
Abstract
Polycystic kidney disease (PKD) is a Mendelian renal disease characterised by the development of cysts and progressive decline in kidney function, leading to kidney failure. Although genetic testing can provide a precise molecular diagnosis of PKD in the majority of cases, 6–13% of patients remain unsolved. Copy number variants (CNVs) are an established pathogenic mechanism in PKD, however detection typically relies on multiplex ligation-dependent probe amplification (MLPA) which is resource intensive and separate to next-generation sequencing (NGS) pipelines. Here, a bioinformatics tool ClinCNV was used to call CNVs from NGS data of 371 people with PKD who had previously undergone short nucleotide variant (SNV) analysis with a standard NGS pipeline. Diagnostic CNVs were confirmed in 13 patients across 7 families, increasing the diagnostic yield from 86.5 to 90.0%. We also tested CNVs as potential disease modifiers. Regression models indicated an association of cystic gene duplication burden to worse kidney survival (HR = 1.56, 95% CI: 1.26, 1.93, adj-p = 0.0004). These models also revealed that duplication burden in genes unrelated to cystic kidney disease associated with the absence of liver cysts, possibly driven by a region containing LRP5L. These results demonstrate the utility of targeted gene panel and exome sequencing for the detection of CNVs in key PKD genes.

Similar content being viewed by others
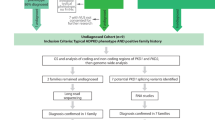


Introduction
Polycystic kidney disease (PKD) is a kidney disorder characterised by the presence of cysts on and enlargement of the kidneys, leading to progressive kidney failure [1]. Extrarenal manifestations can occur with PKD, including hepatic and pancreatic cysts, and intracranial aneurysms [2]. PKD can occur with autosomal dominant (ADPKD) or autosomal recessive (ARPKD) inheritance. Pathogenic variants in PKD1 and PKD2 account for 75–80% and 15–20% of ADPKD cases, respectively [2]. Other genes including IFT140, GANAB, ALG9, and DNAJB11 are known to cause atypical ADPKD phenotypes [3,4,5,6,7,8]. ARPKD is a rare form of PKD, with nearly all suspected cases harbouring PKHD1 mutations [9], or, rarely, mutations in DZIP1L [10]. However, 6–13% of PKD patients lack a genetic diagnosis even after appropriate genetic testing [11,12,13].
The severity of ADPKD is correlated with the type of pathogenic variant. Patients with PKD1-associated disease have a more severe disease outcome compared to those with pathogenic variants in PKD2 [11]. Protein-truncating variants in PKD1 (PKD1-T) are associated with earlier kidney failure relative to PKD1 non-truncating (PKD1-NT) and PKD2 variants (about 18 years or 28 years earlier, respectively) [11,13,14]. Individuals with a pathogenic missense variant in either PKD1 or PKD2 experience less severe disease progression. Missense variants in PKD1 and PKD2 can exhibit variable penetrance and therefore are often classified as variants of uncertain significance (VUS) [15].
Copy number variants (CNVs) are deleted or duplicated segments of DNA, typically defined as larger than 50 bp [16]. CNVs impacting PKD-associated genes are known to cause about 1–5% of genetically-defined PKD cases [12,13,17,18]. In addition to PKD, there are multiple examples of other genomic disorders resulting from a CNV encompassing a renal disease gene. For example, a recurrent deletion on chromosome 16 impacting PKD1 and TSC2 results in the development of both ADPKD and tuberous sclerosis complex [19]. A recurrent 17q12 deletion encompassing HNF1B results in renal abnormalities and cysts, but also neurodevelopmental phenotypes including autism and neuropsychiatric comorbidities [20]. Another recurrent deletion encompassing NPHP1 causes nephronophthisis [21] and can result in Joubert syndrome [22].
As most cases of PKD are caused by short nucleotide variants (SNVs), CNVs are generally only assessed after a negative screen of SNVs. The gold standard for testing for CNVs in PKD is multiplex ligation-dependent probe amplification (MLPA), a resource-intensive, gene-specific, and expensive test [23]. Array-CGH can also be used, but high-resolution arrays are needed for detection of smaller (intragenic) CNVs. Array-CGH and MLPA are both relatively costly compared to NGS and must be analysed in a different workflow to NGS variant detection [12, 24]. This separation of testing workflows may result in increased time to a molecular diagnosis for people with CNV-related PKD, or lack of diagnosis where MLPA or array-CGH is unavailable.
