
Abstract
Synthetic rewriting technologies, encompassing large-scale DNA assembly, transfer, maintenance, and rearrangement, enabled de novo synthesis or large-scale modifications of genomes. While significant progress has been made in model organisms of viruses, bacteria, and unicellular eukaryotes, their development in mammalian cells faces unique challenges. This review summarizes key breakthroughs in synthetic rewriting technologies, including megabase (Mb)-scale assembly of human DNA, yeast-mediated transfer methods, bottom-up human artificial chromosomes (HACs), and genome-scale rearrangement, along with emerging applications in constructing models and decoding genomes for mammals. These tools will expand functional engineering in mammals and deepen mechanistic insights into complex biological systems.
Similar content being viewed by others
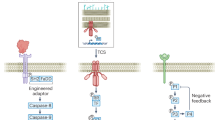

Introduction
Synthetic rewriting technologies have enabled us to perform large-scale modifications of genetic material through synthetic approaches1,2,3,4. This includes technologies for the assembly, transfer, maintenance, and rearrangement of large DNA. These advancements enable the de novo design, synthesis, and reprogramming of entire genomes. Utilizing this technological pipeline can surpass the limitations of natural genomes, allowing for the design of genetic materials independent of native genomic templates and even endowing them with entirely novel functions5.
The use of synthetic rewriting technologies, combined with diverse genomic design strategies, including codon reprogramming6,7,8,9, genome simplification10,11,12,13,14,15,16, and genome expansion17, enables the large-scale, template-free modification of viruses, bacteria, and unicellular eukaryotic cells. These rewritten genomes provide an unprecedented platform for exploring and reshaping complex phenotypes, addressing problems that are difficult to study within existing genetic frameworks. For example, they have facilitated the rapid production of viruses for vaccine development18,19,20, the creation of minimal bacterial cells to advance fundamental cell biology research11,12,21,22, the reprogramming of organisms for incorporating non-standard amino acids6,7,23, and the extensive reorganization of genomes that can be triggered on demand15. Moreover, they offer new insights into several fields, including the functional consequences of structural variants (SVs) at scale24, complex gene regulatory networks25, 3D chromosome topology-function relationships26,27, causal genotype-phenotype mapping28,29,30,31,32, exploring the plasticity of the genome33,34, protecting viral infections35,36,37, and the production of synthetic proteins35,38.
The design and synthesis of synthetic mammalian genomes is considered the next major frontier and challenge in the field of synthetic genomics1,5. This endeavor helps to elucidate the blueprint of life provided by the Human Genome Project (HGP-read) and addresses a range of human health challenges. The field has garnered support from initiatives in the United States, China, the United Kingdom, and other countries, including projects, such as Genome Project-Write (GP-write)39, the Dark Matter Project4, the National Key R&D Program of China, and the Synthetic Human Genome Project (SynHG). These projects aim to accelerate the development of synthetic rewriting technologies, advance the engineering and testing of synthetic mammalian genomes, and establish ethical frameworks. However, mammalian genomes are significantly more complex than those of unicellular organisms, such as Escherichia coli and Saccharomyces cerevisiae. This complexity arises from the vast scale of mammalian genomes and the prevalence of repetitive DNA40,41,42,43,44,45,46. Core technologies still face substantial challenges, as the assembly and transfer of large DNA fragments are often hindered by shearing forces in vitro, leading to cumbersome processes and reducing efficiency. Additionally, the functionality of vectors in mammalian cells is limited, and the difficulty in precise genome integration further presents technological obstacles.
Recent breakthroughs in DNA assembly, transfer, maintenance, and rearrangement technologies have led to significant advancements in synthetic rewriting in mammalian cells. The use of yeast life cycle-based47, haploidization-based17,48, and conjugation-based49,50 assembly techniques has enabled large-scale iterative assembly, greatly improving the efficiency of DNA assembly at the megabase (Mb) -scale and reducing the time required. These methods have successfully assembled various human sequences at the Mb-scale, demonstrating the feasibility of assembling mammalian DNA. Techniques for transferring large DNA into mammalian cells have improved the success rate of DNA transfer by minimizing the presence of ‘naked DNA’51. Additionally, the development of bottom-up approaches for constructing single-copy human artificial chromosomes (HACs)52 and the continuous optimization of integration strategies53,54,55,56,57,58,59 have provided new solutions for the maintenance of large DNA within mammalian cells. Genome rearrangement technologies that do not require whole-genome-scale synthesis offer novel approaches for constructing SV models or simplifying genomes.
