
Abstract
Compared with bulk water, water at polymer interfaces is characterized by fundamentally different behaviors and is partitioned into free, intermediate, and nonfreezing states. These interfacial hydration states influence the initial interactions with proteins and cells, shaping downstream biological responses. Intermediate water plays a key role in controlling protein adsorption/desorption/denaturation and cell adhesion at biointerfaces, thereby contributing to enhanced biocompatibility. We describe a time-resolved investigation of water sorption onto polymer films using attenuated total reflection infrared spectroscopy coupled with data analysis facilitated by machine learning. Our findings highlight the role of intermediate water in modulating interfacial interactions. Furthermore, the intermediate water concept is extended to self-assembled monolayers (SAMs), which serve as well-defined model systems for polymer surfaces. This focus review also provides a comprehensive understanding of direct experimental evidence supporting the “water barrier model” of biocompatibility, starting from synthetic SAMs and progressing toward more complex biomimetic interfaces.
Similar content being viewed by others
Biocompatibility of biomedical devices
Material/protein/cell interactions at the biointerface
Biomaterials and biomedical devices designed for blood-contacting applications have been practically implemented for many years [1,2,3]. Biocompatibility is among the most critical issues in biomaterial/device design, yet the mechanisms of biocompatibility are not understood at the molecular level. This focus review primarily addresses the “biocompatibility” of materials against various biological elements in blood flow systems. Biocompatibility is defined as being synonymous with bioinertness, anti-fouling, and stealth properties. When biomaterials comprising these devices encounter blood, the body’s innate defense mechanisms recognize them as foreign [1,2,3]. Foreign body reactions via blood coagulation on biomaterial surfaces proceed through a sequence of events (Fig. 1): (1) adsorption of water molecules from blood, leading to material hydration; (2) nonspecific adsorption of plasma proteins and conformational changes in adsorbed proteins, exposing cell adhesion sites; (3) platelet adhesion; (4) platelet activation; and (5) subsequent blood cell deposition, fibrin network formation and thrombus formation on blood-contacting materials. Water plays a crucial role in biointerfacial interactions, including protein adsorption and cell adhesion on biomaterials [4,5,6,7,8], yet water is often neglected at the interface between materials and biology [9]. Since the 1970s, some studies have investigated the relationships among hydrated water, hydrated materials, and biocompatibility from various perspectives, such as water content, hydrophilicity, water mobility, hydration energy, water density and orientation, and polymer chain mobility [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24] (Fig. 2). However, the relationship between the state of hydration and biocompatibility has not been fully elucidated.

Initial events at the interface between biological fluids and materials. When biomaterials come into contact with blood/tissues, (1) water molecules immediately adsorb onto their surface. (2) This is followed by protein adsorption, protein restructuring, and denaturation of the adsorbed protein. (3) Cell adhesion. (4) Cell activation. (5) Fibrin formation and thrombus formation on blood-contacting materials

Water States Linking Biocompatibility. Representative articles on the relationship between biocompatibility and water states in terms of the water content, water mobility, hydration energy, water density arrangement, and hydration surface molecular mobility. The concept of intermediate water (IW) can bridge these articles
Role of hydration states in biocompatibility
Materials properties—wettability, surface roughness, and elastic modulus—are critical for influencing protein adsorption and cell adhesion. However, these factors alone do not fully account for the biological responses observed. We hypothesize that the water states around materials are critically important for determining their biocompatibility. Compared with bulk water, water at biointerfaces exhibits fundamentally different behavior. There are three types of distinct water surrounding synthetic and biological molecules (Table 1): (i) strongly bound water, in which the water molecules are strongly bound to materials and with restricted mobility such that the water is unable to freeze—termed nonfreezing water; (ii) loosely bound water, in which the water molecules are close to the surface of biomolecules with less restricted mobility than nonfreezing water; these water molecules can freeze and melt but at temperatures well below 0 °C—termed intermediate water; and (iii) scarcely bound water, in which the water molecules are free from materials and freeze at 0 °C—termed free water. We can experimentally determine these three types of water using calorimetry, spectroscopy, and direct interfacial methods, such as differential scanning calorimetry (DSC), time-resolved in situ attenuated total reflection infrared spectroscopy (ATR-IR), Raman spectroscopy, sum-frequency generation (SFG) spectroscopy, terahertz spectroscopy, dielectric relaxation spectroscopy, soft X-ray emission spectroscopy, quasielastic neutron scattering, solid-state nuclear magnetic resonance (NMR), surface force measurements, and a wide variety of analytical techniques [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50]. Among these, intermediate water was found in hydrated biopolymers and hydrated biocompatible synthetic polymers. Notably, intermediate water prevents the denaturation of proteins and facilitates cell adhesion on biointerfaces [51, 52]. However, the mechanism of how intermediate water is formed near materials is still elusive. Here, we discuss the fundamental concepts for determining the interactions of proteins and cells with hydrated materials along with selected examples corresponding to our recent studies [53, 54] on poly(2-methoxyethyl acrylate) (PMEA), PMEA derivatives, and self-assembled monolayers (SAMs).
Infrared spectroscopy of hydrated materials
Spectroscopic observations are promising techniques for investigating the detailed hydration structures of materials at the functional group level because of their sensitivity to the microscopic environment. Time-dependent attenuated total reflection infrared (ATR-IR) spectroscopic observations during the sorption process of water into a polymer film, as well as the subsequent data analysis based on machine learning, are described herein.
Time-dependent ATR-IR spectroscopy
Water has intense light absorption in the mid-infrared region. The absorption coefficients \(\alpha =4\pi k\left(\lambda \right)/\lambda\) of liquid water for O-H stretching at 3400 cm-1 and that for O-H bending at 1640 cm-1 are approximately 1.3 × 106 m-1 and 2.7 × 105 m-1, respectively, where \(k\left(\lambda \right)\) is an imaginary part of the refractive index as a function of wavelength \(\lambda .\)[55]. Therefore, transmission infrared (IR) spectroscopy for aqueous solutions requires controlling the optical path length to be approximately several micrometers, which can be difficult [56]. However, IR spectroscopy is a promising technique for investigating the molecular structures and interactions of compounds, including water molecules, because it reflects the microscopic environment around their chemical bonds [57,58,59]. Infrared spectroscopic observations of hydrated polymeric materials have also been well discussed [60,61,62,63]. We reported the hydration structures of PMEA and its analogous polymers by means of ATR-IR spectroscopy [64,65,66]. The equilibrium water content for PMEA is only 9.0 wt%, and three different types of hydrated water, namely, nonfreezing water, intermediate water and free water, were identified by differential scanning calorimetry (DSC) below 0 °C [67]. We captured the hydration structure of three different types of hydrated water in PMEA at ambient temperature by using spectroscopic techniques coupled with computational data analyses.
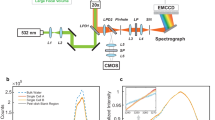
The ATR-IR spectra of dry and wet PMEA, measured using the flow-through cell shown in Fig. 3C, are shown in Fig. 3A[64]. A film sample of PMEA was deposited on a hemispherical internal reflection element (IRE) made of zinc selenide, which has a large refractive index of approximately 2.4 in the mid-infrared region. The thickness of the film sample was adjusted to exceed the penetration depth of the near-field light generated at the IRE/sample interface because of total internal reflection of the incident light. Therefore, the O-H stretching at approximately 3700–3000 cm-1 and the O-H bending at 1640 cm-1 arise from water sorbed into the polymer matrix, excluding information about bulk water in contact with the surface of the polymer film. The spectral waveform of the O-H stretching of water in PMEA looks somewhat different from that of bulk water. This suggests that the ATR-IR spectroscopic observations capture information about the hydration structure of water in the PMEA. Spectral intensities arising from PMEA below 1800 cm-1, excluding the band due to water at 1640 cm-1, decrease after contact with water. This is due to the expansion of the polymer film induced by water sorption.