CNV calling from NGS data is a well-established practise, and tools have been developed for discovering CNVs from targeted sequencing such as targeted gene panel (TGP) or exome sequencing (WES). Most tools that support targeted sequencing utilise read-depth based algorithms, which attempt to identify CNVs through local changes in sequencing depth [25]. Targeted sequencing thus limits the detection of copy-neutral structural variants, which typically require algorithms such as paired-end mapping [26]. PKD1 falls within a segmentally duplicated region [27], which have been reported as challenging for current CNV calling tools [26]. Despite this, the efficacy of calling PKD and renal-disease related CNVs from targeted sequencing data has been demonstrated [15, 28, 29]. However, to date, a large study with a focus on NGS-called CNVs in PKD is lacking.
Here, we set out to assess the effectiveness of an NGS driven pipeline for the identification of CNVs for increasing the diagnostic utility of WES and TGP. Furthermore, we also assess the accuracy of calling CNVs from NGS and investigate the impact of non-diagnostic CNVs in cystic kidney genes on measured clinical outcomes of PKD.
Patients and methods
Patient inclusion criteria
Patients were recruited from Beaumont Hospital, Dublin, Ireland as a part of the Irish Kidney Gene Project (IKGP) [30]. To qualify for inclusion, patients had to be clinically diagnosed with PKD using the unified criteria for ultrasonography diagnosis of ADPKD or having evidence of >5 cysts [31] and have WES or TGP data available. Participants must have undergone CT, MRI or ultrasound imaging of their abdomen to assess for kidney and liver cysts.
During recruitment, patients were given an individual ID and a family ID to allow for segregation analysis of identified variants. Individuals with a known familial connection (i.e. through a pedigree) were assigned the same family ID.
Clinical records were used to gather information on age at kidney failure, height-adjusted total kidney volume (htTKV), and presence or absence of liver cysts. Kidney failure was defined as the need for dialysis or pre-emptive kidney transplantation. HtTKV was estimated by calculating the total kidney volume from imaging data correcting for height of the patient [32]. The occurrence of liver cysts was evaluated based on imaging report by expert clinical radiologist.
Short-read sequencing data
Sequencing of the patients was performed on whole blood as a part of the IKGP. Of the 378 patient cohort, 168 had WES (Twist Human Core Exome with RefSeq kit, sequenced on NovaSeq 6000, 2 x 100 bp reads, “low coverage” batch with average ~101x coverage (s.d. ±30.9x) and “high coverage” batch with average ~117x coverage (s.d. ±27.5)), 100 had TGP (custom Roche SeqCap EZ Choice on MiSeq or NextSeq, 2 x 150 bp reads with average ~222x coverage [17]), and 110 had both WES and TGP. Sequencing data was aligned on the GRCh38 build of the human genome. All patients had previously undergone SNV (SNP and indel) analysis using the GATK4 pipeline as described previously [17].
CNV calling
Both the GATK GermlineCNVCaller and ClinCNV were trialled for CNV calling. While both tools performed similarly, we decided to report the results for ClinCNV due to ease of pipeline execution and flexible parameters. CNVs were called from WES and TGP using ClinCNV version 1.18.3 (ref. 33). To prevent spurious CNV calls, the TGP cohort was analysed as one batch, and the WES cohort was split into two batches, as one WES batch was sequenced at a higher coverage compared to the other WES batches. The annotation of the BED file (containing the sequencing target regions) and calculation of coverage for each sample was done using ngs-bits (https://github.com/imgag/ngs-bits) with default parameters according to the ClinCNV GitHub. The mergeFilesFromFolder.R script was used to merge the coverage files, and the clinCNV.R script was run with –scoreG 50 for balanced sensitivity and –minimumNumOfElemsInCluster 15 to account for a smaller cohort size. The resulting TSV file of CNVs was converted into VCF format using an in-house bash script. All scripts used are available on GitHub (https://github.com/FutureNeuroIE/clincnv-pipeline).
Annotation and Filtering
The VCFs were annotated using the Ensembl Variant Effect Predictor (VEP) version 107 (ref. 34). The StructuralVariantOverlap plugin with the 1000 Genomes Project data was used to annotate a CNV overlap, if the overlap was >80%. The –mane_select flag was used to identify transcripts affected by the CNV.
The 1000 Genomes Project annotation was used to gather the number of rare (allele frequency (AF) < 1%) CNVs per sample, which were then plotted onto a QQ plot to determine if the distribution was normal. Normality was confirmed in all CNV calling batches, which allowed for filtering out outliers via z-scoring. Z-scores were calculated for each sample and those with a score >3 within their own CNV calling run were considered low quality and excluded from further analysis.
