
Abstract
Despite the presence of obligately parthenogenetic (OP) lineages derived from sexual ancestors in diverse phylogenetic groups, the genetic mechanisms giving rise to the OP lineages remain poorly understood. The freshwater microcrustacean Daphnia pulex typically reproduces via cyclical parthenogenesis. However, some populations of OP D. pulex have emerged due to ancestral hybridization and introgression events between two cyclically parthenogenetic (CP) species D. pulex and D. pulicaria. These OP hybrids produce both subitaneous and resting eggs parthenogenetically, deviating from CP isolates where resting eggs are produced via conventional meiosis and mating. This study examines the genome-wide expression and alternative splicing patterns of early subitaneous versus early resting egg production in OP D. pulex isolates to gain insight into the genes and mechanisms underlying this transition to obligate parthenogenesis. Our differential expression and functional enrichment analyses revealed a downregulation of meiosis and cell cycle genes during early resting egg production, as well as divergent expression patterns of metabolism, biosynthesis, and signaling pathways between the two reproductive modes. These results provide important gene candidates for future experimental verification, including the CDC20 gene that activates the anaphase-promoting complex in meiosis.
Similar content being viewed by others


Introduction
The existence of obligately parthenogenetic eukaryotic lineages that have entirely abandoned sexual reproduction has long fascinated evolutionary biologists. The transition from sexual reproduction to obligate parthenogenesis is phylogenetically widespread, having occurred independently in most multicellular taxa (Bell 1982; Neiman et al. 2014). Various cytogenetic manifestations of obligate parthenogenesis (i.e., modifications of meiosis) have been well described in the literature, including automictic parthenogenesis, apomictic parthenogenesis, gynogenesis, and hybridogenesis (Stenberg and Saura 2009; Vrijenhoek 1998). However, the molecular mechanisms and genes underlying these cytogenetic modifications are poorly understood (Ferree et al. 2006; King and Hurst 2010; Riparbelli et al. 2005; Suomalainen et al. 1987).
Obligate parthenogenesis can originate through multiple evolutionary routes. Spontaneous mutations in genes involved in meiosis or reproduction could lead to the loss of sexual reproduction as in monogonont rotifers (Serra and Snell 2009). Contagious parthenogenesis could result due to the spread of asexuality conferring elements as in the pea aphid (Jaquiéry et al. 2014), and parasite-induced parthenogenesis could occur, for example, in wasps infected by Wolbachia (Simon et al. 2003). The most common route, interspecific hybridization, could disrupt meiosis due to genetic incompatibilities between parental species, resulting in the loss of sex (Vrijenhoek 1998; White et al. 1977). Among vertebrates, there are currently around 100 known parthenogenetic lineages of amphibians, reptiles, and fish that are interspecific hybrids (Avise 2008; Avise 2015; Dawley and Bogart 1989; Neaves and Baumann 2011). For invertebrates, the occurrence of hybrid parthenogens have been demonstrated in snails (Johnson and Bragg 1999), crustaceans (Innes and Hebert 1988), and many insects (Schwander et al. 2011; Stenberg and Lundmark 2004; White et al. 1977).
This widespread occurrence of obligate parthenogens with a hybrid ancestry suggests that understanding meiotic modifications associated with hybridization may provide insight into the origin of obligately parthenogenetic (OP) species. In this study, we investigate the origin of obligate parthenogenesis in the cladoceran microcrustacean, Daphnia pulex, commonly found in freshwater habitats in North America. Previous studies have shown that obligately parthenogenetic Daphnia pulex originated due to hybridization and introgression events between two cyclically parthenogenetic (CP) sister species, D. pulex and D. pulicaria (Xu et al. 2013, 2015). This hypothesis is supported by genome-wide association studies showing microsatellite and SNP alleles exclusively associated with OP D.pulex originated in D. pulicaria (Tucker et al. 2013; Xu et al. 2013, 2015). To date, no reproductive mode tests performed on D. pulicaria isolates have revealed OP lineages (Heier and Dudycha 2009). Having diverged about 800,000–2,000,000 years ago (Colbourne and Hebert 1996; Cristescu et al. 2012; Omilian and Lynch 2009), CP D. pulex and D. pulicaria, members of the D. pulex species complex, are morphologically similar (Brandlova et al. 1972), yet can be distinguished due to inhabiting very different environments and by species-specific microsatellite markers or allozyme loci such as the lactate dehydrogenase (Ldh) locus (Cristescu et al. 2014).
The main difference between the cyclically parthenogenetic parental species and obligately parthenogenetic hybrids is how they reproduce. Under favorable environmental conditions such as high food abundance and low population density, both CP and OP Daphnia females (Fig. 1A, B) reproduce asexually, producing diploid subitaneous eggs through a modified meiosis. In this modified meiosis, during meiosis I, bivalents align at the metaphase plate; however, cell division is arrested before the onset of anaphase I, after which each half-bivalent moves back to the metaphase plate and sister chromatids rearrange (Hiruta et al. 2010). Thus, during modified meiosis I, there is no segregation of homologous chromosomes and no cytokinesis resulting in daughter cells. Next, meiosis II proceeds normally and results in diploid eggs (Ojima 1958; Zaffagnini and Sabelli 1972; Hiruta et al. 2010). In the absence of rare events such as ameiotic recombination, conversion, and mutation (Omilian et al. 2006; Xu et al. 2011), all asexually produced progenies, males and females alike, are genetically identical to the mother.

A Cyclically parthenogenetic and B obligately parthenogenetic life cycles in Daphnia. C Early subitaneous egg and D early resting egg production as determined by the color and size of the ovaries (red circles).
Once the environment deteriorates (e.g., low food abundance, high population density), some subitaneous eggs can develop into males through environmental sex determination (Gorr et al. 2006; Tatarazako et al. 2003). The CP Daphnia females switch to produce haploid eggs (usually two) through meiosis, which upon fertilization by sperm become resting embryos. In contrast, OP D. pulex females produce chromosomally unreduced resting eggs without mating through a modified meiosis. Previous cytological observations have shown that the parthenogenesis producing subitaneous eggs in OP and CP isolates is highly similar to the parthenogenesis producing resting eggs in OP isolates (Zaffagnini and Sabelli 1972). The resulting resting eggs from OP isolates (without fertilization) are deposited into a protective case (i.e., ephippium) and remain dormant until environmental conditions turn favorable for hatching. As environmental conditions drive the alteration of reproductive modes in Daphnia, we argue that transcriptomic changes due to environmental variation may play a fundamental role and provide insight into the origin of obligate parthenogenesis.