ATR-IR Analysis of Water States in PMEA. A ATR-IR spectra of dry and wet PMEA, B time-dependent ATR-IR spectra obtained during a sorption process of liquid water (adapted from Morita et al., Langmuir 2007;23:3750-61, © 2007 American Chemical Society, with permission), C a schematic illustration of the in situ ATR-IR flow-through cell, D three-component spectra of water in PMEA deconvoluted by MCR, A nonfreezing water, intermediate water and C free water
To clarify the hydration structures of PMEA in detail, time-dependent ATR-IR spectra during the sorption process of water into the PMEA film were observed (Fig. 3B) [64]. The spectral waveform of O-H stretching in the 3700–3000 cm-1 region changes gradually with increasing spectral intensity. This is evidence of a change in the water structure in the PMEA with increasing water content. In the case of water vapor sorption into PMEA, all of the sorbed water is nonfreezing water [67], and the spectral waveform of the nonfreezing water has a relatively high wavenumber contribution of approximately 3600 cm-1[64].
Machine learning using spectroscopic data
To clarify the overlapping spectral waveforms attributed to the three different types of water in PMEA, multivariate curve resolution (MCR) and two-dimensional correlation spectroscopy (2DOS) have been applied to the time-dependent ATR-IR spectra in the O-H stretching region [68, 69]. The three types of waveforms of water in the PMEA deconvoluted by the MCR are shown in Fig. 3D. Nonfreezing water has a higher wavenumber contribution of approximately 3600 cm-1, which corresponds to the same waveform of water vapor sorbed into the PMEA (Fig. 3D, A). The spectral waveform of free water is somewhat similar to that of bulk water, with a broad peak at approximately 3400–3200 cm-1 (Fig. 3D, C). Intermediate water has a characteristic spectral waveform in the O-H stretching region, with a maximum peak at approximately 3400 cm-1 and few contributions at approximately 3200 cm−1 (Fig. 3D, B). Note that the spectral contribution at approximately 3200 cm-1 is generally observed in the ice Ih, which has a hexagonal crystal structure because of hydrogen bonds among water molecules. These findings suggest that compared with free water or bulk water, intermediate water forms a small water cluster with a locally formed ice Ih structure in the liquid phase. The characteristic waveform for intermediate water, featuring a remarkable peak at approximately 3400 cm-1 and a smaller contribution at approximately 3200 cm-1, appears only on the surface of a self-assembled monolayer (SAM) modified on a surface-enhanced infrared absorption (SEIRA) substrate, which suppresses nonspecific protein adsorption and cell adhesion [70]. These results imply that a surface hydration structure similar to that of the intermediate water in the PMEA is responsible for the material function in the SAM. Crucially, DSC measurements of the SAM are challenging, and SEIRA spectroscopic observations enabled evaluation of the hydration structure on the SAM surface, which is similar to that of the intermediate water in the PMEA.
In the fingerprint region below 1800 cm-1, which was attributed mainly to the polymer chain, a characteristic peak splitting was identified at 1727 cm-1 and assigned to C = O stretching. The positive lower wavenumber contribution at 1712 cm-1 is due to hydrogen-bonded C = O, whereas the negative higher wavenumber contribution at 1727 cm-1 is due to C = O being free from hydrogen bonding. Our kinetic analysis [64] revealed that the increase in the relaxation time of hydrogen-bonded C = O is associated with that of the spectral waveform attributed to the nonfreezing water deconvoluted by MCR. Quantum chemical calculations also support the observed vibrational frequencies of the C = O・・・HO type of hydrogen bonding interaction [58]. Furthermore, 85.6% of the nonfreezing water in PMEA exhibited C = O・・・HOH・・・O = C-type hydrogen bonding interactions, i.e., one water molecule hydrated to two carbonyl groups. This type of water hardly crystallizes among water molecules even below –100 °C because of the strong interactions with the polymer side chains as a bridge structure.
A higher wavenumber shift of the band at 1155 cm-1 arising from the methoxy moiety was also found [64]. The relaxation time of the shift is in good agreement with the intensity variation of the intermediate water deconvoluted by MCR [68]. These findings suggest that the intermediate water hydrated to the methoxy moiety in the PMEA side chain terminal has a relatively smaller water cluster than the free water or bulk water does.
The hydration structure of PMEA described above was obtained by analyzing a large number of spectra acquired by time-dependent ATR-IR spectroscopy during the sorption process of water using several kinds of computational data analyses, including unsupervised machine learning and quantum chemical calculations. Recently, this interpretation has been supported by molecular dynamics (MD) simulations [71]. Furthermore, a phase separation structure in hydrated PMEA consisting of a water-rich phase and a water-poor or polymer segment-rich phase was found in a study using a model monomer for the PMEA of 2-methoxyethyl acetate (MEAc) [72]. Sum-frequency generation (SFG) [73] and atomic force microscopy (AFM) [74] studies revealed that the hydration structure of the water/PMEA interface is concentrated approximately four times greater than that of the polymer matrix because of inhomogeneous phase separation [72].
Surface force measurements of hydrated materials
The intermediate water concept is also valid for explaining the biocompatibility of self-assembled monolayers (SAMs), which are characterized by their high molecular packing density and relatively rigid, ordered structures. This section systematically reviews the direct experimental evidence that validates the water barrier model, beginning with foundational synthetic SAMs and extending to more complex biomimetic interfaces.
Water barriers near bioinert self-assembled monolayers (SAMs)
The most direct and compelling evidence for the existence and function of a water barrier on anti-fouling SAMs has been obtained through surface force measurements [75,76,77,78,79,80,81]. Seminal studies on canonical bioinert systems, such as SAMs terminated with oligo(ethylene glycol) (OEG) or zwitterionic sulfobetaine (SB) moieties, have utilized colloidal probe atomic force microscopy (AFM) to quantify the interaction forces between two such surfaces in physiological environments (Fig. 4). These experiments, conducted in phosphate-buffered saline (PBS), revealed a long-range repulsive force that is absent on bioadhesive surfaces such as methyl-terminated SAMs, which instead show attractive hydrophobic interactions. This water-induced repulsion is characterized by a range of approximately 4–6 nm. The nature and range of this force were found to correlate directly with the resistance of SAMs to protein adsorption and their overall blood compatibility, providing the first strong evidence of a water-mediated barrier [50].

Chemical structures of the thiols used to fabricate the SAMs in this work (upper). SEM image of a colloidal probe for force measurements (lower left) and surface force–distance curves measured between the SAMs in PBS buffer (lower right). The range of the repulsive force was 4 to 6 nm, and half of the range (2 to 3 nm) was considered to be the thickness of the interfacial water inducing repulsion
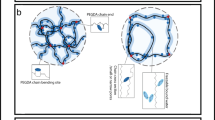
Definitive validation of this model was achieved through visualization of the hydration structure with frequency-modulation AFM (FM-AFM). This 3D high-resolution imaging method was employed to examine the interface of a bioinert mixed-charge (MC) SAM in PBS (Fig. 5). The findings demonstrated a highly ordered layer of interfacial water with an approximate physical thickness of 2 nm. This observation constitutes the first direct visual evidence of a structured hydration layer that was hypothesized to generate a repulsive force [82].