This review highlights the emerging field of rewriting mammalian genomes, providing an overview of key breakthroughs in core enabling technologies related to the large DNA assembly-transfer-maintenance-rearrangement technological pipeline (Fig. 1). These advancements, from a technological breakthrough perspective, have provided unprecedented capabilities for the design and de novo synthesis of mammalian DNA. The field is rapidly transitioning from theoretical frameworks to practical engineering applications, thereby accelerating progress in disease modeling and enabling a range of novel biomedical applications. These developments represent a new generation of rewriting tools in synthetic biology, possessing transformative potential to reshape the future landscape of genomic engineering.

a Mb-scale DNA and repetitive DNA assembly techniques. Rectangles with different colors represent the assembled fragments. Pink triangles represent the repetitive DNA. b Technologies for the transfer of large DNA to mammalian cells. The left section displays the yeast-mediated transfer techniques, the right section displays the bacteria-mediated transfer techniques, and the bottom section displays the microcell-mediated transfer techniques. And the purple ring and the blue circle respectively represent the cell surface proteins and the envelope proteins. c Methods for stable maintenance of exogenous large DNA in mammalian cells. The top section displays large DNA integration into the genome. The bottom section displays human/mammalian artificial chromosomes (HACs/MACs). The circle represents exogenous large DNA. d Genome rearrangement techniques. Blue, uniform cells indicate cells without rearrangement, while colorful cells represent cells that have undergone rearrangement.
Large DNA and repetitive DNA assembly techniques
Large DNA assembly is a crucial technique for de novo genome synthesis. Compared to bacterial and yeast genomes, mammalian genomes are approximately two orders of magnitude larger and contain high-identity duplicated sequences40. In the T2T-CHM13, segmental duplications account for approximately 218 Mb, primarily located in centromeric regions and five short arms of human chromosomes41. These differences highlight the greater complexity of mammalian genomes, which significantly complicates the assembly of large mammalian DNA. This necessitates the development of assembly techniques for handling large and repetitive DNA (Fig. 2). DNA sequences smaller than 100 kb can typically be assembled in vitro using methods, such as restriction enzyme ligation60,61 and Gibson assembly62. In contrast, large DNA is susceptible to shear force in vitro, which is usually assembled in vivo through homologous recombination, with E.coli and S.cerevisiae serving as the primary hosts. Furthermore, the complex repetitive DNA poses challenges to the assembly strategies based on homologous recombination. The emergence of new technologies has provided new ideas for the construction of repetitive DNA63,64.

a Iterative assembly techniques of Mb-scale DNA in E. coli. Rectangles with different colors represent the assembled fragments. i. The schematic of the BASIS (bacterial artificial chromosome (BAC) stepwise insertion synthesis) technique. ii. The schematic of CALBIA (conjugation-associated linear-BAC iterative assembling) technique. b Iterative assembly techniques of Mb-scale DNA in S. cerevisiae. Rectangles with different colors represent the assembled fragments. i. The schematic of YLC-assembly (large DNA assembly via yeast life cycle) technique. The pink and blue circles represent yeast spores. ii. The schematic of HAnDy (haploidization-based DNA assembly and delivery in yeast) technique. c Assembly techniques for repetitive DNA. The blue arrow represents the repeat unit. i. The schematic of RCA combined with TAR (rolling circle amplification combined with transformation-associated recombination) technique. ii. The schematic of CAT (construction of a repetitive array through transformation) technique. iii. The schematic of Goldengate technique. The orange rectangles represent the restriction enzyme recognition sites. iv. The schematic of BioBrick technique. The rectangles represent the restriction enzyme recognition sites. v. The schematic of AE (amplification editing) technique. The red line represents the target sequence, and the yellow shape represents nCas9 protein and the blue shape represents the reverse transcriptase.