Attempting to uncover the molecular basis of parthenogenesis, previous work by Xu et al. (2015) functionally annotated the 647 asexual-specific SNPs introgressed from D. pulicaria into the D. pulex genome and identified 206 protein-coding genes associated with obligate parthenogenesis. Of these genes, 52 were predicted to have general or unknown functions and the analysis showed no functional enrichment for any pathway. The authors identified some candidate genes affecting meiosis, the cell cycle and DNA replication; however, they were unable to distinguish between causal genes or spurious associations (Xu et al. 2015). Building on this work, other studies have examined genome-wide expression differences between reproductive stages and modes to elucidate the role of gene expression changes in the origin of OP Daphnia. Xu et al. 2022 examined the genome-wide expression patterns during early resting egg production between various OP D. pulex and parental CP D. pulex and D. pulicaria isolates (Fig. 2A). The results showed that early resting egg production in OP D. pulex is associated with a downregulation of many meiosis and cell cycle genes, and an upregulation of metabolic and biosynthesis genes compared to early resting egg production in CP isolates (Xu et al. 2022). These results suggest that the origin of obligate parthenogenesis from meiosis is likely caused by the downregulation of key meiosis and cell cycle genes, and an upregulation of metabolism. Interestingly, when comparing the gene expression patterns between early subitaneous egg and early resting egg production within individual CP D. pulex and D. pulicaria isolates (Fig. 2B), meiosis and cell cycle genes were also found to be downregulated while metabolic and biosynthesis genes were upregulated in early subitaneous egg development (i.e., parthenogenesis) compared to resting egg production (i.e., meiosis) in both species (Huynh et al. 2021). Together, the results from these two transcriptomic studies strongly suggest that parthenogenesis in both OP (resting egg production) and CP (subitaneous egg production) isolates are associated with a downregulation of meiosis and cell cycle genes and an upregulation of metabolic and biosynthesis genes, which may play essential roles in triggering parthenogenesis. Therefore, it has been hypothesized that the origin of obligate parthenogenesis in Daphnia involves the use of existing ameiotic germline cell division pathways (normally used in the production of asexual, subitaneous eggs) in the production of resting eggs (Xu et al. 2022).

A Regulation of genes during early resting egg production between OP D. pulex and CP D. pulex and D. pulicaria isolates. B Regulation of genes during early resting egg production and early subitaneous egg production within CP isolates. C and within OP D. pulex isolates.
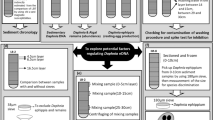
To further test this hypothesis, this work examines the genome-wide gene expression variation differentiating early subitaneous vs. early resting egg parthenogenesis within hybrid OP D. pulex isolates (Fig. 2C). We hope that these data will illuminate the genetic basis of the cytological modifications occurring in modified meiosis I, providing insight into the origin of obligate parthenogenesis. Furthermore, we set out to identify patterns of alternative splicing in the two reproductive modes to understand whether alternative splicing plays a role in the origin of obligate parthenogenesis.
Materials and methods
Daphnia isolates
A total of three obligate parthenogenetic (OP) Daphnia pulex isolates (DB4-4, K09, and Main 348-1) were used in this study. These isolates were previously collected from Texas, Ontario, and Maine, respectively (Supplementary Table S1) and have been maintained in the lab as clonal cultures. Initiated from a single female, each isolate has been kept as an asexually reproducing line in artificial lake water (Kilham et al. 1998) under a 16:8 light/dark cycle at 18 °C and fed with the green algae, Scenedesmus obliquus, twice a week.
Animal tissue collection
For each OP D. pulex isolate, experimental animals were maintained in the same environmental conditions for two generations to minimize maternal effects, which could significantly impact gene expression. Then, one-day-old neonates were continuously collected from each isolate and grown until sexual maturity in the same environmental conditions described above. Sexually mature animals were examined daily under a light microscope to collect females engaging in early subitaneous eggs and early resting eggs production. Early subitaneous egg production is characterized by a thin clear, or greenish ovary extending along the gut with oil droplets visible. In contrast, early resting egg production is characterized by a small milky brown ovary starting to develop along the end of the gut (Fig. 1C, D). For each isolate, three replicates of each reproductive mode were collected (15–20 whole-body individuals) for RNA extraction.
RNA extraction and sequencing
RNA of all samples was extracted using the Promega (Madison, WI, USA) SV Total RNA Isolation kit, following the manufacturer’s instructions. RNA concentration was measured using a Qubit 4.0 Fluorometer (Thermo Scientific, Waltham, MA, USA). RNA integrity was checked with a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA sequencing libraries were prepared by Novogene Corporation Inc. (Sacramento, CA, USA) using standard Illumina sequencing library protocol. Each library was sequenced on an Illumina NovaSeq6000 platform with at least 20 million 150-bp paired-end reads. The raw RNA sequence data were deposited at NCBI SRA under PRJNA847604.
Sequencing quality control and mapping
The software package FastQC (Andrews 2010) was used to examine the quality of the raw reads. Because of no observed adapter contamination, reads were mapped directly to the D. pulex reference genome PA42 3.0 (Ye et al. 2017) using STAR aligner (Dobin et al. 2013) with default parameters. SAMtools (Li et al. 2009) was used to remove reads that mapped to multiple locations in the genome, and the program featureCounts (Liao et al. 2014) was used to obtain the raw counts for expressed genes in each sample.
Differential gene expression analysis
We performed differential expression (DE) analysis using DESeq2 v.1.34.0 (Love et al. 2014) in R Studio Team (2020). Differentially expressed genes were determined for early subitaneous vs early resting egg development for each isolate individually to investigate for differential expression within each Daphnia isolate and by pooling all the isolates to establish commonalities. The Wald negative binomial test using the design formula ~ Reproductive_mode for individual isolates and ~ Isolate + Reproductive_mode for pooled isolates were utilized in DEseq2. P values were adjusted for multiple testing using the Benjamini-Hochberg method as implemented in DEseq2, and genes were considered significantly differentially expressed if they had an adjusted p value <0.05 and a fold-change >1.5 and <−1.5 for upregulation and downregulation, respectively. Mapped read counts were normalized using the regularized log (rlog) transformation function to generate the PCA and volcano plot.
