
Abstract
Risk exists throughout medicine. Understanding health system pressure points permits implementation of controls for risk reduction. The literature lacks a systematic approach to risk evaluation in Clinical Genetics. We aimed to develop Clinical Genetics-specific risk assessment tools to prospectively monitor risk. A retrospective review of 115 cases with identified adverse events or near misses in Clinical Genetics in Ireland was used to design a process map to define the steps where risk occurs across the patient journey through clinical genetics. We piloted a clinical audit form using the process map to capture risk event frequency. The draft process map and audit form were trialled (2022–2023) in 5 other European clinical genetics centres for validity and usability, and 2 re-audited in 2024 to assess utility. Using narrative summaries from the case review, we modified the national health risk severity scoring for clinical genetics use. The design of the risk process map, risk frequency audit and severity assessment align with Failure Modes and Effect Analysis methodology. Adverse events occurred in >3% of appointments in 4 of 6 centres (range 0.8–20.3%). High frequency failure modes varied by centre and included consent, sample processing, and patient discussion. Re-audit results reflected interventions introduced since initial audit. We propose these tools provide a standardized approach to discussing systematic risk in clinical genetics, and can be used to prospectively monitor adverse events, allowing controls to be put in place, reducing risk, thereby improving quality of service.
Similar content being viewed by others


Introduction
The Irish Health Service Executive (HSE) defines risk as ‘the effect of uncertainty on our objectives. Risk management is therefore about how we manage those uncertainties to give us the best chance of success in meeting our objectives.’ [1]. It is fundamental to proactively manage, anticipate, and respond to risk ensuring safe delivery of care [1,2,3]. Risk management measures can lead to quality improvement, defined as the combined and unceasing efforts of healthcare professionals, patients, and families to make changes that will lead to better patient outcomes, better experiences of care, and continued development and supported staff [4].
What is risk in clinical genetics? The World Health Organisation describes Clinical Genetics as a preventative specialty [5]. As such, the consequences or impact of any risk event may not be evident for years or generations. Notably, in Clinical Genetics, compared to other specialities, the impact of an adverse event may extend beyond the individual to impact the family [6]. In addition to minor harms, severe harms in Clinical Genetics include a recurrence of an inherited condition that might have been avoided had the family been informed of the risk, the occurrence of a cancer that might have been prevented, or disability due to a delay in medical treatment or dietary restrictions. More recently, misinterpretation of complex genetic test results has resulted in high-profile cases of harm because of interventions that were subsequently shown not to be indicated, e.g., terminations of pregnancy [7] or prophylactic mastectomies for DNA variants that were later interpreted as benign [8]. The risk of severe adverse events has been used as a reason to call for the regulation of genetic counsellors [9]. Tools have been developed to assess quality (good care) in clinical genetics and genetic counselling based on service structures, process of care and health and patient outcomes [10], but these are quality and not risk (lack of safe care) assessment measures.
Members of our research team have a longstanding interest in risk factors in clinical genetics practice such as staffing and length of wait to appointment [11, 12]. However, these were evaluations of service delivery on practice and did not examine the patient journey. In order to meet our aim of assessing and describing risk across the patient journey through clinical genetics, we needed to determine the most appropriate methodology to do so. With respect to practice, types and magnitude of risks in prenatal diagnosis have been described [13], as has the pathway of clinical referral and first visit in a genetics clinic [14]. In genetics laboratory risk analysis, Claxton et al. [15] describe prospective risk assessment of the laboratory process of genetic variant testing by failure modes and effects analysis (FMEA). Donohue et al. [7] provide a narrative exploration of challenges of genetic test misinterpretation and the resulting clinical impacts captured in a survey of 360 American genetics providers. A literature review of the subject showed few publications related to risk across the patient journey in clinical genetics.
In contrast, Clinical Genetics Laboratories have systematic, dedicated process risk assessment for each laboratory process as part of Quality Management Schemes [16] and ISO compliance such as ISO 15189 [17]. Considering the complexity of genomic testing, including request, reporting, and consent requirements, many laboratories have developed process maps specific to sample processing and reporting as part of Internal Quality Control [18]. Our interest lies in describing the clinical genetics pre- and post-test activity that ‘sandwiches’ the genetics laboratory activity.