For the remaining samples, CNVs were filtered for genes in the PanelApp cystic kidney disease v4.6 list and for overlap with the MANE select transcript. We identified high quality CNV calls by filtering for the following ClinCNV quality information: qvalue < 0.05, loglikelihood:no_of_regions ratio >5, and potential_AF < 0.1. CNV calls were excluded if they occurred in highly polymorphic regions in the GRCh38 build of the human genome, specifically the major histocompatibility complex (MHC) region on chromosome 6 (chr6:28,510,120-33,480,577) and the leucocyte receptor complex (LRC) region on chromosome 19 (chr19:54,025,634-54,910,145).
Validation of CNVs and estimation of ClinCNV sensitivity and specificity
CNVs called by ClinCNV in PKD1 or PKD2 were validated using SALSA MLPA Probemixes P351 PKD1 and P352 PKD1-PKD2. In total, 50 samples had MLPA testing (Leipzig, Germany); sixteen individuals with a suspected PKD1/PKD2 CNV were chosen for MLPA (with at least one person per family sent for testing), and 34 individuals were chosen at random, prioritising unsolved samples and an equal number of samples with WES or TGP available to assess the accuracy of each NGS type in detecting PKD1 or PKD2 CNVs. Array-CGH was conducted on 5 samples using an Agilent 180k array. Similarly, the samples chosen for array-CGH were prioritised for unsolved samples as well as those containing large >50 kb CNVs for detection by the array.
For array-CGH, sensitivity calculations were restricted to high quality CNV calls, defined as those spanning >10 probes and >50 kb covering at least one coding exon from any gene from the sequenced data. Sensitivity was calculated as (True Positive)/(True Positive + False Negative) for array and MLPA results, and specificity was calculated as (True Negative)/(True Negative + False Positive) for MLPA results.
Classification of CNVs
CNVs impacting cystic kidney genes that passed quality criteria (see “Methods”: Annotation and Filtering) were assigned ACMG classifications based on the CNV ACMG guidelines [35]. We considered CNVs classified as “likely pathogenic” or “pathogenic” by ACMG guidelines as “diagnostic”. CNVs classified as non-pathogenic or CNVs where only one allele of a recessive gene was impacted were deemed “non-diagnostic”.
Regression analyses
Presence of CNV analysis
The association between clinical outcomes (age at kidney failure, presence of liver cysts, and htTKV) and an additional CNV was tested using regression models for each outcome, correcting for sex and diagnostic variant type i.e., PKD1-T, PKD1-NT, and PKD2. Individuals with a diagnostic PKD1 or PKD2 CNV deletion were coded as a PKD1-T or PKD2 variant, respectively. The presence of a non-diagnostic CNV in a cystic kidney gene was coded as 0 or 1, with 1 being the existence of a quality non-diagnostic CNV call in a cystic kidney gene. R (version 4.1.2) was used to conduct regression model analyses. The appropriate regression model was used depending on the type of data tested: Cox proportional hazards model (coxph()) for age at kidney failure, a logistic regression model (glm()) for presence of liver cysts, and a linear regression model (lm()) for htTKV (in mL/m). Assumptions of the models were tested. The diagnostic variant variable violated the proportional hazards assumption in the Cox model, which was corrected for with strata() in R. Cluster robust standard errors were calculated using coeftest() from the lmtest R package (or cluster() from the survival package for the Cox model), clustering by family ID.
CNV burden analysis
CNV burden analysis focused on the subset of participants with WES (n = 278). CNV burden was calculated as the total length in 100 kb of the CNVs in each respective group, which was calculated for exome-wide, cystic kidney gene regions, and non-cystic kidney gene regions. Cystic kidney gene burden was calculated as the total length of CNVs impacting genes in the PanelApp Cystic kidney disease gene list, and the non-cystic gene burden was calculated by totalling together all other CNVs that did not impact any cystic kidney genes. Statistical significance was determined based on Bonferroni corrected p value < 0.05 (correcting for 3 regions × 3 CNV types = 9 tests, significance threshold p < 0.006) within each series of tests, correcting for diagnostic variant type and sex as previously. Assumptions of the models were tested and corrected for as previously.
Power analysis
Study power was calculated using the powerMediation.VSMc.logistic() function in the powerMediation package in R, with alpha = 0.05. The correlation variable (corr.xm) was calculated by taking the R-squared value from a linear regression testing the burden variable as the outcome, with sex and diagnostic variant as covariates.