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway and GO term enrichment analysis
To investigate the biological relevance of the differentially expressed genes, we performed a functional enrichment analysis using the R package topGO (Alexa and Rahnenfuhrer 2021) and GO terms from the D. pulex transcriptome (Ye et al. 2017). The default algorithm, weight01, was used along with the Fisher exact test, and GO terms were considered significantly enriched if the p value was less than 0.05. Our script is available at https://github.com/Marelize007/RNAseq_obligate_parthenogenesis.
We also examined whether any KEGG pathways were enriched for differentially expressed genes. We queried 18,440 gene sequences from the D. pulex transcriptome (Ye et al. 2017) in the KEGG Automatic Annotation Server (KAAS) using the GHOSTX program (Moriya et al. 2007). A set of 10, 135 genes were assigned a KO (KEGG ortholog) number, and from these, 6282 were assigned to a KEGG pathway map. Hypergeometric tests with Holm-Bonferroni corrected p values (p value < 0.05) were used to identify enriched pathways (script available at: https://github.com/Marelize007/RNAseq_obligate_parthenogenesis). The upregulated and downregulated genes were analyzed separately to increase the power to detect biologically relevant pathways (Hong et al. 2014). The Gene Annotation Easy Viewer (GAEV) (Huynh and Xu 2019) was used to visualize the functional pathways that each gene is mapped to.
Alternative splicing analysis
Alternative splicing events were identified using the software rMATS v4.1.1 (Shen et al. 2014). Reads mapped to both exons and splice junctions were used to detect the following events: SE (skipped exon), A5SS (alternative 5′ splice site), A3SS (alternative 3′ splice site), RI (retained intron), and MXE (mutually exclusive exons). An SE event occurs when an entire exon including its flanking introns is spliced out. A5SS and A3SS events may result in the inclusion or exclusion of different parts of exons, while entire introns are retained during RI events. During MXE events only one out of two exons is spliced into the resulting mRNA (Pohl et al. 2013; Wang et al. 2015). Genes were considered to be differentially spliced if at least four uniquely mapped reads supported the events, reads had a minimum anchor length of 10 nucleotides, the Benjamini-Hochberg adjusted p value < 0.05, and the difference in exon inclusion level (Δ|ψ|) > 5% (Shen et al. 2014; Suresh et al. 2020). Similar to the differential expression analysis, this analysis was completed for each isolate to identify within-isolate differences, as well as by pooling the isolates together to obtain a comprehensive overview of the splicing differences between the early subitaneous and early resting egg production. Chi-squared tests were performed using R in Rstudio to test for significant (p < 0.05) over- and under-representation of splicing events within each isolate and the pooled sample.
Results
Data quality
Three biological replicates were collected for each isolate during early subitaneous egg and early resting egg production, leading to a total of 18 RNA-seq samples. An average of 26.4 million (SD = 5 million) raw reads were sequenced per sample. Our quality check using FastQC revealed no issues related to read quality or adapter contamination. On average 93% (SD = 2%) of the reads uniquely mapped to the D. pulex reference genome were retained for differential gene expression and alternative splicing analysis (Supplementary Table S2).
Differential expression analysis
A principal component analysis (PCA) was performed to visualize the grouping of samples. The first two principal components accounted for 47 and 17% of the total variance, respectively. The first principal component (PC1) is likely due to differences between the early subitaneous and early resting egg production, whereas the second principal component (PC2) could be due to clonal variance (Fig. 3A). The same separation pattern for PC1 and PC2 was seen among individual isolates (Supplementary Fig. S1). These results strongly suggest that our data captured the transcriptional differences between the early subitaneous and early resting egg production and some inter-clonal differences.

A PCA plot based on all samples. B Volcano plot of genes differentially expressed during early subitaneous egg production for the pooled sample. C Number of genes upregulated and downregulated during early subitaneous egg production in each sample. D Venn diagram showing the number of shared differentially expressed genes between early subitaneous and early resting egg production. OP represents the set of differentially expressed genes from the pooled transcriptomic analysis.
To increase statistical power and obtain a comprehensive overview of the main differences between early subitaneous and early resting egg production contributing to PC1, we performed a pooled analysis comparing the reproductive modes across all three isolates. A total of 3263 genes were significantly differentially expressed (p.adjust < 0.05), with 1771 genes upregulated and 1492 genes downregulated in early subitaneous egg production compared to early resting egg production (Fig. 3B, C, Supplementary Table S3).
To reveal lineage-specific differences and commonalities, the transcriptomes between the two reproductive modes were compared within each isolate. For DB4-4, K09, and M348, a total of 1116, 2591, and 3942 genes were differentially expressed between the two reproductive modes, respectively (Supplementary Table S3). Of these genes, 534, 1299, and 2125 were upregulated and 582, 1292, and 1817 were downregulated during early subitaneous egg production for DB4-4, K09, and M348, respectively (Fig. 3C). Additionally, 475 genes were shared among these isolates and the pooled analysis, with a common set of 168 genes upregulated and 307 genes downregulated for each sample. Furthermore, 99, 1483, 712, and 428 genes were uniquely differentially expressed in DB4-4, M348, K09, and the pooled analysis, respectively (Fig. 3D).
KEGG pathway enrichment
A KEGG pathway enrichment analysis comparing the two reproductive modes within each isolate and in the pooled data revealed that the onset of early subitaneous egg production compared to early resting egg production is associated with the upregulation of meiosis and cell cycle genes, as well as the upregulation of genes mapped to sugar, lipid, and hormone metabolic pathways (the same set of genes were downregulated in early resting egg production). On the other hand, downregulated genes in early subitaneous egg production (i.e., upregulated genes in early resting egg production) were mainly enriched in various metabolic, biosynthesis, and signaling pathways (Fig. 4A). Specifically, of the genes upregulated during early subitaneous egg production obtained from the pooled analysis, 576 were mapped to KEGG pathways and were enriched in 28 pathways (p.adjust < 0.05, Supplementary Table S4). The most notable of the pathways enriched with upregulated genes included the Hedgehog signaling pathway, which plays a vital role in embryonic development by coordinating cell proliferation, coordination, and migration (Carballo et al. 2018). Additionally, the cell cycle, oocyte meiosis, and progesterone-mediated oocyte maturation pathways were all significantly enriched for upregulated genes in early subitaneous egg production (i.e., downregulated genes in resting egg production). Significantly enriched sugar and lipid metabolic pathways included starch and sucrose metabolism, galactose metabolism, and sphingolipid metabolism (Fig. 4A). KEGG pathways maps for the cell cycle, oocyte meiosis, and progesterone-mediated oocyte maturation pathways were produced using the software GAEV (Huynh and Xu 2019) and can be viewed in Supplementary Figs. 2–4. The same enrichment pattern for the Hedgehog signaling pathway, cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, and various sugar and lipid metabolic pathways observed in the pooled analysis was also identified within individual isolates (Fig. 4B).

