nanostructures (Fig. 4a). The acquired SAED patterns allowed the measurement of the d-spacings of the crystals, which matched the crystal structure of samples determined with XRD experiments (Supplementary Fig. 7 and Supplementary Table 1). These nanorods’ corresponding high-resolution TEM (HRTEM) analysis allowed the visualization of the structure of samples in real space and recorded distance of the well-defined lattice fringes, a hallmark of highly crystalline materials, matched nicely with d-spacing measured by performing the SAED experiments (Fig. 4b). Interestingly, the SAED analysis also unveils a preferential growth direction for the α-MoO3 nanobelts on the [001] axis (Fig. 4c, d). This anisotropic crystal structure of α-MoO3 is likely responsible for this favored orientation. In this structure, distorted [MoO6] octahedra share corners to form sheets, and pairs of these sheets connect via edges along the [001] direction. These bilayers then weakly stack upon each other via van der Waals forces along the [010] axis. Elemental composition analysis of the samples in TEM-EDS mode revealed high-purity crystals with a composition close to the stoichiometric numbers (see Supplementary Fig. 8f). Furthermore, the STEM-EDS hyperspectrum imaging showed a uniform distribution of Mo and O elements through the regions of the particles (Fig. 4e), which implies a uniform crystal and, hence, the uniform corresponding emergent properties of the samples. As depicted in Fig. 4e, Elemental analysis mapping offers a powerful visualization of the α-MoO3 sample’s homogeneity. This technique reveals a uniform distribution of elements throughout the material, signifying the absence of sulfur or other impurities. This observation strongly supports the high purity of the synthesized α-MoO3. STEM-EELS (Fig. 5) reveals the dielectric properties of synthesized samples. Figure 5c, d shows the real and imaginary parts of the dielectric function, which have been obtained from the bulk and surface regions of a particle shown by the dark-field STEM (DF-STEM) image in Fig. 5a, b. Kramer–Kronig analysis (KKA) of the energy loss function (ELF) from bulk and surface regions (Fig. 6b–d) showed a significant enhancement in the density of states (DOS) at the surface in the visible light range. This suggests increased visible light absorption at the surface, potentially due to surface strain.

SEM Images of (a) MoS2, (b, c) MoO3 of different magnifications. d, e Optical microscope images Demonstrating the macroscopic scale of the synthesized α-MoO3 crystals (f–h) Optical images showcasing the crystal nature and the reflection of white light if the OM. i AFM topography of low-thickness MoO3 crystal. j Conductive-AFM of the same crystal.

a TEM low magnification of MoO3, (b) SAED of MoO3, (c, d) HR-TEM of MoO3 with FFT as inset, (e) Elemental mapping of MoO3.

a DF-STEM of MoO3, (b–d) Dielectric properties of MoO3, (e–g) Strain Properties of MoO3.

The mining process of Molybdenum Ores to MoS2 and MoO3.
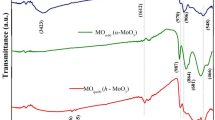
surface regions. The results revealed a significant enhancement in the surface-region DOS in the visible light range of energies compared to the bulk region’s DOS. These results imply that the surface regions of the particles absorb more energy in the visible light range than the bulk. The enhancement in DOS at the surface regions can be linked to the presence of strain in these regions, as shown in the strain analysis of the particles, presented in Fig. 5e–g, showing a normal strain in the surface regions, which may be correlated with the enhanced surface DOS and altered dielectric properties. The FTIR spectrum shown in Supplementary Fig. 9a further confirmed the α-MoO3 phase. The peaks are primarily located below 14,000 cm−1 on the low-energy side of the spectrum. The peak at 989 cm−1 for the α-MoO3 sample is attributed to the terminal Mo=O stretching vibration, indicating the layered orthorhombic α-MoO3 phase49,50. A strong peak at 815 cm−1 is assigned to the stretching vibrations of the O(3) atoms in the Mo-O-Mo units in the orthorhombic α-MoO3 structure51. The band at 563 cm−1 is due to the bending vibrations of the oxygen atom (O(2)) linked to three metal atoms ν(O – 3Mo) for the α-MoO3 sample52. Additionally, a peak around 470 cm−1 could not be detected due to instrumental limitations, but it is thought to be due to the stretching mode of the terminal Mo–O bond53,54. The absence of other significant peaks like 3400 cm−1 for water molecules further confirms the high purity of the α-MoO3 sample. Thermogravimetric analysis (TGA) was performed to investigate the α-MoO3 sample’s thermal stability (Supplementary Fig. 9b). The results demonstrate remarkable thermal stability up to 700 °C, indicating their robustness for high-temperature applications. The TGA curve shows no significant decomposition stages up to this point. However, at around 790 °C, there is a dramatic % weight loss of about 40%, resulting from the sublimation of MoO355.
This work vs state-of-the-art techniques
Molybdenum is naturally found with other elements, including several molybdenum-bearing minerals. However, the one with the most commercial significance is molybdenite (MoS₂), a natural mineral form of molybdenum disulfide56,57.