Our process of risk assessment tool development best aligns with Failure Modes and Effects Analysis. FMEA differs from hospital clinical risk reporting in that it prospectively examines a pathway to identify risks; while hospital risk reporting retrospectively examines adverse events and/or near-misses to explore the cause and severity of risk the occurred [19]. While both approaches allow for risk identification to mitigate risk leading to service improvements, FMEA can proactively recognize risk, may prevent risk [19] and fits within hospital quality improvement programs [20].
FMEA is well aligned to the aim of our project, as methodology is based on the creation of a risk process map, followed by consideration of risk frequency and severity to define a risk score and identify risks [15, 19,20,21]. FMEA methodology involves 5 steps [19, 21]:
-
1.
Define the high-risk process.
-
2.
Assemble of a multidisciplinary team including subject experts and risk management experts.
-
3.
Graph or map the process, with each step having a risk of an adverse event or near miss (called a failure mode).
-
4.
Conduct a hazard analysis for each step in the process, by calculating the frequency and the severity (impact) of the failure mode. ‘How often do we see a failure at this step, and how severe is the impact?’.
-
5.
Define action measures to mitigate the most hazardous risks.
FMEA step 4 of hazard analysis [19] dovetails with the Irish HSE Incident Management Framework [22] and Risk Assessment Tool [23] which use a Traffic Light Risk Assessment system of hazard analysis. The Traffic Light risk assessment system uses a risk matrix of frequency and impact to grade risks as green (low-), amber (medium-) or red (high-risk) [23]. This tool can be used both prospectively and in risk review of incidents [24]. In order to align our FMEA analysis with this existing HSE framework for risk analysis, we adopted the Traffic Light Risk Assessment system in step 4 of our FMEA analysis.
The HSE Risk Assessment Tool [23] considers risk impact categories: harm to a person (patient/staff/public), service user experience, business/service disruption, loss of trust/confidence, organizational outcomes/objectives, compliance requirements, financial harm to health service, environmental harm, and health service strategic programme/project harm. Eight of these harms are generic to all specialties, however, “harm to person” can be specialty-specific. The HSE Risk Assessment tool describes ‘harm to person’ in terms of physical injury (no first aid required to loss of life) or psychological damage (no impaired psychosocial functioning to permanent psychosocial impairment). While this aligns well with surgical harms for in-patient use, it does not completely describe the harm severities of clinical genetics being a preventive speciality [5] and the familial nature of clinical genetic harms [6].
This study outlines our creation of a framework of process mapping and hazard analysis to produce tools for risk analysis in clinical genetics.
Methodology
Scope
The scope of FMEA analysis was defined as risks to the patient moving through outpatient Clinical Genetics to obtain genetic testing and/or genetic diagnosis, including receiving genetic testing through mainstream (i.e., non-genetic) clinics and subsequently attending Clinical Genetics for interpretation of results. The methodology for process map development is outlined as the framework described by Antonacci et al. [25].
Research teams
The overall study design was conceived by the core research team and included a Consultant Clinical Geneticist (SAL) and two European Registered Genetic Counsellors (AJW and DL), all of whom had undertaken Clinical Risk training with national risk management agencies, a MSc trained research assistant (DB) and a patient representative, VMcG who as a trained engineer has an understanding of risk processes. AJW is also a PhD molecular genetics scientist with extensive experience in laboratory quality and processes.
External participating Clinical Genetics teams (Oxford (HS and MB), Newcastle (MB2), Oulu (OK and JM), Northern Ireland (GR), and Dolj (IS)) were recruited through the European Reference Network for Rare Malformations (ERN ITHACA) and through the British Society of Genetic Medicine in 2022. The participating Risk experts were recruited by direct contact from the Principal Investigator and included a hospital risk manager (DB2), a member of the Health Service Executive’s Enterprise Risk team (EK) and a senior clinical risk manager and a clinical risk advisor with the State Claims Agency (NC and SK) which advises and assists State authorities with risk management services and resolves third-party claims against State authorities.
See Fig. 1 for an overview of methodology used for FMEA tool development.

