
Abstract
Folic acid plays a crucial role in cellular regulation and metabolism, commonly included in dietary supplements. Despite this, its involvement in colorectal cancer (CRC), particularly in metabolic pathways and immune evasion, remains poorly understood. We developed the FMRG_score system using machine learning algorithms on TCGA and GEO data to assess modification patterns influencing clinical outcomes and immune characteristics in CRC. The system’s reliability was validated across multiple external immunotherapy cohorts. We examined associations between FMRG-related features and clinical traits, mutation profiles, biological functions, immune infiltration, therapy responses, and drug sensitivities. By integrating in vitro and in vivo experiments with bioinformatics, we built a nine-gene risk model linked to folate metabolism for CRC prognosis. Notably, CYP26A1, a key gene in the model, was upregulated in CRC tissues, promoting cell proliferation, migration, invasion, and contributing to an immunosuppressive tumor microenvironment. Significant differences in clinical traits, immune infiltration, checkpoint expression, therapy response, and drug sensitivity were observed between risk groups. This folate-based scoring system provides a novel tool for evaluating CRC prognosis, tumor microenvironment, and response to immunotherapy. We also propose CYP26A1 as a potential oncogene in CRC, offering new therapeutic insights.
Similar content being viewed by others


Introduction
CRC is the third most common malignancy. It is also the second deadliest cancer worldwide [1]. CRC is 1.5 times more common in men than in women, with most cases occurring after the age of 50 [2]. However, CRC incidence has been increasing in countries with a rising human development index, especially in those under age 50 [3]. By 2035, the number of deaths from colon cancer and rectal cancer is projected to rise by 60.0% and 71.5%, respectively, due to population growth and aging [4]. Research definitively shows that early diagnosis and precise treatment of CRC significantly improve patient survival rates [5, 6]. Recent advances in molecular biology have facilitated the delineation of critical genetic pathways that play a role in colorectal carcinogenesis [7, 8]. These pathways include the adenomatous polyposis coli (APC) pathway [9], the microsatellite instability (MSI) pathway [10], and the CpG island methylator phenotype (CIMP) pathway [11]. Understanding the intricate mechanisms of these pathways can provide valuable insights for developing targeted therapeutic strategies to combat colorectal cancer.
In recent years, the rapid advancement of immune checkpoint inhibitor (ICI)-based immunotherapy has brought about a promising new era in anticancer treatment. ICIs enhance the antitumor immune response by disrupting the signaling of key immunosuppressive proteins like programmed cell death 1 (PD-1), programmed cell death-1 ligand 1 (PD-L1), and cytotoxic T lymphocyte antigen 4 (CTLA-4) [12,13,14]. Some patients with melanoma, non-small-cell lung cancer (NSCLC), and other cancers show sustained responses [15,16,17,18]. Immunotherapy using ICI in CRC patients has shown promising safety and efficacy outcomes. Approved immunotherapeutic agents for advanced CRC treatment include pembrolizumab, nivolumab, and ipilimumab [19]. In 2015, Le et al. discovered that the anti-PD-1 drug pembrolizumab exhibited a notably higher response rate in patients with the DNA mismatch repair–deficient (dMMR)/microsatellite instability-high (MSI-H) molecular subtype of CRC [20]. This finding implies the potential benefits of anti-PD-1 therapy for patients with both CRC and non-CRC dMMR tumors. ICIs are effective in treating dMMR/microsatellite-stable (MSS) CRC due to the elevated mutational load, abundance of neoantigens, and increased immune cell presence within this tumor subtype [21, 22]. Significantly, MSS-pMMR patients make up a substantial portion of CRC patients [23]. Hence, it is essential to actively explore new biomarkers and innovative approaches for early detection and treatment to understand the underlying mechanisms of varied treatment responses in current colorectal cancer research.
The relationship between folate metabolism and CRC has been extensively studied due to folate’s crucial role in DNA methylation, repair, and synthesis processes [24,25,26,27,28]. Folate, a- vitamin B naturally present in foods, and its synthetic form folic acid, commonly utilized in supplements and food fortification, play a vital role in cell production and upkeep, especially during stages of accelerated cell division and growth, such as infancy and pregnancy [29, 30]. Based on these pivotal functions, it has been hypothesized by researchers that sufficient folate intake has the potential to mitigate DNA alterations that might serve as precursors to cancer development, particularly in the colon and rectum [31]. Recent studies and reviews suggest a complex relationship between folate intake, folate status, and CRC risk. A systematic review and meta-analysis examining the impact of folic acid supplement intake and total folate intake on CRC risk found no significant effect of folic acid supplements in randomized controlled trials. Despite these findings, the effect of folate status, as measured by red blood cell folate content, on CRC risk was not significant, indicating that the relationship may depend on the form of folate consumed and other individual factors [32]. A study found that high folic acid intake accelerates methionine cycling in cancerous tissues in vivo, potentially contributing to the development of hepatocellular carcinoma [33]. Recent evidence suggests that the relationship between high plasma levels of folate and CRC risk may not be straightforward [34,35,36]. For example, one study observed a significant decrease in CRC risk among women in the highest quartile of plasma folate levels [37]. In contrast, another study identified an increased risk of CRC associated with high plasma folate levels over a follow-up period [35]. These inconsistencies underscore the challenges inherent in accurately assessing the impact of folate on CRC risk. The relevance of genetic polymorphisms in folate metabolism genes, particularly in genes such as MTHFR that code for enzymes in folate metabolism, should be noted [38]. However, the impact of folate metabolism on TME characteristics and clinical outcomes of CRC patients is still uncertain.
We systematically analyzed the underlying effects of folate metabolism in CRC in this study. Through our analysis, we constructed a folate metabolism-related signature to predict the prognostic outcomes, TME characteristics, and immunotherapy response in CRC patients. Our findings not only revealed the paramount role of folate metabolism in the complex heterogeneity of CRC but also indicated its potential to enhance individualized management strategies for CRC patients. This study also marked the first instance of proposing the potential protumor function of Cytochrome P450 Family 26 Subfamily A Member 1 (CYP26A1) in CRC, laying the groundwork for further exploration of this promising molecular target in CRC.
Materials and methods
Data collection
The study included CRC patients from TCGA (n = 585) with available RNA-seq data (FPKM format converted to TPM), complete clinical annotations, and survival data. External validation cohorts (GSE39582, GSE17536, GSE38832, GSE17537) were selected based on the availability of comparable expression and survival data. Exclusion criteria included missing survival information, poor-quality sequencing data, and non-adenocarcinoma CRC subtypes. Normal adjacent tissues were excluded from survival analysis but included for differential expression comparisons. The training cohort included the transcriptome data and clinical information of 585 patients with colorectal adenocarcinoma (COAD and READ) were retrieved from the Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/repository). Validation sets were obtained from GSE39582 (579 patients), GSE17536 (177 patients), GSE38832 (122 patients) GSE17537 (55 patients) expression profiles from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). Single-cell RNA sequencing (scRNA-seq) data for colorectal adenocarcinomas were obtained from the GSE161277 and GSE205506 databases, which include 13 and 19 CRC samples, respectively. 410 folate metabolism-related genes (FMRGs) were retrieved from GeneCards (https://www.genecards.org/). The complete gene details are displayed in Table S1. The RNA-seq sample expression levels were converted from fragments per kilobase of transcript per million mapped reads (FPKM) to transcripts per million (TPM), and then log2(TPM + 1) was calculated. Somatic mutation data, copy number variation (CNV) files, and tumor mutation burden (TMB) data of CRC patients were retrieved from the TCGA database.