Assembly techniques for Mb-scale DNA
E. coli serves as an assembly host, benefiting from a shorter assembly cycle compared with S. cerevisiae. However, early DNA assembly in E. coli relied on the inefficient RecA-mediated recombination65. The advent of λ-red recombination 66and other related phage-encoded recombination systems67,68 significantly improved recombination efficiency in E. coli. Despite these advancements, the assembly of large-scale DNA remains challenging. The process of importing and exporting DNA for subsequent rounds of assembly is particularly susceptible to shear forces in vitro, which can lead to prolonged cycle times and hinder the assembly of complete constructs. To address the challenges associated with assembling DNA at the Mb-scale, several systems have been developed to overcome these obstacles. Replicon excision enhanced recombination (REXER)9,69 utilizes the CRISPR/Cas9 system to cleave episomal plasmids in vivo, generating target double-stranded DNA (dsDNA) to facilitate recombination mediated by the λ-Red system. This approach circumvents the challenges associated with delivering long dsDNA fragments into cells. The iterative extension of REXER, known as genome stepwise interchange synthesis (GENESIS)9,69, enables the stepwise replacement of genomic DNA with synthetic DNA. Through the use of REXER and GENESIS technologies, a high-fidelity replacement of approximately 0.5 Mb of synthetic DNA in the E. coli genome can be achieved6. Subsequent conjugative-based assembly enabled the synthesis of a 4 Mb synthetic genome. Based on REXER, conjugation coupled with programmed excision for enhanced recombination (CONEXER)49 combines universal spacers and conjugation-based DNA input, simplifying the workflow by circumventing the issues associated with variable CRISPR/Cas9 cleavage efficiencies at different loci and the cumbersome electroporation process. This approach can be extended to the bacterial artificial chromosome (BAC) stepwise insertion synthesis (BASIS) method49, which utilizes conjugation-based transfer for seamless, iterative assembly of Mb-scale DNA using BACs (Fig. 2a. i). With this method, a 1.1 Mb region of the human genome within chromosome 21 can be successfully assembled. The ∆recA E. coli was used for multiple rounds of CONEXER, establishing a rapid and continuous genome synthesis (CGS) method. This approach enabled the replacement of 0.5 Mb of the genome in just 10 days, without the need for sequencing at each step, thereby saving both time and cost. In addition, another conjugation-based assembly strategy, termed conjugation-associated linear-BAC iterative assembling (CALBIA), was developed (Fig. 2a. ii). This method innovatively employed the prokaryotic telomere system TelN-tos to generate linear BACs. Through a single crossover of homologous recombination, thereby facilitating the efficient assembly of large DNA. This approach enabled the assembly and stable maintenance of a 2.1 Mb human DNA, demonstrating sustained genetic stability within the host for up to three days50. To enhance the stability of assembled extrachromosomal large DNA in E. coli, functional replication modules were designed and incorporated into Mb-scale plasmids70.
Due to its efficient homologous recombination capability, S.cerevisiae has become a commonly used platform for large DNA assembly. Previous strategies were mainly based on stepwise assembly, where the initial fragments were assembled into intermediates, linearized in vitro, and then gradually transferred into S. cerevisiae to achieve the assembly of Mb-scale DNA. The assembly of a 1.08 Mb Mycoplasma mycoides genome from eleven 100 kb sequences was completed using yeast spheroplast transformation-associated recombination (TAR)21. In the Synthetic Yeast Genome Project (Sc2.0), the switching auxotrophies progressively for integration (SwAP-in) method was mainly employed to assemble synthetic chromosome fragments to replace wild-type chromosome fragments15,71,72,73,74. The procedure of these methods is straightforward, while the need for multiple time-consuming in vitro steps during large DNA assembly reduces the overall efficiency of obtaining the desired large DNA. Parallel assembly strategies combined with multiple iterative methods have significantly reduced the time required for Mb-scale DNA assembly. The meiotic recombination-mediated (MRA) parallel assembly approach exploits the potential for crossover interchanges of sister chromatids during meiosis in diploids to address the time-consuming issue of stepwise replacement of large DNA, facilitating the assembly of the final stages of the synthetic yeast chromosomes synXII (0.98 Mb) 75and synIV (1.45 Mb)26. Cas9-facilitated homologous recombination assembly (CasHRA)76, assembly via yeast life cycle (YLC-assembly)47, and haploidization-based DNA