
Abstract
Recently, the Tohoku Medical Megabank Organization released whole-genome allele frequencies of single-nucleotide variants and indels from approximately 60,000 individuals from the general Japanese population in the Tohoku region (60KJPN). Here we analyzed the 60KJPN dataset for BRCA1/BRCA2 variants and compared them with the previous version, 54KJPN, to ascertain the frequency of hereditary breast and ovarian cancers in the general Japanese population. We hope that these results will contribute to strategies for cancer prevention.
Similar content being viewed by others

In a previous paper, we analyzed the whole-genome sequence data of approximately 54,000 individuals from the general population of the Tohoku area in Japan (54KJPN); these data were released by the Tohoku Medical Megabank Organization (ToMMo)1,2. We comprehensively examined pathogenic and truncating variants in BRCA1/BRCA2 and their frequency3. Recently, ToMMo released an updated data version, including approximately 60,000 individuals (60KJPN). Therefore, we also updated our analysis with 60KJPN and compared it with that of 54KJPN.
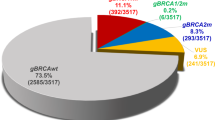
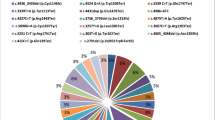
To confirm the frequencies of the pathogenic variants, we analyzed ToMMo-60KJPN data (https://jmorp.megabank.tohoku.ac.jp/downloads/tommo-60kjpn-20240904-af_snvindelall) in the same manner as in the previous paper3,4. Compared with those in 54KJPN, the number of unique variants of BRCA1 and BRCA2 increased from 7711 and 6320 to 8427 and 6885, respectively, in 60KJPN. Among the variants, 44 in BRCA1 and 66 in BRCA2 were predicted to be pathogenic or likely pathogenic according to the ClinVar criteria (20250421), and 4 in BRCA1 and 8 in BRCA2 were newly identified in 60KJPN (Table 1 and Supplementary Tables 1 and 2). Furthermore, among the truncating variants unclassified in ClinVar, 3 in BRCA1 and 14 in BRCA2 were classified as pathogenic on the basis of the ClinGen ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) BRCA1 and BRCA2 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1.2.05, and 3 in BRCA2 were newly identified in 60KJPN (Table 1 and Supplementary Tables 1 and 2). The pathogenicity of the three BRCA1 variants included in 54KJPN changed to likely pathogenic according to the ClinVar update (Table 1). The allele frequencies were more than 1.5 times greater for five pathogenic variants, BRCA1:c.68_69del (p.E23Vfs*17), BRCA2:c.3859_3860del (p.N1287*), c5722_5723del (p.L1908Rfs*2), c.6405_6409del (p.N2135Kfs*3) and c.6715G>T (p.E2239*), in 60KJPN than in 54KJPN. Therefore, we calculated carrier frequencies for BRCA1 and BRCA2 by summing all allele counts, assuming each variant occurs independently, assuming each variant occurs independently. Given the very low allele frequencies, the likelihood that an individual carries more than one variant was considered negligible. The estimated carrier frequency in 60KJPN was 2.668 × 10−3 (160/119,860.5) for BRCA1, 3.219 × 10−3 (193/119,807.6) for BRCA2, and 5.879 × 10−3 for BRCA1 or BRCA2 with ClinVar-classified pathogenic variants. The frequency increased to 2.735 × 10−3 (164/119,859.3) for BRCA1, 3.436 × 10−3 (206/119,814.2) for BRCA2, and 6.161 × 10−3 for BRCA1 or BRCA2 with pathogenic variants, including ENIGMA-classified variants.
In the analysis of 60KJPN, a total of 15 pathogenic variants in BRCA1/BRCA2 were newly identified, and all estimated carrier frequencies were higher than those in 54KJPN, even though the sample size increased by only 6000 individuals (12,000 alleles) from 54KJPN to 60KJPN. These results suggest the possibility of other rare pathogenic variants in the general Japanese population.