Construction and validation of FMRGs risk signature
First, we analyzed the genes related to folate metabolism and the differential genes from TCGA, leading to the identification of 138 genes. To identify key prognostic genes among the 138 folate metabolism-related candidates, we performed univariate Cox regression, selecting 19 genes with p < 0.05. LASSO Cox regression with 10-fold cross-validation was applied to refine the model, ultimately identifying 9 genes that provided the most robust prognostic prediction. To further ensure statistical rigor, multivariate Cox regression was conducted, confirming these genes as independent prognostic factors. Genes with borderline significance (p > 0.05) or high collinearity were excluded to optimize model performance. The FMRG_score was constructed using LASSO Cox regression, which selected the optimal gene set and assigned coefficients based on 10-fold cross-validation to prevent overfitting. The λ penalty parameter was optimized to retain the most predictive genes while eliminating less significant ones. The final coefficients were obtained from the LASSO model and further validated through multivariate Cox regression to ensure their independent prognostic value. The FMRG_score for each patient was computed as a weighted sum of gene expression levels using these coefficients, and patients were classified into high- and low-risk groups based on the median score. The FMRG_score was calculated as follows:
In the training set, 585 patients were stratified into low-risk and high-risk groups based on the median risk score, which was calculated using the risk coefficient (Coefi) and expression of each gene (Expi). Subsequently, Kaplan-Meier survival analysis was conducted on the two groups to assess their survival outcomes. The predictive performance of the signature was evaluated using receiver operating characteristic (ROC) curves. Kaplan–Meier survival curves were plotted, and log-rank tests were performed to assess the statistical significance of the observed differences in survival between the two risk groups. The effectiveness of the prediction model was further validated in three independent GEO datasets (GSE38832, GSE39582, GSE17537) through survival analysis and calculation of the area under the curve (AUC) in ROC analysis.
Relationship between risk groupings and clinical characteristics
The study analyzed clinical factors such as age, gender, and TNM stage. Differences in prognostic outcomes were assessed using Kaplan-Meier analysis in R software with the “survival” and “survminer” packages [39].
Development and validation of a nomogram scoring system
To determine the independent prognostic values and the predictive efficacy of the FMRG_score in predicting the survival of CRC patients, we conducted univariable and multivariate Cox regression analyses for better clinical practice. Furthermore, we investigated the relationship between the FMRG_score and various clinical characteristics. Additionally, in order to improve the prognostic accuracy of our model, a nomogram was developed utilizing the risk score, T stage, and N stage as independent prognostic factors to estimate the probability of overall survival (OS) at 2-, 4-, and 6-years. Subsequently, the predictive efficacy was compared between the FMRG_score and different clinical pathological factors based on AUC.
Functional and pathway enrichment analysis
To identify specific biological pathways enriched between the high- and low-risk groups, we performed a comprehensive analysis using various techniques. Initially, we conducted Gene Ontology (GO) analysis (Supplementary Table S8) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Supplementary Table S7) to understand the functions of the screened candidate genes and related pathways. Following this, we utilized the “ClustProfiler” and “GSVA” packages for further elucidation [40, 41]. Additionally, we employed Gene Set Variation Analysis (GSVA) (Supplementary Table S10) to identify variations in gene sets. Subsequently, Gene Set Enrichment Analysis (GSEA) was utilized to identify enriched pathways or gene sets based on differential expression results between the high-risk and low-risk groups (Supplementary Table S9). Moreover, GSVA (Supplementary Table S11) and correlation analysis between Hallmark pathway activities and the FMRG risk score were conducted to explore potential pathways associated with the identified signature.
Exploration of the immune landscape in distinct risk groupings
The immune characteristics of 585 CRC samples were assessed by evaluating the scores of tumor microenvironment (TME) cells using the single-sample gene set enrichment analysis (ssGSEA) algorithm [42]. To analyze the relative proportion of 22 immune cells within the CRC samples, CIBERSORT (https://cibersort.stanford.edu) was employed. The CIBERSORT algorithm was executed using R software. By leveraging the 585 samples gene expression matrix and the provided gene expression feature set of the 22 immune cell subtypes from the official website, simulation calculations were iterated 1000 times to derive the relative composition ratio of the 22 immune cells in each sample. Subsequently, the immune score and ESTIMATE score of each patient were assessed utilizing the R package of estimate [43]. Statistical significance was assessed using the Wilcoxon rank-sum test.
Prediction of response to immunotherapy
Initially, the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm was utilized to evaluate the potential disparities in treatment responses between high- and low-risk groups [44]. A higher TIDE score was found to be associated with reduced treatment efficacy, thereby highlighting a negative correlation between the TIDE score and treatment effectiveness. Furthermore, data on immunotherapy was gathered from various datasets including the IMvigor210 dataset for urothelial carcinoma (UC), the TCGA dataset, GSE17536 dataset, GSE39582 dataset, and GSE38832 dataset for CRC. Subsequently, within each dataset, the FMRG_score was calculated to predict responses to immunotherapy.
Mutation and drug susceptibility analysis
To explore the differences in therapeutic effects of chemotherapeutic drugs between high- and low-risk groups of CRC patients, we first generated the mutation annotation format (MAF) from the TCGA database using the “maftools” R package. Subsequently, we calculated the TMB score for each patient within these groups. Additionally, we determined the semi-inhibitory concentration (IC50) values of commonly used chemotherapeutic drugs for treating CRC by utilizing the “pRRophetic” package.
Single-cell RNA sequencing analysis
The scRNA-seq analysis was conducted using the “Seurat” R package for data processing, including quality control steps where cells expressing 200–7000 genes were retained, and cells with more than 20% mitochondrial gene expression were excluded. The cells were classified into eight primary types using t-SNE dimensionality reduction. The “inferCNV” package was used to infer copy number variations (CNVs) from single-cell RNA sequencing data by comparing gene expression profiles to a reference dataset. This approach allowed identification of regions with CNV alterations. Based on these CNV patterns, the malignancy score was calculated to classify cells as either malignant or non-malignant, reflecting tumor characteristics. The “AUCell” algorithm was employed to assess gene set activity at the single-cell level. Cell-to-cell communication networks were analyzed using “CellChat,” focusing on receptor-ligand interactions. Additionally, the “Monocle 2” algorithm was used to construct pseudotime trajectories to track the dynamic functional changes in FDRG-related signatures.
Cell culture and reagents
The human colon carcinoma cell line HCT116, RKO and MC38 were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). All cell lines were authenticated by monitoring cell vitality, mycoplasma contamination, and short tandem repeat profiling. Cells were maintained in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. Culture media and supplements were obtained from BasalMedia (Shanghai, China).
DNA constructs, transfection, and viral transduction
CYP26A1 cDNA in penter vector with C terminal Flag and His tag was purchased from Vigene Bioscience. Short hairpin RNAs (shRNAs) targeting human CYP26A1 (shCYP26A1) in GIPZ lentiviral vector and corresponding control constructs were purchased from Dharmacon. The detailed information of expression vectors for molecular cloning is provided in Supplementary Table S14. shRNAs in lentiviral expression vectors were transfected into HEK293T together with packaging plasmid mix using Neofect DNA transfection reagent. Supernatants were collected after 48 h of transfection and used for infecting cells in the presence of 8 mg/mL of polybrene. After 24 h of infection, cells were selected with 2 mg/mL of puromycin (Cayman, Ann Arbor, USA) for 1–2 weeks.