A Distribution of log2 fold-change for up- and downregulated genes in the enriched KEGG pathways during early subitaneous egg production. B Heatmap illustrating a subset of up- and downregulated KEGG pathways during early subitaneous egg production across all replicates.
Of the downregulated genes for early subitaneous egg production (i.e., upregulated genes in resting egg production) obtained from the pooled analysis, 449 were mapped to KEGG pathways and were enriched in 12 pathways (p.adjust < 0.05, Supplementary Table S5). These pathways included metabolic and biosynthesis pathways such as various N-glycan, O-glycan, and glycosphingolipid biosynthesis, as well as the GnRH, Hippo, and calcium signaling pathways (Fig. 4A). The GnRH signaling pathway is a regulator of the reproductive system (Kraus et al. 2001), while the Hippo signaling pathway controls organ size and development (Boopathy and Hong 2019).
Collectively, these results suggest that the upregulation of meiosis and cell cycle genes and genes mapped to various sugar and lipid metabolic pathways are associated with early subitaneous egg production. On the opposite, the downregulation of meiosis and cell cycle genes characterizes early resting egg production. Additionally, early subitaneous egg production is associated with a downregulation of genes mapped to various metabolic and biosynthesis pathways including N-glycan, O-glycan, and glycosphingolipid biosynthesis, as well as the GnRH, Hippo, and calcium signaling pathways, whereas all of these pathways are enriched with upregulated genes in early resting egg production.
GO term enrichment analysis
Our GO term enrichment analysis further corroborated the idea that early subitaneous egg production is associated with an upregulation of meiosis and cell cycle genes and genes mapped to various sugar and lipid metabolic processes, whereas the downregulation of these genes is true of early resting egg production. Upregulated genes in early subitaneous egg production (i.e., downregulated genes in resting egg production) revealed enrichment for GO terms associated with carbohydrate metabolic process (p value = 0.00014), oogenesis (p value = 0.0016), lipid transport (p value = 0.0022), trehalose hydrolysis (p value = 0.0058), regulation of mitotic cell cycle phase transition (p value = 0.021) and cellular carbohydrate biosynthetic process (p value = 0.040), Supplementary Table S6). In line with the KEGG pathway analysis, our GO term enrichment analysis revealed a downregulation of various metabolic processes in early subitaneous egg production (also meaning upregulation in early resting egg production), including arginine metabolic process (p value = 0.0065) and proline metabolic process (p value = 0.0096), cell differentiation (p value = 0.021), protein glycosylation (p value = 9 × 10−7), and other biosynthetic and signaling processes (Supplementary Table S7). Again, this pattern of regulation observed during early subitaneous egg production was observed within each isolate (Supplementary Tables S8–S13).
Alternatively spliced genes
Alternatively spliced transcripts were identified between the two reproductive modes within each isolate and using a pooled splicing approach to investigate how alternative splicing distinguishes between early subitaneous egg and early resting egg production. Across the pooled samples, 293 differentially spliced transcripts (FDR corrected p value < 0.05) were identified between early subitaneous egg and early resting egg production. For the individual isolates, 397, 466, and 515 differentially spliced transcripts were identified for DB4-4, M348, and K09, respectively (Fig. 5A, Supplementary Table S14). Across all samples analyzed, 60 differentially spliced transcripts were shared (Fig. 5B). A GO term enrichment analysis of the differentially spliced transcripts between early subitaneous and early resting egg production shared by at least two samples (total transcripts = 213) showed enrichment for various metabolic and biosynthetic processes including the arginine metabolic process (p value = 0.043) and prostaglandin metabolic process (p value = 0.018, Fig. 5C, Supplementary Table S15–S18). Additional annotation also revealed the gene UBE2I (UBC9), Ubiquitin Conjugate Enzyme E2 I, to be differentially spliced among all samples. Previous work has shown that UBE2I may fulfill essential roles in regulating gene expression during oocyte growth and maturation (Ihara et al. 2008).

A Number of differentially spliced transcripts between early subitaneous and early resting egg production for all samples. B Venn diagram showing the number of differentially spliced genes between early subitaneous egg and early resting egg production. OP represents the set of differentially spliced genes from the pooled analysis. C Significantly enriched GO terms of differentially spliced genes. D Composition of differentially spliced genes.
Furthermore, analyses of the alternative splicing events using single isolates and the pooled samples revealed that skipped exon (SE) events were most abundant, totaling 729 (35%) events across all comparisons, followed by 417 (20%) RI events, 350 A5SS events (17%), 345 A3SS events (17%) and 235 MXE events (11%) (Fig. 5D, Supplementary Table S14). Lastly, we tested for over- and under-representation of splicing type among transcripts differentially spliced between early subitaneous and early resting egg production for each isolate and the pooled splicing analysis. The DB4-4 isolate showed an over-representation of A3SS and RI events, while K09 showed an over-representation of RI and SE events (chi-squared test p < 0.05). For the pooled splicing analysis, there was a significant under representation of A3SS and SE events (chi-squared test p < 0.05), whereas no significant over-representation of splicing events was identified.
Genes of interest
With early subitaneous egg production showing significant upregulation of meiosis and cell cycle genes (same genes downregulated in early resting egg production), we compiled a list of consensus genes mapped to the following significant pathways: Hedgehog signaling pathway, cell cycle, oocyte meiosis, and progesterone-mediated oocyte maturation pathway. Consensus genes had to be shared by all 3 of the individual isolates or two of the individual isolates and the pooled analysis (Fig. 6). These consensus genes mainly consisted of cell cycle regulators such as AURKA, which regulates spindle formation and controls chromosome segregation (Blengini et al. 2021), various cyclins including CCNA (cyclin A2), CCNB2 (cyclin B2), CCNB3 (cyclin B3), CLB, and CDC20, CDC4, REC8L, and SMC1. Upregulation across all three isolates and the pooled analysis was observed for paralogs of CDC20 (cell division cycle 20), which activates the anaphase-promotion complex/cyclosome (APC/C) to initiate sister chromatid separation (Jin et al. 2010), and paralogs of REC8L, a gene coding the meiosis-specific component of the cohesion complex, which regulates the separation of sister chromatids and homologous chromosomes (Ward et al. 2016). Additionally, CDC4 (cell division control protein 4), essential for the transition from G1 to S phase, the onset of anaphase, and the transition from G2 to M phase (Goh and Surana 1999), showed upregulation in all three isolates and the pooled sample, with DB4-4, M348 and the pooled sample (OP) having an average log2 fold-change > 2.