Development of clinical genetics process map, audit form, and impact scale development from clinical case review of adverse and near-miss events, aligned with FMEA methodology.
Information sources
-
1.
Clinical genetics charts with risk events. This was a selective, purposeful sampling of genetic charts captured retrospectively by SAL over a 3-year period in Clinical Genetics in Children’s Health Ireland. Charts were identified when they contained adverse events (that had been reported to the Hospital Clinical Risk department, Laboratory reporting risk systems, and/or Data Protection Office for data breaches), or contained near-miss events where significant time had been taken by Clinical Genetics staff to prevent the clinical situation escalating to an adverse event.
-
2.
A concurrent survey of Rare Disease families, which was undertaken as part of this research grant [26] captured the impact of harm on families, confirming the harm that can occur. This informed the development of the impact severity scale.
Data for the 115 genetic charts with risk were captured in Excel. Hospital ID, diagnosis, recurrence risk, waiting time from referral to first appointment, age category of proband (fetal/neonate/child/adult), and speciality of referrer were recorded. Cases were reviewed to determine all proximate cause(s) of the adverse event or near miss. All cases were independently reviewed by 2 or more members of the core research team, each case was discussed amongst the team until consensus on proximate causes of the risk was reached. The harm to the proband or family was recorded as a narrative summary statement.
Creation of a process map
All proximate causes of risks identified were organized in sequential order to create a 22-step process map of the patient journey through clinical genetics or genetic testing, based on clinical knowledge of the core research team. The process map was plotted in Biorender.
Creation of a risk frequency audit form
The frequency of missteps (failure modes) could not be established from the purposeful sampling of 115 risk charts, so an audit form for prospective ascertainment was developed. This incorporated the process map, with questions regarding the demographics of the reporter, a broad de-identified disease category, a 1-sentence summary of the nature of the event, and whether the risk occurred in genetics or a mainstream clinic. A 6-week audit period was chosen based on the researchers’ professional estimate of adverse event frequency. The full clinical audit instructions and data collection form can be found in Supplemental Fig. 1.
A prospective audit was undertaken at Clinical Genetics, CHI-Crumlin, Dublin, to trial the audit form’s usability and assess the frequency of events. Members of the clinical team completed one audit form for each appointment where an adverse event or near miss was identified. The total number of outpatient clinic appointments within the Centre in the audit period was obtained from the administrative team. At the end of the 6-week period, audit data were compiled in Excel for analysis. Frequency for each step along the map was calculated as the number of events per total number of appointments in the audit period.
Following data analysis for the CHI-Crumlin audit, a feedback meeting was held with the CHI Clinical Genetics team. The research team provided results on the frequency of events observed, and a general discussion about the perception of risk was held. Feedback was sought on the completeness and composition of the process map and the ease of use and wording of the audit form. The research team incorporated the team’s feedback into the process map and audit form.
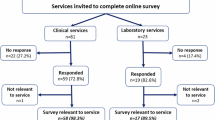
The audit was rolled out to the 5 participating clinical genetics centres. Characteristics of the participating centres, including catchment, scope of patients included in audit, and type of clinical staff on team, are shown in Table 1. Belfast and Oxford produced an electronic version of the audit form for their own use. Centres provided the number of outpatient genetics appointments given during the audit period. All 5 centres provided Excel spreadsheets of results to the core research team. Feedback meetings were held with each centre upon audit completion to determine redundant or missing steps in the process map. They were also asked if the audit form was simple and time-efficient to complete, and whether a summary of the findings permitted them to reflect on risk within their clinical practice. The frequency of events at each step was calculated as the number of events per the total number of appointments in the audit period.
Adaptation of impact (Severity) score
In parallel to the development of the process map and audit, the research team engaged with Risk experts in order to understand how adverse events are managed on a national basis and how this could be applied to a clinical genetics’ context. The impact of risk on patients and their families from the 115-case review as well as from the public survey on clinical genetics and genetic testing, was reviewed within the core research team. Generalized and anonymized versions of risks identified in the case review and public survey were discussed with Risk experts. While originally consideration was given to adaptation of all categories of the risk assessment tool [1, 23], it was decided that most categories (such as harm to the environment, financial, reputational harm to the health service) did not differ from other medical specialties. The risk severity levels on the Irish public health service risk assessment tool [1, 23] for the category ‘Harm to person (Service User, Patient, Staff & Public)’ were adapted into a Clinical Genetics-specific tool by the team by considering the individual and family-specific outcomes particular to Clinical Genetics at different severity levels.