CNVRuler region analysis
CNVRuler [36] was used to define CNV regions and to conduct a logistic regression to determine the association between the CNV regions and the clinical outcome. The “CNVR” method was used to group the CNVs into CNV regions. The gain/loss option was selected to determine the association separately between duplications and deletions.
Gene ontology term enrichment analysis
A gene set enrichment analysis was carried out using the Gene Ontology Consortium GO enrichment analysis tool [37] with the GO aspect “biological process” selected. Genes impacted by duplications in non-cystic regions were acquired from the VEP annotations (1599 genes). Genes impacted by any CNV were also acquired similarly (2431 genes). The list of non-cystic genes impacted by duplications was run in the GO enrichment analysis tool against all annotated human genes (20,580 genes, provided by the tool), as well as the genes impacted by any CNV in our cohort.
Results
Clinical description of study cohort
Three hundred and seventy-eight patients met the study inclusion criteria (see “Methods”), of which 371 (across 213 families) passed the CNV quality control criteria (see “Methods”) and were carried forward as the cohort for analysis.
The median age for kidney survival of these 371 patients was 49 years, with 64% of the cohort having reached kidney failure at last follow up. Liver cyst data was available for 90.8% of the cohort, of which 78.6% had imaging-confirmed liver cysts. 86.5% of patients had a genetic diagnosis using standard SNV NGS analysis, with a PKD1 diagnostic variant present in 77.3% of cases. A clinical breakdown of the cohort is provided in Table 1.
Diagnostic CNVs in PKD1 and PKD2
ClinCNV-called CNVs were first evaluated for pathogenic deletions in PKD1 and PKD2. Seven families were identified with a heterozygous deletion in either gene (Table 2). In these families, twelve affected individuals were positive for a deletion using ClinCNV, and one affected individual was negative. To confirm ClinCNV deletions, MLPA was conducted on at least two affected individuals per family, which confirmed all deletions detected by ClinCNV. The affected individual with a negative ClinCNV result was positive for a diagnostic CNV by MLPA, confirming that all 13 individuals harboured a pathogenic CNV in PKD1 or PKD2. All deletions were classified as likely-pathogenic/pathogenic by ACMG criteria [35]. The identification of CNVs with this workflow increased the diagnostic yield from 86.5% (for SNVs only) to 90.0% (when CNVs were included).
When comparing results from ClinCNV and MLPA, some discrepancies were noted in the exons impacted by the called CNV. For example, in family PED1, ClinCNV called a deletion of exons 27–30 in PKD1. MLPA of this family indicated a smaller deletion, limited to exons 29–30. This is because the MLPA probemix lacked a probe for exon 28, and the drop in sequencing coverage observed in IGV suggests the deletion occurred after the probe for exon 27 (Fig. 1). Similarly, visualisation of the sequencing in IGV for individual PED2-I1 showed that their PKD1 deletion breakpoints can be resolved to exon 15, and just after exon 39 (Supplementary Fig. S1). However, MLPA reported a deletion from exons 14–35, and ClinCNV reported a deletion from exons 16–39 (Table 2).

A Pedigree of Family PED1 indicating individuals with sequencing data. B Visualisation in IGV showing a control, i.e. an unrelated individual with no deletion, and the 3 individuals in PED1 with the PKD1 deletion in exons 27–30. Approximate location of probes from the MLPA probemix are denoted by the red triangles on the exon track. The beginning of the deletion (teal arrows) can be seen by the sudden drop in read coverage within exon 27.
Expanding the search for CNVs in other cystic kidney disease genes
Having established the utility of ClinCNV for identifying pathogenic CNVs in PKD1 and PKD2, calling of CNVs was expanded to other cystic kidney disease associated genes (see “Methods”: Annotation and Filtering). Such CNVs were identified in 21 individuals, all of which harboured a previously identified diagnostic PKD variant in addition to the CNV (Supplementary Table S1). Four patients had two additional CNVs, three of which had a PKD1 duplication detected by ClinCNV and verified by MLPA. The majority (22/25, 88%) of the non-diagnostic CNVs found were duplications. Five non-diagnostic CNVs span the entirety of the impacted gene, all affecting NPHP1. Nearly all additional CNVs were classified as variants of uncertain significance (VUSs) according to ACMG criteria. The exception to this was a deletion in TMEM231, deemed not diagnostic due to its heterozygous genotype, where the gene is disease-causing under an autosomal recessive model [38]. Hereafter we refer to these additional CNVs as “non-diagnostic”.