Chemical structure (upper left) and molecular packing configuration (upper right) of MC-SAMs. The molecular packing (in-plane imaging) (bottom left) and hydration structure (cross-sectional imaging) (bottom right) of interfacial water near a bioinert MC-SAM, as observed using frequency-modulated atomic force microscopy in PBS buffer. In the cross-sectional view, there was a region that induced a repulsive force to the AFM probe with a thickness of approximately 2 nm
Key insights were obtained when the physical thickness of the hydration layer was compared with its functional interaction range. The approximately 2 nm layer observed via FM-AFM represents the most tightly bound and structurally organized water molecules in the primary and secondary hydration shells of the SAM. However, the colloidal probe AFM detected a repulsive force at a much greater distance of 4–6 nm. This finding indicates that the influence of the SAM extends far beyond the physically bound water layer. The structured primary layer acts as a template, inducing some order in the surrounding water molecules and forming an extended hydrogen-bonding network. Therefore, the measured repulsive force reflects the energy needed to compress and disrupt this entire extended hydration region, not just the ~2 nm layer. Recognizing the difference between the physical thickness and the effective functional range of the water barrier is essential for understanding how these surfaces affect long-range biological interactions.
Water barriers in biomimetic systems
Chang et al. demonstrated this principle using zwitterionic peptide-based SAMs [82]. They fabricated SAMs composed of repeating dipeptide units of glutamic acid-lysine (EK) and aspartic acid-lysine (DK), which are the most abundant zwitterionic pairs found on the surfaces of proteins in the human body [83,84,85]. The biological performance of these surfaces significantly differed: the EK and DK SAMs exhibited excellent resistance to both protein adsorption and platelet adhesion, whereas the ER (R: arginine) and DR SAMs significantly fouled (Fig. 6). This dramatic difference in biological response was perfectly mirrored by the interfacial forces measured with AFM. The bioinert EK and DK SAMs generated a powerful, long-range repulsive force with an onset distance of 8–9 nm. In contrast, the bioadhesive ER and DR SAMs produced only weak, short-range repulsion with an onset of less than 4–5 nm.

(upper) Amino acid sequences of the peptides used to fabricate zwitterionic peptide SAMs. (lower left) Amounts of fibrinogen adsorbed on the peptide SAMs. (lower right) Approaching surface force–distance curves obtained with the peptide SAMs. Identical SAMs were fabricated on both the probe and the substrate. All the curves were measured in phosphate-buffered saline (PBS)
Although all four peptide SAMs are hydrophilic, with static water contact angles ranging from 27° to 34°, simple macroscopic wettability does not predict their bioinertness. The key difference lies in the nature of the repulsive force. The decay length of the repulsion for the fouled ER and DR SAMs in PBS was measured to be approximately 0.7 nm, closely matching the theoretical Debye length of the buffer (0.745 nm). This suggests that the force is mainly standard electrostatic double-layer repulsion. In contrast, the decay length for the anti-fouling EK and DK SAMs was approximately 1.8 nm, much longer than the Debye length, indicating a non-DLVO, water-mediated interaction. This finding shows that the ability to establish a strong, long-range hydration barrier depends heavily on the molecular structure of the amino acid side chains—specifically, how water interacts with the primary amine of lysine versus the guanidinium group of arginine. Recent molecular dynamics simulations support this, confirming that zwitterionic polymers create a robust hydration energy barrier that resists protein adsorption and that this ability is precisely tuned by molecular parameters such as the charge separation distance and dipole moment [86].
A similar demonstration of water-mediated interaction control was reported by Kanayama et al. using gold nanoparticles functionalized with double-stranded DNA (dsDNA) (Fig. 7a) [87]. The study revealed that the colloidal dispersion stability of these nanoparticles under high-salt conditions, where electrostatic repulsion is screened, is determined entirely by the pairing status of the outermost terminal base pair (Fig. 7b). Nanoparticles coated with dsDNA terminated in complementary base pairs spontaneously aggregated. This aggregation is driven by attractive hydrophobic interactions between the DNA ends, a phenomenon known as blunt-end stacking. In contrast, nanoparticles functionalized with dsDNA terminated in mismatched, unpairing base pairs remained stably dispersed under identical high-salt conditions. This macroscopic difference in behavior, governed by a single molecular detail, suggests that fundamentally different interfacial forces are at play.

Surface Forces of Hairpin DNA SAMs. a Hairpin structure-folded DNA strands were anchored on the gold surface through Au–S bond formation, resulting in the formation of homogenous dsDNA layers. b (upper) Base sequences and folded structures of hpDNA-SH. (lower) Photographs of the C- and M-DNA − GNP dispersions in sodium phosphate buffer (10 mM, pH 7.4) containing various concentrations of NaCl at 25 °C. c Approaching surface force–distance curves obtained with the DNA SAMs. Identical SAMs were fabricated on both the probe and the substrate. All the curves were measured in sodium phosphate buffer (10 mM, pH 7.4) containing various concentrations of NaCl
To elucidate the mechanism behind this terminal-specific behavior, surface force measurements were conducted between two dsDNA-coated surfaces using colloidal probe AFM (Fig. 7c). The results provide a direct link between the interfacial forces and the observed colloidal stability. For surfaces with complementary dsDNA, the interaction force transitioned from repulsive to strongly attractive as the salt concentration increased, with the attraction beginning at a distance of approximately 8 nm. These findings confirm that salt-facilitated blunt-end stacking is the driving force for aggregation [87, 88]. Conversely, for surfaces with mismatched dsDNA, a persistent, long-range repulsive force was measured, with an onset of approximately 8 nm. This repulsion was attributed to a hydration force, arising from the greater number of potential water-binding sites on the unpaired terminal nucleobases than on the fully paired ones. This robust hydration barrier prevents the nanoparticles from making contact, thus ensuring their colloidal stability and serving as a powerful example of how a single, discrete molecular change can fundamentally alter macroscopic interfacial forces by modulating the local hydration environment.
The consistent observation of non-DLVO repulsive forces across various anti-fouling systems from synthetic OEG and zwitterionic SAMs to biomimetic peptide and DNA monolayers indicates a common underlying physical cause. The “intermediate water concept” provides a solid theoretical foundation for explaining these experimental results. A key aspect of this model is that a high proportion of intermediate water at an interface is strongly linked to excellent blood compatibility and anti-fouling properties. In this framework, the long-range force measured by AFM can be viewed as the physical manifestation of this intermediate water layer. Although less ordered than the primary hydration shell, this layer has a unique hydrogen-bonding network different from that of bulk water, and compressing or displacing it requires a significant amount of energy [79, 89]. This idea establishes a crucial link between the microscopic state of interfacial water, measurable mesoscopic forces, and the broader biological effects associated with protein and cell resistance.
Concluding remarks and future perspective
Hydration water can be classified into three types: free water (scarcely bound water), intermediate water (loosely bound water), and nonfreezing water (tightly bound water). Among these, intermediate water was found in hydrated biopolymers and hydrated biocompatible synthetic polymers. Time-dependent ATR-IR spectroscopy combined with machine learning revealed the detailed hydration structures of PMEA at the functional group level. The results highlight the power of spectroscopic approaches to link molecular-level hydration to macroscopic material properties. We found that intermediate water is a key indicator of the biocompatibility of material surfaces under physiological conditions. The amount of intermediate water is influenced by the type of functional groups, local polymer configuration, and polymer chain mobility. Intermediate water also plays a key role in the bioinertness of SAMs and biomimetic surfaces. Long-range, water-mediated repulsive forces prevent protein adsorption, platelet adhesion, and overall biocompatibility. Molecular details, such as amino acid side chains or DNA terminal pairing, critically influence the strength and range of these hydration barriers. This water barrier model provides a unified framework for designing biocompatible interfaces.