Results
Tools development
The 115 charts reviewed had a time from referral to appointment ranging from 0 to 39 months. 65/115 charts were related to pregnancies that occurred since referral: consultand requested an expedited appointment because pregnant, consultand declared a pregnancy at appointment, or consultand attended appointment, and pedigree analysis revealed the birth of subsequent children since referral. Of note, 3 pregnant women were affected by an autosomal dominant condition with a significant risk to their offspring. 41/115 had urgent genetic testing in order to resolve a genetic diagnosis to facilitate prenatal testing.
The remaining 50/115 charts were unrelated to pregnancy at the time of appointment –11 of these were cancer genetics. 10/115 charts were regarding recurrence for the death of a fetus or baby with a possible genetic syndrome. A sample of de-identified narrative summaries from the charts is shown in Table 2 – some of the vignettes contain more than one adverse event or near miss.
The process map developed from analysis of the cases has 22 steps spanning five stages: (1) patient and family history assessment (2) clinical management of genetic testing (3) sample processing and analysis (4) result transmission (5) result discussion, as shown in Fig. 2.

The process map describes the 22 key steps where risk occurs if care is not optimally managed. The map spans 5 sections of clinical and laboratory activity.
The final adapted version of the ‘Harm to a person (Service User, Patient, Staff & Public)’ severity table is shown as part of the risk assessment tool adapted for clinical genetics use, encompassing all steps in the traffic light system (Supplemental Fig. 2).
Feedback was solicited on the process map and audit form from the 6 clinical teams. All teams found the wording of the process map clear and did not identify any redundant or missing steps. Centres with developed IT infrastructure translated the form into a computerized version for ease of data capture.
Audit
The results of the audit of all centres are shown in Table 3. The number of appointments with risk events ranged from 0.8% to 20.3%. The percentage of appointments with risk, where there was a failure at more than one step occurred ranged from 20.0% to 54%. Oulu noted a few risks compared to all other centres, all observed at the start of the process map. Risk events occurring at the start were also common in Oxford. Events during the sample processing section were common in Dublin, Belfast, Oxford, and Newcastle. Transmission of reports were an issue in Dublin and Belfast. Adverse events were common during the results discussion period in Dublin, Belfast, and Oxford. In Dublin, most risk was attributable to long waiting lists, IT deficiencies, gaps in non-geneticists’ genomic knowledge resulting in sub-optimal consent or misinterpretation of test reports, and wrong testing being ordered by mainstream clinicians due to the limited access to clinical genetic and laboratory expertise.
Re-audit
Two units, Dublin and Oxford, were re-audited in 2024. In Dublin, 57 appointments of 671 had risk events (8.5%). In Oxford, 42 appointments of 1448 had risk events (2.9%), a 55% reduction.
Discussion
The 22-step process map of the patient journey through clinical genetics is novel. Donohue et al. [7], whose qualitative survey examined a shorter journey surrounding genetic testing, identified 6 of the same steps: correct test ordered, communication between laboratory and provider; clear wording of report; correct interpretation; genotype to phenotype correlation; and clear explanation of testing results. Although Donohue et al. [7] did not order the steps, they cited them all as contributing factors to the misinterpretation of genetic testing with resulting patient impact.
While all centres that took part in the research identified adverse events, some of the variances noted can be explained by differing practices and staff roles. The difference in percentage of appointments with risk events identified between centres may relate to where in the pathway those staff completing forms are active. The reduced capture of laboratory events by Dublin, Belfast, Craiova, and Oulu was noted, where clinical teams used external laboratories, leading to less engagement with laboratory processes.