Cell viability and colony formation assays
Cell proliferation assays were performed using Cell Counting Kit-8 (CCK-8) (Dojindo, Shanghai, China) according to the manufacturer’s instructions. The absorbance was measured at a wavelength of 450 nm (A450). For colony-formation assays, cells were grown onto 6-well plate at a density of 2000 cells/well for 14 days with media replacement every 3 days. Cells were stained with 1% crystal violet and the number of survival colonies was counted.
Western blotting
Proteins from cell lines and tissues were extracted using RIPA lysis buffer (Thermo Fisher Scientific, USA) on ice. After centrifugation (15,000 × g, 10 min), protein concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific, USA). Protein samples were separated on PAGE gels (Epizyme Biomedical Technology, Shanghai, China) and transferred to 0.22 µm Immobilon PVDF membranes (Millipore Sigma, USA). Membranes were blocked with 5% milk and incubated with primary antibodies overnight at 4 °C. Secondary antibodies (anti-Rabbit IgG) were applied at room temperature for 1 h, and immunoreactivity was detected using an ECL system (Share-bio, Shanghai, China). Primary antibodies used: pan-kla (#PTM-1401RM, PTMBio, Hangzhou, China) at 1:1000. Secondary antibody dilution was 1:5000.
Transwell migration assays and Matrigel invasion assays
Cell migration and invasion were assessed using transwell chambers (Corning Biocoat, Tewksbury, USA). For invasion assays, the Matrigel (BD Biosciences) was diluted 1:8 (dilution ratio determined based on the MMP expression levels of the cells) and applied to coat the upper surface of the chamber membrane. The coated chambers were incubated at 37 °C for 30 min to allow the Matrigel to polymerize and hydrated prior to use. For both assays, cells were resuspended in DMEM containing 1% FBS and loaded at a density of 5 × 104 cells per well onto the upper well of chambers, while growth medium containing 10% FBS was placed in the lower chamber as a chemoattractant. After 24 h incubation, cells on the upper surface were gently removed with a cotton swab, and cells that had migrated or invaded to the lower surface were fixed with methanol and stained with 1% crystal violet solution. The number of cells was counted under a light microscope with a magnification of 100. All assays were conducted in triplicate and repeated at least three times.
RNA isolation and quantitative reverse transcription-PCR
Total RNA was isolated from cell lines and tissue samples using Trizol reagent (Invitrogen), Reverse transcription was performed using a PrimeScript RT reagent Kit (TaKara). The resultant cDNA was subjected to quantitative real-time PCR (qPCR) using SYBR Premix Ex Taq (Tli RNaseH Plus) (Takara) on an Eppendorf Mastercycler ep realplex4 instrument. Primer sequences for qPCR are listed in Supplementary Table S15. All reactions were performed in triplicate. The data is present as mean ± SD.
In vitro T cell activation and tumor cells-T cells coculture model
PBMCs from healthy donors were treated with RBC lysis buffer and sorted using CD8 A antibody by flow cytometry. After activation with anti-CD3 (BioLegend, 5 μg/ml) and anti-CD28 (BioLegend, 2.5 μg/ml) at the indicated concentrations and cultured in media containing IL-2 (PeproTech, 10 ng/ml), the cells were used in co-culture models. T cells (2 × 105 cells/well) were seeded in the top chamber of a transwell (pore size: 0.4 µm), while tumor cells, with or without CYP26A1 knockdown, were seeded in the bottom chamber for indirect co-culture. In the direct co-culture model, T cells and tumor cells were mixed directly at a 1:4 ratio without using a chamber. Following co-culture, T cells from both models were subjected to surface marker and intracellular cytokine staining and analyzed by FACS.
Subcutaneous tumor model construction
MC38 colon cancer cells were cultured in DMEM medium supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 incubator. Cells were harvested during the logarithmic growth phase and resuspended in PBS at a density of 5 × 105 cells/100 μL. Female C57BL/6 nude mice (6–8 weeks old, 18–22 g) were housed in specific pathogen-free conditions with free access to food and water. A total of 12 mice were evenly divided into two groups. Mice were anesthetized with isoflurane, and the cell suspension was injected subcutaneously into the right hind leg of each mouse using a sterile syringe. Tumor growth was monitored every 3 days by measuring length (L) and width (W) with electronic calipers. Tumor volume was calculated using the formula: Volume = (L × W²)/2. The endpoint was set when the tumor volume in any group reached 1500 mm³. After 3 weeks, mice were euthanized, and tumors were excised, weighed, and processed for further analysis.
Flow cytometry analysis of tumor cells from subcutaneous tumor model
Tumor cells from subcutaneous tumor model were resuspended to a density of 1 × 106 cells/100 μL. The cells were first blocked with Brilliant Violet 510™ anti-mouse CD4 (100449, BioLegend), Alexa Flour 700 anti-mouse FOXP3 (126422, BioLegend), Brilliant Violet 421™ anti-mouse CD8a (100753, BioLegend), APC anti-mouse IFN-γ(505810, BioLegend), PerCP/Cyanine5.5 anti-human/mouse Granzyme B Recombinant (372212, BioLegend) or Brilliant Violet 605™ anti-mouse Ki-67 (652413, BioLegend) purchased from BioLegend (USA). After incubation of 20 min, cells were washed and terminated with 1 mL PBS, and the stained cells were detected by flow cytometry (Accuri™ C6 CSampler, BD Biosciences, San Jose, CA, USA).
Statistical analysis
All experiments were replicated at least three times, and the results are presented as mean ± standard deviation (SD). For comparisons between two groups, unpaired Student’s t tests were used for normally distributed variables, while Mann–Whitney U tests were employed for non-normally distributed variables. For multiple group comparisons, one-way ANOVA (parametric) or Kruskal–Wallis tests (nonparametric) were performed as appropriate. Pearson’s or Spearman’s correlation analyses were conducted to evaluate linear relationships between variables. Survival curves were generated using the Kaplan-Meier method, and differences between groups were assessed using the log-rank test. ROC curves were generated and area under the AUC values were calculated to evaluate predictive performance. Statistical analyses were performed using SPSS version 23.0, R software (version 4.3.2), and GraphPad Prism 9. p values < 0.05 were considered statistically significant.
Results
Construction and validation of the prognostic FMRG_score
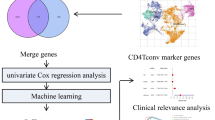
The flowchart for this article was shown in Fig. 1. Omics data from TCGA and GEO were used to develop the FMRG score, explore biological mechanisms, and identify CYP26A1 as a carcinogen.

Omics data were initially generated and validated using TCGA and GEO datasets. Following this, a risk score model for FMRGs was established, demonstrating its potential for prognosis. The biological mechanisms, changes in immune infiltration, and therapeutic implications (including immunotherapy and drug therapy) of FMRGs were subsequently explored. Finally, CYP26A1 was identified as a carcinogenic factor in colorectal cancer, with its role further validated by experimental evidence.