The flowchart presented in Fig. 6 illustrates the streamlined synthesis processes of our novel microwave method compared to SOTA. Both approaches share the same initial four stages—crushing, grinding, flotation, and leaching—from the mined molybdenum ore to molybdenite (MoS₂) formation. As described in detail in the supporting information file, these processes are crucial for extracting and purifying molybdenite, but there is no variation between the methods in these preliminary steps58,59,60,61. The key distinction between the two approaches emerges after molybdenite (MoS₂) is formed. In this work, molybdenite is directly used as the starting material without requiring additional processing. In contrast, in conventional SOTA methods, multiple subsequent processes are required for MoO₃ crystals synthesis. Specifically, molybdenite is roasted at high temperatures (around 500 °C)61, followed by chemical treatments and calcination to produce pure molybdenum powder62. This purified molybdenum is the starting material for various synthesis techniques such as CVD, PVD, and laser ablation. After roasting, further chemical treatments are applied to convert the molybdenite into ammonium heptamolybdate (AHM), the starting material for other commonly used SOTA methods. The takeaway from Fig. 6 is that this work offers an alternative approach to synthesizing MoO3 crystals by bypassing additional steps and intermediate compounds. Directly utilizing molybdenite as the starting material and employing microwave synthesis eliminates energy-intensive processes such as roasting (500 °C)63,64, chemical treatments, and calcination at temperatures ranging from 500–1000 °C. The exclusion of these cumbersome processes significantly streamlines the synthesis route, giving this work a distinct advantage over conventional techniques. Figure 7 quantitatively compares our microwave synthesis with eight common SOTA methods across seven key Criteria: Scalability (SC), Crystal Length (CL), Production Rate (PR), Energy efficiency (EE), Simplicity (SM), (1/CO2), which is kilograms of CO2 emissions equivalent. Data were collected through analysis of Supplementary Table 3 for (CL, PR, SM, SC, and 1/Cost), and by conducting Life Cycle Assessment (LCA) using SimaPro for (EE and 1/CO2). It is a standard unit used to quantify the impact of different greenhouse gases (GHGs) on climate change in terms of the equivalent amount of carbon dioxide (CO2) that would have the same global warming potential (GWP) (Supplementary Fig. 17) and, (1/Cost) is a cost calculation for the machinery involved into making MoO3 crystals for each method.

(Hydrothermal[S9-S95], Spray Pyrolysis[S96- S114], Laser Ablation[S115- S122], Sol-gel[S123- S131], Solution Combustion[S132-S139], Electrodeposition[S140-S146], PVD[S147-S162], and CVD[S163-S175]).
When comparing methods based on energy efficiency -measured by the electricity consumed to produce 1 g of MoO3 in kWh- Microwave Synthesis is the most energy-efficient, consuming only 0.5 kWh. Solution combustion consumes around 4 kWh, while Electrodeposition uses about 19 kWh. Spray pyrolysis requires approximately 15 kWh. Sol-gel and laser ablation each consumes around 30kWh.
Hydrothermal processes need about 35 kWh. CVD and PVD are the least energy-efficient, consuming 70 and 60 kWh, respectively. The production rates for each method were approximately as follows: microwave synthesis was 10 min, which is the least time consumed to produce MoO3 crystals among SOTA methods. Meanwhile, hydrothermal ranged from 8 to 20 h, with an average of 12 h being the most common duration. Spray pyrolysis averaged 30 min for spraying and 1 h for evaporation. Electrodeposition required a couple of minutes for deposition and an additional 2 h for annealing. Sol-gel needed around 10 h for gel formation and subsequent calcination. Solution combustion took a couple of minutes to mix and approximately 1 h for combustion. Laser Ablation averaged 30 min for irradiation of the molybdenum target. CVD and PVD required around 3 h to reach the desired temperature, maintain it, and then evacuate to collect the product. Scalability, measured by the time needed to produce 1 g of MoO3, is another crucial critera. For this criteria, the Microwave Synthesis method needed only 1 h to produce 1 gram of MoO3, which offers a potentially scalable approach to produce MoO3 crystals. Solution Combustion required around 2.5 h. The Sol-gel method and hydrothermal processes each needed around 20 to 25 h. CVD and PVD methods required 20 to 30 h to produce 1 gram. For techniques like laser ablation, spray pyrolysis, and electrodeposition, typically used to create thin films, scalability was assessed by the time required to produce milligrams per square centimeter. It was determined that to make 1 gram of MoO3, electrodeposition would take around 80 h, laser ablation would take about 300 h, and spray pyrolysis would take approximately 600 h. These high times are reasonable given that these methods are not typically used for large-scale production but for creating thin films on substrates. Simplicity is another essential criteria, evaluated based on the number of steps in the process, the complexity of the machinery involved, the number of chemicals used, and how easy these chemicals are to handle. Microwave Synthesis is a simple and easy method that requires only a microwave. Solution combustion also scores high on simplicity, needing minimal chemicals and machinery, as it only involves burning the chemicals. Electrodeposition and spray pyrolysis require fewer chemicals but involve handling specific machinery like a potentiostat and spray nebulizer. Hydrothermal and Sol-gel methods are relatively easy to handle but require a moderate amount of chemicals to initiate the reaction. Laser Ablation is more complex, involving complex machinery and numerous safety concerns. CVD and PVD are the most complicated, requiring many steps and handling a sensitive and controlled environment. The cost calculations are based on the machinery cost and energy consumed during the process. Microwave Synthesis emerged as the most cost-effective method. Solution Combustion followed, ranging from 500 to 1000 USD, while the Sol-gel method ranged between 1000 to 2000 USD. Electrodeposition methods fell within the range of 1500 to 2500 USD. Hydrothermal processes cost between 500 and 1000 USD, whereas spray pyrolysis ranges from 10,000 to 25,000 USD. Laser Ablation method is significantly more costly, ranging between 10,000 to 100,000 USD. CVD and PVD methods were the most expensive, with machinery costs ranging from 100,000 to 250,000 USD. These cost assessments provide insights into the financial feasibility of each method, influencing their practicality and adoption in various applications. The final criteria crystal length is determined by SEM images from the references, and distinct averages emerge. Hydrothermal processes exhibit an average of several micrometers to 200 micrometers, while sol-gel and solution combustion methods typically result in crystal lengths in the range of several micrometers. Electrodeposition, spray pyrolysis, and Laser ablation produce crystals measured in nanometers, whereas CVD and PVD methods yield the longest crystals, with PVD achieving lengths of up to a few mm to cm. In terms of crystal length, Microwave Synthesis can reach up to 7 mm, comparable to other advanced methods like CVD and PVD; being compared to CVD and PVD in crystal synthesis is an advantage, making it a viable alternative without going through the complexity of these methods. To demonstrate the electrical properties and practical utility of the synthesized α-MoO3 crystals, we fabricated and characterized Metal-Interlayer-Oxide-Semiconductor (MIOS) memristive devices.
