
Abstract
Purpose
Genomic studies have demonstrated the necessity of ethnicity-specific population data to ascertain variant pathogenicity for disease diagnosis and treatment. This study examined the carrier prevalence of treatable inherited disorders (TIDs), where early diagnosis of at-risk offspring can significantly improve clinical outcomes.
Methods
Existing exome/ genome sequencing data of 831 Singaporeans were aggregated and examined for disease causing variants in 104 genes associated with 80 TIDs.
Results
Among the 831 Singaporean participants, genomic variant filtering and analysis identified 1 in 18 individuals (6%) to be carriers amongst one of 13 TIDs. Citrin deficiency and Wilson disease had the highest carrier frequency of 1 in 41, and 1 in 103 individuals, respectively. The pathogenic variants associated with citrin deficiency were 24 times more prevalent in our local cohorts when compared to Western cohorts.
Conclusion
This study demonstrates the value of a population specific genomic database to determine true disease prevalence and has enabled the discovery of carrier frequencies of treatable genetic conditions specific to South East Asian populations, which are currently underestimated in existing data sources. This study framework can be adapted to other population groups and expanded to multiple genetic conditions to inform health policies directing precision medicine.
Similar content being viewed by others


Introduction
Precision medicine, through the use of genomics, offers a golden opportunity to improve the overall health of the population.1 One key element of precision medicine is practice of preventative health, which can be facilitated by predicting individuals who are at risk of developing genetically linked diseases. Expanding on this theme would include carrier screening, or the practice of identifying individuals at risk of bearing children with genetic diseases. Since the initiation of screening for cystic fibrosis in Caucasian and Ashkenazi Jewish populations.2 carrier screening, especially for ethnicity specific variants, has grown to include screening for haemoglobinopathies in individuals of South East Asian, Mediterranean and African ancestry, Tay Sachs disease in Ashkenazi Jewish populations and spinal muscular atrophy in all ethnic groups.3 The availability of genomic datasets from a large, relatively healthy population, such as the Exome Aggregation Consortium (ExAC),4 offers an opportunity to expand carrier screening beyond these few diseases by estimating the carrier frequencies of a larger number of disorders. For example, an analysis of 23,453 individuals for 108 genetic disorders revealed 24% were carriers for at least one disorder and 5% were carriers for multiple disorders.5 Similarly, all 100 participants in the MedSeq project were carriers for a median of 2 variants (range 1 to 7) associated with a recessive condition.6
However, lack of genomics data from non-European populations, represented by less than 20% of genomic data, is a major hurdle towards progressing precision medicine worldwide.4,7 Genomics data forms a critical backbone, upon which precision medicine can be practiced, and paucity of data from such under-represented populations can lead to misdiagnoses and mismanagement.8 To contribute to the expansion of genomic diversity, a joint collaboration between Singapore Health Services (SinghHealth) and its affiliated academic institute, Duke-NUS, established the SinghHealth Duke-NUS Institute of Precision Medicine (PRISM) and aggregated genomes of “healthy” individuals from South East Asia, known as the Singapore Exome Consortium (SEC).
Inherited disorders, some of which are treatable, affect 2–3% of all live births worldwide and result in social and financial burden on the family and society.9,10 Amongst these, 81 have been associated with intellectual disability (ID) of variable severity and onset,10 and in ~20% of the patients, early diagnosis and treatment leads to significant improvement of IQ and related developmental scores. Appropriate medical intervention has also demonstrated improvement of neurological and systemic clinical manifestation as well as behavioural and psychiatric disturbances. As such, we refer to this group of 81 disorders as treatable inherited disorders (TIDs). Examples of some of these TIDs (and their associated treatment) include phenylketonuria (dietary phenylalanine restriction +/− tetrahydrobiopterin (BH4) supplement), Wilson disease (penicillamine) and biotinidase deficiency (biotin supplements). Individually these TIDs are rare, however, the collective burden of TIDs is estimated to be significant from a health care perspective, but the extent of this has not been studied previously. Moreover, although 60% of TIDs can be picked up on screening blood and urine tests, either as newborn or at the time of presentation, some of these conditions, such as citrin deficiency or late onset ornithine transcarbamoylase deficiency, may present with non-specific symptoms and remain undetected at the time of birth. Symptom-specific conditions such as in arginine: glycine amidinotransferase (AGAT) deficiency or Niemann-Pick disease type C, require a ‘single test per single disease’ approach and therefore may not be diagnosed in a timely fashion leading to delays in treatment.11
Taking into consideration the genetic differences between populations of different ancestral backgrounds there may be regional or geographical biases in their prevalence for certain disorders. With this, we aimed to review our SEC cohort for the carrier frequency of diseases associated with TIDs and identify specific diseases, which may be at uniquely higher prevalence in our population, with the purpose of guiding public health policies for carrier and newborn screening using genomic testing.