To construct the FMRG_score, we identified 138 target genes through differential analysis of CRC data from TCGA, intersecting with genes associated with folate metabolism (Fig. 2A and Supplementary Tables S1 and 2). We chose to conduct additional analysis on the TCGA cohort to extract a prognostic signature for FMRG. Initially, a univariable Cox regression analysis revealed 19 candidate genes that were found to be associated with CRC, with a p value < 0.05 (Supplementary Tables S3 and 4). To delve deeper into the characteristics of folate metabolism in CRC patients, we utilized LASSO Cox regression to determine the optimal λ value, leading to the identification of 9 key genes (Supplementary Tables S5 and 6). The mRNA expressions of the 9 key genes in tumor and normal specimens were assessed using the TCGA-CRC dataset. Notably, AHCY, PLK1, WNT5A, CYP26A1, BDNF and DRD4 exhibited upregulation, while NAT1, CD36 and GSTM1displayed downregulation in tumor (Fig. 2D). Five of the risk genes (NAT1, WNT5A, CYP26A1, BDNF and DRD4) were identified as independent predictive factors (Table 1). The locations of these 9 risk genes on chromosomes were visualized in Fig. 2I. We displayed the relationship between these nine risk genes and patient OS prognosis through the TCGA CRC dataset (Fig. S1). This comprehensive model integrates the predictive power of individual genes to provide a more precise tool for assessing CRC prognosis. The risk-score model was determined using the following equation:

A Veen plots of folate metabolism-related genes and DEGs in TCGA-CRC. B LASSO coefficient profiles and partial likelihood deviance plot for gene selection. C Time-dependent ROC curves at 2, 4, and 6 years, illustrating the prognostic performance of the FDRG risk model in the TCGA dataset. D Boxplot comparing the expression levels of key genes in normal vs. tumor tissues. E Kaplan–Meier analysis of OS in high- vs. low-risk groups (p = 4.49e−07). F, G Risk score distribution, survival status, and scatterplot of patients based on risk score. H Heatmap showing the expression levels of selected genes in high- and low-risk groups. I Circos plot visualizing the chromosomal locations of genes in the FMRG signature.
Patients were divided into two groups based on their FMRG_score: those with a score lower than the median were categorized as low-risk (n = 292), while those with a score higher than the median were classified as high-risk (n = 293). The distribution plot showed that as FMRG_score increased, survival times decreased and recurrence rates increased (Fig. 2F, G). The Kaplan–Meier survival curves indicated that patients with low scores had a significantly better OS compared to those with high scores (log-rank test, p < 0.001; Fig. 2E). Furthermore, the 2-, 4-, and 6-year survival rates predicted by the FMRG_score were reflected in the AUC values of 0.7026, 0.7027, and 0.6927, respectively (Fig. 2C). Clustering heatmaps revealed that CYP26A1, DRD4, GSTM1, CD36, and BDNF were more prominent in the high-risk group, while AHCY, PLK1, NAT1, and WNT5A were more prevalent in the low-risk group (Fig. 2H). To validate the prognostic performance of the FMRG_score, OS was calculated across three external validation groups (GSE38832, GSE39582, GSE17537) (Fig. S2). Patients were also stratified into low- or high-risk groups based on the formula utilized for the training set. Survival analysis was conducted, revealing a significantly better prognosis in the low-risk group compared to the high-risk group (log rank; p < 0.05). Assessment of the 2-, 4-, and 6-year prognostic prediction classification efficiencies demonstrated that the FMRG_score maintained relatively high AUC values. These results indicate that the FMRG_score exhibits excellent ability to predict the survival outcomes of CRC patients. We combined clinical characteristics with the FMRG_score to create a nomogram predicting the 2-, 4-, and 6-year OS of CRC patients based on the strong relationship between the FMRG_score system and patient prognosis (Fig. S3A). The calibration plot showed excellent agreement between the predicted overall OS using the nomogram and the actual OS of CRC patients at 2, 4, and 6 years (Fig. 3A).

A Calibration curves for the nomogram showing good agreement between predicted and observed 2-, 4-, and 6-year OS. B Forest plots from univariate Cox regression analyses demonstrating that the risk score is an independent predictor of OS. C–G Boxplots comparing the distribution of risk scores across stages (I–IV), T stage (T1–T4), N stage (N0–N2) and M stage(M0–N1) between low- and high-risk groups. H–J Kaplan–Meier survival curves comparing OS in different groups stratified by stage and age. Patients in the high-risk group exhibit worse survival in all comparisons. K Oncoprints showing the distribution of key mutations in high-risk.
Correlation between clinical characteristics and the FMRGs-related predictive signature
Univariable and multivariate Cox regression was used to explore the relationship between the FMRG_score and clinical characteristics (Figs. 3B and S3B), and the results showed that FMRG_score and M stage could be used as independent prognostic factors (HR = 2.177, 95% CI = 1.628, 2.912, p < 0.001; HR = 2.455, 95% CI = 1.508, 3.998, p < 0.001, respectively). Kaplan–Meier analyses were conducted across subgroups stratified by distinct clinical factors to comprehensively validate the predictive reliability of the FMRG_score. Our findings revealed that patients with a low FMRG_score tended to have a lower TNM stage, whereas those with a higher FMRG_score were more likely to present with an advanced TNM stage (Fig. 3C). Specifically, the high-risk group had a smaller proportion of patients with stages I and II relative to the low-risk group, and a higher proportion of patients with stages III and IV. Our results showed that the FMRG_score increased with advancing stages and was significant in all components except for patients at stages I and II, where no significant difference was observed (Fig. 3D–G). This highlights the important role of the FMRG_score in predicting clinical stage. Stratified survival analyses showed that the prognostic outcomes, especially OS, for colorectal cancer patients varied significantly between the high and low FMRG-Groups, particularly in advanced TNM stages (Figs. 3H and S3C). At the same time, patients with high FMRG_score had a worse OS in both young and elderly patients (Fig. 3I, J), indicating the generalization of the FMRG_score. To gain insights into the biological processes linked to the poor survival in the high-risk group and explore the molecular implications of transcriptomic and genetic variances between high- and low-risk groups, we delved into the genomic heterogeneity of the FMRGs model within the TCGA cohort. Our examination of the mutation landscape of FMRGs aimed to uncover the specific genetic alterations associated with the high-risk group and further elucidate the underlying mechanisms contributing to the differential survival outcomes observed. Our study revealed that the high-risk group had more mutations in the genes APC, TP53, TTN, and K-RAS, which may have enhanced their cancer-promoting characteristics (Fig. 3K).
Landscape of biological characteristics of FMRGs-related signature
Functional enrichment analyses were conducted to gain valuable insights into biological processes, molecular functions, and cellular components (Fig. 4). KEGG analysis showed that the high-risk group were considerably enriched in pro-tumor pathways like “Wnt signaling pathway” and “PPAR signaling pathway” (Fig. 4A). In recent years, the role of the tumor microenvironment in cancer development has garnered increasing attention. Newer studies have highlighted the emerging significance of the nervous system as a key factor in promoting tumor growth. Innervation plays a crucial role in the growth of various tumors, as neurons can establish tumor-nerve synapses with tumor cells. Through these signaling mechanisms, typical oncogenic signaling pathways are often activated, consequently fostering tumor growth [45]. GO analysis explained several neural-related sets that may be key to tumor progression (Fig. 4B). The results of the GSEA analysis highlighted that the high-risk group was significantly enriched in sets that promote tumor progression and metastasis such as “ECM RECEPTOR INTERACTION”, while the low-risk group was enriched in “CITRATE CYCLE TCA CYCLE” and “PYRUVATE METABOLISM” (Fig. 4C, D). The results of GSVA analysis also suggested that the high-risk group was enriched in cancer-promoting pathways, while the low-risk group was enriched in cancer-suppressor-related pathways (Fig. 4E, F).