Despite not capturing laboratory events, 1 in 10 appointments in Dublin had an adverse event, and nearly half of those had risk at more than one step in the pathway. The majority of risk was attributable to: (1) long waiting lists, (leading to a pregnancy occurring whilst waiting for existing child to be seen, death of proband on the waiting list or critical sample being discarded before appointment), (2) IT deficiencies, (poor inter-operability between hospitals resulting in convoluted processes to request tests by mainstream and clinical genetics clinicians (adding time to the already limited time to review and discuss patients) test reports not being accessible or tests being duplicated unnecessarily (3) insufficient access to clinical genetics expertise, for example, to support mainstream clinician with queries regarding complicated test reports such as imprinting, resulting in mis-information being given to the family by the mainstream team, (4) gaps in non-geneticists’ genomic knowledge including informed consent when testing, resulting in secondary findings being reported and the family not realising they had consented to this.
Whilst some of these critical steps could be specific to Dublin, as for the example the lack of a test directory, (present in England), others risks are shared within most centres, such as lack of sufficient specialist staff to support mainstream clinicians in the early steps of testing and limited clinical genetics time to support mainstream colleagues and the limitations due to poor interoperability of IT systems.
In Craiova, only one clinician recorded risks, and these were noted throughout the patient journey. All other centres recorded adverse events by multiple team members. The small numbers of adverse events recorded in Newcastle were thought to reflect staff time pressures precluding filling in the audit form. In Dublin, the Principal Investigator (SAL) captured and logged adverse events raised by any staff members at clinical meetings. Belfast suggested under-reporting of adverse events, and a high percentage identified with multiple risks due to staff only recognizing risk when > 1 step failed across the process map. The high percentage of declared risk events in all centres where more than 1 step failed suggests the possibility that staff are only recognizing or recording appointments where the most severe harm occurs.
Oxford recorded many events at sample receipt, but they have a dedicated genomic practitioner team whose goal is to facilitate the consenting process and complete and collate paperwork and samples. Oxford has recognized the need for a quality check process at receipt of sample, allowing any concerns regarding consent to be flagged, demographic errors to be fixed, and allowing clinical questions to be managed with the referring physician early in the testing process, ensuring the correct test is done for genetic tests requested by mainstream teams. Although Oxford recorded many of these sample receipt events, the presence of the genomic practitioner corrected the event so that there were fewer events downstream in the pathway. No other participating centre has this system in place, although a recent review describes the implementation of genetic counsellors into genomic test ordering review as both cost-saving and beneficial to patients [27]. Some adverse events noted by Oxford may have been picked up within laboratory systems in the other units and not have been recorded by the clinical teams elsewhere.
Oulu identified only five events over 6 weeks. In comparison to the other 5 centres, they have limited involvement of mainstream clinicians in genetic testing. For straightforward referrals, they have developed lean processes where genetic nurses have a central role, coordinating the majority of genetic testing, including consent and test requisitions. Interestingly, all events in Oulu occurred early in the clinical pathway suggesting the genetic nurses are capturing events early on and reducing the number of adverse events later in the pathway. Most of the other centres do not have these same pre-test control systems in place, apart from laboratory scientist gatekeeping. It suggests that some downstream adverse events occurring in Dublin, Belfast, Newcastle, and Craiova may result from a lack of early pathway controls.
Following the first audit, Dublin adjusted triage priorities to ensure referrals with critical samples were seen promptly, and the team educated mainstream clinicians regarding European guidelines on secondary findings [28], and a reduction in these referrals was noted on re-audit. The number of adverse events stayed static in Dublin, although the severity of the risk was reduced (e.g., no loss of critical samples or referrals for secondary findings were noted on repeat audit). Between audit and re-audit, Oxford introduced proactive training and education of mainstream clinicians, encouraging the pathway for Genomic Practitioners to complete the records of the discussion with the patients. These initiatives resulted in a reduction in adverse events, particularly at the receipt of the sample. This change of process may explain the reduction in adverse events noted on re-audit. However, the repeat audit in Oxford noted reduced engagement by staff in audit form completion, which may also have been a factor.
Risk reducing practices that could be implemented in other centres to help reduce risk include: (1) specialised staff to support early steps in the patient pathway, ensuring optimum consent etc, (2) robust IT infrastructure and connectivity ensuring timely access to reports, and robust IT infrastructure and administrative support to plan and record Multi-Disciplinary Teams outcomes (3) a genetic testing directory and adequate resource for laboratory staff to facilitate clinical and laboratory gatekeeping, (4) sufficient dedicated clinical genetics time in their job plans to address mainstream clinicians’ enquiries and MDTs for complex results to support colleagues with interpretation and communication of complex genomic results.