Application for alpha molybdenum trioxide
MoO3 has been investigated as a compelling active material for various memristive devices due to its diverse redox states and tunable oxygen vacancy concentrations65,66,67,68. In this section, we demonstrate an enhanced memristive effect in a Si/MoO3/Al2O3/Cu structure (details in the Experimental Section), leveraging the structural and electrical properties of the synthesized MoO3 crystals. Figure 8 illustrates the fabricated MIOS device structure: (a) Top-view optical micrograph showing a device with a ~ 623 nm thick MoO3 crystal. (b) Schematic cross-sectional diagram of the device stack (Si/MoO3/Al2O3/Cu), and (c) AFM topography of the MoO3 crystal surface after Al2O3 and Cu deposition, confirming the crystal’s morphology is preserved during device fabrication. Electrical characterization (details in Experimental Section) of the fabricated MIOS devices reveal clear and robust bipolar resistive switching, as evidenced by the current-voltage (I-V) characteristics in Fig. 9a, which is expected in Transition Metal Oxides (TMO)-based memristors. Notably, Our MIOS devices incorporating the α-MoO3 exhibited remarkably low SET (~ −2V) and RESET (~2 V) voltages even with a relatively thick (~623 nm) MoO3 crystal. This low voltage operation is a significant advantage for energy-efficient memory applications and is not typically observed in MoO3 memristors of this thickness. Control devices fabricated without a MoO3 layer (p + + Si/Al2O3/Cu) showed significantly inferior memristive performance, exhibiting higher switching voltages, smaller switching windows, and a low resistance state (LRS), highlighting the crucial role of the MoO3 crystals. Detailed inspection of the I-V log-log plots in Fig. 9b shows the charge transport mechanisms governing the resistive switching. In the LRS, the I-V curves show a linear ohmic relationship with a slope close to unity (|I| ∝ |V|α, α ≈ 1.06 in negative bias and α ≈ 1.05 in positive bias). This ohmic behavior in the LRS leads to an increased concentration of mobile oxygen vacancies acting as donors at the Al2O3/MoO3 interface66,67. On the other hand, in the high resistance state (HRS), especially under positive bias (after RESET), the I-V characteristics deviate from linearity, showing a slope of approximately 1.8 ( | I| ∝ |V | 1.8). This non-linear space-charge-limited current (SCLC) and trapped-charge-limited current (TCLC) in HRS suggests that the current is limited by space charge effects and available trap states within the MoO3, resulting from a reduced carrier concentration after the Forming and follow-up RESET processes68. This transition from ohmic conduction in the LRS to SCLC in the HRS supports a non-filamentary, interfacial switching mechanism69,70, where the barrier at the MoO3/Al2O3 interface is dynamically modulated66,71. Moreover, we propose that this modulation arises from the field-assisted migration of oxygen vacancies within the MoO3 crystal (Fig. 9d)6,7. Since pristine α-MoO3 crystals possess low intrinsic oxygen vacancy concentrations, an initial electroforming step (Supplementary Figs. 13–15) was crucial to generate and optimally localize the necessary oxygen vaScancies at the Al2O3/MoO3 interface. This electroforming process electrochemically reduces Mo6+ ions, creating mobile oxygen vacancies and facilitating subsequent redox reactions at optimal sites along the crystal interface during SET and RESET operations72. Endurance testing (Fig. 9c) demonstrates stable and repeatable bipolar switching over 33 cycles (for a device with a ~ 511 nm thick MoO3 crystal), with minimal degradation in the on/off ratio and switching voltages. The inset of Fig. 9c shows the voltage pulse train used for the measurements with 2 ms pulse durations. The proposed switching mechanism is illustrated via energy band diagrams in Fig. 9d, showing the electric field-induced drift and diffusion of these generated oxygen vacancies during the forming process within the MoO3 layer. During SET (negative bias), oxygen vacancies migrate towards the Al2O3 interface, forming localized conductive regions responsible for the high resistance state. During RESET (positive bias), oxygen vacancies move away, rupturing these regions and restoring the high resistance state. The Al2O3 layer acts as a crucial interface, controlling the nucleation and localization of these conductive regions and contributing to the observed low-voltage operation71,73.

a Top-view optical micrograph of a fabricated Si/MoO3/Al2O3/Cu structure with a ~ 623 nm thick MoO3 crystal. b Schematic cross-sectional diagram of the structure stack. c Atomic force microscopy (AFM) image showing the topography of the MoO3 crystal after Al2O3 and Cu deposition.

