
Abstract
An episignature is a genome-wide DNA methylation pattern that is specific to each syndrome or etiologic gene. Episignature analysis helps to diagnose patients with variants of uncertain significance (VUS), but this requires positive methylation datasets from patients with a definitive diagnosis. Here we provide methylation datasets of Rubinstein–Taybi syndrome at the individual patient level, which have not been published before. This dataset increases the likelihood of determining the function of the VUS.
Similar content being viewed by others
Congenital neurodevelopmental disorders are often caused by pathogenic variants in genes encoding the epigenetic machinery, which consists of writers, erasers, readers and remodelers that play roles in the modification, demodification, recognition of DNA or histone proteins, and chromatin remodeling, respectively1. At the same time, pathogenic variants in the genes of the epigenetic machinery have been found to cause gene-specific genome-wide DNA methylation patterns, called episignatures, that are detectable in the peripheral blood of affected individuals2. Moreover, in some genes, episignatures differ between the loci of frameshift, nonsense or missense variants, examples of which include CREBBP and EP300. The CREB-binding protein (CBP), encoded by CREBBP, and its paralog E1A-associated protein (p300), encoded by EP300, are involved in histone acetylation and transcriptional regulation. Pathogenic variants in exons 30/31 of CREBBP or EP300 are etiologic for Menke–Hennekam syndromes (MKHK) 1,2 (Online Mendelian Inheritance in Man (OMIM) nos. 618332 and 618333)3, whereas variants that produce a null allele or a functional change in the catalytic domain of either protein cause Rubinstein–Taybi syndrome (RSTS) 1,2 (OMIM nos. 180849 and 613684)4. RSTS is a multiple congenital anomaly syndrome characterized by intellectual disability, postnatal growth failure, microcephaly, broad thumbs and dysmorphic facial features. However, none of the patients with MKHK with developmental delay or intellectual disability have the broad or angulated thumbs or the characteristic facial features of RSTS3,4,5. The episignatures of RSTS and MKHK also differ, as do the phenotypes2, thereby indicating that these two syndromes differ at the cellular level as a consequence of epigenetic regulation6. The diagnostic guidelines for RSTS reported by an international consensus group have described molecular diagnostic pathways, including DNA methylation episignature analysis, for uncertain results of exome or genome sequencing in patients with nonclassical RSTS phenotypes7. Episignatures reveal the molecular dysregulation caused by congenital genetic disorders, which can aid in determining the pathogenicity of variants of uncertain significance (VUS)8. Pathogenicity determination for VUS using episignatures is performed using supervised classification models. One of the most widely used classifiers for differentiating cases and controls based on methylation data is support vector machines (SVM)9. Having identified support vectors using training sets consisting of episignatures from controls and cases with verified pathogenic variants, new cases with VUSs are classified using Platt-modified methods with a probability score between 0 and 1, indicating control and pathogenicity, respectively. Episignatures with scores above 0.50 or 0.75, which depend on the authors, for VUS are classified as pathogenic variants based on DNA methylation analysis8,10,11,12.
So far, there have been no reports of raw DNA methylation β values for the episignature probe sets of RSTSs in individual patients and, consequently, researchers are unable to construct support vectors for classifying individual participants with VUSs within CREBBP or EP300. Although previously published mean methylation β values of RSTS episignature probe sets obtained from 30 patients, together with mean β values for a further 33 syndromes11, can be utilized as positive controls for clustering analysis, this information is insufficient for SVM analysis. However, providing raw data, such as those for BAFopathy10, CHARGE13, Kabuki13, Sotos14, Williams15 and Wolf–Hirschhorn8 syndromes, increases the likelihood of diagnosing pathogenicity of VUS. In addition, DNA methylation is reversible in response to environmental factors and is influenced by nearby nucleotide sequences that affect DNA binding factors' accessibility. Moreover, sharing array data could provide opportunities to establish more robust episignature probe sets that are stable and independent of genetic background.