One participant noted that, when working in a high-risk environment, staff may become complacent about adverse events. Complacency with risk does not make the patient environment safe. Risk management departments encourage staff to conduct risk assessments and report incidents; however, over-reporting of adverse events can lead to ineffective feedback limiting the ability for risk mitigation and improvement [29]. This can leave staff demoralised and further decrease the likelihood of effective risk reporting and resolution. Our audit tools may allow centres to prospectively identify systematic risk and target the most severe risk areas pertinent to their centre for risk mitigation to improve quality, rather than wait for adverse events to occur and report them retrospectively.
Shebl et al. [30] discuss four types of validity with respect to risk assessment tools (face, content, criterion, and construct validity). We have established face validity – an independent, subjective assessment of whether the tools meet their stated aim – by the opinion of the participating clinical teams and risk experts. Content validity – whether the tools contain the domains they intend to measure – was confirmed by independent clinical teams testing to audit tool and providing feedback about the process map and the audit’s ability to capture perceived risk in their clinical service. The content validity of the impact table was confirmed as many of the themes of ‘harm to a patient’ identified by the research team mirror those identified in our patient survey [26], and align with perceived consequences of errors (incorrect diagnosis, incorrect treatment, stress for patient and family, and ethical concerns) described by Donohoe et al. [7]. Criterion validity could not be assessed due to the lack of a comparable measure, as hospital clinical and laboratory risk registers capture adverse events but not near-miss data. Construct validity, whether the measurement tool (risk matrix) measures the concept of the magnitude of risk, was not assessed. However, the integration of HSE’s traffic-light risk assessment tools [23] increases acceptance of the measure and competence in its use.
Once published, future consideration will be given to the use of the risk assessment tools in the Republic of Ireland and in the South-West of Britain.
A limitation of this study is the possibility that the process map is specific to European clinical genetic processes. Expansion of this research would be to trial these tools in private clinical genetics, clinical genetics outside of Europe, and mainstream clinical teams where genetic testing is routinely used. Reproducibility of the genetics-specific risk assessment severity matrix could be demonstrated by a trial with a different dataset of cases with risk identified would be warranted.
In conclusion, each discipline understands its own specialty-specific adverse events. By developing specialty-specific risk assessment tools and reminding staff to log adverse events, it becomes clearer where in the patient journey risk is occurring and where action is needed. We propose that this is a robust risk assessment tool that should allow specific risk areas to be identified and result in a more focused approach to risk mitigation. It provides a framework to help healthcare providers outside of genetics appreciate all the steps that are necessary for safe patient care. This process map and risk assessment tool may (1) allow genetic teams to prospectively identify risk areas in their practice and put controls in place if system errors are identified (2) be used to investigate serious adverse events that require a retrospective review, (3) be used as an investigative tool to manage patient complaints and (4) teach clinical staff about the steps needed for safe and ethical genetic testing. Introduction of this tool on a rolling basis could allow the evaluation of risk mitigation strategy implementation as part of a quality management system.
Data availability
The data that support the findings of this study are not openly available due to the possibility of identification, as cases reviewed were families with rare diseases. These are available from the corresponding author upon reasonable request. Data are located in controlled access data storage at University College Dublin.
References
Health Service Executive [HSE]. Enterprise Risk Management Policy and Procedures. HSE, 2023. Available at: https://www2.healthservice.hse.ie/organisation/national-pppgs/hse-enterprise-risk-management-policy/ Accessed 28 May 2025.
Park SJ, Sharp AL. Improving health and health care efficiency through risk management. J Hosp Manag Health Policy. 2019;3:9–11.
La Russa R, Ferracuti S. Clinical Risk Management: As Modern Tool for Prevention and Management of Care and Prevention Occupational Risk. Int J Environ Res Public Health. 2022;19:831 https://doi.org/10.3390/ijerph19020831.