a Current-voltage (IV) characteristics of a Si/MoO3/Al2O3/Cu structure with a ~ 623 nm thick MoO3 crystal, showing the 1st and 5th voltage sweep cycles compared to a control Si/Al2O3/Cu structure without MoO3. b Log-log plots of IV curves for five sweep cycles, highlighting the ohmic and SCLC/TCLC regimes during the SET and RESET states. c Endurance performance of a device with a ~ 511 nm thick MoO3 crystal over 33 switching cycles, with the inset showing the voltage pulse train. d Schematic energy band diagrams illustrating the proposed oxygen vacancy migration mechanism responsible for resistive switching. During SET (negative bias), oxygen vacancies drift towards the Al2O3 interface, increasing conductivity. During RESET (positive bias), vacancies drift away, restoring high resistance.
The low SET and RESET voltages achieved in our MoO3-based structures, despite the substantial MoO3 thickness, represent a notable advancement. While MoO3 has been extensively explored for resistive switching72,74,75, achieving such low voltage operation in devices with easily synthesized, hundreds of nanometers thick MoO3 crystals are unconventional and desirable for low-power memory applications. Compared to the Al2O3-based structures, which often rely on charge trapping or tunneling mechanisms76, our MoO3 devices exhibit bipolar resistive switching indicative of ionic migration, offering a distinct operational pattern with potentially superior endurance characteristics after further optimization. Supplementary Table S4 benchmarks our devices against reported MoO3 and other high‑k oxide-based memristors. Despite a much thicker switching layer, our operating voltages are lower than those for sputtered and CVD MoO3 [S178, S180, S181, S183] and comparable to solution-processed MoO3 [S176]. The devices also show ON/OFF current ratios and endurance similar to reported MoO3 [S176] and Al2O3 [S181] memristors, highlighting the excellent performance achieved with our microwave-synthesized MoO3 crystals. This performance enhancement underscores the potential of utilizing high-quality MoO3 crystals with a proper tunnel barrier to realize more reliable, cost- and energy-efficient memristive switching. Future research should focus on in-site spectroscopic investigations (e.g., XPS, TEM-EELS) to directly characterize the oxygen vacancy dynamics and Mo oxidation state changes at the Al2O3/MoO3 interface during resistive switching. Further systematic optimization of the Al2O3 interface (e.g., thickness variation, exploring alternative high-k dielectrics like HfO2 or TiO2) could lead to even greater improvements in device performance, potentially unlocking the potential of these devices for emerging applications in energy-efficient neuromorphic computing and advanced in-sensor processing technologies77,78.
Conclusion
In summary, this work demonstrates microwave synthesis as a remarkably efficient and eco-friendly approach for producing high-quality metal oxides, exemplified by synthesizing crystalline α-MoO3. Compared to state-of-the-art methods, our approach offers significant advantages in simplicity, production rate (achieving MoO3 crystals in just 2 min, ~50–600x faster), scalability (1 g/h, 2.5–600x better), energy efficiency (0.5 kWh/g, 8-140x better), and reduced CO2 emissions (~10–150x less). The resulting α-MoO3 exhibits long crystals up to 8 mm in length, compared to complex processes such as CVD and PVD, showcasing the potential of microwave synthesis in achieving high-quality materials with desirable properties. Furthermore, we have shown that integrating these high-quality MoO3 crystals with an Al2O3 barrier in MIOS structures enables stable, low-voltage bipolar switching driven by oxygen vacancy dynamics. Finally, this microwave synthesis method, combined with the demonstrated memristor performance, positions a-MoO3 as a promising material for energy-efficient next-generation memory and neuromorphic computing applications.
Experimental section
Chemicals and reagents
MoS2 powder (average size of less than 5 μm), and isopropyl alcohol (C3H8O, 99%) were purchased from Sigma-Aldrich. All the chemicals used in the experiments were used without further purification.
Characterization and measurements
Samples were irradiated in a microwave (1000 watts) and 2.45 GHz. The crystallinity of the samples was measured using X-ray diffraction (XRD Bruker D2 phaser). Raman analysis of the starting materials and the exfoliated samples were performed by Renishaw spectrometer using 532 nm laser (2.33 eV) excitation and 50x objective lens. The laser power was kept below 1 mW to prevent sample damage. 20 spectra were recorded (each one at a different location) for each sample to create statistical data for the samples. Morphology of the samples was done by scanning electron microscopy ((SEM) Nova NanoSEM 650, beam resolution 0.8 nm). XPS spectra were recorded using a Thermo Scientific K-Alpha spectrometer. Samples were analyzed at the nanoscale using a TEM of model Titan ThemisZ equipped with double aberration correctors, high-throughput X-ray energy dispersive spectroscopy (EDS), and electron energy loss spectroscopy (EELS) detectors. The analysis was carried out by operating the microscope at an accelerating voltage of 300 kV. The morphology of the samples was analyzed using bright-field TEM (BF-TEM). The structure was investigated using selected area electron diffraction (SAED) and high-resolution TEM (HRTEM). Whereas the elemental composition of the samples was determined using EDS in TEM mode. Moreover, the elemental composition maps or distributions were generated by processing the EDS datasets acquired in synchronized scanning transmission electron microscopy (STEM) mode. The samples ‘ dielectric properties and density of states (DOS) were probed using the low-loss EELS, also in the STEM mode. The presence of strain at the edges or surface of the samples was investigated by carrying out the four-dimensional hyperspectral datasets acquired in synchronization of STEM mode and micro-diffraction mode, labeled as the 4DSTEM technique. The elemental composition and the corresponding elemental maps were acquired and processed in ThermoFisher Software called Velox v2.05. Whereas the rest of the TEM analysis-related datasets were acquired and subsequently processed in Gatan Microscopy Suite (GMS) of version 3.5 from Gatan, Inc. TGA analysis was done using STA 449 F5 Jupiter. UV-Vis was done using specord 50 plus. FTIR was done using NiCOLET iS50. UV-vis spectroscopic data was obtained using an Ocena Insight DH-2000 spectrometer utilizing a deuterium lamp. The absorption and transmission spectra were recorded in the 180–600 nm wavelength range. The integration time was set to automatic, and an average of 20 spectra was obtained.
