
Abstract
Cockayne syndrome (CS) is a progressive multisystem disorder characterized by growth failure, microcephaly, developmental delay, and photosensitivity. The characteristic symptoms appear during early childhood in most patients with CS. Herein, we report a mild case of CS with a novel start-loss variant in ERCC8 that did not show the characteristic symptoms of CS during early childhood and exhibited sudden growth failure after the age of 10 years.
Similar content being viewed by others


Cockayne syndrome (CS) (OMIM #216400 and #133540) is a progressive multisystem disorder characterized by growth failure, microcephaly, developmental delay, and photosensitivity. CS is typically caused by biallelic loss-of-function variants of ERCC8 (CS type A) or ERCC6 (CS type B). Although there are no specific disease-modifying therapies for CS, early diagnosis and management of complications are desirable because CS exhibits various systemic symptoms. Most patients show significant growth failure by the age of 2 years. Additionally, it is accompanied by characteristics, such as microcephaly, developmental delay, cutaneous photosensitivity, cataracts, sensorineural hearing loss, enamel hypoplasia, and enophthalmia, making it possible to diagnose the disease1,2. However, the clinical diagnosis is often challenging in patients with atypical or mild phenotypes; therefore, comprehensive genetic analysis is needed for diagnosis3. Herein, we report a mild case of CS that presented with sudden growth failure after 10 years of age. In this patient, whole-exome sequencing (WES) identified a novel start-loss variant combined with a known complex rearrangement involving exon 4 of ERCC8.
The patient was a 12-year-old female. She was born at 38 weeks of gestation and her height, weight, and head circumference were 45.5 cm (−1.2 standard deviation [SD]), 2774 g (0.2 SD), and 31.5 cm (−1.1 SD), respectively, at birth. The patient had no family history of neuromuscular disease or developmental delay. She had a mild developmental delay at 3 years of age and then was diagnosed with autism spectrum disorder and mild intellectual disability at 4 years of age. At 12-year-old, she was first noted to have a short stature at a school checkup and tremors, which led to her referral to our hospital. At the initial examination, she was 135 cm in height (−2.5 SD) and 35 kg in weight (−1.0 SD), and her growth failure became apparent at around 10 years of age (Fig. 1A–C). She also had mild ataxia and kinetic tremors. She had mild enophthalmia but did not have enamel hypoplasia, areflexia, or spasticity. Computed tomography (CT) of the head revealed bilateral striatal calcifications and magnetic resonance imaging (MRI) of the head revealed cerebellar atrophy (Fig. 1D and E). Despite the absence of growth spurt and pubertal signs, the radial epiphyseal line was closed on carpal radiography. Blood tests revealed normal levels of insulin-like growth factor 1, intact parathyroid hormone, and thyroid hormone (thyroid-stimulating hormone, free triiodothyronine, and free thyroxine). Gonadal hormone (luteinizing hormone, follicle-stimulating hormone, and estradiol) levels were equivalent to those of adult females.

A–C Height, weight, and head circumference of this patient compared to Japanese girls with mean and SD values20. D Head CT image showed calcification of the striatum. E Head MRI (T2 weighted) showed cerebellar atrophy. F The audiogram showed moderate bilateral sensorineural hearing loss. BC bone conduction, AC air conduction.