A Bar plot showing enriched Gene Ontology (GO) terms in the high- and low-risk groups. B Bar plot showing enriched KEGG terms in the high- and low-risk groups. C, D Gene Set Enrichment Analysis (GSEA) plots for key biological processes. E Heatmap displaying expression of genes involved in the significantly enriched pathways across the two groups. F Bar plot showing the top enriched pathways ranked by GSVA score, highlighting metabolic and signaling pathways.
scRNA‑seq data processing and analysis of FMRGs-related signature
The scRNA-seq data was analyzed with the “Seurat” R program, which included quality control measures such as screening for cells exhibiting low expression levels in the range of 200–7000 genes, and ensuring that mitochondrial genes accounted for no more than 20% of the total gene count. To define TME cell populations of CRC, we identified and visualized 8 main cell types using the T-distributed Stochastic Neighbor Embedding (tSNE) (Fig. 5A). We visualized the annotated cell clusters and found a significant decrease in T and B cells and a slight increase in plasma cells in the tumor compared to normal tissue (Fig. 5B). The “AUCell” R program, which evaluates gene set activity, was used to assign FMRGs-related signatures to individual cells (Fig. 5C). Our study revealed that epithelial cells and macrophages exhibited higher FMRG scores compared to other cell types. Specifically, epithelial cells showed a greater abundance of FMRG scores (Fig. 5D). As a result, we classified epithelial cells into two groups based on their FMRG scores—high and low—for further analysis (Fig. S4A). We analyzed the communication network between the nine cells, and results showed that T cells and the FMRG scores—high group had the most receptor-ligand communication with other cells (Fig. 5E). To explore the differential signaling pathways in the FMRG scores—high group and FMRG scores—low group, we further analyzed the signaling patterns of these nine cells (Fig. 5F). Our results suggest that epithelial cells with high expression of FMRG score are significantly enriched in some oncogenic pathways, such as “TGF”, “WNT” and “EGF”. To uncover dynamic functional changes of FMRGs-related signature, we adopted the Monocle 2 algorithm to chronologically order FMRGs-related signature in pseudotime and indicate their trajectories (Fig. S4B, C). We next investigated the transcriptional changes of FMRGs-related signature associated with trajectory and found that NAT1 was significantly enriched early in development, BDNF, DRD4, CD36 and WNT5A were progressively enriched as development progressed, and CYP26A1, AHCY and PLK1 were significantly enriched at the terminal end of development (Fig. S4D).

A t-SNE plot visualizing distinct cell populations within the tumor microenvironment, identifying eight main cell types, including epithelial cells, T cells, macrophages, and fibroblasts. B Histogram of the proportions of different subpopulations of cells. C Score distribution of FDRG expression in various cells, showing clear separation of high and low scoring groups. D Violin plot illustrating the distribution of FDRG scores across different cell types. E Cell-cell communication network analysis, highlighting interaction patterns between different cell types. F Heatmap showing incoming signaling pathways among cell types, with TGF, WNT, and EGF signaling predominantly activated in certain cell groups.
The FMRG_score reshape the immune cell infiltration landscape
Recent research has highlighted the critical role of the inflammatory environment in the development of CRC, particularly focusing on the activation status and interactions of immune and stromal cells with tumor cells [46]. The varying composition of the TME impacts the effectiveness of immunotherapy. We examined the immune profiles of CRC patients across different FMRG groups to better understand this. We analyzed 22 different types of immune cells in CRC patients with high and low FMRG scores. Significantly heightened infiltrations were observed in the high FMRG group for central memory CD4 T cell, CD56dim natural killer cell, Macrophages, Myeloid-derived suppressor cells (MDSC), natural killer cell, plasmacytoid dendritic cell, and T follicular helper cell. Conversely, diminished infiltrations were noted for activated CD4 T cell and type 2 T helper cell within the same group (Fig. 6A). A correlative analysis between the abundance of immune cell types and FMRG scores was further depicted in Fig. S5A. A high FMRG_score was closely associated with a high stromal score, as well as a high estimate score (Fig. 6B). We examined how the nine genes in the proposed model were related to the abundance of immune cells. Our findings showed significant correlations between most immune cells and seven of the genes (Fig. S5B). Using the TIDE score, we found a notable rise in the Dysfunction score and TIDE score in the high-risk group (Fig. S5C–E), indicating an increased risk of immune escape in high-risk patients, which may reduce the efficacy of ICI therapy. We also predicted immunotherapy efficacy using TIDE, which was validated using the IMvigor cohort, TCGA CRC group, GSE17536, GSE39582, and GSE38832, and we found that the higher the FMRG score, the more insensitive the patient may be to immunotherapy leading to ineffectiveness of immunotherapy (Fig. 6C–G). The sensitivity of patients in the low-risk and high-risk groups to chemotherapeutic agents currently used for treating colorectal cancer or undergoing clinical trials was evaluated (Fig. S6). The IC50 values of cisplatin and gemcitabine were notably lower in patients with low FMRG scores, potentially offering more precise clinical treatment guidance. These findings collectively indicate an association between FMRG and drug sensitivity. Emerging evidence suggests that folate metabolism is intricately linked to multiple oncogenic pathways, extending beyond its well-established role in nucleotide biosynthesis. Folate and its derivatives contribute to one-carbon metabolism, which is essential for DNA methylation, redox homeostasis, and amino acid metabolism, thereby influencing cellular responses to a broad spectrum of chemotherapeutic agents [47]. For example, DNA synthesis inhibitors such as 5-fluorouracil and gemcitabine exert their cytotoxic effects by targeting thymidylate synthase or inhibiting ribonucleotide reductase, respectively [48]. Furthermore, folate metabolism has been linked to kinase signaling pathways, including mTOR and Wnt, which regulate tumor cell proliferation and survival [49]. This broad metabolic influence may explain why FMRG_score remains predictive across diverse chemotherapy agents, supporting its potential as a versatile biomarker. Future studies should further validate its role in stratifying patients for folate-targeted therapies and other chemotherapeutic strategies.

A Abundance of immune cell types in different FMRG Groups. Wilcoxon rank-sum test was used to determine statistical significance between high- and low-risk groups. B Differences in the immune, stromal, and ESTIMATE scores between the two FMRG Groups. Wilcoxon rank-sum test was used to determine statistical significance between high- and low-risk groups. C–G Boxplot showing a significant difference in risk scores between responders and non-responders. Wilcoxon rank-sum test was used to determine statistical significance between high- and low-risk groups.