MIOS devices were fully fabricated in a class 1000 cleanroom, and fabrication started with transferring the MoO3 crystals onto cleaned and O2 plasma-treated p + + silicon (0.002-0.003 Ω.cm) substrates using a PDMS stamp to maintain their structural integrity. Subsequently, a 4 nm thick Al2O3 layer was deposited via atomic layer deposition (ALD) as a uniform, high-k oxide, pinhole-free barrier. Finally, a 100 nm thick Cu top electrode (TE) was thermally evaporated and patterned through a shadow mask carefully aligned over the MoO3 crystals.
Electrical characterizations were conducted using a probe station and parameter analyzer under ambient conditions in a dark box, with the p + + Si substrate serving as the grounded bottom electrode (BE) and a gold-coated tungsten probe applying bias to Cu TE.
References
Ruta, F. L. et al. Surface plasmons induce topological transition in graphene/α-MoO3 heterostructures. Nat. Commun. 13, 3719, https://doi.org/10.1038/s41467-022-31477-z (2022).
Zhang, Q. et al. Unidirectionally excited phonon polaritons in high-symmetry orthorhombic crystals. Sci. Adv. 8, eabn9774, https://doi.org/10.1126/sciadv.abn9774 (2022).
Choi, D. K. et al. Highly efficient, heat dissipating, stretchable organic light-emitting diodes based on a MoO3/Au/MoO3 electrode with encapsulation. Nat. Commun. 12, 2864, https://doi.org/10.1038/s41467-021-23203-y (2021).
Cong, S. et al. Diverse adsorption/desorption abilities originating from the nanostructural morphology of VOC gas sensing devices based on molybdenum trioxide nanorod arrays. Adv. Mater. Interfaces 3, 1600252, https://doi.org/10.1002/admi.201600252 (2016).
Puebla, S. et al. Strain tuning MoO3 vibrational and electronic properties. Npj 2D Mater. Appl. 8, 1–9, https://doi.org/10.1038/s41699-024-00442-3 (2024).
Imtiaz, S. et al. Electrocatalysis on separator modified by molybdenum trioxide nanobelts for lithium–sulfur batteries. Adv. Mater. Interfaces 5, 1800243, https://doi.org/10.1002/admi.201800243 (2018).
Martinez-Garcia, A. et al. High rate and durable, binder free anode based on silicon loaded MoO3 nanoplatelets. Sci. Rep. 5, 10530, https://doi.org/10.1038/srep10530 (2015).
Li, S., Shao, C., Liu, Y., Tang, S. & Mu, R. Nanofibers and nanoplatelets of MoO3 via an electrospinning technique. J. Phys. Chem. Solids 67, 1869–1872, https://doi.org/10.1016/j.jpcs.2006.04.017 (2006).
Huang, L. et al. Three-dimensional MoO3 nanoflowers assembled with nanosheets for rhodamine B degradation under visible light. Mater. Res. Bull. 108, 38–45, https://doi.org/10.1016/j.materresbull.2018.08.036 (2018).
Zhao, D. et al. Facile synthesis of MoO3 nanospheres and their application in water treatment. Mater. Lett. 256, 126648, https://doi.org/10.1016/j.matlet.2019.126648 (2019).
Yan, B. et al. Orientation controllable growth of MoO3 nanoflakes: micro-raman, field emission, and birefringence properties. J. Phys. Chem. C 113, 20259–20263, https://doi.org/10.1021/jp907602w (2009).
Scanlon, D. O. et al. Theoretical and experimental study of the electronic structures of MoO3 and MoO2. J. Phys. Chem. C 114, 4636–4645, https://doi.org/10.1021/jp9093172 (2010).
Qu, Q., Zhang, W.-B., Huang, K. & Chen, H.-M. Electronic structure, optical properties and band edges of layered MoO3: a first-principles investigation. Comput. Mater. Sci. 130, 242–248, https://doi.org/10.1016/j.commatsci.2017.01.014 (2017).
Andron, I. et al. Photochromic behavior of ZnO/MoO3 interfaces. ACS Appl. Mater. Interfaces 12, 46972–46980, https://doi.org/10.1021/acsami.0c13335 (2020).
Ivanova, T., Gesheva, K. A., Popkirov, G., Ganchev, M. & Tzvetkova, E. Electrochromic properties of atmospheric CVD MoO3 and MoO3–WO3 films and their application in electrochromic devices. Mater. Sci. Eng. B 119, 232–239, https://doi.org/10.1016/j.mseb.2004.12.084 (2005).
Hu, G. et al. Topological polaritons and photonic magic angles in twisted α-MoO3 bilayers. Nature 582, 209–213, https://doi.org/10.1038/s41586-020-2359-9 (2020).
Morita, M. et al. Chromogenic amorphous MoO3–x nanosheets and their nanostructured films for smart window applications. ACS Appl. Nano Mater. 4, 8781–8788, https://doi.org/10.1021/acsanm.1c01428 (2021).
Zheng, W. et al. Improvement of the thermoelectric properties of a MoO3 monolayer through oxygen vacancies. Beilstein J. Nanotechnol. 10, 2031–2038, https://doi.org/10.3762/bjnano.10.199 (2019).
Alex, K. V., Silva, J. P. B., Kamakshi, K. & Sekhar, K. C. Ternary heterostructures based on BaTiO3/MoO3/Ag for highly efficient and reusable photocatalytic applications. Adv. Mater. Interfaces 10, 2201948, https://doi.org/10.1002/admi.202201948 (2023).