In this study, we examined the DNA methylation β values of the episignature probes for RSTSs (RSTS1 (CREBBP) and RSTS2 (EP300))2,11, of eight Japanese patients diagnosed with RSTS1 and one diagnosed with RSTS2 (Table 1). The experimental protocol was approved by the Committee for Ethical Issues at National Center for Child Health and Development (nos. 2020-326 and 2022-183). Written informed consent was obtained from the patients or guardians. All participants showed classical RSTS phenotypes: three harbor nonsense variants at the 5′ end of the catalytic domain, one harbors a small frameshift nonsense Delins variant, one harbors a single-nucleotide substitution at the splicing donor site, one harbors a small deletion at the exon–intron junction, one harbors a large deletion from exon 24 to the transcription termination site of CREBBP (NM_004380.3) and two harbor a large deletion from the neighboring gene to exon 2 of CREBBP (Fig. 1A). The final two of the aforementioned individuals are monozygotic twins (RSTS1_7, _8)16. DNA from peripheral blood cells was converted by bisulfite treatment and processed using Illumina Infinium EPIC bead chip arrays as previously described8. We used publicly available raw data files from the EPIC array, which were collected from blood cells of healthy children in various environments as a control group for congenital disorders. Very few files were obtainable (Supplementary Table 1). Our data were combined and co-processed with the above data. Briefly, methylated and unmethylated signal intensities were normalized using R version 4.3.1 and ENmix package version 1.38.1, with background and dye bias corrections. β Values were calculated by dividing methylated signals by (methylated + unmethylated) signals. β Values of the published episignature probe sets for RSTSs, RSTS1 and RSTS2 in the participants are presented in Supplementary Tables 2–4. Hierarchical clustering based on Manhattan distance using the average method distinguished the nine RSTS patients from controls corresponding to the β values of each of the RSTS and RSTS1 episignature probe sets. The RSTS2 probe set distinguished only the RSTS2_1 patient across a great distance (Fig. 1B)2,11. Multidimensional scaling based on scaling the pairwise Euclidean distances between samples revealed the common DNA methylation changes in the eight RSTS1 patients when focusing on the β values of the RSTS1 probe set, which was clearer than multidimensional scaling using the RSTS probe set. Furthermore, the β values for the RSTS2 probe set clearly classified the single RSTS2 patient from the RSTS1 patients and controls. These results reconfirmed that episignatures are specific for the CREBBP and EP300 pathogenic variants. Furthermore, none of the control subjects without congenital disorders exhibited an RSTS episignature, even when exposed to various environmental and/or physical conditions.

A The variants loci of participants in CREBBP or EP300 in this study. B The participants (RSTS1 and RSTS2) were classified by DNA methylation of each probe set for RSTS episignatures using hierarchical clustering (left) or multidimensional scaling (right). Control data were obtained from publicly available data. Cont_ob, obese or overweight children in GSE193730; Cont_20wks and Cont_Excs, 20 wks after usual lifestyle and exercise intervention of Cont_ob, respectively, from GSE193730; Cont_normal and Cont_obesity, normal control and simple obesity children, respectively, from GSE221864; Cont_DON_l and Cont_DON_h, low and high exposure deoxynivalenol group children, respectively, from GSE180534.
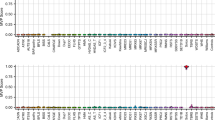
Among our samples, RSTS1_3 harbors an intron variant in CREBBP (NM_004380.3:c.1676+2T>G). The corresponding locus is a well-established conserved nucleotide in the splicing donor site that influences mRNA splicing. Although this variant has not been previously reported in ClinVar17, in silico predictions, dbscSNV and MaxEntScan, identify this variant as a pathogenic variant18,19. Having removed RSTS1_3, training was performed using datasets from the remaining seven RSTS1 patients, and we obtained a RSTS1 probability of 0.985 in RSTS1_3 by test (Fig. 2A). The variant in RSTS1_3 was also classified as ‘pathogenic’ based on episignature analysis. High probability scores (>0.9) were also confirmed in the other seven RSTS1 patients by performing leave-one-out cross-validation (Fig. 2B). Meanwhile, we obtained a RSTS1 probability of 0.542 in RSTS2_1, which is lower than the border when 0.75 is adopted8. These results again showed that the episignatures between CREBBP and EP300 pathogenic variants are indeed different. To examine whether the batch effects between the publicly available controls and our original data are involved in the results, we calculated the RSTS1 probability in a patient with Prader–Willi syndrome (PWS), which was assessed by the same batch as the nine RSTSs in this study, and previously reported 28 congenital disease cases8 (Fig. 2A). All disease cases showed RSTS1 probability scores of less than 0.3, which were much lower than the threshold of 0.75. The SVM model in this study worked accurately.

A The SVM was trained by the seven RSTS1 patients and publicly available 150 samples from control children. Testing set composed of RSTS1 with an intron variant (RSTS1_3), RSTS2, PWS and 28 congenital disease cases8 (20 Wolf–Hirschhorn syndromes (WHS), three Rauch–Steindl syndrome (RAUST), one CHARGE syndrome (CHARGE), two Kabuki syndrome (KS) and two Sotos syndrome (Sotos)) was calculated to determine the probability of RSTS1. B The probability of RSTS1 in each RSTS1 patient was calculated by leave-one-out cross-validation.