CYP26A1 plays a carcinogenic role in CRC
We validated the FMRGs-related signature by our own sequencing data (Fig. 7A). A protein-protein interaction (PPI) network among the FMRGs-related signature was constructed from the STRING database, in which CYP26A1 interacts most with other proteins (Fig. 7B and Supplementary Table S12). We performed a follow-up analysis of CYP26A1, which we verified in the GSE161277 dataset to be highly expressed in tumor tissues (Fig. 7C). The CYP26A1 gene encodes a member of the cytochrome P450 superfamily of enzymes crucial for various biological processes, such as drug metabolism, the synthesis of cholesterol, steroids, and other lipids [50]. In the context of cancer, CYP26A1 plays a significant role in the metabolism of retinoic acid (RA), a key metabolite of vitamin A that governs cell proliferation, differentiation, and apoptosis [51, 52]. Aberrant expression of CYP26A1 has been linked to the pathogenesis and progression of various tumors by affecting RA levels and modulating RA signaling pathways [53]. Studies have shown that increased CYP26A1 expression can diminish RA levels in cells, thereby promoting tumorigenesis by facilitating cell proliferation and maintaining a less differentiated cell state [54]. Conversely, the downregulation of CYP26A1 in specific cancers can elevate RA concentrations, potentially inhibiting tumor growth and inducing apoptosis [55]. Interestingly, in our sequencing data of tumor primary and metastatic foci from 8 pairs of patients, we found that CYP26A1 was highly expressed in metastatic foci. Additionally, analysis of TCGA CRC data indicated an association between elevated CYP26A1 expression and poor OS in stage III and IV patients (Fig. 7D, E). We performed GSEA analysis of CYP26A1 using our own data, and we found that some pro-oncogenic pathways were significantly enriched when CYP26A1 was highly expressed, whereas pathways related to immunity and autophagy were enriched when CYP26A1 was expressed, also suggesting that CYP26A1 functions as an oncogene (Fig. 7F and Supplementary Table S13). To further explore the relationship between CYP26A1 and the TME of CRC patients, single-cell RNA sequencing data were analyzed from 19 patients with d-MMR/MSI-H CRC treated with the neoadjuvant PD-1 blockade [56] (Fig. 8A). Significant reduction of malignant cells accompanied by an increase in CD4+ T cells, CD8+ T cells and B cells in the CYP26A1 low-expression group (Fig. 8B, C) and CYP26A1 expression negatively correlated with the number of CD8+ T cells (Fig. 8D). Based on classical markers [57, 58], CD8+ T cells were reclassified into five subpopulations: exhausted T cells (Tex), effector T cells (Teff), tissue resident memory T cells (Trm), naive T cells, and memory T cells (Fig. 9E). In CRCs with low CYP26A1 expression, the proportion of Tex was significantly reduced, accompanied by a notable increase in Trm, while no significant changes were observed in Teff (Fig. 8F, G). In addition, we observed that CYP26A1 expression was higher in non-pathological complete response (pCR) patients, suggesting that CYP26A1 were associated with immunotherapy non-response (Fig. 8H).

A Boxplot comparing the expression levels of key genes in normal vs. tumor tissues. B Protein-protein interaction networks functional enrichment analysis. C Dot plot displaying CYP26A1 expression in normal versus tumor tissues from GSE161277, highlighting elevated CYP26A1 expression in tumor tissues. D Boxplot of CYP26A1 expression in primary and metastatic foci. E Kaplan–Meier OS survival analysis comparing high- and low-CYP26A1 groups in stage III–IV patients (p = 0.035). F Gene Set Enrichment Analysis (GSEA) plots demonstrate enrichment of key pathways in CYP26A1 high and low groups.

A–C UMAP visualization of immune cell clustering based on CYP26A1 expression. Clusters of immune cells, including CD8+ T cells, macrophages, and fibroblasts, are colored to reflect high and low CYP26A1 expression, indicating distinct immune cell subpopulations based on CYP26A1 levels. D Box plot shows T cell ratio according to different CYP26A1 expression. E UMAP embedding of CD8+ T cell subtypes stratified. F, G Proportional bar plots and Re-clustering analysis of T cell subpopulations based on CYP26A1 levels reveals functional differences, including a shift in activation status, with CYP26A1_high groups exhibiting higher CD8+ effector T cells. H Dot plot of CYP26A1 expression in CRC tumors from individuals treated with the neoadjuvant PD-1 blockade. Mean expression is shown as color and is standard scaled (binarized), whereas dot size represents the fraction of samples with expression (pCR n = 15; non-pCR n = 4).

A Immunohistochemical analysis of CRC tissue microarrays showing significantly elevated CYP26A1 protein levels in CRC tissues compared to normal tissues. B qPCR quantification from our institutional database confirming higher CYP26A1 mRNA expression in primary colon tumors. C Kaplan–Meier OS analysis demonstrating poor overall survival in CRC patients with high CYP26A1 expression (log-rank p = 0.025). D Proportion of recurrence in CRC patients, with higher recurrence rates observed in those with elevated CYP26A1 expression. E Kaplan–Meier analysis of immunotherapy outcomes showing that high CYP26A1 expression is associated with reduced OS with anti-PD1 therapy (HR = 1.52, 95% CI 1.16–1.98; p = 0.0022) and inferior PFS with anti-CTLA4 therapy (HR = 2.24, 95% CI 1.27–3.95; p = 0.0041). F Western blot and qPCR validation of CYP26A1 knockdown in HCT116 and RKO cells using lentiviral shCYP26A1 vectors. G CCK-8 assays indicating reduced cell proliferation in CYP26A1-depleted HCT116 and RKO cells. H Transwell migration and invasion assays showing reduced migration and invasion in CYP26A1-depleted cells compared to controls. Data are shown as mean ± SD. Statistical significance: p < 0.05 (*), p < 0.001 (**), p < 0.001 (***), p < 0.0001 (****).
CYP26A1 is upregulated in CRC, promotes colon cancer cell proliferation, enhances colorectal cancer cell migratory and invasive potential
Our study investigates CYP26A1’s role in CRC progression and immune modulation, revealing its prognostic significance. Immunohistochemical analysis of tissue microarrays showed markedly elevated CYP26A1 protein levels in CRC versus normal tissues (Fig. 9A), corroborated by qPCR quantification of mRNA from our institutional database demonstrating increased expression in primary colon tumors (Fig. 9B). Clinically, elevated CYP26A1 expression correlated with poorer OS (log-rank p = 0.025) and higher recurrence rates (Fig. 9C, D), establishing its prognostic value.
To further examine the prognostic value of CYP26A1 expression, we performed Kaplan–Meier survival analysis in ICB-treated cancer patients [59]. Notably, CYP26A1 expression demonstrated predictive potential for immunotherapy outcomes: high-expressing patients showed reduced overall survival with anti-PD1 therapy (HR = 1.52, 95% CI 1.16–1.98; p = 0.0022) and inferior progression-free survival with anti-CTLA4 treatment (HR = 2.24, 95% CI 1.27–3.95; p = 0.0041) compared to low-expression counterparts (Fig. 9E).
Furthermore, to explore the biological function of CYP26A1 in colon cancer, we stably depleted endogenous CYP26A1 in HCT116 and RKO cells (Fig. 9F) by infecting them with lentiviral vectors expressing shCYP26A1. The expression status of CYP26A1 in these stable cell lines was validated by Western blot (WB) and qPCR. CCK-8 assays showed that knockdown of CYP26A1 suppressed cell proliferation in HCT116 and RKO cells (Fig. 9G). Given the invasive and metastatic nature of colon cancer cells, we next investigated the effect of CYP26A1 on the invasive and metastatic phenotype of these cells. Transwell migration and invasion assays revealed that shCYP26A1-infected HCT116 and RKO cells displayed reduced migratory and invasive abilities compared to shNC-infected cells (Fig. 9H).
The role of CYP26A1 in the tumor immune microenvironment and its potential as a therapeutic target in CRC
Additionally, this study demonstrates that CYP26A1 critically regulates CD8+ T cell function and shapes the tumor immune microenvironment. Knockdown of CYP26A1 significantly enhanced CD8+ T cell activity in vitro, evidenced by increased granzyme B, IFN-γ, and TNF-α secretion compared to shNC controls (*p < 0.01, **p < 0.001; Fig. 10A). In MC38 xenograft models, tumors with shCYP26A1-1 (selected for optimal knockdown efficiency) exhibited markedly suppressed growth, showing reduced volume from week 2 and significantly lower endpoint tumor weight (**p < 0.001; Fig. 10B–D). Mechanistically, CYP26A1 depletion remodeled the tumor microenvironment by increasing CD4+/CD8+ T cell infiltration while decreasing immunosuppressive Tregs (FOXP3+) (Fig. 10E, F), elevating the CD8+/Treg ratio (Fig. 10G), and boosting CD8+ T cell cytotoxicity (enhanced granzyme B/IFN-γ; Fig. 10H, I) and proliferation (Ki-67+ cells; Fig. 10J). These findings establish CYP26A1 as a therapeutic target in colorectal cancer by enhancing antitumor immunity through T cell functional activation, Treg suppression, and microenvironment reprogramming.