Shahzad, R. F. et al. Designing molybdenum trioxide and hard carbon architecture for stable lithium-ion battery anodes. Adv. Mater. Interfaces 11, 2400258, https://doi.org/10.1002/admi.202400258 (2024).
Brezesinski, T., Wang, J., Tolbert, S. H. & Dunn, B. Ordered mesoporous α-MoO3 with iso-oriented nanocrystalline walls for thin-film pseudocapacitors. Nat. Mater. 9, 146–151, https://doi.org/10.1038/nmat2612 (2010).
Cai, P. et al. An ultraviolet-deposited MoO3 film as anode interlayer for high-performance polymer solar cells. Adv. Mater. Interfaces 7, 1901912, https://doi.org/10.1002/admi.201901912 (2020).
Kumar, R. et al. Growth of MoS2–MoO3 hybrid microflowers via controlled vapor transport process for efficient gas sensing at room temperature. Adv. Mater. Interfaces 5, 1800071, https://doi.org/10.1002/admi.201800071 (2018).
Huang, P.-R., He, Y., Cao, C. & Lu, Z.-H. Impact of lattice distortion and electron doping on α-MoO3 electronic structure. Sci. Rep. 4, 7131, https://doi.org/10.1038/srep07131 (2014).
Kedves, E.-Z. et al. Dependence of cationic dyes’ adsorption upon α-MoO3 structural properties. Appl. Surf. Sci. 573, 151584, https://doi.org/10.1016/j.apsusc.2021.151584 (2022).
Ramirez, I. J. Synthesis of h-MoO3 by vacuum drying and its structural and electrochemical characterisation. Mater. Lett. 57, 1034–1039 (2003).
Mariotti, D., Lindström, H., Bose, A. C. & Ostrikov, K.(K. en). Monoclinic β-MoO3 nanosheets produced by atmospheric microplasma: application to lithium-ion batteries. Nanotechnology 19, 495302, https://doi.org/10.1088/0957-4484/19/49/495302 (2008)..
Song, J., Ni, X., Gao, L. & Zheng, H. Synthesis of metastable h-MoO3 by simple chemical precipitation. Mater. Chem. Phys. 102, 245–248, https://doi.org/10.1016/j.matchemphys.2006.12.011 (2007).
Sen, S. K. et al. Structural and optical properties of sol-gel synthesized h-MoO3 nanorods treated by gamma radiation. Nano Express 1, 020026, https://doi.org/10.1088/2632-959X/aba4f8 (2020).
da Silva Júnior, M. G. et al. A brief review of MoO3 and MoO3-based materials and recent technological applications in gas sensors, lithium-ion batteries, adsorption, and photocatalysis. Materials 16, 7657, https://doi.org/10.3390/ma16247657 (2023).
Sharma, R. et al. Synthesis & material properties of α-MoO3 nanoparticles. Mater. Today Proc. 48, 683–686, https://doi.org/10.1016/j.matpr.2021.08.092 (2022).
Han, Y., Rheem, Y., Lee, K.-H., Kim, H. & Myung, N. V. Synthesis and characterization of orthorhombic-MoO3 nanofibers with controlled morphology and diameter. J. Ind. Eng. Chem. 62, 231–238, https://doi.org/10.1016/j.jiec.2017.12.063 (2018).
Sreedhara, M. B., Matte, H. S. S. R., Govindaraj, A. & Rao, C. N. R. Synthesis, characterization, and properties of few-layer MoO3. Chem. –Asian J. 8, 2430–2435, https://doi.org/10.1002/asia.201300470 (2013).
Anjaneyulu, R. B., Mohan, B. S., Naidu, G. P. & Muralikrishna, R. Visible light enhanced photocatalytic degradation of methylene blue by ternary nanocomposite, MoO3/Fe2O3/rGO. J. Asian Ceram. Soc. 6, 183–195, https://doi.org/10.1080/21870764.2018.1479011 (2018).
Rahman, M.dH., Chowdhury, E. H. & Hong, S. High temperature oxidation of monolayer MoS2 and its effect on mechanical properties: a ReaxFF molecular dynamics study. Surf. Interfaces 26, 101371, https://doi.org/10.1016/j.surfin.2021.101371 (2021).
Reidy, K. et al. Atomic-scale mechanisms of MoS2 oxidation for kinetic control of MoS2/MoO3 interfaces. Nano Lett. 23, 5894–5901, https://doi.org/10.1021/acs.nanolett.3c00303 (2023).
Garadkar, K. M. et al. MoS2: preparation and their characterization. J. Alloy. Compd. 487, 786–789, https://doi.org/10.1016/j.jallcom.2009.08.069 (2009).
Bouzidi, A. et al. Effect of substrate temperature on the structural and optical properties of MoO3 thin films prepared by spray pyrolysis technique. Mater. Sci. Eng. B 97, 5–8, https://doi.org/10.1016/S0921-5107(02)00385-9 (2003).
Ali, S. & Farrukh, M. A. Effect of calcination temperature on the structural, thermodynamic, and optical properties of MoO3 nanoparticles. J. Chin. Chem. Soc. 65, 276–288, https://doi.org/10.1002/jccs.201700163 (2018).
Viršek, M., Jesih, A., Milošević, I., Damnjanović, M. & Remškar, M. Raman scattering of the MoS2 and WS2 single nanotubes. Surf. Sci. 601, 2868–2872, https://doi.org/10.1016/j.susc.2006.12.050 (2007).