There are 60 and 34 intron variants in CREBBP and EP300, respectively, that are classified as either ‘Conflicting classifications of pathogenicity’ or ‘Uncertain significance’ in ClinVar17. The detection of intron variants with ‘uncertain significance’ will be further enhanced by whole-genome analysis. The β values of the episignatures published by this study could contribute to identifying the pathogenicity of these intron variants, as well as missense VUS. Furthermore, with respect to CREBBP and EP300 in particular, the loci of the variants in which domain are essential for phenotypes and episignatures3,6. The data obtained in this study could provide an opportunity to apply episignature analysis in classifying the variants of RSTS or MKHK.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3560, https://doi.org/10.6084/m9.figshare.hgv.3563, https://doi.org/10.6084/m9.figshare.hgv.3566, https://doi.org/10.6084/m9.figshare.hgv.3569, https://doi.org/10.6084/m9.figshare.hgv.3572, https://doi.org/10.6084/m9.figshare.hgv.3575, https://doi.org/10.6084/m9.figshare.hgv.3578, https://doi.org/10.6084/m9.figshare.hgv.3581.
References
Bjornsson, H. T. The Mendelian disorders of the epigenetic machinery. Genome Res. 25, 1473–1481 (2015).
Levy, M. A. et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. Hum. Genet. Genom. Adv. 3, 100075 (2022).
Menke, L. A. et al. CREBBP mutations in individuals without Rubinstein–Taybi syndrome phenotype. Am. J. Med. Genet. A 170, 2681–2693 (2016).
Rubinstein, J. H. & Taybi, H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am. J. Dis. Child. 105, 588–608 (1963).
Hennekam, R. C. M. Rubinstein–Taybi syndrome. Eur. J. Hum. Genet 14, 981–985 (2006).
Haghshenas, S. et al. Menke–Hennekam syndrome; delineation of domain-specific subtypes with distinct clinical and DNA methylation profiles. Hum. Genet. Genom. Adv. 5, 100287 (2024).
Lacombe, D. et al. Diagnosis and management in Rubinstein–Taybi syndrome: first international consensus statement. J. Med. Genet. 61, 503–519 (2024).
Kawai, T. et al. Loss of function in NSD2 causes DNA methylation signature similar to that in Wolf–Hirschhorn syndrome. Genet. Med. Open 2, 101838 (2024).
Haghshenas, S., Bhai, P., Aref-Eshghi, E. & Sadikovic, B. Diagnostic utility of genome-wide DNA methylation analysis in Mendelian neurodevelopmental disorders. Int. J. Mol. Sci. 21, 9303 (2020).
Aref-Eshghi, E. et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin–Siris and Nicolaides–Baraitser syndromes. Nat. Commun. 9, 4885 (2018).
Aref-Eshghi, E. et al. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 Mendelian neurodevelopmental disorders. Am. J. Hum. Genet. 106, 356–370 (2020).
Choufani, S. et al. An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome. Am. J. Hum. Gene.t 109, 1867–1884 (2022).
Butcher, D. T. et al. CHARGE and Kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet. 100, 773–788 (2017).
Choufani, S. et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 6, 10207 (2015).
Strong, E. et al. Symmetrical dose-dependent DNA-methylation profiles in children with deletion or duplication of 7q11.23. Am. J. Hum. Genet. 97, 216–227 (2015).
Kosaki, R. et al. Monozygotic twins of Rubinstein–Taybi syndrome discordant for glaucoma. Am. J. Med. Genet. A 155a, 1189–1191 (2011).
ClinVar (NCBI, accessed 7 July 2025); https://www.ncbi.nlm.nih.gov/clinvar/
Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
Eng, L. et al. Nonclassical splicing mutations in the coding and noncoding regions of the ATM Gene: maximum entropy estimates of splice junction strengths. Hum. Mutat. 23, 67–76 (2004).
Acknowledgements
We thank the patients and guardians for participating in this study. This work was supported by generous donations, the Japan Agency for Medical Research and Development (AMED) under the grant numbers 25ek0109760s0202 (T. Kaname), 25ek0109672s01 (K.H.) and 25ek0109672s03 (K.N.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kawai, T., Aoki, T., Nakabayashi, K. et al. DNA methylation data from Japanese patients with Rubinstein–Taybi syndrome. Hum Genome Var 12, 27 (2025). https://doi.org/10.1038/s41439-025-00332-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41439-025-00332-0