This concept focuses on the presence and amount of intermediate water—a type of water common to both biopolymers functioning in aqueous environments and biocompatible synthetic polymers of nonbiological origin—and serves as a guiding principle for understanding and developing polymer properties and functions at the molecular level [90,91,92]. In the rational design of next-generation polymeric interfaces under complex biological conditions based on the intermediate water concept, we have demonstrated that subtle, systematic changes in molecular architecture can profoundly influence the bulk and surface properties and biological performance of polymers [7, 8, 20, 21, 52,53,54, 93,94,95,96,97,98,99,100,101]. For example, we successfully synthesized a series of several model polymers with identical elemental compositions but with the ester carbonyl group systematically repositioned along the side chain [53]. Our investigation revealed a clear relationship between the position of the carbonyl group, the resulting hydration states, the polymer dynamics under hydrated conditions, and the biocompatibility. A key finding is that compared with PMEA, poly[(2-acetoxyethyl)vinyl ether], in which the carbonyl group is located at the terminus of the side chain, is more resistant to platelet adhesion [53]. This superior performance is correlated with its unique hydration properties, including the largest amount of intermediate water, as quantified via DSC. The rational design of biocompatible polymers remains a significant challenge. Our work contributes a fundamental design principle, showing that precise control over the placement of a single functional group can be used to tune critical polymer properties that govern biological response. We believe these findings, which link a specific molecular architectural parameter to macroscopic physicochemical properties and ultimately to biological performance, will be of substantial interest in the fields of polymer chemistry, polymeric biomaterials science, and biomedical device engineering for novel biomedical interventions.
Traditionally, the development of biomaterials has relied largely on empirical screening strategies. In recent years, however, the introduction of the intermediate water concept has begun to redefine this paradigm. By providing a physiochemically grounded framework for characterizing biomaterial–water interactions, this approach enables more precise, quantitative, and rational screening of biomaterials, thereby improving both the efficiency and reliability of early-stage material selection. In fact, research and development are underway on molecular designs that achieve a balance between multiple functions, including not only the suppression of nonspecific adsorption and the prevention of fouling and adhesion but also the properties required in various fields where materials are used in aqueous environments—such as environmental, energy, electrical, electronic, mechanical, food, and agricultural applications. These functions include ionic conductivity, electrical properties, toughness, temperature responsiveness, biodegradability, selective adhesion to cancer cells, drug delivery systems, protein stabilization and antiaggregation properties, and pharmacological efficacy [102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141].
References
Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Biomaterials Science: An Introduction to Materials in Medicine. 3rd ed. Amsterdam: Academic Press/Elsevier; 2012.
Brash JL, Horbett TA, Latour RA, Tengvall P. The blood compatibility challenge. Part 2: Protein adsorption phenomena governing blood reactivity. Acta Biomater. 2019;94:11–24.
Ishihara K. Biomimetic polymers with phosphorylcholine groups as biomaterials for medical devices. Proc Jpn Acad Ser B. 2024;100:579–606.
Tsuruta T. On the role of water molecules in the interface between biological systems and polymers in special issue on the occasion of the 90th birthday of Prof. T. Tsuruta. J Biomater Sci Polym Ed. 2010;21:1827–48.
Tanaka M, Motomura T, Kawada M, Anzai T, Kasori Y, Shiroya T, et al. Blood compatible aspects of poly(2-methoxyethyl acrylate) (PMEA): Relationship between protein adsorption and platelet adhesion on PMEA surface. Biomaterials. 2000;21:1471–81.
Tanaka M, Mochizuki A. Clarification of the Blood Compatibility Mechanism by Controlling the Water Structure at the Blood-Poly(meth)acrylate Interface. J Biomater Sci Polym Ed. 2010;21:1849–63.
Tanaka M, Sato K, Kitakami E, Kobayashi S, Hoshiba T, Fukushima K. Design of biocompatible and biodegradable polymers based on intermediate water concept. Polym J. 2015;47:114–21.
Tanaka M, Kobayashi S, Murakami D, Aratsu F, Kashiwazaki A, Hoshiba T, et al. Design of Polymeric Biomaterials: The “Intermediate Water Concept. Bull Chem Soc Jpn. 2019;92:2043–57.
Dargaville BL, Hutmacher DW. Water as the often neglected medium at the interface between materials and biology. Nat Commun. 2022;13:4222–31.
Bruck SD. Aspects of three types of hydrogels for biomedical applications. J Biomed Mater Res. 1973;7:387–404.
Andrade JD, Lee HB, Kim JhonMS, Hibbs SW. JB Jr. Water as a biomaterial. Trans Am Soc Artif Intern Organs. 1973;19:1–7.
Ratner BD, Hoffman AS, Hanson SR, Harker LA, Whiffen JD. Blood-compatibility–water-content relationships for radiation-grafted hydrogels. J Polym Sci Polym Symp. 1979;66:363–75.
Yamada-Nosaka A, Ishikiriyama K, Todoki M, Tanzawa H. 1H-NMR studies on water in methacrylate hydrogels. J Appl Polym Sci. 1990;39:2443–52.
Lu DR, Lee SJ, Park K. Calculation of solvation interaction energies for protein adsorption on polymer surfaces. J Biomater Sci Polym Ed. 1992;3:127–47.
Israelachvili J, Wennerström H. Role of hydration and water structure in biological and colloidal interactions. Nature. 1996;379:219–25.
Feldman K, Hähner G, Spencer ND, Harder P, Grunze M. Probing resistance to protein adsorption of oligo(ethylene glycol)-terminated self-assembled monolayers by scanning force microscopy. J Am Chem Soc. 1999;121:10134–41.
Kataoka K, Ito H, Amano H, Nagasaki Y, Kato M, Tsuruta T, et al. Minimized platelet interaction with poly(2-hydroxyethyl methacrylate-block-4-bis(trimethylsilyl)methylstyrene) hydrogel showing anomalously high free water content. J Biomater Sci Polym Ed. 1998;9:111–29.
Ishihara K, Nomura H, Mihara T, Kurita K, Iwasaki Y, Nakabayashi N. Why do phospholipid polymers reduce protein adsorption? J Biomed Mater Res. 1998;39:323–30.
Morisaku T, Watanabe J, Konno T, Takai M, Ishihara K. Hydration of phosphorylcholine groups attached to highly swollen polymer hydrogels studied by thermal analysis. Polymer. 2008;49:4652–7.
Tanaka M, Mochizuki A, Ishii N, Motomura T, Hatakeyama T. Study of blood compatibility with poly(2-methoxyethyl acrylate): Relationship between water structure and platelet compatibility in poly(2-methoxyethyl acrylate-co-2-hydroxyethyl methacrylate). Biomacromolecules. 2002;3:36–41.
Tanaka M, Mochizuki A. Effect of water structure on blood compatibility: Thermal analysis of water in poly(meth)acrylate. J Biomed Mater Res A. 2004;68:684–95.
Berglin M, Andersson M, Sellborn A, Elwing H. The effect of substrate molecular mobility on surface-induced immune complement activation and blood plasma coagulation. Biomaterials. 2004;25:4581–90.