Validation of CNVs and Estimation of ClinCNV Sensitivity and Specificity
With the identification of CNVs in cystic kidney genes, MLPA and array-CGH were employed to assess the accuracy of ClinCNV calls. First, we assessed the sensitivity and specificity of ClinCNV in detecting CNVs in PKD1 and PKD2 in the cohort. To this end, we MLPA-tested 50 samples; 34 suspected negative and 16 suspected positive, including those with the diagnostic PKD1/PKD2 deletions (see “Diagnostic CNVs in PKD1 and PKD2” above) (Supplementary Table S9). Results indicated a 62.5% sensitivity and 100% specificity of ClinCNV in detecting PKD1 or PKD2 CNVs when considering calls from the combined WES and TGP datasets. WES sensitivity and specificity was 61.90% and 100%, respectively, and TGP sensitivity and specificity was 63.6% and 100% respectively. Discordance between MLPA and ClinCNV was mainly due to PKD1 duplications (8/9 CNVs), which seems to be driving the lower sensitivity. The other discordance was due to a small single-exon PKD2 deletion, which is at the lower limits of detection by read-depth based callers [25]. The sensitivity when assessing only deletions is 90.9%, compared to assessing only duplications which is 42.9%.
To assess the performance of ClinCNV on CNVs larger than 50 kb, 5 samples with qualifying CNVs (see “Methods”) were tested using array-CGH. This analysis suggested 83% (5/6 CNVs) sensitivity of ClinCNV for regions fulfilling the criteria outlined in “Methods” (Supplementary Table S2).
Impact of CNVs in cystic kidney genes on clinical outcomes
To determine the extent to which CNVs act as modifiers of PKD, we applied regression models to investigate the association between CNVs and (1) age at kidney failure, (2) presence or absence of liver cysts, and (3) htTKV. As these regressions required adding the diagnostic variant type (PKD1-T, etc., see “Methods”) as a covariate, only ADPKD individuals were tested, as the ADPKD variant type groups had sufficient numbers for testing. Two types of regression models were conducted: (1) testing the presence of a CNV in a cystic kidney gene as a covariate and (2) testing the burden of CNVs (in 100 kb) as a covariate. Burden was quantified across the exome, stratifying into cystic and non-cystic regions, to determine whether the amount of the exome that the CNVs covered was associated with ADPKD outcomes (see “Methods”).
The presence of a CNV in a cystic kidney gene did not associate with any clinical outcomes (Supplementary Table S3). Duplication burden in cystic genes associated with worse kidney survival (HR = 1.56, 95% CI: 1.26, 1.93, adj-p = 0.0004), with this model having 43.0% power to detect this effect. Additionally, regression models indicated a significant association of exome-wide duplication burden with the presence of liver cysts (OR = 0.82; 95% CI: 0.73–0.91, adj-p = 0.006) (Fig. 2). Stratification into cystic and non-cystic regions showed that the association was driven by the burden of duplications impacting non-cystic genes. Power analysis indicated this model had 95% power to detect an effect of the observed size.

The effect sizes for the burden of each CNV type are shown for age at kidney failure (n = 254) (A), htTKV (n = 95) (B), and presence of liver cysts (n = 228) (C). No data is available for cystic deletion burden as there were too few observations with cystic deletion burden to create reliable models. CNV burden is stratified by region (exome-wide, cystic kidney genes only, non-cystic kidney genes) and CNV type (all CNVs, deletions only, duplications only). The adjusted p-value is the value after Bonferroni correction.
The non-cystic duplication burden result prompted us to investigate further if there were certain regions of the genome that were driving the observed effect. We therefore conducted gene set enrichment analysis to determine if there was overrepresentation of certain GO terms, comparing the non-cystic genes impacted by duplications to (1) all annotated human genes (20,580 genes) and (2) the list of genes impacted by any type of CNV in our samples (2431 genes). In both cases, keratinisation (GO:0031424) was the most significantly enriched GO term (p < 0.005) (Supplementary Tables S4 and S5). We then implemented the CNVRuler tool [36] to determine if a specific CNV region was driving this association. Although no associations to liver cysts remained significant after Bonferroni correction, the top region (OR = 0.13; 95% CI: 0.04–0.42, Bonferroni corrected p = 0.052) (Supplementary Table S8) was a duplication that contained exons 5-8 of LRP5L (chr22:25,351,794-25,360,092), a pseudogene of LRP5, a monogenic cause of polycystic liver disease [39]. This region occurred in 7/39 (17.9%) individuals without liver cysts and 6/189 (3.2%) individuals with liver cysts. A nucleotide BLAST indicated the pseudogene LRP5L had 90.5% similarity to 31% of LRP5. According to InterProScan (accession: IPR000033), the hypothetical protein for LRP5L contained low-density lipoprotein receptor class B repeats, which encode for beta-propeller protein domains that are important for the function of its parent gene, LRP5 [39].