A Knockdown of CYP26A1 in CRC cell lines significantly enhances CD8+ T cell function in vitro, as indicated by increased secretion of granzyme B, IFN-γ, and TNF-α compared to shNC controls (Created with BioRender.com). B Western blot and qPCR confirming successful CYP26A1 knockdown in MC38 cells. C, D MC38 xenograft model showing reduced tumor growth and significantly lower tumor weight in CYP26A1-depleted tumors (shCYP26A1-1) compared to shNC controls (**p < 0.001). E Depletion of CYP26A1 increases CD4+/CD8+ T cell infiltration while reducing immunosuppressive Tregs (FOXP3+ cells). F Flow cytometry analysis revealing decreased Treg populations (FOXP3+CD4+) in CYP26A1-depleted tumors. G Elevated CD8+/Treg ratio in CYP26A1-depleted tumors compared to controls. H, I Enhanced CD8+ T cell cytotoxicity, indicated by increased granzyme B and IFN-γ expression in CYP26A1-depleted tumors. J Increased CD8+ T cell proliferation (Ki-67+) in CYP26A1-depleted tumors. Data are shown as mean ± SD. Statistical significance: p < 0.05 (*), p < 0.001 (**), p < 0.001 (***), p < 0.0001 (****).
Discussion
In this study, we developed and validated an FMRG_score to predict prognosis, TME characteristics, and response to immunotherapy in CRC. Our integrative analysis, which combines large-scale datasets from TCGA and GEO with in vitro experiments, underscores the critical role of folate metabolism in CRC heterogeneity. It identifies key molecular pathways associated with immune evasion and treatment resistance. Notably, this study is the first to propose CYP26A1 as a potential oncogene in CRC, highlighting its significance as a biomarker and therapeutic target.
The FMRG_score effectively stratified patients into high- and low-risk groups, revealing significant survival differences. Patients with a high FMRG_score exhibited a poor prognosis, which correlated with increased activation of pro-tumor pathways and immunosuppressive cell infiltration in the TME, including a higher prevalence of MDSCs and exhausted T cells. These findings suggest that folate metabolism plays a pivotal role in shaping the immune landscape of CRC, potentially mediating immune escape mechanisms that reduce the efficacy of ICIs. This aligns with existing literature on the complex interactions between metabolic reprogramming and immune regulation in cancer progression.
One of the most striking findings of this study is the identification of CYP26A1 as a novel oncogenic driver in CRC. CYP26A1, a member of the cytochrome P450 family, is known for its role in retinoic acid metabolism, which regulates cellular proliferation, differentiation, and apoptosis. Our analysis revealed that high CYP26A1 expression was significantly associated with advanced tumor stage, poor overall survival, and an immunosuppressive TME. These results are consistent with previous reports linking aberrant retinoic acid metabolism to tumorigenesis. Furthermore, our single-cell RNA sequencing data demonstrated a correlation between high CYP26A1 expression and reduced infiltration of effector CD8+ T cells, reinforcing its potential role in immune evasion. These findings open new avenues for targeting CYP26A1 in CRC, either through direct inhibition or by modulating retinoic acid signaling to enhance immune response.
However, it is not without limitations. A primary limitation is that the majority of our findings were derived from bioinformatic analyses. While these analyses are powerful, they necessitate further validation in in vivo systems to confirm their clinical relevance. Although we used dataset-specific median risk scores for stratification in external validation, this approach was chosen to ensure independent analysis within each cohort and to minimize potential biases from applying a threshold derived from a single dataset. Notably, our results consistently demonstrated that the FMRG_score effectively stratifies patients into distinct prognostic groups across multiple independent datasets, supporting its robustness and clinical relevance. We acknowledge that defining a fixed cut-off value could improve the model’s applicability for individual patient risk assessment in clinical settings. Future studies will explore the use of a standardized threshold, potentially derived from a larger pooled cohort or optimized using time-dependent ROC analysis. This refinement will further enhance the translational utility of FMRG_score while maintaining its predictive accuracy across diverse patient populations. Although we successfully demonstrated the tumor-suppressive function of CYP26A1 in vitro and in vivo, it is imperative that future studies prioritize the use of clinical samples and clinical treatment cohorts to verify these results in more physiologically relevant environments. Despite utilizing comprehensive public datasets, the clinical utility of the FMRG_score in predicting treatment outcomes requires validation through prospective clinical trials. Additionally, further mechanistic studies are essential to elucidate the precise role of CYP26A1 in CRC pathogenesis and to assess its potential as a therapeutic target. We recognize that certain aspects, such as cell-based drug response validation and clinical treatment cohort analyses, present challenges that are difficult to address within the scope of this manuscript. We have acknowledged these limitations and outlined plans to explore these areas in future studies. Future research should also investigate how folate metabolism interacts with other metabolic pathways, influencing cancer progression and therapy resistance.
Data and code availability
All sequencing data generated in this paper can be accessed from GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The data that support the results of current study are available on TCGA (https://portal.gdc.cancer.gov/) and GEO websites (https://www.ncbi.nlm.nih.gov/geo).
Change history
31 December 2025
The original online version of this article was revised: In this article fig. 9 has been updated.
10 February 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41435-025-00374-y
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Murphy N, Ward HA, Jenab M, Rothwell JA, Boutron-Ruault M, Carbonnel F, et al. Heterogeneity of colorectal cancer risk factors by anatomical subsite in 10 European countries: a multinational cohort study. Clin Gastroenterol Hepatol. 2019;17:1323–31.
Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145–64.
Araghi M, Soerjomataram I, Jenkins M, Brierley J, Morris E, Bray F, et al. Global trends in colorectal cancer mortality: projections to the year 2035. Int J Cancer. 2019;144:2992–3000.
Aguiar Junior S, Oliveira MMD, Silva DRME, Mello CALD, Calsavara VF, Curado MP. Survival of patients with colorectal cancer in a cancer center. Arq Gastroenterol. 2020;57:172–7.
Vega P, Valentín F, Cubiella J. Colorectal cancer diagnosis: pitfalls and opportunities. World J Gastro Oncol. 2015;7:422–33.
Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16:713–32.
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507.
Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67.
Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–62.
Wu J, Zhang L, Kuchi A, Otohinoyi D, Hicks C. CpG site-based signature predicts survival of colorectal cancer. Biomedicines. 2022;10:3163.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86.
Weiner LM. Cancer immunology for the clinician. Clin Adv Hematol Oncol. 2015;13:299–306.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18:155.
O’Brien M, Paz-Ares L, Marreaud S, Dafni U, Oselin K, Havel L, et al. Pembrolizumab versus placebo as adjuvant therapy for completely resected stage IB-IIIA non-small-cell lung cancer (PEARLS/KEYNOTE-091): an interim analysis of a randomised, triple-blind, phase 3 trial. Lancet Oncol. 2022;23:1274–86.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34.
Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112:1421–7.
Zhao W, Jin L, Chen P, Li D, Gao W, Dong G. Colorectal cancer immunotherapy-recent progress and future directions. Cancer Lett. 2022;545:215816.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Guidoboni M, Gafà R, Viel A, Doglioni C, Russo A, Santini A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol. 2001;159:297–304.