Zheng J, Li L, Tsao H-K, Sheng Y-J, Chen S, Jiang S. Strong repulsive forces between protein and oligo(ethylene glycol) self-assembled monolayers: A molecular simulation study. Biophys J. 2005;89:158–66.
Seo J-H, Kakinoki S, Inoue Y, Nam K, Yamaoka T, Ishihara K, et al. The significance of hydrated surface molecular mobility in the control of the morphology of adhering fibroblasts. Biomaterials. 2013;34:3206–14.
Hatakeyama T, Kishi A, Tanaka M. Comparison of measurement techniques for the identification of bound water restrained by polymers. Thermochim Acta. 2012;532:159–63.
Shiomoto S, Inoue K, Higuchi H, Nishimura SN, Takaba H, Tanaka M, et al. Characterization of hydration water bound to choline phosphate-containing polymers. Biomacromolecules. 2022;23:2999–3008.
Kojima C, Suzuki Y, Ikemoto Y, Tanaka M, Matsumoto A. Comparative study of PEG and PEGylated dendrimers in their eutectic mixtures with water analyzed using X-ray diffraction and infrared spectroscopy. Polym J. 2023;55:63–73.
Miwa Y, Ishida H, Tanaka M, Mochizuki A. H-2-NMR and C-13-NMR study of the hydration behavior of poly(2-methoxyethyl acrylate), poly(2-hydroxyethyl methacrylate) and poly(tetrahydrofurfuryl acrylate) in relation to their blood compatibility as biomaterials. J Biomater Sci Polym Ed. 2010;21:1911–24.
Miwa Y, Ishida H, Saito H, Tanaka M, Mochizuki A. Network structures and dynamics of dry and swollen poly(acrylate)s. Characterization of high- and low-frequency motions as revealed by suppressed or recovered intensities (SRI) analysis of C-13 NMR. Polymer. 2009;50:6091–9.
Morita S, Tanaka M. Effect of sodium chloride on hydration structures of PMEA and P(MPC-r-BMA). Langmuir. 2014;30:10698–703.
Kitano H, Tada S, Mori T, Takaha K, Gemmei-Ide M, Tanaka M, et al. Correlation between the structure of water in the vicinity of carboxybetaine polymers and their blood-compatibility. Langmuir. 2005;21:11932–40.
Ikemoto Y, Harada Y, Tanaka M, Nishimura S-n, Murakami D, Kurahashi N, et al. Infrared spectra and hydrogen-bond configurations of water molecules at the interface of water-insoluble polymers under humidified conditions. J Phys Chem B. 2022;126:4143–51.
Saleh MA, Higuchi Y, Shiomoto S, Anada T, Hishida M, Tanaka M. Effect of zwitterionic monomer polymerization on water dynamics: a molecular dynamics simulation study supported by differential scanning calorimetry and terahertz spectroscopy. Polym J. 2025;57:1127–39.
Hishida M, Anjum R, Anada T, Murakami D, Tanaka M. Effect of osmolytes on water mobility correlates with their stabilizing effect on proteins. J Phys Chem B. 2022;126:2466–75.
Ye S, Morita S, Li G, Noda H, Tanaka M, Uosaki K, et al. Structural changes in poly(2-methoxyethyl acrylate) thin films induced by absorption of bisphenol A. An infrared and sum frequency generation (SFG) study. Macromolecules. 2003;36:5694–703.
Hirata T, Matsuno H, Kawaguchi D, Hirai T, Yamada NL, Tanaka M, et al. Effect of local chain dynamics on a bioinert interface. Langmuir. 2015;31:3661–7.
Hirata T, Matsuno H, Kawaguchi D, Yamada NL, Tanaka M, Tanaka K. Effect of interfacial structure on bioinert properties of poly(2-methoxyethyl acrylate)/poly(methyl methacrylate) blend films in water. Phys Chem Chem Phys. 2015;17:17399–405.
Hirata T, Matsuno H, Kawaguchi D, Yamada NL, Tanaka M, Tanaka K. Construction of a blood-compatible interface based on surface segregation in a polymer blend. Polymer. 2015;78:219–24.
Nishimura S-n, Kurahashi N, Shiomoto S, Harada Y, Tanaka M. Effects of hydration water on bioresponsiveness of polymer interfaces revealed by analysis of linear and cyclic polymer-grafted substrates. Soft Matter. 2024;20:9454–63.
Higaki Y, Masuda T, Shiomoto S, Tanaka Y, Kiuchi H, Harada Y, et al. Pronounced cold crystallization and hydrogen bonding distortion of water confined in microphases of double zwitterionic block copolymer aqueous solutions. Langmuir. 2024;40:19612–18.
Murakami D, Yamazoe K, Nishimura S-n, Kurahashi N, Ueda T, Miyawaki J, et al. Hydration mechanism in blood-compatible polymers undergoing phase separation. Langmuir. 2022;38:1090–98.
Hirata T, Matsuno H, Kawaguchi D, Inutsuka M, Hirai T, Tanaka M, et al. Dynamics of a bioinert polymer in hydrated states by dielectric relaxation spectroscopy. Phys Chem Chem Phys. 2017;19:1389–94.
Kanamaru T, Araki M, Takahashi R, Fujii S, Shikata T, Murakami D, et al. First observation of the hydration layer around polymer chain by scattering and its relationship to thromboresistance: dilute solution properties of PMEA in THF/water. J Phys Chem B. 2021;125:7251–61.
Nakada M. Low-temperature behaviors, cold crystallization, and glass transition in poly(vinylpyrrolidone) aqueous solution. J Phys Chem B. 2023;127:10556–63.
Kishi A, Tanaka M, Mochizuki A. Comparative study on water structures in polyHEMA and polyMEA by XRD-DSC simultaneous measurement. J Appl Polym Sci. 2009;111:476–81.
Fujii Y, Tominaga T, Murakami D, Tanaka M, Seto H. Local dynamics of the hydration water and poly(methyl methacrylate) chains in PMMA networks. Front Chem. 2021;9:728738.
Kikuchi T, Tominaga T, Murakami D, de Souza NR, Tanaka M, Seto H. Detailed dynamical features of the slow hydration water in the vicinity of poly(ethylene oxide) chains. J Chem Phys. 2024;160:064902.
Tominaga T, Hishida M, Murakami D, Fujii Y, Tanaka M, Seto H. Experimental evidence of slow mode water in the vicinity of poly(ethylene oxide) at physiological temperature. J Phys Chem B. 2022;126:1758–67.
Higaki Y, Toyama H, Masuda T, Kobayashi S, Tanaka M. Microphase separation of double-hydrophilic poly(carboxybetaine acrylate)-poly(2-methoxyethyl acrylate) block copolymers in water. Polym J. 2023;55:1357–65.
Hayashi T, Tanaka Y, Koide Y, Tanaka M, Hara M. Mechanism underlying bioinertness of self-assembled monolayers of oligo(ethyleneglycol)-terminated alkanethiols on gold: Protein adsorption, platelet adhesion, and surface forces. Phys Chem Chem Phys. 2012;14:10196–206.
Nishida K, Anada T, Tanaka M. Roles of interfacial water states on advanced biomedical material design. Adv Drug Deliv Rev. 2022;186:114310.
Nishimura S-n, Tanaka M. The Intermediate Water Concept for Pioneering Polymeric Biomaterials: A Review and Update. Bull Chem Soc Jpn. 2023;96:1052–70.
