
Abstract
Biallelic IGFALS variants lead to acid‒labile subunit (ALS) deficiency characterized by growth hormone resistance with or without delayed puberty. Here, we report a prepubertal boy with a homozygous 2-amino acid deletion within the fourth N-glycosylation motif (c.1103_1108del, p.N368_S370delinsT) associated with parental consanguinity. He showed short stature consistent with ALS deficiency. This case expands the mutation spectrum of IGFALS to include the elimination of only one N-glycosylation motif of ALS.
Similar content being viewed by others
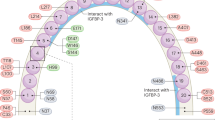


IGFALS on 16p13.3 encodes the acid-labile subunit (ALS). ALS forms a ternary complex with insulin-like growth factors (IGFs) and IGF-binding proteins (IGFBPs) and thereby regulates the half-life of IGFs in the circulation1. ALS plays a critical role in the growth-promoting effects of growth hormone (GH)1. The ALS protein contains seven N-glycosylation motifs with a consensus 3-amino acid sequence (N-X-S/T)2,3,4. These motifs mediate the formation of the ternary complex2,3,4. To date, more than 60 patients with ALS deficiency resulting from biallelic IGFALS variants have been reported1. Most of these patients exhibited moderate short stature due to GH resistance with or without delayed puberty, although several patients achieved low‒normal adult height1,5,6. Salient endocrine findings of these patients were low levels of IGF-I and IGFBP-3 and normal or elevated levels of GH1,5. Known IGFALS variants associated with ALS deficiency include several substitutions and indels widely distributed in the coding region (Fig. 1)1,6,7,8,9,10,11,12,13,14,15,16,17,18. However, only two of these variants (p.S87L and p.N580del) caused changes in the consensus amino acid sequence of the N-glycosylation motifs7,16. The pathogenicity of these two variants remains uncertain. This is because the p.S87L variant was coupled with an additional variant in IGF1R, and the p.N580del variant was present only in a heterozygous state7,16. In fact, Janosi et al. reported that site-directed mutagenesis to replace the asparagine residue at any of the N-glycosylation motifs with an alanine residue did not alter the in vitro binding activity of ALS to the IGF/IGFBP binary complex2.

Missense substitutions, indels, and protein-truncating variants are indicated by thin, broken, and thick arrows, respectively. The seven N-glycosylation sites are shown in blue. Variants within N-glycosylation motifs are shown in red. The 2-amino acid deletion in the present case is boldfaced. Brown boxes and the green structure indicate cysteine-rich regions and leucine-rich repeats, respectively.
Here, we report a patient who carried a homozygous 2-amino acid deletion within the fourth N-glycosylation motif of ALS. The patient was a Japanese boy born from a first-cousin couple at 39 weeks and 5 days of gestation. His body weight and length at birth were 2342 g (−2.6 SD) and 47 cm (−1.2 SD), respectively. During childhood, his growth pattern followed the −2.5 SD growth curve of Japanese boys. At 5.2 years of age, he visited our clinic for the evaluation of short stature. Physical examinations showed no phenotypic abnormalities except for short stature (96.6 cm, −2.7 SD). He had no signs of pubertal sexual development. His bone age was 1.5 years younger than his chronological age. Endocrine examinations revealed high basal levels of GH (8.6–22.5 ng/ml; reference range, ≤ 2.5 ng/ml) and a low IGF-I level (17 ng/ml; reference range, 44–193 ng/ml). Arginine stimulation yielded a prominent GH response (82.0 ng/ml; reference range, ≥ 6.0 ng/ml). The levels of thyroid hormones and gonadotropins were within the reference ranges for age-matched boys. The heights of his father and mother were 165 cm (−1.0 SDS) and 148 cm (−1.9 SDS), respectively. The height of his 9-year-old sister was −2.0 SDS.