Average log2 fold-change for genes upregulated during early subitaneous egg production (downregulated in resting egg production) which mapped to the cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, and Hedgehog signaling pathway. OP represents the pooled sample.
Discussion
In this study, we investigated the transcriptomic signatures between two modes of parthenogenesis in three obligately parthenogenetic Daphnia pulex isolates to gain insight into the genetic modifications critical to the emergence of obligate parthenogenesis. In comparison to previous studies, our results provide a more nuanced view of the origin of obligate parthenogenesis in Daphnia.
As stated in the Introduction, previous studies on the transcriptomics of different reproductive modes in Daphnia (Xu et al. 2022; Huynh et al. 2021) suggest that the initiation of parthenogenesis (in subitaneous egg or resting egg production) is associated with an upregulation of metabolic and biosynthesis genes and a downregulation of meiosis and cell cycle genes, leading to the hypothesis that the origin of obligate parthenogenesis in Daphnia is due to the extension of parthenogenesis occurring in subitaneous egg production into resting egg production (Xu et al. 2022). This hypothesis also draws support from the highly similar cytological observations between the parthenogenesis of subitaneous eggs and resting eggs in Daphnia (Zaffagnini and Sabelli 1972).
Expression of meiosis and cell cycle genes
In the context of this hypothesis, our comparative transcriptomic analysis of the two modes of parthenogenesis (early subitaneous vs. resting egg) in OP D. pulex isolates revealed new insights into the genetic modifications associated with obligate parthenogenesis. Our differential gene expression analyses and functional enrichment analyses clearly show that in OP Daphnia the parthenogenesis in resting egg production, which distinguishes OP from CP Daphnia, is associated with reduced expression of meiosis and cell cycle genes compared with the subitaneous, parthenogenetic egg production. Therefore, despite the highly similar cytological modifications of parthenogenesis in subitaneous egg and resting egg production compared to normal meiosis, the expression requirement for meiosis and cell cycle genes is likely different for the successful execution of these two reproductive stages. Parthenogenesis in resting egg production in OP Daphnia may only be achieved with a reduced expression of meiosis and cell cycle genes compared to subitaneous egg production. For the transition from cyclical parthenogenesis to obligate parthenogenesis in Daphnia (i.e., meiosis converted to parthenogenesis in resting egg production), substantially reduced expression of genes involved in meiosis and cell cycle seems to be critical, which could be due to introgression of CP D. pulicaria alleles into the CP D. pulex genomic background (Fig. 7, Xu et al. 2022). We also note that the reduced expression of meiosis and cell cycle genes in resting egg production could be attributed to the fact that a single female Daphnia produce a maximum of two resting eggs versus multiple subitaneous eggs. Some expression differences between subitaneous and resting egg parthenogenesis can also be attributed to different developmental rates between the two types of eggs. The growth and maturation of subitaneous eggs have been previously described (Hiruta et al. 2010). However, very little is known about the growth and maturation of parthenogenetic resting eggs.

Summary of pathway regulation across various modes of reproduction in OP and CP Daphnia.
Divergent expression of metabolism and biosynthesis genes
Interestingly, our analyses identify a divergent expression pattern of metabolism and biosynthesis pathways in resting egg production vs. subitaneous egg production in OP Daphnia. This is worthy of attention because altering the amino acid composition through diet can induce Daphnia to enter different reproductive modes (Fink et al. 2011; Koch et al. 2011), which suggests an important role of metabolism and biosynthesis in regulating the reproduction of Daphnia.
In CP D. pulex and D. pulicaria (Huynh et al. 2021), the parental species of OP Daphnia, resting egg production is predominantly associated with the downregulation of metabolic and biosynthesis genes. However, the results in this study clearly show that resting egg production in OP Daphnia is associated with both the downregulation and upregulation of genes enriched in some specific metabolism and biosynthesis pathways. The identified divergent expression of these pathways may contain the genetic regulators of reproductive modes and indicate the varying metabolic requirements of different reproductive modes in OP Daphnia.
Metabolism and biosynthesis differences between two reproductive modes
The metabolic and biosynthesis genes upregulated during early subitaneous egg production, i.e., downregulated during early resting egg production, were enriched in various sugar metabolic pathways including galactose, starch and sucrose metabolism, sphingolipid metabolism, steroid hormone biosynthesis and progesterone-mediated oocyte maturation (Fig. 4A, B). We suggest that a few of these pathways are important for reproductive regulation in crustaceans and arthropods. For example, in the study by Varki et al. (2015), it was shown that sphingolipids play a vital role in growth factor signaling and morphogenesis in arthropods, and changes in sphingolipid abundance could impact cell proliferation, apoptosis, senescence, and differentiation (Hannun and Obeid 2002). Moreover, in the brine shrimp, Artemia franciscana, that shares reproductive similarities with CP Daphnia (Nambu et al. 2004), Kojima et al. (2013) showed that sphingolipids involved in signaling and signal transduction pathways might be vital in determining which reproductive mode ensue in A. franciscana (Kojima et al. 2013). Thus, it is likely that sphingolipid metabolism may be critical in initiating and differentiating the two reproductive modes in OP Daphnia isolates.
We also note that steroid hormones previously described in crustaceans include ecdysteroids, which play an essential role in regulating the molting cycle, as well as vertebrate-type steroids such as estrogen and progesterone which fulfill functions in vitellogenesis and ovarian development (Lafont and Mathieu 2007; Liu et al. 2019; Nakagawa and Henrich 2009). The divergent expression pattern of the steroid hormone biosynthesis pathway and progesterone-mediated oocyte maturation pathway in our study indicates that the two reproductive modes in OP Daphnia have differential requirement for the level of steroid hormones. It would be of great interest for future studies to address whether any of the differentially expressed genes in these two pathways drive the initiation of different reproductive modes or their differential expression is a consequence of different reproductive modes.