Kobayashi S, Okazaki Y, Tanaka M. Impact of side chain carbonyl group position on polymer hydration, dynamics, and antithrombotic activity: A systematic study of structure-property relationships. Macromolecules. 2025;58:8821–32.
Cho IS, Shiomoto S, Yukawa N, Tanaka Y, Huh KM, Tanaka M. The role of intermediate water in enhancing blood and cellular compatibility of chitosan-based biomaterials. Langmuir. 2025;41:8301–11.
Hale GM, Querry MR. Optical constants of water in the 200-nm to 200-μ m wavelength region. Appl Opt. 1973;12:555–63.
Tajiri T, Morita S, Ozaki Y. Time-resolved conformational analysis of poly(ethylene oxide) during the hydrogelling process. Polymer. 2011;52:5560–6.
Ohno K, Okimura M, Akai N, Katsumoto Y. The effect of cooperative hydrogen bonding on the oh stretching-band shift for water clusters studied by matrix-isolation infrared spectroscopy and density functional theory. Phys Chem Chem Phys. 2005;7:3005–14.
Morita S, Kitagawa K, Ozaki Y. Hydrogen-bond structures in poly(2-hydroxyethyl methacrylate): Infrared spectroscopy and quantum chemical calculations with model compounds. Vibr Spectrosc. 2009;51:28–33.
Morita S. Hydrogen-bonds structure in poly(2-hydroxyethyl methacrylate) studied by temperature-dependent infrared spectroscopy. Front Chem. 2014;2:1–5.
Yarwood J, Sammon C, Mura C, Pereira M. Vibrational spectroscopic studies of the diffusion and perturbation of water in polymeric membranes. J Mol liq. 1999;80:93–115.
Ide M, Mori T, Ichikawa K, Kitano H, Tanaka M, Mochizuki A, et al. Structure of water sorbed into poly (MEA-co-HEMA) films as examined by atr-ir spectroscopy. Langmuir. 2003;19:429–35.
Tajiri T, Morita S, Ozaki Y. Hydration mechanism on a poly(methacrylic acid) film studied by in situ attenuated total reflection infrared spectroscopy. Polymer. 2009;50:5765–70.
Morita S, Kitagawa K. Temperature-dependent structure changes in nafion ionomer studied by pcmw2d ir correlation spectroscopy. J Mol Struct. 2010;974:56–9.
Morita S, Tanaka M, Ozaki Y. Time-resolved in situ atr-ir observations of the process of sorption of water into a poly(2-methoxyethyl acrylate) film. Langmuir. 2007;23:3750–61.
Tanaka M, Hayashi T, Morita S. The roles of water molecules at the biointerface of medical polymers. Polym J. 2013;45:701–10.
Tanaka M, Morita S, Hayashi T. Role of interfacial water in determining the interactions of proteins and cells with hydrated materials. Colloids Surf B Biointerfaces. 2021;198:111449.
Tanaka M, Motomura T, Ishii N, Shimura K, Onishi M, Mochizuki A, et al. Cold crystallization of water in hydrated poly(2-methoxyethyl acrylate) (pmea). Polym Inter. 2000;49:1709–13.
Tanabe A, Morita S, Tanaka M, Ozaki Y. Multivariate curve resolution analysis on the multi-component water sorption process into a poly(2-methoxyethyl acrylate) film. Appl Spectrosc. 2008;62:46–50.
Morita S, Tanaka M, Noda I, Ozaki Y. Phase angle description of perturbation correlation analysis and its application to time-resolved infrared spectra. Appl Spectrosc. 2007;61:867–72.
Sekine T, Asatyas S, Sato C, Morita S, Tanaka M, Hayashi T. Surface force and vibrational spectroscopic analyses of interfacial water molecules in the vicinity of methoxy-tri (ethylene glycol)-terminated monolayers: Mechanisms underlying the effect of lateral packing density on bioinertness. J Biomater Sci Polym Ed. 2017;28:1231–43.
Shikata K, Kikutsuji T, Yasoshima N, Kim K, Matubayasi N Revealing the hidden dynamics of confined water in acrylate polymers: Insights from hydrogen-bond lifetime analysis. J. Chem. Phys. 2023;158:174901.
Morita S, Tanaka M, Kitagawa K, Ozaki Y. Hydration structure of poly(2-methoxyethyl acrylate): Comparison with a 2-methoxyethyl acetate model monomer. J Biomater Sci Polym Ed. 2010;21:1925–35.
Li GF, Ye S, Morita S, Nishida T, Osawa M. Hydrogen bonding on the surface of poly(2-methoxyethyl acrylate). J Am Chem Soc. 2004;126:12198–9.
Murakami D, Nishimura S-n, Tanaka Y, Tanaka M. Observing the repulsion layers on blood-compatible polymer-grafted interfaces by frequency modulation atomic force microscopy. Biomater Adv. 2022;133:112596.
Chang R, Asatyas S, Lkhamsuren G, Hirohara M, Mondarte EAQ, Suthiwanich K, et al. Water near bioinert self-assembled monolayers. Polym J. 2018;50:563–71.
Hayashi T. Prof. George whitesides’ contributions to self-assembled monolayers (sams): Advancing biointerface science and beyond. Chemistry-Switzerland. 2025;7:9–24.
Hayashi T, Hara M. Nonfouling self-assembled monolayers: Mechanisms underlying protein and cell resistance. Cur Phy Chem. 2011;1:90–8.
Hayashi T, Latag GV. Self-assembled monolayers as platforms for nanobiotechnology and biointerface research: Fabrication, analysis, mechanisms, and design. ACS Appl Nano Mater. 2025;8:8570–87.
Maeda S, Chikami S, Song S, Balois-Oguchi MV, Katase A, Latag GV, et al. Gap-controlled infrared absorption spectroscopy: A unique interface-sensitive spectroscopy based on the combination of linear spectroscopy and multivariate curve resolution. Anal Chem. 2025;97:20156–63.
Sekine T, Asatyas S, Sato C, Morita S, Tanaka M, Hayashi T. Surface force and vibrational spectroscopic analyses of interfacial water molecules in the vicinity of methoxy-tri(ethylene glycol)-terminated monolayers: Mechanisms underlying the effect of lateral packing density on bioinertness. J Biomater Sci Polym Ed. 2017;28:1231–43.
Sekine T, Tanaka Y, Sato C, Tanaka M, Hayashi T. Evaluation of factors to determine platelet compatibility by using self-assembled monolayers with a chemical gradient. Langmuir. 2015;31:7100–5.
Araki Y, Sekine T, Chang R, Hayashi T, Onishi H. Molecular-scale structures of the surface and hydration shell of bioinert mixed-charged self-assembled monolayers investigated by frequency modulation atomic force microscopy. RSC Adv. 2018;8:24660–4.
Hu G, Moon J, Hayashi T. Protein classes predicted by molecular surface chemical features: Machine learning-assisted classification of cytosol and secreted proteins. J Phys Chem B. 2024;128:8423–36.
Moon J, Hu G, Hayashi T. Application of machine learning in the quantitative analysis of the surface characteristics of highly abundant cytoplasmic proteins: Toward ai-based biomimetics. Biomimetics. 2024;9:162.
White AD, Nowinski AK, Huang WJ, Keefe AJ, Sun F, Jiang SY. Decoding nonspecific interactions from nature. Chem Sci. 2012;3:3488–94.
Cui Z, Wang Y, Zhang L, Qi H. Zwitterionic peptides: From mechanism, design strategies to applications. ACS Appl Mater Interfaces. 2024;16:56497–518.