We performed molecular analyses. This study was approved by the Institutional Review Board Committee at the National Center for Child Health and Development. Written informed consent was obtained from the parents of the patient. Genomic DNA samples were obtained from the peripheral leukocytes of the patient and his parents. We performed mutation screening for 16 major growth genes (ACAN, CDKN1C, FGFR3, GH1, GHR, GHRHR, GHSR, GNAS, IGF1, IGF1R, IGF2, IGFALS, JAK2, NPR2, SHOX, and STAT5B) using a next-generation sequencer panel (Kazusa DNA Research Institute, Kisarazu, Japan). The methods were described previously16. As a result, we identified a homozygous 6-bp deletion (NM_004970.3: c.1103_1108del, p.N368_S370delinsT) in exon 2 of IGFALS. No pathogenic variants were detected in the other genes examined. Sanger sequencing confirmed that the patient had a homozygous IGFALS variant, and his parents were heterozygous carriers (Fig. 2a). The IGFALS variant was not found in the Human Genome Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), the Genome Aggregation Database (https://gnomad.broadinstitute.org/), or the Japanese Multi Omics Reference Panel (https://jmorp.megabank.tohoku.ac.jp). This variant was classified as a variant of uncertain significance (VUS) based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines (PM2, PM4, PP4)19. This variant caused a 3-amino acid deletion and a 1-amino acid insertion in the fourth N-glycosylation motif of ALS (Fig. 2a). Comparative genomic hybridization of the patient using a CGH + SNP array (SurePrint G3 Human Genome Microarray, 2 × 400 k format; Agilent Technologies, Santa Clara, CA, USA) detected loss of heterozygosity (LOH) regions on 10 different chromosomes (Fig. 2b). The total size of the LOH regions was ~187 Mb. The LOH region on chromosome 16 encompassed IGFALS (Fig. 2b). Array-based CGH identified no pathogenic copy number variations.

a Sanger sequencing showing the c.1103_1108del, p.N368_S370delinsT variant. The parents of the patient were heterozygous carriers. The 6-bp deletion was located within the fourth N-glycosylation motif (N-L-S). b The results of comparative genomic hybridization using a SNP + CGH array. The gray-shaded areas depict regions of loss of heterozygosity (LOH). The LOH region on chromosome 16 included IGFALS.
The boy carried a hitherto unreported variant of IGFALS. The homozygosity of this rare variant can be ascribed to parental consanguinity; the patient carried large LOH regions on 10 chromosomes. The total size of the LOH regions (187 Mb, 6.05% of the entire genome) was similar to the theoretical size of LOH regions in a child born to first-cousin parents (6.25%)20. Of note, the heights of the patient (~−2.6 SD) and his heterozygous parents (−1.0 SD and −1.9 SD) were comparable to those of previously reported individuals with biallelic and monoallelic pathogenic variants of IGFALS, respectively (biallelic variants, −2.46 ± 1.07 SD and monoallelic variants, −1.13 ± 1.08 SD)1. Moreover, the endocrine data of our patient, such as high GH levels and a low IGF-1 level, were indicative of ALS deficiency5. In addition, he had no pathogenic variants in other major growth genes. These results indicate that his phenotype results from the IGFALS variant. The patient may develop delayed puberty in the future because this is a common manifestation of ALS deficiency6.
The IGFALS variant in the patient was a 6-bp deletion. This in-frame deletion is unlikely to cause gross conformational changes or mRNA decay; however, this deletion disrupted one of the seven N-glycosylation motifs of ALS. The phenotype of our patient implies that the loss of the fourth N-glycosylation motif abolishes ALS function. Consistent with this, 3D structure prediction suggested that the asparagine residue at the 368 position mediates the binding between ALS and the binary complex4. These results highlight the clinical importance of the N-glycosylation motifs of ALS. In conclusion, this study expands the mutation spectrum of IGFALS to include 2-amino acid deletion within an N-glycosylation motif.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3412.
References
Domené, S. & Domené, H. M. The role of acid-labile subunit (ALS) in the modulation of GH-IGF-I action. Mol. Cell Endocrinol. 518, 111006 (2020).