Li, H. et al. From bulk to monolayer MoS2: evolution of Raman scattering. Adv. Funct. Mater. 22, 1385–1390, https://doi.org/10.1002/adfm.201102111 (2012).
Chakraborty, B., Matte, H. S. S. R., Sood, A. K. & Rao, C. N. R. Layer-dependent resonant Raman scattering of a few layer MoS2. J. Raman Spectrosc. 44, 92–96, https://doi.org/10.1002/jrs.4147 (2013).
Siciliano, T., Tepore, A., Filippo, E., Micocci, G. & Tepore, M. Characteristics of molybdenum trioxide nanobelts prepared by thermal evaporation technique. Mater. Chem. Phys. 114, 687–691, https://doi.org/10.1016/j.matchemphys.2008.10.018 (2009).
Diaz-Droguett, D. E., El Far, R., Fuenzalida, V. M. & Cabrera, A. L. In situ-Raman studies on thermally induced structural changes of porous MoO3 prepared in vapor phase under He and H2. Mater. Chem. Phys. 134, 631–638, https://doi.org/10.1016/j.matchemphys.2012.03.042 (2012).
Wang, L., Li, H. & Xue, Z. Synthesis of h-MoO3 nanorods and h-/α-MoO3 composites and their photocatalytic performance. Trans. Nonferrous Met. Soc. China 33, 2155–2167, https://doi.org/10.1016/S1003-6326(23)66250-8 (2023).
Lee, S.-H. et al. Raman spectroscopic studies of electrochromic a-MoO3 thin films. Solid State Ion-. 147, 129–133, https://doi.org/10.1016/S0167-2738(01)01035-9 (2002).
Yang, X., Tang, H., Zhang, R., Song, H. & Cao, K. Synthesis of high-quality crystalline α-MoO3 nanobelts. Cryst. Res. Technol. 46, 409–412, https://doi.org/10.1002/crat.201100061 (2011).
Sun, H. L. et al. One-step synthesis of centimeter-size alpha-MoO3 with single crystallinity. Appl. Surf. Sci. 476, 789–795, https://doi.org/10.1016/j.apsusc.2019.01.169 (2019).
Zakharova, G. S. et al. MoO3−δ nanorods: synthesis, characterization and magnetic properties. Solid State Sci. 9, 1028–1032, https://doi.org/10.1016/j.solidstatesciences.2007.07.022 (2007).
Chen, Y. et al. Single-crystalline orthorhombic molybdenum oxide nanobelts: synthesis and photocatalytic properties. CrystEngComm 12, 3740, https://doi.org/10.1039/c000744g (2010).
Wongkrua, P., Thongtem, T. & Thongtem, S. Synthesis of h- and α-MoO3 by refluxing and calcination combination: phase and morphology transformation, photocatalysis, and photosensitization. J. Nanomater. 2013, 1–8, https://doi.org/10.1155/2013/702679 (2013).
Xia, T., Li, Q., Liu, X., Meng, J. & Cao, X. Morphology-controllable synthesis and characterization of single-crystal molybdenum trioxide. J. Phys. Chem. B 110, 2006–2012, https://doi.org/10.1021/jp055945n (2006).
Guzman, G., Yebka, B., Livage, J. & Julien, C. Lithium intercalation studies in hydrated molybdenum oxides. Solid State Ion. 86–88, 407–413, https://doi.org/10.1016/0167-2738(96)00338-4 (1996).
Sen, S. K. et al. Characterization and antibacterial activity study of hydrothermally synthesized h-MoO3 nanorods and α-MoO3 nanoplates. BioNanoScience 9, 873–882, https://doi.org/10.1007/s12668-019-00671-7 (2019).
Hou, X. et al. Single-crystal MoO3 micrometer and millimeter belts prepared from discarded molybdenum disilicide heating elements. Sci. Rep. 8, 16771. https://doi.org/10.1038/s41598-018-34849-y (2018).
Aleksandrov, P. V. et al. Molybdenum recovery from molybdenite concentrates by low-temperature roasting with sodium chloride. Int. J. Miner. Process. 161, 13–20, https://doi.org/10.1016/j.minpro.2017.02.007 (2017).
Molybdenum Processing. https://www.imoa.info/molybdenum/molybdenum-processing.php.
Yi, G. et al. Recent progress on research of molybdenite flotation: a review. Adv. Colloid Interface Sci. 295, 102466, https://doi.org/10.1016/j.cis.2021.102466 (2021).
Huang, W., Tang, H., Cao, Y., Liu, R. & Sun, W. Separation of molybdenite from chalcopyrite with thiolactic acid depressant: flotation behavior and mechanism. Trans. Nonferrous Met. Soc. China 33, 3157–3167, https://doi.org/10.1016/S1003-6326(23)66324-1 (2023).
Yuan, D., Cadien, K., Liu, Q. & Zeng, H. Separation of talc and molybdenite: challenges and opportunities. Miner. Eng. 143, 105923, https://doi.org/10.1016/j.mineng.2019.105923 (2019).
Wang, L., Zhang, G., Dang, J. & Chou, K. Oxidation roasting of molybdenite concentrate. Trans. Nonferrous Met. Soc. China 25, 4167–4174, https://doi.org/10.1016/S1003-6326(15)64067-5 (2015).
Shalchian, H., Birloaga, I., Moghaddam, M. B., Nasiri, H. & Vegliò, F. “A hydrometallurgical process flowsheet for recovering MoO3 from Molybdenite. Hydrometallurgy 228, 106355, https://doi.org/10.1016/j.hydromet.2024.106355 (2024).