Materials and methods
Participant recruitment
Participants for this research were obtained from cohorts of pre-existing studies. These studies were institutional ethics review board approved genomics projects. Informed consent was obtained from the eligible individual (or parent/ legal guardian for minors). As part of the research protocol, the genomic data of these individuals was de-identified and analysed in a cumulative manner.
Genomic sequencing
Genomic sequencing, either exome or genome sequencing, was performed on leucocyte derived DNA from consented individuals. Sequencing was performed as per manufacturer’s protocols on Illumina sequencers (HiSeq 2000/ HiSeq 2500 or HiSeq X).
Gene panel
A pre-determined list of genes associated with 81 TIDs as defined by van Karnebeek et al.10 was analysed. This gene list contains 71 autosomal recessive conditions, six X-linked conditions, one autosomal dominant disorder, one mitochondrial disorder, one condition described with both X-linked and autosomal recessive inheritance and one condition described with both autosomal recessive and dominant inheritance. As our bioinformatic pipeline only targeted nuclear coded genes, we excluded genes associated with mitochondrial inheritance and analysed variants in 104 genes associated with 80 disorders as listed in Table S2. All of the listed disorders have medical interventions, which for the majority of the disorders, are affordable, non-invasive and safe.10 Furthermore, although the majority of interventions are based on single case reports or expert opinion without critical appraisal, they are initiated as standard of care in routine clinical practice once a diagnosis is made.10
Bioinformatic analysis
Genomic data, in the form of FASTQ, was processed through an established bioinformatic pipeline to generate variant calling format (gVCF) files. The gVCF files were then combined to create the Singapore Exome Consortium (SEC). Variants were quality filtered to exclude false positives according to standard thresholds (quality scores > 30, coverage > 10 ×, and absence of clustered variants within a window size of 10 variants). From variants that passed this threshold, we extracted variants in each of the genes in our gene list. We then annotated the variants using ANNOVAR12 to include information regarding the gene, genetic change, protein change, type of pathogenic variant (PV) (frameshift, nonsense, nonsynonymous, splicing, and synonymous); prediction of the variant from multiple algorithms (Polyphen-2, SIFT, likelihood ratio test and MutationTaster2), allele frequencies in different databases (Exome Sequencing Project, dbSNP, 1000 Genomes, Complete Genomics, ExAC, and our in-house database of common variants (present in >5% of the population)), and annotation of variants in the clinical mutation database, ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk).
Filtering and classification of variants
Variants occurring in one of the genes from our gene list and identified as either (a) “pathogenic”, “probably pathogenic”, or “untested” in ClinVar, or (b) novel (absent or rare in public databases) protein truncating PV (insertions—deletions (indels), stopgain, stoploss or disruption of an essential splice site) were selected for further analysis. As the evidence for pathogenicity of some of the variants listed in the clinical mutation databases as pathogenic may be lacking,13,14 we reviewed the primary literature regarding each of the filtered variants and reclassified them as per the ACMG guidelines on variant classification.13 A literature search for each variant was manually conducted using PubMed, disease specific mutation databases (Table S3) and an in-house mutation database, when available. Only variants that were classified as pathogenic were considered for the final analysis.
Quality control
The aligned sequence read of each exome sample was reviewed to ensure that there was adequate coverage for each of the 104 genes (Table S2). Insufficiently covered samples were excluded when calculating the allele frequency of any particular variant.
Results
Characteristics of the participants
The study participants comprised of 831 individuals with no known pre-existing health conditions or intellectual disability. The participants were aggregated from three existing cohorts: KK Women’s and Children’s Hospital (KKH), Singapore Eye Research Institute (SERI) and National Heart Centre Singapore (NHCS) (Table 1). The average age of the cohort was 50.8 years (S.D. 13.67 years, range 7 to 84 years). The majority of the SEC cohort was of Chinese ethnicity (91%).