Kanayama N, Sekine T, Ozasa K, Kishi S, Nyu T, Hayashi T, et al. Terminal-specific interaction between double-stranded DNA layers: Colloidal dispersion behavior and surface force. Langmuir. 2016;32:13296–304.
Sekine T, Kanayama N, Ozasa K, Nyu T, Hayashi T, Maeda M. Stochastic binding process of blunt-end stacking of DNA molecules observed by atomic force microscopy. Langmuir. 2018;34:15078–83.
Ang A, Maeda S, Chikami S, Hayashi T. Analysing the correlation between the water’s OH stretching band and its hydrogen bonding configurations by machine learning. Phys Chem Chem Phys. 2025;27:21083–97.
Hatakeyama T, Tanaka M, Hatakeyama H. Studies on bound water restrained by poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC): Comparison of the polysaccharides-water systems. Acta Biomater. 2010;6:2077–82.
Hatakeyama T, Tanaka M, Hatakeyama H. Thermal properties of freezing bound water restrained by polysaccharides. J Biomater Sci Polym Ed. 2010;21:1865–75.
Kumar A, Sood A, Agrawal G, Thakur S, Thakur VK, Tanaka M, et al. Polysaccharides, proteins, and synthetic polymers based multimodal hydrogels for various biomedical applications: A review. Int J Biol Macromol. 2023;247:125606.
Suzuki T, Konishi H, Suzuki A, Katsumata T, Fukuda Y, Miyamoto K, et al. Role of intermediate water in reducing postsurgical intrapericardial adhesion. Surg Today. 2025;55:847–56.
Tsujimoto H, Uehara H, Yoshida M, Nishio K, Furuta T, Inui T, et al. Different hydration states and passive tumor targeting ability of polyethylene glycol-modified dendrimers with high and low PEG density. Mater Sci Eng C. 2021;126:112159.
Fujiura K, Naito M, Tanaka Y, Tanaka M, Nakanishi Y, Ejima H, et al. Development of stealth nanoparticles coated with poly(2-methoxyethyl vinyl ether) as an alternative to poly(ethylene glycol). J Appl Polym Sci. 2024;141:e55044.
Toyokawa Y, Kobayashi S, Tsuchiya H, Shibuya T, Aoki M, Sumiya J, et al. A fully covered self-expandable metallic stent coated with poly(2-methoxyethyl acrylate) and its derivative: In vitro evaluation of early-stage biliary sludge formation inhibition. Mater Sci Eng C. 2021;120:111386.
Ishizawa T, Makino N, Kakizaki Y, Matsuda A, Toyokawa Y, Ooyama S, et al. Biosafety of a novel covered self-expandable metal stent coated with poly(2-methoxyethyl acrylate) in vivo. PLoS ONE. 2021;16:e0257828.
Nomura M, Yokoyama Y, Yoshimura D, Minagawa Y, Yamamoto A, Tanaka Y, et al. Simple detection and culture of circulating tumor cells from colorectal cancer patients using poly(2-methoxyethyl acrylate)-coated plates. Int J Mol Sci. 2023;24:3949.
Kobayashi S, Sugasaki A, Yamamoto Y, Shigenoi Y, Udaka A, Yamamoto A, et al. Enrichment of cancer cells based on antibody-free selective cell adhesion. ACS Biomater Sci Eng. 2022;8:4547–56.
Nishimura S-n, Nishida K, Shiomoto S, Tanaka M. Surfactant-free suspension polymerization of hydrophilic monomers with an oil-in-water system for the preparation of microparticles toward the selective isolation of tumor cells. Mater Adv. 2022;3:5043–54.
Nishida K, Nishimura S-n, Tanaka M. Selective accumulation to tumor cells with coacervate droplets formed from a water-insoluble acrylate polymer. Biomacromolecules. 2022;23:1569–80.
Lee W, Kobayashi S, Nagase M, Jimbo Y, Saito I, Inoue Y, et al. Nonthrombogenic, stretchable, active multielectrode array for electroanatomical mapping. Sci Adv. 2018;4:eaau2426.
Lee W, Jeong SH, Lim YW, Lee H, Kang J, Lee H, et al. Conformable microneedle pH sensors via the integration of two different siloxane polymers for mapping peripheral artery disease. Sci Adv. 2021;7:eabi6290.
Huang JJ, Lin CH, Tanaka Y, Yamamoto A, Luo SC, Tanaka M Manipulation of surface hydration states by tuning the oligo(ethylene glycol) moieties on PEDOT to achieve platelet-resistant bioelectrode applications. Adv Mater Interfaces. 2022;22:2200707.
Araki T, Yoshida F, Uemura T, Noda Y, Yoshimoto S, Kaiju T, et al. Long-term implantable, flexible, and transparent neural interface based on Ag/Au core–shell nanowires. Adv Healthcare Mater. 2019;8:1900130.
Nishida K, Ikura R, Yamaoka K, Urakawa O, Konishi T, Inoue T, et al. Relation between the water content and mechanical properties of hydrogels with movable cross-links. Macromolecules. 2024;57:7745–54.
Park J, Ueda T, Kawai Y, Araki K, Kido M, Kure B, et al. Simultaneous control of the mechanical properties and adhesion of human umbilical vein endothelial cells to suppress platelet adhesion on a supramolecular substrate. RSC Adv. 2022;12:27912–7.
Osaki M, Yonei S, Ueda C, Ikura R, Park J, Yamaguchi H, et al. Mechanical properties with respect to water content of host-guest hydrogels. Macromolecules. 2021;54:8067–76.
Okada M, Xie SC, Kobayashi Y, Yanagimoto H, Tsugawa D, Tanaka M, et al. Water-mediated on-demand detachable solid-state adhesive of porous hydroxyapatite for internal organ retractions. Adv Healthcare Mater. 2024;13:2304616.
Okada M, Hara E, Kobayashi D, Kai S, Ogura K, Tanaka M, et al. Intermediate water on calcium phosphate minerals: Its origin and role in crystal growth. ACS Appl Bio Mater. 2019;2:981–6.
Mabrouk M, Beherei HH, Tanaka Y, Tanaka M. Sol-gel silicate glass doped with silver for bone regeneration: Antibacterial activity, intermediate water, and cell death mode. Biomater Adv. 2022;138:212965.
Mabrouk M, Beherei HH, Shiomoto S, Tanaka Y, Osama L, Tanaka M. Effect of titanium-doped bioactive glass on poly(2-hydroxyethyl methacrylate) hydrogel composites: Bioactivity, intermediate water, cell proliferation, and adhesion force. Ceram Int. 2023;49:13469–81.
Mabrouk M, Beherei HH, Tanaka Y, Tanaka M. Investigating the intermediate water feature of hydrated titanium containing bioactive glass. Int J Mol Sci. 2021;22:8038.
Nishimura S-n, Nishida K, Ueda T, Shiomoto S, Tanaka M. Biocompatible poly(N-(ω-acryloyloxy-n-alkyl)-2-pyrrolidone)s with widely-tunable lower critical solution temperatures (LCSTs): a promising alternative to poly(N-isopropylacrylamide). Polym Chem. 2022;13:2519–30.
Koguchi R, Jankova K, Tanaka M. Fluorine-containing bio-inert polymers: Roles of intermediate water. Acta Biomater. 2022;138:34–56.
Sonoda T, Kobayashi S, Tanaka M. Periodically functionalized linear polyethylene with tertiary amino groups via regioselective ring-opening metathesis polymerization. Macromolecules. 2021;54:2862–72.