On the other hand, the metabolic and biosynthesis genes downregulated during early subitaneous egg production, i.e., upregulated during early resting egg production, showed enrichment for various metabolic pathways including arginine metabolism, glycosphingolipid biosynthesis, parathyroid hormone synthesis, secretion, and action (Fig. 4A, B). Previous work on the inducibility of resting eggs in CP Daphnia isolates have revealed that supplementing specific dietary amino acids such as arginine could promote subitaneous egg production and suppress resting egg production, suggesting their important roles in the switch between parthenogenesis and sexual reproduction in CP Daphnia (Fink et al. 2011; Koch et al. 2011). Intriguingly, out results show the opposite association pattern where the downregulation of genes in the arginine metabolic pathway is associated with early subitaneous egg production and their upregulation is associated with resting egg production (Supplementary Table S7). It is likely that the upregulated or downregulated expression of the genes in arginine pathway is not equivalent to the increased or reduced availability of arginine to Daphnia. On the other hand, it remains to be tested whether the availability of arginine has a different impact on reproductive mode between OP and CP Daphnia.
Furthermore, the parathyroid hormone synthesis, secretion, and action pathway fulfil functions in regulating the concentrations of calcium, phosphate and active vitamin D metabolites (Murray et al. 2005). The upregulation of this pathway indicates another aspect of the special metabolic requirements or consequence for resting egg production in OP Daphnia. Notably, because most metabolic pathways were downregulated in early subitaneous egg production and upregulated during early resting egg production, these upregulated metabolic pathways in resting egg production are worthy of special attention in future studies.
Signaling pathways
We note that resting egg and subitaneous egg production in OP Daphnia are also associated with the divergent expression of genes enriched in some important signaling pathways, which may contain some potential genetic triggers of the two distinct reproductive modes. For example, the evolutionarily conserved Hedgehog signaling pathway was enriched with upregulated genes during early subitaneous egg production, and downregulated genes during early resting egg production (Fig. 4A, B). This pathway plays an essential role in cell patterning, embryogenesis, and development (Carballo et al. 2018; Niyaz et al. 2019).
Signaling pathways downregulated during early subitaneous egg production and upregulated during early resting egg production provides insight into the transcriptomic signature of resting egg production where OP Daphnia shows a major phenotypic modification. These signaling pathways include the calcium signaling pathway, GnRH signaling pathway, and the Hippo signaling pathway (Fig. 4A, B). Calcium in Daphnia is vital for growth, molting, and ephippia formation (Giardini et al. 2015). In contrast to subitaneous egg production, during resting egg formation, Daphnia undergo two molting cycles and form an ephippium containing a thick layer of calcium phosphate enclosing the resting eggs (Gerrish and Cáceres 2003). This suggests that the calcium requirement for resting egg production is higher than for subitaneous egg production. Calcium additionally fulfills vital functions in the GnRH signaling pathway through regulating the secretion of the gonadotropins: luteinizing hormone (LH) and follicle stimulating hormone (FSH), both playing a vital role in embryogenesis and reproduction (Haisenleder et al. 2003; Marques et al. 2022). Lastly, the Hippo signaling pathway which promotes apoptosis and inhibits cell proliferation was also upregulated in early subitaneous egg production and downregulated in early resting egg production (Sherbet 2017).
Alternative splicing
Our alternative splicing analysis between early subitaneous and early resting egg production revealed 60 differentially spliced transcripts shared by all samples (Fig. 5B). Further annotation of the shared differentially spliced transcripts revealed UBE2I (UBC9), Ubiquitin Conjugating Enzyme E2 I, as a gene of interest. Work done in mouse oocytes revealed that disrupted meiotic maturation and defects in spindle organization resulted from the inhibition of UBE2I in fully grown oocytes, while overexpression of UBE2I caused a stimulation of transcription in meiotically incompetent oocytes (Ihara et al. 2008; Yuan et al. 2014). These results suggest that isoforms of UBE2I, and thus sumoylation, may play an essential role in regulating gene expression during oocyte growth and maturation (Ihara et al. 2008).
Additionally, our GO term enrichment analysis of alternatively spliced transcripts shared by at least two samples showed enrichment for various metabolic and biosynthetic processes, including the arginine metabolic process (Fig. 5C). The enrichment in the arginine metabolic process indicates that alternative splicing may contribute to the regulation of transcript abundance in different reproductive stages in Daphnia.
Candidate genes for obligate parthenogenesis
Lastly, we compiled a list of candidate genes mapped to the cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, and Hedgehog signaling pathway for future investigation (Fig. 6). A gene of particular interest on this list introgressed from D. pulicaria, and previously identified as playing an important role in parthenogenesis is CDC20 (Xu et al. 2022). During oocyte meiosis, CDC20, a subunit responsible for activating the anaphase-promoting complex/cyclosome (APC/C), promotes progression from metaphase to anaphase via the destruction of cyclin B1 and securin (Jin et al. 2010). Studies in mice have shown that a reduction in CDC20 increased the average time from metaphase entry to the onset of anaphase (Jin et al. 2010), while a lack of CDC20 caused metaphase arrest (Li et al. 2007). This metaphase arrest due to a deficiency in CDC20 was also observed in bovine oocytes (Yang et al. 2014) and budding yeast (Lim et al. 1998). It will be of great interest to investigate the role of CDC20 in the parthenogenesis of Daphnia by manipulating its gene expression (e.g., overexpression).
Data availability
The raw reads for this study are deposited at NCBI SRA PRJNA847604.
References
Avise J (2008) Clonality: the genetics, ecology, and evolution of sexual abstinence in vertebrate animals. Oxford University Press, USA
Avise JC (2015) Evolutionary perspectives on clonal reproduction in vertebrate animals. Proc Natl Acad Sci USA 112(29):8867–8873
Andrews S (2010) FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Alexa A, Rahnenfuhrer J (2021) topGO: Enrichment Analysis for Gene Ontology. R package version 2.46.0.
Bell G (1982) The masterpiece of nature: the evolution and genetics of sexuality. University of California Press, Berkeley, CA
Blengini CS, Ibrahimian P, Vaskovicova M, Drutovic D, Solc P, Schindler K (2021) Aurora kinase A is essential for meiosis in mouse oocytes. PLOS Genet 17:e1009327
Boopathy GTK, Hong W (2019) Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Frontiers in Cell and Developmental Biology, 7.