Fatheema, J. et al. A comprehensive investigation of MoO3 based resistive random access memory. RSC Adv. 10, 19337–19345, https://doi.org/10.1039/D0RA03415K (2020).
Sawa, A. Resistive switching in transition metal oxides. Mater. Today 11, 28–36, https://doi.org/10.1016/S1369-7021(08)70119-6 (2008).
Bharathi, M. et al. Effect of Ag doping on bipolar switching operation in molybdenum trioxide (MoO3) nanostructures for non-volatile memory. J. Alloy. Compd. 862, 158035, https://doi.org/10.1016/j.jallcom.2020.158035 (2021).
Xu, Y. et al. High-performance MoOx /n-Si heterojunction NIR photodetector with aluminum oxide as a tunneling passivation interlayer. Nanotechnology 32, 275502, https://doi.org/10.1088/1361-6528/abf37c (2021).
Noby, S. Z. et al. Oxygen vacancies in oxidized and reduced vertically aligned α-MoO3 nanoblades. Mater. Adv. 3, 3571–3581, https://doi.org/10.1039/D1MA00678A (2022).
Paparoni, F. et al. Unraveling oxygen vacancy formation, dynamics and site distribution in MoO3-x thin films. Mater. Chem. Phys. 333, 130317, https://doi.org/10.1016/j.matchemphys.2024.130317 (2025).
Lü, Z. et al. Ohmic contact and space-charge-limited current in molybdenum oxide modified devices. Phys. E Low.-Dimens. Syst. Nanostruct. 41, 1806–1809, https://doi.org/10.1016/j.physe.2009.07.003 (2009).
Wang, Z., Huang, H. & Guo, X. Memristive devices with multiple resistance states based on the migration of protons in α-MoO3/SrCoO2.5 stacks,. Adv. Electron. Mater. 7, 2001243, https://doi.org/10.1002/aelm.202001243 (2021).
Liu, D. & Robertson, J. Oxygen vacancy levels and interfaces of Al2O3. Microelectron. Eng. 86, 1668–1671, https://doi.org/10.1016/j.mee.2009.03.012 (2009).
Qin, J. et al. Improved resistive switching performance and mechanism analysis of MoO3 nanorods based memristors. Mater. Today Commun. 36, 106770, https://doi.org/10.1016/j.mtcomm.2023.106770 (2023).
Shen, S., Liu, Y., Gordon, R. G. & Brillson, L. J. Impact of ultrathin Al2O3 diffusion barriers on defects in high-k LaLuO3 on Si. Appl. Phys. Lett. 98, 172902, https://doi.org/10.1063/1.3583462 (2011).
Rahman, F. et al. Reversible resistive switching behaviour in CVD grown, large area MoOx. Nanoscale 10, 19711–19719, https://doi.org/10.1039/C8NR04407D (2018).
Rasool, A., Amiruddin, R., Mohamed, I. R. & Kumar, M. C. S. Fabrication and characterization of resistive random access memory (ReRAM) devices using molybdenum trioxide (MoO3) as switching layer. Superlattices Microstruct. 147, 106682, https://doi.org/10.1016/j.spmi.2020.106682 (2020).
Goul, R. et al. Atomic-scale tuning of ultrathin memristors. Commun. Phys. 5, 260, https://doi.org/10.1038/s42005-022-01037-4 (2022).
S. A. S & R. V. Review of memristor based neuromorphic computation: opportunities, challenges and applications. Eng. Res. Express 6, 032203 2024, https://doi.org/10.1088/2631-8695/ad6662 (2024).
Kim, S. J., Kim, S. & Jang, H. W. Competing memristors for brain-inspired computing. iScience 24, 101889, https://doi.org/10.1016/j.isci.2020.101889 (2021).
Acknowledgements
This work was supported by Khalifa University of Science and Technology under the project FSU-2023-010, 2D Materials for Space Applications. The authors gratefully acknowledge the financial and technical support provided by Khalifa University, as well as the facilities and resources made available by the Advanced Research and Innovation Center (ARIC), Research and Innovation Center for 2D Materials (RIC-2D) and INTRATOMIC. The authors thank the King Abdulla University of Science and Technology (KAUST) CoreLab facilities for allowing them to use electron microscopes to carry out the presented TEM analysis.
Author information
Authors and Affiliations
Contributions
R.E. and Y.A. contributed to the conceptualization and experimental design. R.E., S.S., I.A., and D.A. were responsible for the experimental fabrication and characterization of the materials. A.R. and A.N. were responsible for fabricating and analyzing the device. R.E., Y.A., H.B., and A.N. performed the formal analysis and data curation. R.E. wrote the original manuscript. Y.A., A.N., and H.B. supervised this study, including the revisions. Y.A. was responsible for acquiring the funds. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
A provisional patent application directed to this technology (63/571,840) has been filed by Khalifa University, but it is currently unlicensed. The inventors listed are Rami Elkaffas, Fahad Al Absi, and Yarjan Abdul Samad. The other authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. This study did not involve human participants, personal data, human tissue, or live vertebrate animals. Accordingly, approval from a human research ethics committee or an animal ethics committee was not required under institutional and national regulations. As no human participants were involved, informed consent to participate was not required.
Use of large language model (ChatGPT)
The authors used ChatGPT to rephrase and correct spelling and grammar errors to ensure a clearer depiction of the work. The authors ensured that the final manuscript agreed with the technical and scientific knowledge.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elkaffas, R., Rezk, A., Shajahan, S. et al. Microwave synthesis of gram scale millimeter size layered transition metal oxide crystals. NPG Asia Mater 18, 11 (2026). https://doi.org/10.1038/s41427-026-00637-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41427-026-00637-8