Overman MJ, Lonardi S, Wong KYM, Lenz H, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Ganesh K. Optimizing immunotherapy for colorectal cancer. Nat Rev Gastroenterol Hepatol. 2022;19:93–4.
Kim JW, Jeon YJ, Jang MJ, Kim JO, Chong SY, Ko KH, et al. Association between folate metabolism-related polymorphisms and colorectal cancer risk. Mol Clin Oncol. 2015;3:639–48.
Du W, Li W, Lu R, Fang J. Folate and fiber in the prevention of colorectal cancer: between shadows and the light. World J Gastroenterol. 2010;16:921–6.
Kherbek H, Daoud R, Soueycatt T, Soueycatt Y, Ali Z, Ehsan J, et al. The relationship between folic acid and colorectal cancer; a literature review. Ann Med Surg. 2022;80:104170.
Jang MJ, Kim JW, Jeon YJ, Chong SY, SP Hong, Hwang SG, et al. Polymorphisms of folate metabolism-related genes and survival of patients with colorectal cancer in the Korean population. Gene. 2014;533:558–64.
Bouras E, Kim AE, Lin Y, Morrison J, Du M, Albanes D, et al. Genome-wide interaction analysis of folate for colorectal cancer risk. Am J Clin Nutr. 2023;118:881–91.
McNulty H, Pentieva K. Folate bioavailability. Proc Nutr Soc. 2004;63:529–36.
Iyer R, Tomar SK. Folate: a functional food constituent. J Food Sci. 2009;74:R114–22.
Hubner RA, Houlston RS. Folate and colorectal cancer prevention. Br J Cancer. 2009;100:233–9.
Moazzen S, Dolatkhah R, Tabrizi JS, Shaarbafi J, Alizadeh BZ, de Bock GH, et al. Folic acid intake and folate status and colorectal cancer risk: a systematic review and meta-analysis. Clin Nutr. 2018;37:1926–34.
Li J, Yang H, Lei M, Zhu W, Su Y, Li K, et al. Dietary folate drives methionine metabolism to promote cancer development by stabilizing MAT IIA. Signal Transduct Target Ther. 2022;7:192.
Ma J, Stampfer MJ, Giovannucci E, Artigas C, Hunter DJ, Fuchs C, et al. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res. 1997;57:1098–102.
Van Guelpen B, Hultdin J, Johansson I, Hallmans G, Stenling R, Riboli E, et al. Low folate levels may protect against colorectal cancer. Gut. 2006;55:1461–6.
Otani T, Iwasaki M, Sasazuki S, Inoue M, Tsugane S. Plasma folate and risk of colorectal cancer in a nested case-control study: the Japan Public Health Center-based prospective study. Cancer Causes Control. 2008;19:67–74.
Kato I, Dnistrian AM, Schwartz M, Toniolo P, Koenig K, Shore RE, et al. Serum folate, homocysteine and colorectal cancer risk in women: a nested case-control study. Br J Cancer. 1999;79:1917–22.
Weile J, Kishore N, Sun S, Maaieh R, Verby M, Li R, et al. Shifting landscapes of human MTHFR missense-variant effects. Am J Hum Genet. 2021;108:1283–1300.
Rich JT, Neely JG, Paniello RC, Voelker CCJ, Nussenbaum B, Wang EW. A practical guide to understanding Kaplan-Meier curves. Otolaryngol Head Neck Surg. 2010;143:331–6.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Yu G, Wang L, Han Y, He Q. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–7.
Wang W, Chen Z, Hua Y. Bioinformatics prediction and experimental validation identify a novel cuproptosis-related gene signature in human synovial inflammation during osteoarthritis progression. Biomolecules. 2023;13.
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–59.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Shi DD, Guo JA, Hoffman HI, Su J, Mino-Kenudson M, Barth JL, et al. Therapeutic avenues for cancer neuroscience: translational frontiers and clinical opportunities. Lancet Oncol. 2022;23:e62–74.
Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol. 2021;21:653–67.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83.
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–71.
Thatcher JE, Isoherranen N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin Drug Metab Toxicol. 2009;5:875–86.
He W, Wang X, Chen M, Li C, Chen W, Pan L, et al. Metformin reduces hepatocarcinogenesis by inducing downregulation of Cyp26a1 and CD8(+) T cells. Clin Transl Med. 2023;13:e1465.
Osanai M, Sawada N, Lee G. Oncogenic and cell survival properties of the retinoic acid metabolizing enzyme, CYP26A1. Oncogene. 2010;29:1135–44.
Alshafie GA, Walker JR, Curley RWJ, Clagett-Dame M, Highland MA, Nieves NJ, et al. Inhibition of mammary tumor growth by a novel nontoxic retinoid: chemotherapeutic evaluation of a C-linked analog of 4-HPR-glucuronide. Anticancer Res. 2005;25:2391–8.
Osanai M, Lee G. Increased expression of the retinoic acid-metabolizing enzyme CYP26A1 during the progression of cervical squamous neoplasia and head and neck cancer. BMC Res Notes. 2014;7:697.
Lavudi K, Nuguri SM, Olverson Z, Dhanabalan AK, Patnaik S, Kokkanti RR. Targeting the retinoic acid signaling pathway as a modern precision therapy against cancers. Front Cell Dev Biol. 2023;11:1254612.
Li J, Wu C, Hu H, Qin G, Wu X, Bai F, et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell. 2023;41:1152–69.
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–72.
Liu C, Yu H, Huang R, Lei T, Li X, Liu M, et al. Radioimmunotherapy-induced intratumoral changes in cervical squamous cell carcinoma at single-cell resolution. Cancer Commun (Lond). 2022:1407–11.
Győrffy B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation. 2024;5:100625.
Acknowledgements
Thanks for the help of the CRC tissues form Fudan University Shanghai Cancer Center and thanks for the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) Database for sharing a large amount of data. The graphic figure was created with BioRender.com.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (grant no. 82373359); Shanghai Science and Technology Innovation Action Plan (grant no. 24SF1901700); CSCO Cancer Research Fund (grant nos. Y-HR2020MS-0298, Y-pierrefabre202102-0066); Chinese Young Breast Experts Research project (grant no. CYBER-2021-001); Beijing Science and Technology Innovation Medical Development Foundation Key Project (grant no. KC2022-ZZ-0091-6); Shanghai Youth Technological Talent Program—Yangfan Program (grant no. 21YF1408200).
Author information
Authors and Affiliations
Contributions
YF and TZ designed the studies, conducted most of the experiments and data analysis, and wrote the draft manuscript. YZ, YX, MX, CZ, YX, and YF conducted some of the molecular experiments. DD and JZ were involved in the overall design of the study and the revision of the final manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Declaration
All authors have made significant contributions to the conception, design, analysis, and/or interpretation of the work presented in this manuscript. Furthermore, all authors have thoroughly reviewed and approved the final version of the manuscript. They fully agree with its content and the decision to submit it for consideration.
Ethical approval
This study was approved by the Research Ethical Committee of Fudan University Shanghai Cancer Center (FUSCC, No. 1612167-18). All animal experiments were conducted in accordance with the protocol approved by the Research Ethical Committee of FUSCC.
Informed consent
All authors have read and approved the final version of the manuscript and agree to its publication. Informed consent was obtained from all participants involved in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Y., Zhou, T., Zheng, Y. et al. Folate metabolism-associated CYP26A1 is a clinico-immune target in colorectal cancer. Genes Immun 26, 376–393 (2025). https://doi.org/10.1038/s41435-025-00342-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41435-025-00342-6