Discussion
In this study, we applied ClinCNV to NGS data to call CNVs across cystic kidney genes in a cohort of 371 PKD patients. ClinCNV and subsequent testing by MLPA revealed PKD1 or PKD2 deletions in 13 individuals, increasing our diagnostic yield from 86.5% to 90.0%, with high specificity (100%) but lower sensitivity (62.5%) in comparison with MLPA. Additionally, CNV burden testing indicated an association of duplications in genes unrelated to cystic kidney disease to a lower likelihood of liver cysts, which may in part be driven by a duplication affecting LRP5L.
WES and TGP data are routinely used for molecular diagnosis of PKD and while a causal variant is found for most patients, 13.5% of our cohort remained unsolved when using only SNV analysis. The identification of pathogenic variants is important for management of disease, determining likely prognosis, and aiding in the patient’s family planning [40]. It is known that approximately 1–5% of patients have ADPKD caused by CNVs in key genes such as PKD1 and PKD2, yet the identification of these variants relies on a separate clinical pipeline requiring an expensive MLPA test.
The utilisation of a bioinformatics-based CNV detection tool with existing NGS data allowed the detection of disease-causing CNVs in PKD1 or PKD2. This has both clinical and cost implications, as the NGS data generated for these patients can be used for both SNV analysis as well as CNV detection, confirming the diagnosis for these patients at a similar specificity to MLPA. This contributes to maximising the utility of NGS data generated for each patient and improves efficiency in achieving a molecular diagnosis. The CNV caller struggled with small (<1 exon) deletions, which has been highlighted by other read-depth based CNV callers [41]. This emphasises the importance of having multiple family members with PKD sequenced, as the CNV caller was able to detect other family members who harboured a single-exon deletion. The probes available in any given MLPA probemix should be considered, as highlighted by Fig. 1. MLPA suggests that the deletion occurs in exons 29–30, when visualisation of the sequencing data clearly shows the breakpoint of the CNV occurring in exon 27; this is due to the start position of the deletion occurring after the location of the probe, and the lack of a probe in exon 28. Therefore, CNV detection using NGS data may also be beneficial in cases where sufficient probe coverage is not available in MLPA, or an MLPA kit is not available for the gene of interest. Since the completion of this study, we have discovered an additional 2 patients with diagnostic CNVs in PKD1 or PKD2 in our clinical cohort by utilising this pipeline, demonstrating the applicability of this workflow.
The majority (22/25) of additional non-diagnostic CNVs found in cystic kidney genes were duplications. Dosage sensitivity is a known pathogenic mechanism for specific genes. But for duplications impacting other genes, or where the breakpoints are intragenic, it can be more challenging to infer pathogenicity. Intragenic duplications may disrupt the reading frame of a gene as most occur tandem in direct orientation to the original locus [42]. However, in most cases it is unclear whether a normal allele is preserved, as was the case in this study. As a result, intragenic duplications are classified as pathogenic only if they occur within established haploinsufficiency genes [35]. The results of our regression analysis indicated an association of cystic gene duplications to worse kidney survival. This may suggest that while these duplications are not pathogenic or disease-causing in our patients, they may compound the effect of a diagnostic PKD variant on kidney survival. However, the power analysis suggests we may not have been adequately powered to detect this effect, so replication in a larger cohort will be necessary.
The CNV burden analyses also suggested that duplication burden in the exome is associated with a significantly lower likelihood of the presence of liver cysts, and a GO term enrichment analysis suggested that genes involved in keratinisation may be driving this association. CNVRuler indicated a duplicated region impacting the gene LRP5L associates with the absence of liver cysts, though this does not survive multiple testing. Examples exist of CNV burden associating with disease phenotypes [43, 44], but generally burdens are associated with increased risk of disease. “Protective” CNVs in specific regions have been identified in other diseases, but the exact mechanism of protectiveness is relatively unclear [45, 46]. LRP5L is a pseudogene of LRP5, a key gene involved in polycystic liver disease [39]. LRP5L is predicted to produce a protein at the transcript level according to UniProt, and is not an OMIM disease gene, unlike its parent gene LRP5. LRP5 is involved in the Wnt signalling cascade, which has been shown previously to be involved in keratinisation [47]. Keratins have been noted in their protective effects in the liver, helping hepatocytes cope with stress and injury [48]. LRP5L has been identified in keratinocytes as well [49]. Most strikingly, the parent gene LRP5 has been postulated to be involved in liver cyst development in ADPKD patients previously [50]. The lack of knowledge about LRP5L limits us from drawing any further conclusions; but, speculating, if LRP5L does in fact translate into a protein, and encodes similar functional protein domains to its parent gene, this may explain the effect seen in our results. Replication of these results in a larger cohort is most likely necessary.