Brandlova J, Brandl Z, Fernando CH (1972) The Cladocera of Ontario with remarks on some species and distribution. Can J Zool 50(11):1373–1403
Carballo GB, Honorato JR, de Lopes GPF, de Sampaio e Spohr TCL (2018) A highlight on Sonic hedgehog pathway. Cell Commun Signal 16(1):11
Colbourne JK, Hebert PDN (1996) The systematics of North American Daphnia (Crustacea: Anomopoda): a molecular phylogenetic approach. Philos Trans R Soc Lond Ser B: Biol Sci 351(1337):349–360
Cristescu ME, Constantin A, Bock DG, Cáceres CE, Crease TJ (2012) Speciation with gene flow and the genetics of habitat transitions. Mol Ecol 21(6):1411–1422
Cristescu ME, Demiri B, Altshuler I, Crease TJ (2014) Gene expression variation in duplicate lactate dehydrogenase genes: do ecological species show distinct responses? PLoS ONE 9(7):e103964
Dawley RM, Bogart JP (1989) Evolution and ecology of unisexual vertebrates. University of the State of New York, State Education Department, New York State Museum
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21
Ferree PM, McDonald K, Fasulo B, Sullivan W (2006) The origin of centrosomes in parthenogenetic hymenopteran insects. Curr Biol: CB 16(8):801–807
Fink P, Pflitsch C, Marin K (2011) Dietary Essential Amino Acids Affect the Reproduction of the Keystone Herbivore Daphnia pulex. PLOS ONE 6(12):e28498
Gerrish GA, Cáceres CE (2003) Genetic versus environmental influence on pigment variation in the ephippia of Daphnia pulicaria. Freshw Biol 48(11):1971–1982
Giardini JL, Yan ND, Heyland A (2015) Consequences of calcium decline on the embryogenesis and life history of Daphnia magna. J Exp Biol 218(Pt 13):2005–2014.
Gorr TA, Rider CV, Wang HY, Olmstead AW, LeBlanc GA (2006) A candidate juvenoid hormone receptor cis-element in the Daphnia magna hb2 hemoglobin gene promoter. Mol Cell Endocrinol 247(1–2):91–102
Goh PY, Surana U (1999) Cdc4, a protein required for the onset of S phase, serves an essential function during G(2)/M transition in Saccharomyces cerevisiae. Mol Cell Biol 19(8):5512–5522
Haisenleder DJ, Ferris HA, Shupnik MA (2003) The calcium component of gonadotropin-releasing hormone-stimulated luteinizing hormone subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology 144(6):2409–2416
Hannun YA, Obeid LM (2002) The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J Biol Chem 277(29):25847–25850
Heier CR, Dudycha JL (2009) Ecological speciation in a cyclic parthenogen: Sexual capability of experimental hybrids between Daphnia pulex and Daphnia pulicaria. Limnol Oceanogr 54(2):492–502
Hiruta C, Nishida C, Tochinai S (2010) Abortive meiosis in the oogenesis of parthenogenetic Daphnia pulex. Chromosome Res 18(7):833–840
Hong G, Zhang W, Li H, Shen X, Guo Z (2014) Separate enrichment analysis of pathways for up- and downregulated genes. J R Soc Interface 11(92):20130950
Huynh TV, Hall AS, Xu S (2021) The transcriptomic signature of cyclical parthenogenesis (p. 2021.09.27.461985). bioRxiv.
Huynh T, Xu S (2019) Gene annotation easy viewer (GAEV): integrating KEGG’s gene function annotations and associated molecular pathways. F1000Research 7:416
Ihara M, Stein P, Schultz RM (2008) UBE2I (UBC9), a SUMO-conjugating enzyme, localizes to nuclear speckles and stimulates transcription in mouse oocytes. Biol Reprod 79(5):906–913
Innes DJ, Hebert PDN (1988) The origin and genetic basis of obligate parthenogenesis in daphnia pulex. Evolution 42(5):1024–1035
Jaquiéry J, Stoeckel S, Larose C, Nouhaud P, Rispe C, Mieuzet L et al. (2014) Genetic control of contagious asexuality in the pea aphid. PLOS Genet 10(12):e1004838
Jin F, Hamada M, Malureanu L, Jeganathan KB, Zhou W, Morbeck DE et al. (2010) Cdc20 is critical for meiosis I and fertility of female mice. PLOS Genet 6(9):e1001147
Johnson SG, Bragg E (1999) Age and polyphyletic origins of hybrid and spontaneous parthenogenetic campeloma (gastropoda: viviparidae) from the southeastern United States. Evolution 53(6):1769–1781
Kilham SS, Kreeger DA, Lynn SG, Goulden CE, Herrera L (1998) COMBO: A defined freshwater culture medium for algae and zooplankton. Hydrobiologia 377(1):147–159
King KC, Hurst GD (2010) Losing the desire: Selection can promote obligate asexuality. BMC Biol 8(1):101
Koch U, Martin-Creuzburg D, Grossart HP, Straile D (2011) Single dietary amino acids control resting egg production and affect population growth of a key freshwater herbivore. Oecologia 167(4):981–989
Kojima H, Tohsato Y, Kabayama K, Itonori S, Ito M (2013) Biochemical studies on sphingolipids of Artemia franciscana: Complex neutral glycosphingolipids. Glycoconj J 30(3):257–268
Kraus S, Naor Z, Seger R (2001) Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch Med Res 32(6):499–509
Lafont R, Mathieu M (2007) Steroids in aquatic invertebrates. Ecotoxicol (Lond, Engl) 16:109–130
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The sequence alignment/map format and SAMtools. Bioinforma (Oxf, Engl) 25(16):2078–2079
Li M, York JP, Zhang P (2007) Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol Cell Biol 27(9):3481–3488
Liao Y, Smyth GK, Shi W (2014) featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinforma (Oxf, Engl) 30(7):923–930
Lim HH, Goh PY, Surana U (1998) Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol 8(4):231–237
Liu M, Pan J, Dong Z, Cheng Y, Gong J, Wu X (2019) Comparative transcriptome reveals the potential modulation mechanisms of estradiol affecting ovarian development of female Portunus trituberculatus. PLoS ONE 14(12):e0226698
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550
Marques P, Skorupskaite K, Rozario KS, Anderson RA, George JT (2022) Physiology of GnRH and Gonadotropin Secretion. In K. R. Feingold et al., (Eds) Endotext. MDText.com, Inc
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M (2007) KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185.