Characteristics of the genomic data and variants
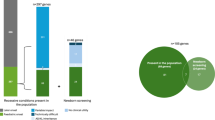
Focusing on protein coding regions in the 104 genes associated with TIDs, a mean of 3676 single-nucleotide variants (SNVs) and 379 indel variants were detected per sample. Collectively, 4343 unique variants occurring at an allele frequency of less than 1% were detected amongst the 831 individuals. Upon filtering, we identified 90 variants for further curation - 35 were reported as “pathogenic” or “probably pathogenic” or “untested” in ClinVar (as reported on 19 May 2017) and 55 variants were novel protein-truncating variants (29 indels, 15 splicing alteration variants, 10 stop gain and 1 stop loss) (Fig. 1). We then reviewed the primary literature and re-classified these 90 variants as per ACMG variant classification guidelines - 20 as pathogenic (Table 2) and 25 as likely pathogenic (Table S4). The remaining 45 variants were classified as either variants of unknown significance (VUS, n = 39) or likely benign (n = 6) (Table S4). Some of the variants were found in more than one individual, and collectively, the 45 pathogenic and likely pathogenic variants were observed in 71 individuals.

Filtering of variants
Carrier frequency detection
While we detected 45 pathogenic/ likely pathogenic variants, to determine the carrier frequency, we took a conservative approach and included only pathogenic variants (and excluded likely pathogenic variants) as disease causing alleles. The 20 pathogenic variants were detected in 46 unrelated individuals (6% or 1 in 18) in the SEC cohort. These variants were associated with 13 of the 80 treatable forms of intellectual disability as listed in Table 2. All of these conditions are known to follow an autosomal recessive pattern of inheritance. No individual in the SEC cohort was found to be homozygous or compound heterozygous for the pathogenic variants in any of the 80 disorders, nor carry more than one TID causing allele.
Allele frequency comparison between SEC and ExAC
Overall, the disease causing alleles associated with the 13 disorders were found to be more frequent in our SEC cohort by comparison to their corresponding overall allele frequencies in the ExAC data set. This was particularly evident in some conditions such as citrin deficiency, where correlating to the total allele frequency in ExAC demonstrated that variants SLC25A13 p.Arg285fs and c.615 + 5G > A were 24 times more common in our local population (Table 2). Although the occurrence of our local alleles more closely resembled those of East Asian ethnicity in ExAC, certain alleles were more frequent in our local population in comparison to East Asian individuals in ExAC, including ATP7B (associated with Wilson disease) p.T784M (12 times more common), MMAHC (associated with cobalamin C deficiency) p.R132X (6 times more common) and p.R161Q (6 times more common), MUT (associated with methylmalonic acidemia) c.1677-1G > A (6 times more common) and TH (associated with tyrosine hydroxylase deficiency) p.R202H (6 times more common) (Table 2). In addition, a number of reportedly pathogenic variants, such as OXCT1 (associated with SCOT deficiency) p.T58M, were seen in homozygous states in healthy individuals, likely representing misclassification of these variants in ClinVar and/or HGMD (Table S4 and Fig. S1).
Discussion
The SinGapore Incidental Finding (SGIF) study group was set up to develop a formalized framework to understand the prevalence of genetic conditions in our community. In our previous analysis of 377 individuals, we estimated the prevalence of asymptomatic individuals with incidental findings related to adult onset dominant monogenic disorders at 2%.15 We have since developed an exomic data reference bank which, to date, contains sequences of 831 individuals. In this study, we chose TIDs as a model as, despite the rarity of these disorders, their association with ID and availability of treatment make them clinically significant.
Our study found that 1 in 18 individuals, or 6% of the population, carried a PV associated with the risk of having an offspring with these disorders. Five of the 81 TIDs had more than one carrier in our local population (the cumulative prevalence of these 5 disorders was 1 in 23 individuals). The newborn screening program for inborn errors of metabolism in Singapore is based on tandem mass spectrometry and currently screens for 42 disorders, 24 of which are TIDs.16 However as only three of the five most prevalent conditions identified from our analysis are included, this suggests a potential role for genetic based screening as an adjunct tool to identify additional at risk individuals within the local population.