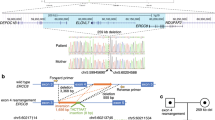
Trio-based WES was performed using the xGen® Exome Research Panel v2 (IDT, Iowa, USA) after written informed consent was obtained from the parents in accordance with the Kyoto University Review Committee and Ethics Committee. The imported library was sequenced using DNBSEQ-G400 (MGI Tech, Shenzhen, China). WES identified a novel heterozygous start-loss variant [NM_000082: c.1A>T, p.M1?] in ERCC8. The same variant was identified in the patient’s mother. Furthermore, PCR with published primers that detect a rearrangement in exon 4 of ERCC8, which is frequently found in Japanese patients with CS, revealed that the patient and the father have the rearrangement4,5. The maternal-derived variant was absent in the gnomAD, HGVD, and 8.3KJPN. This variant was predicted to be deleterious by CADD (score, 18.4; https://cadd.gs.washington.edu/), deleterious by SIFT (score, 0; https://sift.bii.a-star.edu.sg), disease-causing by Mutation Taster (probability, 0.99; http://www.mutationtaster.org), and pathogenic by PoStaL, a machine learning-based prediction tool for the pathogenicity of start-loss variants (score, 0.689; https://github.com/a-tkt/PoStaL). The pathogenicity of this variant was evaluated according to the 2015 American Society for Medical Genetics and Genomics (ACMG) guidelines and was classified as pathogenic (PVS1, PM2, PM3, PP3, and PP4)6. She was diagnosed with CS caused by compound heterozygous ERCC8 variants. After diagnosis, an examination to check for complications revealed sensorineural hearing loss that the family was unaware of and started using bilateral hearing aids (Fig. 1F). A detailed interview revealed that she had mild photosensitivity and received proper guidance throughout her life. Visual impairment and renal dysfunction, which can be complicated by Cockayne syndrome, have not been seen, and the patient continues to be monitored for these findings. Although vision issues and renal dysfunction, which can be complicated in patients with CS, have not been observed, the patient is still under follow-up.
CS is classified as CS type 1 (CS1), in which symptoms such as growth failure and developmental delay appear within the first 2 years of birth; CS type 2 (CS2), a more severe form in which symptoms appear from birth; and CS type 3 (CS3), a mild or delayed form in which the first symptoms appear after 3−4 years of birth2. The overall incidence of CS is estimated to be 2.7/1 million people in Japan and Western Europe, of which CS3 is very rare (6.4–12.5%)5,7,8. The patient was classified as CS3 because the characteristic CS symptoms did not become apparent until the patient was in her teens. Our patient followed the same course as other patients with CS3. Her severity score for CS was 15 at 0-year-old, 14 at 3-year-old, 12 at 12-year-old, and 10 at 14-year-old. The severity score for CS encompasses five key items: weight and height, head circumference, neurosensorial symptoms, autonomy and motor development, and communication skills. The severity score for CS is evaluated on a 4-point scale from 0 to 3 for each item, with a total possible score of 15 points. A lower score indicates a more severe condition9.
Growth failure is one of the most characteristic symptoms seen in most patients with CS. CS1 shows microcephaly at 2-month-old and growth failure from 5 to 22-month-old; CS2 shows microcephaly at birth and growth failure from birth to 3-month-old1. Although there are few detailed reports on the clinical manifestations of CS3, some patients show growth failure before 2-year-old, while others show no growth failure at all8,10,11. However, there have been no cases of sudden growth failure after 10-year-old, as observed in the present case.
Although it has been assumed that there is no clear correlation between phenotype and genotype in CS, missense variants in ERCC8 have recently been reported to result in milder phenotypes than protein-truncated variants2,8. In Japan and other East Asian regions, patients with CS frequently exhibit exon 4 rearrangement variants in ERCC8. This variant was considered the founder variant. Many patients who are homozygous for this founder variant present with typical CS and severe intellectual disability. Additionally, patients with CS with compound heterozygosity involving the founder variant and other pathogenic variants have been reported in these regions (Table 1). Due to the small number of cases, it was difficult to identify a clear relationship between the types of variants and their phenotypes. However, in this case, some of the patients had mild intellectual disability.
Currently, in the ACMG guidelines, the pathogenicity of start-loss variants is considered very strong evidence of pathogenicity (PVS1), as well as other protein-truncated variants, such as nonsense and frameshift variants6. However, it has recently been pointed out that the pathogenicity of start-loss variants may be overestimated and that variants are not necessarily deleterious12. Translation initiation can occur at ATG sites located downstream of the native ATG site or even at non-ATG sites13,14. Therefore, a start-loss variant may not be deleterious if it has an in-frame ATG site near the original ATG or if the shortening site is not highly conserved among species12,15. Interestingly, a previously reported case of CS with a start-loss variant of ERCC8 [c.2T>A, p.M1?] also showed no characteristic symptoms until 3-year-old and the phenotype was mild, with only slight ataxia and mental language developmental delay at 13-year-old8,16. Patients with a start-loss variant of ERCC8 may exhibit relatively mild symptoms similar to those in the present case. However, the actual translation initiation site of ERCC8 with the start-loss variant has not yet been identified. It is unclear whether the resulting truncated protein retains partial function; therefore, further studies are required.