Murray TM, Rao LG, Divieti P, Bringhurst FR (2005) Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl-terminal ligands. Endocr Rev 26(1):78–113
Nakagawa Y, Henrich VC (2009) Arthropod nuclear receptors and their role in molting. FEBS J 276(21):6128–6157
Nambu Z, Tanaka S, Nambu F (2004) Influence of photoperiod and temperature on reproductive mode in the Brine shrimp, Artemia franciscana. J Exp Zool Part A, Comp Exp Biol 301(6):542–546
Neaves WB, Baumann P (2011) Unisexual reproduction among vertebrates. Trends Genet 27(3):81–88
Neiman M, Sharbel TF, Schwander T (2014) Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. J Evolut Biol 27(7):1346–1359
Niyaz M, Khan MS, Mudassar S (2019) Hedgehog signaling: an achilles’ heel in cancer. Transl Oncol 12(10):1334–1344
Omilian AR, Cristescu MEA, Dudycha JL, Lynch M (2006) Ameiotic recombination in asexual lineages of Daphnia. Proc Natl Acad Sci USA 103(49):18638–18643
Omilian AR, Lynch M (2009) Patterns of Intraspecific DNA Variation in the Daphnia Nuclear Genome. Genetics 182(1):325–336
Ojima Y (1958) A cytological study on the development and maturation of the parthenogenetic and sexual eggs of Daphnia pulex. Kwansei Gakuin Univ Ann Stud 6:123–171
Pohl M, Bortfeldt RH, Grützmann K, Schuster S (2013) Alternative splicing of mutually exclusive exons—A review. Biosystems 114(1):31–38
Riparbelli MG, Tagu D, Bonhomme J, Callaini G (2005) Aster self-organization at meiosis: A conserved mechanism in insect parthenogenesis? Developmental Biol 278(1):220–230
RStudio Team (2020) RStudio: Integrated Development for R. RStudio, PBC, Boston, MA URL http://www.rstudio.com/
Schwander T, Henry L, Crespi BJ (2011) Molecular evidence for ancient asexuality in timema stick insects. Curr Biol 21(13):1129–1134
Serra M, Snell TW (2009) Sex loss in monogonont rotifers. In: I Schön, K Martens, P van Dijk, editors. Lost Sex. Dordrecht: Springer. p. 281-294
Shen S, Park JW, Lu Z, Lin L, Henry MD, Wu YN et al. (2014) rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci USA 111(51):E5593–E5601
Sherbet GV (2017) Chapter 7—Hippo Signalling in Cell Proliferation, Migration and Angiogenesis. In G. V. Sherbet (Ed.), Molecular Approach to Cancer Management (pp. 65–80). Academic Press
Simon JC, Delmotte F, Rispe C, Crease T (2003) Phylogenetic relationships between parthenogens and their sexual relatives: The possible routes to parthenogenesis in animals. Biol J Linn Soc 79(1):151–163
Stenberg P, Lundmark M (2004) Distribution, mechanisms and evolutionary significance of clonality and polyploidy in weevils. Agric For Entomol 6(4):259–266
Stenberg P, Saura A (2009) Cytology of Asexual Animals. In I. Schön, K. Martens, & P. Dijk (Eds.), Lost Sex: The Evolutionary Biology of Parthenogenesis (pp. 63–74). Springer Netherlands
Suomalainen E, Saura A, Lokki J (1987) Cytology and evolution in parthenogenesis. CRC Press
Suresh S, Crease TJ, Cristescu ME, Chain FJJ (2020) Alternative splicing is highly variable among Daphnia pulex lineages in response to acute copper exposure. BMC Genomics 21(1):433
Tatarazako N, Oda S, Watanabe H, Morita M, Iguchi T (2003) Juvenile hormone agonists affect the occurrence of male. Daphnia Chemosphere 53(8):827–833
Tucker AE, Ackerman MS, Eads BD, Xu S, Lynch M (2013) Population-genomic insights into the evolutionary origin and fate of obligately asexual Daphnia pulex. Proc Natl Acad Sci USA 110(39):15740–15745
Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M et al. (2015) Essentials of Glycobiology (3rd ed.). Cold Spring Harbor Laboratory Press.
Vrijenhoek R (1998) Animal Clones and Diversity. Bioscience 48:617–628
Wang Y, Liu J, Huang B, Xu YM, Li J, Huang LF et al. (2015) Mechanism of alternative splicing and its regulation (Review). Biomed Rep. 3(2):152–158
Ward A, Hopkins J, Mckay M, Murray S, Jordan PW (2016) Genetic Interactions Between the Meiosis-Specific Cohesin Components, STAG3, REC8, and RAD21L. G3 Genes|Genomes|Genetics, 6(6):1713–1724
White MJ, Contreras N, Chency J, Webb GC (1977) Cytogenetics of the parthenogenetic grasshopper Warramaba (formerly Moraba) virgo and its bisexual relatives. II. Hybridization studies. Chromosoma 61(2):127–148
Xu S, Huynh TV, Snyman M (2022) The transcriptomic signature of obligate parthenogenesis. Heredity 128(2):132–138
Xu S, Innes DJ, Lynch M, Cristescu ME (2013) The role of hybridization in the origin and spread of asexuality in Daphnia. Mol Ecol 22(17):4549–4561
Xu S, Omilian AR, Cristescu ME (2011) High rate of large-scale hemizygous deletions in asexually propagating Daphnia: omplications for the evolution of sex. Mol Biol Evolution 28:335–342
Xu S, Spitze K, Ackerman MS, Ye Z, Bright L, Keith N et al. (2015) Hybridization and the origin of contagious asexuality in Daphnia pulex. Mol Biol Evolution 32(12):3215–3225
Yang WL, Li J, An P, Lei AM (2014) CDC20 downregulation impairs spindle morphology and causes reduced first polar body emission during bovine oocyte maturation. Theriogenology 81(4):535–544
Ye Z, Xu S, Spitze K, Asselman J, Jiang X, Ackerman MS et al. (2017) A new reference genome assembly for the microcrustacean Daphnia pulex. G3: Genes, Genomes, Genet 7(5):1405–1416
Yuan YF, Zhai R, Liu XM, Khan HA, Zhen YH, Huo LJ (2014) SUMO-1 plays crucial roles for spindle organization, chromosome congression, and chromosome segregation during mouse oocyte meiotic maturation. Mol Reprod Dev 81(8):712–724
Zaffagnini F, Sabelli B (1972) Karyologic observations on the maturation of the summer and winter eggs of Daphnia pulex and Daphnia middendorffiana. Chromosoma 36(2):193–203
Acknowledgements
This work is supported by NIH grant R35GM133730 to SX. We would like to thank two anonymous reviewers for their constructive suggestions and the Xu lab members for their helpful discussions.
Author information
Authors and Affiliations
Contributions
SX designed the study. MS performed the tissue collection, molecular work, and data analyses. SX and MS wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Louise Johnson.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Snyman, M., Xu, S. Transcriptomics and the origin of obligate parthenogenesis. Heredity 131, 119–129 (2023). https://doi.org/10.1038/s41437-023-00628-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41437-023-00628-3