In summary, our results demonstrate how WES and TGP data can be used to successfully call CNVs, both diagnostic and non-diagnostic. This has potential clinical and cost impacts, as NGS data performed previously for SNP and indel analysis can be repurposed for CNV detection in PKD patients.
Data availability
The dataset analysed in this study are not publicly available due to the privacy required of patient data, but are available from the corresponding author on reasonable request.
Code availability
The code used to generate the copy number variant calls from the targeted gene panel sequencing and exome sequencing is available at: https://github.com/FutureNeuroIE/clincnv-pipeline.
References
Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–30.
Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019;393:919–35.
Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription–factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305.
Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, et al. ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol. 2019;30:2091–102.
Cordido A, Besada-Cerecedo L, García-González MA. The genetic and cellular basis of autosomal dominant polycystic kidney disease-A primer for clinicians. Front Pediatr. 2017;5:279.
Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. 2018;102:832–44.
Reddy BV, Chapman AB. A patient with a novel gene mutation leading to autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2017;12:1695–8.
Senum SR, Li YSM, Benson KA, Joli G, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet. 2022;109:136–56.
Szabó T, Orosz P, Balogh E, Jávorszky E, Máttyus I, Bereczki C, et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr Nephrol. 2018;33:1713–21.
Hartung EA, Guay-Woodford LM. DZIP1L defines a new functional zip code for autosomal recessive PKD. Nat Rev Nephrol. 2017;13:519–20.
Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–13.
Audrézet MP, Cornec-Le Gall E, Chen JM, Redon S, Quéré I, Creff J, et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239–50.
Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–60.
Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27:1861–8.
Chang AR, Moore BS, Luo JZ, Sartori G, Fang B, Jacobs S, et al. Exome sequencing of a clinical population for autosomal dominant polycystic kidney disease. JAMA. 2022;328:2412–21.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16:172–83.
Benson KA, Murray SL, Senum SR, Elhassan E, Conlon ET, Kennedy C, et al. The genetic landscape of polycystic kidney disease in Ireland. Eur J Hum Genet. 2021;29:827–38.
Wilson EM, Choi J, Torres VE, Somlo S, Besse W. Large Deletions in GANAB and SEC63 Explain 2 Cases of Polycystic Kidney and Liver Disease. Kidney Int Rep. 2020;5:727–31.
Rijal JP, Dhakal P, Giri S, Dahal KV Case Report: Tuberous sclerosis complex with autosomal dominant polycystic kidney disease: a rare duo. BMJ Case Rep [Internet]. 2014 Dec;2014. Available from: /pmc/articles/PMC4275747/
Moreno-De-Luca D, Mulle JG, Kaminsky EB, Sanders SJ, Myers SM, Adam MP, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am J Hum Genet. 2010;87:618–30.
Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, et al. Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet. 2000;66:778–89.
Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DWW, et al. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57.
Schönauer R, Baatz S, Nemitz-Kliemchen M, Frank V, Petzold F, Sewerin S, et al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet Med. 2020;22:1374–83.
Gabrielaite M, Torp MH, Rasmussen MS, Andreu-Sánchez S, Vieira FG, Pedersen CB, et al. A comparison of tools for copy-number variation detection in germline whole exome and whole genome sequencing data. Cancers. 2021;13:6283.
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinforma. 2013;14:S1.
Bogdanova N, Markoff A, Gerke V, McCluskey M, Horst J, Dworniczak B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics. 2001;74:333–41.
Claus LR, Ernst RF, Elferink MG, van Deutekom HWM, van der Zwaag B, van Eerde AM. The importance of copy number variant analysis in patients with monogenic kidney disease. Kidney Int Rep. 2024;9:2695–704.
Oh J, Shin JI, Lee K, Lee C, Ko Y, Lee JS. Clinical application of a phenotype-based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clin Genet. 2021;99:236–49.
Elhassan EAE, Murray SL, Connaughton DM, Kennedy C, Cormican S, Cowhig C, et al. The utility of a genetic kidney disease clinic employing a broad range of genomic testing platforms: experience of the Irish Kidney Gene Project. J Nephrol. 2022;35:1655–65.
Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. JASN. 2009;20:205.
Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012;7:479.
Demidov G, Sturm M, Ossowski S ClinCNV: multi-sample germline CNV detection in NGS data. bioRxiv. 2022;2022.06.10.495642.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med J Am Coll Med Genet. 2020;22:245–57.
Kim JH, Hu HJ, Yim SH, Bae JS, Kim SY, Chung YJ. CNVRuler: a copy number variation-based case–control association analysis tool. Bioinformatics. 2012;28:1790–2.
Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, Ebert D, et al. The Gene Ontology knowledgebase in 2023. Genetics. 2023;224:iyad031.
Srour M, Hamdan FF, Schwartzentruber JA, Patry L, Ospina LH, Shevell MI, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet. 2012;49:636–41.
Cnossen WR, te Morsche RHM, Hoischen A, Gilissen C, Chrispijn M, Venselaar H, et al. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc Natl Acad Sci USA. 2014;111:5343–8.
Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6:197–206.
Moreno-Cabrera JM, del Valle J, Castellanos E, Feliubadaló L, Pineda M, Brunet J, et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur J Hum Genet. 2020;28:1645–55.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–20.
Auwerx C, Jõeloo M, Sadler MC, Tesio N, Ojavee S, Clark CJ, et al. Rare copy-number variants as modulators of common disease susceptibility. Genome Med. 2024;16:5.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Hardwick RJ, Ménard A, Sironi M, Milet J, Garcia A, Sese C, et al. Haptoglobin (HP) and Haptoglobin-related protein (HPR) copy number variation, natural selection, and trypanosomiasis. Hum Genet. 2014;133:69–83.
Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: Characterization, ethnic distribution and evolutionary implications. Ann Med. 2006;38:592–604.
Kosumi H, Watanabe M, Shinkuma S, Nohara T, Fujimura Y, Tsukiyama T, et al. Wnt/β-catenin signaling stabilizes hemidesmosomes in keratinocytes. J Invest Dermatol. 2022;142:1576–86.e2.
Ku NO, Strnad P, Zhong BH, Tao GZ, Omary BM. Keratins let liver live: mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology. 2007;46:1639.
Toulza E, Mattiuzzo NR, Galliano MF, Jonca N, Dossat C, Jacob D, et al. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol. 2007;8:R107.
Cnossen WR, te Morsche RHM, Hoischen A, Gilissen C, Venselaar H, Mehdi S, et al. LRP5 variants may contribute to ADPKD. Eur J Hum Genet EJHG. 2016;24:237–42.
Acknowledgements
This publication has emanated from research supported in part by a research grant from Research Ireland under Grant number 18/CRT/6214 and BioMarin Pharmaceutical Inc. The authors acknowledge the help of Dr. Monika Sigg from BioMarin Pharmaceutical Inc. for aiding the direction of the project. We are grateful for the help of Dr. German Demidov with suggestions for ClinCNV parameters for the CNV calling in this study. We also acknowledge the help of Dr. Ciarán Kelly with utilising the CNVRuler tool. We also acknowledge that this research would not be possible without the participation of the patients and their families.
Funding
This research is supported by a research grant from Research Ireland under Grant number 18/CRT/6214 and funding from BioMarin Pharmaceutical Inc. BioMarin Pharmaceutical Inc, Novato, CA, USA and Research Ireland Centre for Research Training in Genomics Data Science, Dublin, Ireland (grant no. 18/CRT/6214). Open Access funding provided by the IReL Consortium.
Author information
Authors and Affiliations
Contributions
KB, GLC, and PC conceptualised the study design. SH, GLC, and PC prepared the manuscript with review from all authors. SH carried out copy number variant calling and data analysis of the current study. PC and EE gathered the clinical information for the patients included in the study. HG and KC contributed code for the analysis and aided in data interpretation. OT processed the sequencing data and filtered for short variants prior to the current study.
Corresponding author
Ethics declarations
Competing interests
This publication has emanated from research supported by BioMarin Pharmaceutical Inc.
Ethical approval
This study was approved by Beaumont Hospital Ethics Committee under protocol 19/23 and was conducted according to the Declaration of Helsinki. All participants provided informed, written consent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heneghan, S., Elhassan, E.A.E., Ghani, H. et al. An NGS-based investigation of copy number variants in the diagnosis and severity of adult polycystic kidney disease. Eur J Hum Genet (2026). https://doi.org/10.1038/s41431-026-02027-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41431-026-02027-8