Among the disorders identified, citrin deficiency (1 in 41) and Wilson disease (1 in 103) were the most common. Both of these conditions are recessive and present with features of hepatic, neurologic and/or psychiatric symptoms ranging in severity and age of onset. Notably, due to the non-specific phenotype of these conditions, and that newborn screening commenced in Singapore in 2006, it is possible that many affected individuals may be under-diagnosed.17,18,19,20 Citrin deficiency is caused by a PV in the SLC25A13 gene encoding mitochondrial transported citrin.21 Two major phenotypes have been described: neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and childhood to adult onset citrullinemia type II (CTLN2), which can present with neuropsychiatric symptoms.22 Some individuals can present with fulminant liver failure requiring liver transplantation.23 Diagnosis can be confirmed by quantitative amino acid analysis and symptoms in both early and late onset can be treated with a protein-rich, lipid-rich and lactose-free diet.22
Wilson disease has a global carrier frequency of 1 in 90,24 and patients present with hepatic failure and neuropsychiatric symptoms and are successfully treated with copper chelation therapy such as penicillamine. Wilson disease is caused by PV in the ATP7B gene, which results in accumulation of copper in the liver and brain. Diagnosis can be established by biochemical findings of low ceruloplasmin concentrations and presence of ocular Kayser-Fleisher rings. However, these methods do not accurately detect all individuals with Wilson disease25 and diagnosis may be delayed in individuals who present with non-specific symptoms preventing initiation of appropriate treatment.26 Similarly, each of the remaining 78 conditions can be readily managed by interventions such as vitamin supplementation, dietary restrictions or medications such as chelators, emphasising the compelling need and medical actionability for screening for these treatable disorders.10 The timely diagnosis and recognition of the underlying metabolic defect enables clinical management before the symptoms manifest.
This study demonstrates the value of an ethnicity-specific genomic data set to study disease prevalence relevant to the local population. Lazarin et al., 2014, performed a large scale analysis of carrier frequencies targeting a pre-set list of variants from 23,453 individuals of diverse ethnicities.5 In comparison to previously published frequencies, they found discrepancies in several conditions. For example, they detected a carrier frequency of cystic fibrosis to be 1 in 40 in South Asians which had previously been reported as 1 in 118 and an additional under-reporting of carnitine palmitoyltransferase II deficiency in East Asians as 1 in 378, which had been previously reported as rare. Likewise, newborn screening in Singapore has detected a higher incidence of fatty acid oxidation disorders (1 in 6565) in comparison to other Asian population studies in Taiwan, China and Japan (1 in 9300 to 1 in 54,000).16 Without such studies, which are currently underrepresented in Asia, the prioritisation of genetic screening programs to inform public health initiatives is challenging. Such genetic epidemiological studies provide a framework and evidence of genetic risk to prioritise public health initiatives for diagnosis and management of these disorders for a very local, yet internationally applicable, context.
One limitation of the study is the lack of genomic data from the non-Chinese populace of Singapore. Singapore is a multiethnic population comprising of Chinese (76%), Malays (15%), Indians (7%) and other minority ethnicities including Eurasians. Our cohort was predominantly Chinese (91%) and hence underrepresented other ethnicities of Singapore. To address this issue, we have embarked on generating data targeting individuals of Malay and Indian ancestry. Despite this current limitation, this is the first study to report on the prevalence of TIDs in the local population, demonstrating a difference in burden of genetic disorders compared to the Western population. More importantly, the framework provided here can be applied to any country to guide local public health policies, provided there is adequate genomic data, not only in relation to TIDs, but other genetic disorders including monogenic adult onset disorders like hereditary breast and cancer syndromes.15
In conclusion, while the promise of precision medicine is alluring, there is an urgent need for genomic data from populations of underserved countries. With more population-relevant data, healthcare practices can be tailored towards improving preventative care and treatment, taking the fullest advantage of precision medicine technologies to fulfil this promise.
Change history
08 August 2018
At the time of publication the author Jyn Ling Kuan did not have a master's degree; this has now been amended to BSc. This has now been corrected in the PDF and HTML versions of the article.
References
Ashley EA. Towards precision medicine. Nat Rev Genet. 2016;17:507–22.
Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–91.
Committee on Genetics. Carrier Screening for Genetic Conditions. Obstet Gynecol. 2017;129:e41–e55.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Lazarin GA, Haque IS, Nazareth S, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15:178–86.
Vassy JL, Christensen KD, Schonman EF, et al. The Impact of Whole-Genome Sequencing on the Primary Care and Outcomes of Healthy Adult Patients: A Pilot Randomized Trial. Ann Intern Med. 2017.
Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538:161–4.
Manrai AK, Funke BH, Rehm HL, et al. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med. 2016;375:655–65.
Meerding WJ, Bonneux L, Polder JJ, Koopmanschap MA, van der Maas PJ. Demographic and epidemiological determinants of healthcare costs in Netherlands: cost of illness study. BMJ. 1998;317:111–5.
van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. 2012;105:368–81.
van Karnebeek CD, Houben RF, Lafek M, Giannasi W, Stockler S. The treatable intellectual disability APP http://www.treatable-id.org: a digital tool to enhance diagnosis & care for rare diseases. Orphanet J Rare Dis. 2012;7:47.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
McLaughlin HM, Ceyhan-Birsoy O, Christensen KD, et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet. 2014;15:134.