Janosi, J. B., Firth, S. M., Bond, J. J., Baxter, R. C. & Delhanty, P. J. N-linked glycosylation and sialylation of the acid-labile subunit. Role in complex formation with insulin-like growth factor (IGF)-binding protein-3 and the IGFs. J. Biol. Chem. 274, 5292–5298 (1999).
Firth, S. M., Yan, X. & Baxter, R. C. D440N mutation in the acid-labile subunit of insulin-like growth factor complexes inhibits secretion and complex formation. Mol. Endocrinol. 25, 307–314 (2011).
Kim, H. et al. Structural basis for assembly and disassembly of the IGF/IGFBP/ALS ternary complex. Nat. Commun. 13, 4434 (2022).
Argente, J., Tatton-Brown, K., Lehwalder, D. & Pfäffle, R. Genetics of growth disorders-which patients require genetic testing? Front. Endocrinol. (Lausanne) 10, 602 (2019).
Poyrazoğlu, Ş. et al. A novel homozygous mutation of the acid-labile subunit (IGFALS) gene in a male adolescent. J. Clin. Res Pediatr. Endocrinol. 11, 432–438 (2019).
Castilla-Cortazar I., et al. Growth hormone insensitivity: Mexican case report. Endocrinol. Diabetes Metab. Case Rep. 2017, 17-0126 (2017).
Plachy, L. et al. High prevalence of growth plate gene variants in children with familial short stature treated with GH. J. Clin. Endocrinol. Metab. 104, 4273–4281 (2019).
Domené, H. M. et al. Heterozygous IGFALS gene variants in idiopathic short stature and normal children: impact on height and the IGF system. Horm. Res Paediatr. 80, 413–423 (2013).
Wit, J. M. et al. Genetic analysis of short children with apparent growth hormone insensitivity. Horm. Res Paediatr. 77, 320–333 (2012).
Franzoni, A. et al. Novel IGFALS mutations with predicted pathogenetic effects by the analysis of AlphaFold structure. Endocrine 79, 292–295 (2023).
Grandone, A. et al. Clinical features of a new acid-labile subunit (IGFALS) heterozygous mutation: anthropometric and biochemical characterization and response to growth hormone administration. Horm. Res Paediatr. 81, 67–72 (2014).
Yoo, H. J. et al. Whole exome sequencing for a patient with Rubinstein-Taybi syndrome reveals de novo variants besides an overt CREBBP mutation. Int J. Mol. Sci. 16, 5697–5713 (2015).
Kamil, G., Yoon, J. Y., Yoo, S. & Cheon, C. K. Clinical relevance of targeted exome sequencing in patients with rare syndromic short stature. Orphanet J. Rare Dis. 16, 297 (2021).
Kumar, A. et al. Pathogenic/likely pathogenic variants in the SHOX, GHR and IGFALS genes among Indian children with idiopathic short stature. J. Pediatr. Endocrinol. Metab. 33, 79–88 (2020).
Hattori, A. et al. Next-generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocr. J. 64, 947–954 (2017).
He, J. et al. Preliminary investigation into the genetic etiology of short stature in children through whole exon sequencing of the core family. Open Life Sci. 19, 20220853 (2024).
David, A. et al. Acid-labile subunit deficiency and growth failure: description of two novel cases. Horm. Res Paediatr. 73, 328–334 (2010).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Sund, K. L. et al. Regions of homozygosity identified by SNP microarray analysis aid in the diagnosis of autosomal recessive disease and incidentally detect parental blood relationships. Genet. Med. 15, 70–78 (2013).
Acknowledgements
This work was supported by grants from the National Center for Child Health and Development and the Takeda Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doi, H., Kageyama, I., Katoh-Fukui, Y. et al. Homozygous 6-bp deletion of IGFALS in a prepubertal boy with short stature. Hum Genome Var 11, 27 (2024). https://doi.org/10.1038/s41439-024-00285-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41439-024-00285-w