HSE (2024) Quality Improvement Guide and Toolkit. Available at https://assets.hse.ie/media/documents/HSE_Quality_Improvement_Guide_and_Toolkit_2024.pdf [Accessed 11 July 2025].
World Health Organization (2010). Community Genetics Services: report of a WHO consultation on community genetics in low- and middle-income countries. https://iris.who.int/bitstream/handle/10665/44532/9789241501149_eng.pdf. [Accessed 27 July 2025].
Van Riper M. Genetic Testing and the Family. J Midwifery Wom Health. 2005;50:227–33. https://doi.org/10.1016/j.jmwh.2005.02.008.
Donohue KE, Gooch C, Katz A, Wakelee J, Slavotinek A, Korf BR. Pitfalls and challenges in genetic test interpretation: An exploration of genetic professionals experience with interpretation of results. Clin Gen. 2021;99:638–49. https://doi.org/10.1111/cge.13917.
Rashkin M, Kingham K, Lara-Otero K, McKenna M, Villiers J, Mykolas Worthington M, et al. How should we address the inevitable harms from non-negligent variant reclassification in predictive genetic testing? J Gen Couns 2022. https://doi.org/10.1002/jgc4.1638
Shugar AL, Quercia N, Trevors C, Rabideau MM, Ahmed S. Risk for Patient Harm in Canadian Genetic Counseling Practice: It’s Time to Consider Regulation. J Genet Couns. 2017;26:93–104. https://doi.org/10.1007/s10897-016-9983-4.
Chou AF, Norris AI, Williamson L, Garcia K, Baysinger J, Mulvihill JJ. Quality assurance in medical and public health genetics services: a systematic review. Am J Med Genet C Semin Med Genet. 2009;151C:214–34. https://doi.org/10.1002/ajmg.c.30219.
Lynch SA, Borg I. Wide disparity of clinical genetics services and EU rare disease research funding across Europe. J Community Genet. 2016;7:119–26. https://doi.org/10.1007/s12687-015-0256-y.
Bradley L, Lynch SA. Dying to see you? Deaths on a clinical genetics waiting list in the Republic of Ireland; what are the consequences?. J Community Genet. 2021;12:121–7. https://doi.org/10.1007/s12687-020-00491-3.
Anumba DO. Errors in prenatal diagnosis. Best Pr Res Clin Obstet Gynaecol. 2013;27:537–48. https://doi.org/10.1016/j.bpobgyn.2013.04.007.
McCann E, Baines EA, Gray JR, Procter AM. Improving service delivery by evaluation of the referral pattern and capacity in a clinical genetics setting. Am J Med Genet C Semin Med Genet. 2009;151C:200–6. https://doi.org/10.1002/ajmg.c.30223.
Claxton K, Campbell-Allen NM. Failure modes effects analysis (FMEA) for review of a diagnostic genetic laboratory process. Int J Qual Reliab Man. 2017;34:265–22. https://doi.org/10.1108/IJQRM-05-2015-0073.
Deans Z, Ahn JW, Bergbaum A, Colclough K, Dalton A, Dinning H et al. Best Practice Guidelines for Internal Quality Control in Genetic Laboratories. Assoc Clin Genomic Sci. 2015. file:///C:/Users/User/Downloads/_media_10760_iqc_bpg_2015_-_final.pdf [Accessed July 27, 2025].
Ellard S, Morgan S, Wynn SL, Walker S, Parrish A, Mein R, et al. Rare disease genomic testing in the UK and Ireland: promoting timely and equitable access. J Med Genet. 2024;61:1103–12. https://doi.org/10.1136/jmg-2024-110228.
Brady C, Ryan E, Lynch SA, Butler A, Barton DE. Achieving an equitable cross border referral service for genetic testing; findings from an Irish quality perspective P14.082. Eur J Hum Genet. 2016;24 esupp1:329.
Shaqdan K, Aran S, Daftari Besheli L, Abujudeh H. Root-cause analysis and health failure mode and effect analysis: two leading techniques in health care quality assessment. J Am Coll Radio. 2014;11:572–9. https://doi.org/10.1016/j.jacr.2013.10.024.