CS exhibits a broad clinical spectrum ranging from mild to severe. In particular, CS3 is difficult to diagnose because its characteristic growth failure and physical symptoms are often absent until later in childhood. In the cases of sudden growth failure, CS should be cited as a differential diagnosis, and detailed interviews are necessary for photosensitivity and hearing, as well as comprehensive genetic analysis. It will be possible to diagnose CS at an earlier stage, leading to early detection of complications and genetic counseling.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at: https://doi.org/10.6084/m9.figshare.hgv.3441.
References
Baer, S. et al. Growth charts in Cockayne syndrome type 1 and type 2. Eur. J. Med. Genet. 64, 104105 (2021).
Laugel, V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech. Ageing Dev. 134, 161–170 (2013).
Duong, N. T. et al. Cockayne syndrome without UV-sensitivity in Vietnamese siblings with novel ERCC8 variants. Aging 14, 5299–5310 (2022).
Ren, Y. et al. Three novel mutations responsible for Cockayne syndrom group A. Genes Genet. Syst. 78, 93–102 (2003).
Kubota, M. et al. Nationwide survey of Cockayne syndrome in Japan: incidence, clinical course and prognosis. Pediatr. Int. 57, 339–347 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Kleijer, W. J. et al. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair 7, 744–750 (2008).
Calmels, N. et al. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 55, 329–343 (2018).
Spitz, M. A. et al. Diagnostic and severity scores for Cockayne syndrome. Orphanet J. Rare Dis. 16, 63 (2021).
Rapin, I. et al. Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J. Child Neurol. 21, 991–1006 (2006).
Calmels, N. et al. Uncommon nucleotide excision repair phenotypes revealed by targeted high-throughput sequencing. Orphanet J. Rare Dis. 11, 26 (2016).
Takata, A., Hamanaka, K. & Matsumoto, N. Refinement of the clinical variant interpretation framework by statistical evidence and machine learning. Med 2, 611–632 (2021).
Brar, G. A. Beyond the triplet code: context cues transform translation. Cell 167, 1681–1692 (2016).
Ingolia, N. T. Ribosome footprint profiling of translation throughout the genome. Cell 165, 22–33 (2016).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Sugita, K., Takanashi, J., Suzuki, N. & Niimi, H. Comparison of cellular sensitivity to UV killing with neuropsychological impairment in Cockayne syndrome patients. Brain Dev. 13, 163–166 (1991).
Xie, H. et al. A complex intragenic rearrangement of ERCC8 in Chinese siblings with Cockayne syndrome. Sci. Rep. 7, 44271 (2017).
Ting, T. W. et al. Cockayne Syndrome due to a maternally-inherited whole gene deletion of ERCC8 and a paternally-inherited ERCC8 exon 4 deletion. Gene 572, 274–278 (2015).
Wang, X. et al. Molecular spectrum of excision repair cross-complementation group 8 gene defects in Chinese patients with Cockayne syndrome type A. Sci. Rep. 7, 13686 (2017).
Isojima, T., Kato, N., Ito, Y., Kanzaki, S. & Murata, M. Growth standard charts for Japanese children with mean and standard deviation (SD) values based on the year 2000 national survey. Clin. Pediatr. Endocrinol. 25, 71–76 (2016).
Acknowledgements
We would like to thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsuoka, T., Yoshida, T., Kora, K. et al. A mild case of Cockayne syndrome with a novel start-loss variant of ERCC8. Hum Genome Var 11, 39 (2024). https://doi.org/10.1038/s41439-024-00297-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41439-024-00297-6