Jamuar SS, Kuan JL, Brett M, et al. Incidentalome from Genomic Sequencing: A Barrier to Personalized Medicine? EBioMedicine. 2016;5:211–6.
Lim JS, Tan ES, John CM, et al. Inborn Error of Metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): An 8 year journey from pilot to current program. Mol Genet Metab. 2014;113:53–61.
Takeuchi S, Yazaki M, Yamada S, et al. An Adolescent Case of Citrin Deficiency With Severe Anorexia Mimicking Anorexia Nervosa. Pediatrics. 2015;136:e530–534.
McKiernan PJ, Baumann U. Neonatal classical galactosaemia presenting as citrin deficiency. J Hepatol. 2008;49:463. author reply 464
Komatsu M, Yazaki M, Tanaka N, et al. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol. 2008;49:810–20.
Tazawa Y, Kobayashi K, Abukawa D, et al. Clinical heterogeneity of neonatal intrahepatic cholestasis caused by citrin deficiency: case reports from 16 patients. Mol Genet Metab. 2004;83:213–9.
Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 2002;47:333–41.
Saheki T, Inoue K, Tushima A, Mutoh K, Kobayashi K. Citrin deficiency and current treatment concepts. Mol Genet Metab. 2010;100(Suppl 1):S59–64.
Tamamori A, Okano Y, Ozaki H, et al. Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur J Pediatr. 2002;161:609–13.
Das SK, Ray K. Wilson's disease: an update. Nat Clin Pract Neurol. 2006;2:482–93.
Kanwar P, Kowdley KV. Metal storage disorders: Wilson disease and hemochromatosis. Med Clin North Am. 2014;98:87–102.
Millard H, Zimbrean P, Martin A. Delay in Diagnosis of Wilson Disease in Children With Insidious Psychiatric Symptoms: A Case Report and Review of the Literature. Psychosomatics. 2015;56:700–5.
Acknowledgements
We thank the individuals and their families who volunteered for the genomic studies. SSJ is supported by the National Medical Research Council, Singapore (NMRC/CISSP/003/2016). Genomic sequencing of families at KKH was supported by a grant from Biomedical Research Council, Singapore (IAF 311019), NMRC/CNIG/1139/2015, NMRC/CNIG/1142/2015, Genome Institute of Singapore (Project SeqCDS) and NMRC/CG/006/2013 from the National Medical Research Council, Ministry of Health, Republic of Singapore. The genomic sequencing of Chinese adults in SEED was supported by NMRC/CIRG/1407/2014. We would like to thank the Lee Foundation for their grant support for the SingHEART study conducted at National Heart Centre Singapore. This work was supported by core funding from SingHealth and Duke-NUS through their Institute of Precision Medicine (PRISM).
The SinGapore Incidental Finding (SGIF) study group
Vikrant Kumar, Christropher Blöcker, Ryanne Wu, Angeline Lai, Ee Shien Tan, Ivy Ng, Breana Cham, Jiin Ying Lim, Jasmine Goh, Eileen Lim, Charis Lim, Hai Yang Law, Maggie Brett, Siew Ching, NHCS Biobank and Singheart Study, Tien Yin Wong, Aung Tin, John Allen Carson, E-Shyong Tai, Xueling Sim, T2D-GENES consortium, Bruno Reversade, Brygappa Venkatesh, Yik Ying Teo.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
10A full list of members and affiliations appear in Table S1.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Bylstra, Y., Kuan, J.L., Lim, W.K. et al. Population genomics in South East Asia captures unexpectedly high carrier frequency for treatable inherited disorders. Genet Med 21, 207–212 (2019). https://doi.org/10.1038/s41436-018-0008-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41436-018-0008-6
Keywords
This article is cited by
-
Global burden of Wilson disease: a comprehensive evidence synthesis
Orphanet Journal of Rare Diseases (2026)
-
Investigating common mutations in ATP7B gene and the prevalence of Wilson’s disease in the Thai population using population-based genome-wide datasets
Journal of Human Genetics (2025)
-
Information content as a health system screening tool for rare diseases
npj Digital Medicine (2025)
-
Comprehensive analysis of recessive carrier status using exome and genome sequencing data in 1543 Southern Chinese
npj Genomic Medicine (2022)
-
Implementation of genomics in medical practice to deliver precision medicine for an Asian population
npj Genomic Medicine (2019)