Liu HC, Zhang LJ, Ping YJ, Wang L. Failure mode and effects analysis for proactive healthcare risk evaluation: A systematic literature review. J Eval Clin Pr. 2020;26:1320–37. https://doi.org/10.1111/jep.13317.
Taleghani YM, Vejdani M, Vahidi S, Ghorat F, Raeisi AR. Application of prospective approach of healthcare failure mode and effect analysis in the risk assessment of healthcare systems. Eurasia J Biosci. 2018;12:95–104.
HSE (2020) Incident Management Framework https://www.hse.ie/eng/about/who/nqpsd/qps-incident-management/incident-management/hse-2020-incident-management-framework-guidance.pdf Accessed May 12 2025.
Health Service Executive [HSE]. HSE Risk assessment tool. HSE, 2023. https://www.hse.ie/eng/about/who/riskmanagement/risk-management-documentation/hse-enterprise-risk-management-supporting-tools/hse-risk-assessment-tool.pdf, Accessed May 5 2025.
Lemmens SMP, Lopes van Balen VA, Röselaers YCM, Scheepers HCJ, Spaanderman MEA. The risk matrix approach: a helpful tool weighing probability and impact when deciding on preventive and diagnostic interventions. BMC Health Serv Res. 2022;22:218. https://doi.org/10.1186/s12913-022-07484-7.
Antonacci G, Lennox L, Barlow J, Evans L, Reed J. Process Mapping in Healthcare: a systematic review. BMC Health Serv Res. 2021;21:342. https://doi.org/10.1186/s12913-021-06254-1.
Ward AJ, Lambert DM, Butterly D, O’Byrne JJ, McGrath V, Lynch SA. Genetic services survey-experience of people with rare diseases and their families accessing genetic services in the Irish Republic. J Community Genet. 2023;14:583–92. https://doi.org/10.1007/s12687-023-00664-w.
Cook CB, Pistawka C, GenCOUNSEL Study, Elliott AM. The impact of genetic counselor involvement in genetic and genomic test order review: A scoping review. Genet Med. 2025;27:101354 https://doi.org/10.1016/j.gim.2025.101354.
de Wert G, Dondorp W, Clarke A, Dequeker EMC, Cordier C, Deans Z, et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2021;29:365–77. https://doi.org/10.1038/s41431-020-00758-w.
Macrae C. The problem with incident reporting. BMJ Qual Saf. 2016;25:71–5. https://doi.org/10.1136/bmjqs-2015-004732.
Shebl NA, Franklin BD, Barber N. Failure mode and effects analysis outputs: are they valid?. BMC Health Serv Res. 2012;12:150. https://doi.org/10.1186/1472-6963-12-150.
Acknowledgements
Adelaide Health Foundation. European Reference Network ERN-ITHACA. Clinical Genetics Society UK. Julie Egan, Dept of Clinical Genetics, Children’s Health Ireland. Clinical staff in Dublin, Oxford, Oulu, Belfast, Craiova, and Newcastle for facilitating data capture.
Funding
Source of funding received: Adelaide Health Foundation grant R22808.
Author information
Authors and Affiliations
Contributions
SAL: project design, identification of retrospective adverse events, recruitment of participating centres. DB, DL, AW: analysis of retrospective adverse events, adaptation of generic risk matrix. DB, DL: design of 22-step pathway and data collection forms. SAL, HS, MB Oxford, MB, Newcastle, GR, IS, OK, JM: coordinated prospective audit in their respective centres and provided feedback. NC, SK, EK, DB: advised on traffic-light risk evaluation and adaption of risk framework to a Clinical Genetics setting. DB, DL, AG: analysis of prospective events. SAL, DL, AW, HS, MB Oxford, MB Newcastle, GR, OK, JM, VMcG: drafting manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Ethical approval was obtained from the Children’s Health Ireland Research Ethics Committee (GEN/937/21).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lambert, D.M., Stewart, H., Bandiola, M. et al. What is risk in clinical genetics? Designing and piloting tools to evaluate risk in clinical genetics using failure modes and effects analysis. Eur J Hum Genet 34, 505–514 (2026). https://doi.org/10.1038/s41431-025-01961-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01961-3
This article is cited by
-
When clinical genetics turns the risk lens on itself
European Journal of Human Genetics (2025)