Liu S, Kobayashi S, Sonoda T, Tanaka M. Poly(tertiary amide acrylate) copolymers inspired by poly(2-oxazoline)s: Their blood compatibility and hydration states. Biomacromolecules. 2021;22:2718–28.
Liu S, Kobayashi S, Nishimura S-n, Ueda T, Tanaka M. Effect of pendant groups on the blood compatibility and hydration states of poly(2-oxazoline)s. J Polym Sci. 2021;59:2559–70.
Anjum R, Nishimura S, Kobayashi S, Nishida K, Anada T, Tanaka M. Protein stabilization effect of zwitterionic osmolyte-bearing polymer. Chem Lett. 2021;50:1699–702.
Sonoda T, Kobayashi S, Herai K, Tanaka M. Side-chain spacing control of derivatives of poly(2-methoxyethyl acrylate): Impact on hydration states and antithrombogenicity. Macromolecules. 2020;53:8570–80.
Koguchi R, Jankova K, Hayasaka Y, Kobayashi D, Amino Y, Miyajima T, et al. Understanding the effect of hydration on the bio-inert properties of 2-hydroxyethyl methacrylate copolymers with small amounts of amino- or/and fluorine-containing monomers. ACS Biomater Sci Eng. 2020;6:2855–66.
Koguchi R, Jankova K, Tanabe N, Amino Y, Hayasaka Y, Kobayashi D, et al. Controlling the hydration structure with a small amount of fluorine to produce blood compatible fluorinated poly(2-methoxyethyl acrylate). Biomacromolecules. 2019;20:2265–75.
Koguchi R, Jankova K, Tanaka Y, Yamamoto A, Murakami D, Yang Q, et al. Altering the Bio-inert Properties of Surfaces by Fluorinated Copolymers of mPEGMA. Biomater Adv. 2023;153:213573.
Yoshikawa C, Hattori S, Huang C-F, Kobayashi H, Tanaka M. In vitro and in vivo blood compatibility of concentrated polymer brushes. J Mater Chem B. 2021;9:5794–804.
Jankova K, Javakhishvili I, Kobayashi S, Koguchi R, Murakami D, Sonoda T, et al. Hydration states and blood compatibility of hydrogen-bonded supramolecular poly(2-methoxyethyl acrylate). ACS Appl Bio Mater. 2019;2:4154–61.
Sato K, Kobayashi S, Sekishita A, Wakui M, Tanaka M. Synthesis and thrombogenicity evaluation of poly(3-methoxypropionic acid vinyl ester): A candidate for blood-compatible polymers. Biomacromolecules. 2017;18:1609–16.
Kobayashi S, Wakui M, Iwata Y, Tanaka M. Poly(ω-methoxyalkyl acrylate)s: Nonthrombogenic polymer family with tunable protein adsorption. Biomacromolecules. 2017;18:4214–23.
Kobayashi S, Fukuda K, Kataoka M, Tanaka M. Regioselective ring-opening metathesis polymerization of 3-substituted cyclooctenes with ether side chains. Macromolecules. 2016;49:2493–501.
Sato K, Kobayashi S, Kusakari M, Watahiki S, Oikawa M, Hoshiba T, et al. The relationship between water structure and blood compatibility in poly(2-methoxyethyl acrylate) (PMEA) analogues. Macromol Biosci. 2015;15:1296–303.
Fukushima K, Inoue Y, Haga Y, Ota T, Honda K, Sato C, et al. Monoether-tagged biodegradable polycarbonate preventing platelet adhesion and demonstrating vascular cell adhesion: A promising material for resorbable vascular grafts and stents. Biomacromolecules. 2017;18:3834–43.
Nishimura S-n, Ueda T, Murakami D, Tanaka M. Chain-end effect for intermediate water formation of poly(2-methoxyethyl acrylate). Org Mater. 2021;3:214–20.
Nishimura S-n, Ueda T, Kobayashi S, Tanaka M. Silsesquioxane/poly(2-methoxyethyl acrylate) hybrid with both antithrombotic and endothelial cell adhesive properties. ACS Appl Polym Mater. 2020;2:4790–801.
Fukushima K, Hakozaki S, Lang R, Haga Y, Nakai S, Narumi A, et al. Hydrolyzable and biocompatible aliphatic polycarbonates with ether functionalized side chains attached via amide linker. Polym J. 2024;56:431–42.
Hayakawa N, Nishiura M, Anada T, Kobayashi S, Sawada T, Serizawa T, et al. Suspension culture system for isolating cancer spheroids using enzymatically synthesized cellulose oligomers. ACS Appl Bio Mater. 2024;7:306–14.
Kobayashi S, Tanaka M. Design of biomaterials through direct ring-opening metathesis polymerisation of functionalised cyclic alkenes. Mol Syst Des Eng. 2023;8:960–91.
Wen P, Ke W, Dirisala A, Toh K, Tanaka M, Li J. Stealth and pseudo-stealth nanocarriers. Adv Drug Deliv Rev. 2023;198:114895.
Higuchi Y, Saleh MA, Anada T, Tanaka M, Hishida M. Rotational dynamics of water near osmolytes by molecular dynamics simulations. J Phys Chem B. 2024;128:5008–17.
Kawai Y, Park J, Motokawa R, Ikura R, Murayama S, Yamaoka K, et al. Effects of the hydration states of water molecules on the mechanical properties of dual movable crosslinked polymeric gels. ACS Appl Polym Mater. 2025;7:7767–76.
Anada T, Kawahara M, Shimada T, Kuroda R, Okamura H, Setoyama D, et al. A nucleic acid prodrug that activates mitochondrial respiration, promotes stress resilience, and prolongs lifespan. J Am Chem Soc. 2025;147:22161–75.
Kawahara M, Miyazaki K, Anada T, Kobayashi S, Tanaka M Engineering three-dimensional cellular organization by regulating bound water-mediated cell–substrate interactions for disease modeling. ACS Omega. 2025;10:54951–66.
Li J, Toh K, Wen P, Liu X, Dirisala A, Guo H, et al. Steric stabilization-independent stealth cloak enables nanoreactors-mediated starvation therapy against refractory Cancer. Nat Biomed Eng. https://doi.org/10.1038/s41551-025-01534-1 2025.
Acknowledgements
The authors gratefully acknowledge the financial support from JSPS KAKENHI grant numbers 19H05720 22H00591 and 24K22382 (for M.T.), JP24K08531 (for S.M.), JP22H04530 and JP23H04059 (for T.H.) and Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from Japan Science and Technology Agency (JST) Grant Number JPMJTR22T7 (for M.T.). This work was supported by the Japan–Taiwan Exchange Association and the Innovative Science and Technology Initiative for Security (ISTIS), the Acquisition, Technology & Logistics Agency (ATLA) (JPJ013268), Japan (for T.H.). This study was partially supported by the Dynamic Alliance for Open Innovation Bridging Human, Environment, and Materials and the Integrated Research Consortium on Chemical Sciences, CORE Lab (T.H.) and CORE2 A lab (M.T and T.H.).
Author information
Authors and Affiliations
Contributions
M.T., S.M., and T.H. contributed to the conceptualization, planning, writing, and editing.
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tanaka, M., Morita, S. & Hayashi, T. Interfacial water states and the biocompatibility of biomaterials: The role of intermediate water. Polym J 58, 343–356 (2026). https://doi.org/10.1038/s41428-025-01139-0
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41428-025-01139-0