In addition, many variants of uncertain significance (VUSs) were also identified in BRCA1/BRCA2 as rare variants in the 60KJPN dataset; VUSs may represent yet-undetermined, population-specific pathogenic variants. Notably, two recent intriguing reports concerning BRCA2 focused on the DNA-binding domain (DBD), where ClinVar-registered VUSs are enriched6,7. This DBD plays a critical role in the homologous recombination function of BRCA2 through single-stranded DNA binding and RAD51 recruitment, making it highly sensitive to functional disruption by missense or truncating variants. In addition, the moderate size and well-defined structural features of the domain allow feasible saturation genome editing and robust phenotypic readouts in cell-based assays. In their respective studies, Sahu et al. used saturation genome editing in mouse embryonic stem cells, whereas Huang et al. applied a CRISPR–Cas9–based saturation genome editing approach in haploid human cells, together providing a comprehensive, experimentally derived reference for the clinical interpretation of BRCA2 DBD variants. When cross-referenced with variants detected in 60KJPN, among the 63 variants in the DBD classified in ClinVar as VUSs or with conflicting interpretations of pathogenicity, 49 were rated as likely benign or benign in both studies, and 9 were rated as likely benign in either study (Supplementary Table 3). Thus, most of the variants were classified as likely benign or benign. Among the remaining variants, two remained as VUSs in both studies, and three were rated as likely pathogenic in either study; however, no variants were consistently classified as likely pathogenic/pathogenic in both analyses. Because many variants remain classified as VUS under ENIGMA criteria, the impact of saturation genome editing-based functional assessment is particularly substantial. Of note, among ClinVar-unclassified variants, nine were classified as Likely Benign or Benign by ENIGMA criteria, and all of these classifications were concordant with the results of saturation genome editing studies. Furthermore, for variants classified as benign or pathogenic in ClinVar, the results of these analyses revealed nearly identical classifications. Thus, these comprehensive approaches using saturation genome editing have the potential to elucidate the pathogenicity of unresolved VUSs in other domains and genes in future studies.
The allele count of two pathogenic variants, BRCA1:c.4601dup (p.E1535Gfs*39) and BRCA2:c.6666C>G (p.Y2222*), changed from one in 54KJPN to zero in 60KJPN. Inquiries with ToMMo revealed that this change is due to the exclusion of close relatives in each dataset, aimed at preventing an upward shift in allele frequencies caused by the accumulation of related individuals, which may have led to differences in the samples used compared with previous versions. Therefore, care should be taken when evaluating rare variants.
Since ToMMo announced that it has already acquired genome data for approximately 100,000 people, it is expected that the analyzed data will be available in the near future. Expanding the sample size is expected to contribute not only to a more comprehensive understanding of the diversity of BRCA1/BRCA2 pathogenic variants but also to cancer prevention and treatment decisions in Japan.
References
Tadaka, S. et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 6, 28 (2019).
Tadaka, S. et al. jMorp: Japanese Multi-Omics Reference Panel update report 2023. Nucleic Acids Res. 52, D622–D632 (2024).
Idogawa, M. et al. The frequency and pathogenicity of BRCA1 and BRCA2 variants in the general Japanese population. J. Hum. Genet. 69, 225–230 (2024).
Idogawa, M. et al. Renal angiomyolipoma (AML) harboring a missense mutation of TSC2 with copy-neutral loss of heterozygosity (CN-LOH). Cancer Biol. Ther. 21, 315–319 (2020).
Parsons, M. T. et al. Evidence-based recommendations for gene-specific ACMG/AMP variant classification from the ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel. Am. J. Hum. Genet. 111, 2044–2058 (2024).
Sahu, S. et al. Saturation genome editing-based clinical classification of BRCA2 variants. Nature 638, 538–545 (2025).
Huang, H. et al. Functional evaluation and clinical classification of BRCA2 variants. Nature 638, 528–537 (2025).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41439_2026_335_MOESM1_ESM.xlsx (download XLSX )
Supplementary Table 1. Pathogenic variants and truncating variants of BRCA1 and their frequencies in the general Japanese population (ToMMo: 60KJPN).
41439_2026_335_MOESM2_ESM.xlsx (download XLSX )
Supplementary Table 2. Pathogenic variants and truncating variants of BRCA2 and their frequencies in the general Japanese population (ToMMo:60KJPN).
41439_2026_335_MOESM3_ESM.xlsx (download XLSX )
Supplementary Table 3. Pathogenicity evaluation of BRCA2 DBD variants and their frequencies in the Japanese general population (ToMMo: 60KJPN).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mariya, T., Idogawa, M., Saito, T. et al. Updated analysis of pathogenic variants in BRCA1/BRCA2 among the general Japanese population. Hum Genome Var 13, 1 (2026). https://doi.org/10.1038/s41439-026-00335-5
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41439-026-00335-5