assembly and delivery in yeast (HAnDy)17,48 utilize the cleavage capability of CRISPR/Cas9 in assembly technology to release linear DNA and improve Mb-level DNA assembly efficiency. Through CasHRA, the assembled large plasmids were transferred into S. cerevisiae via yeast protoplast fusion, linearized, and greatly improved the assembly efficiency76. The YLC-assembly47 integrates the assembly process into the yeast life cycle, reducing damage to large DNA by eliminating complex and time-consuming in vitro manipulations during large DNA assembly (Fig. 2b. i). For the assembly of DNA to Mb-size, both the colony counts and assembly accuracy remained consistent, suggesting that YLC-assembly could be a method independent of size. Using this method, a 1.26 Mb human IGH region was successfully assembled. HAnDy17,48 enables yeast to bypass the meiotic process for sustained mating using CRISPR/Cas9-mediated genome elimination (Fig. 2b. ii). This facilitates haploidization and significantly reduces the experimental period of the sporulation process. This method has no need for any cumbersome operations in vitro. For each round of assembly, more than 10³ colonies can be easily obtained in the selective medium, while maintaining approximately 60% assembly accuracy. They efficiently assembled a 1.024 Mb synthetic accessory chromosome starting from commercially synthesized fragments of around 6 kb. Notably, the entire assembly process was carried out in yeast, thereby demonstrating the potential of yeast as an efficient platform for assembling Mb-scale DNA (Table 1).
Assembly techniques for repetitive DNA
Repetitive DNA plays an important role in maintaining chromosomal spatial structure and the development of diseases77,78. For example, the centromere and telomere regions of the human genome are composed of various types of repetitive DNA, and the genome is also interspersed with a large number of transposable elements79,80,81. The α-satellite sequence in the centromere region provides binding sites for centromeric proteins (such as CENP-A), which are involved in chromosome segregation and maintaining genomic stability82. The telomere regions contain tandem TTAGGG repeats, which protect the chromosome ends and insulate them from chromosome shortening83. Transposable elements are closely associated with genetic diversity and genomic rearrangement, and play key regulatory roles84. Constructing large synthetic repetitive DNA will contribute to understanding their functions and uncovering the mechanisms. Several methods have been reported for assembling repetitive DNA. Based on the properties of the obtainable products, the methods can be classified into random-size assembly methods and precise-size assembly methods (Table 1 and Fig. 2c).
Rolling circle amplification (RCA) combined with TAR allows for the rapid amplification of a few hundred base pairs repeat units into large DNA arrays up to 140 kb85 (Fig. 2c. i). Introducing specific seed sequences (short repetitive unit sequences) carrying selection markers and using selection pressure can generate random copies of the seed sequences. In the construction of the synthetic yeast chromosome synXII, rDNA seed sequence was inserted at the designated locus, and the formation of new rDNA clusters (100–200 copies) was achieved through successive amplification under increasing concentrations of hygromycin B75,86. The method was named construction of a repetitive array through transformation (CAT) in subsequent studies86 (Fig. 2c. ii). While these methods enable the convenient acquisition of large repetitive DNA, the precise length of the obtained sequences remains uncertain and typically requires further techniques, such as pulsed-field gel electrophoresis and sequencing for confirmation.
For cases where precise repetitive DNA are needed, methods, such as Goldengate87,88, BioBrick89,90,91, and amplification editing (AE)63 can be employed. Both Goldengate and BioBrick rely on restriction endonucleases and ligase to achieve the assembly of repetitive DNA, thereby avoiding the instability of repetitive DNA often caused by homologous recombination. Through the Goldengate method, an sgRNA array was successfully constructed, incorporating 43 copies of repetitive gRNA scaffold and the hU6 promoter, thereby addressing the scarcity of standard biological parts87 (Fig. 2c. iii). By employing multiple rounds of BioBrick ligation, the 2.7 kb natural repeat sequences were successfully arrayed to reach a total length of 86 kb90 (Fig. 2c. iv). It is worth noting that a recently developed AE technique can precisely generate in situ 1Mb-100Mb duplications in mammalian genomes. This technique uses Cas9 nickases (nCas9) to cut different strands, creating flaps. By strategically altering the orientation of the nCas9 recognition site from a PAM-in orientation to a PAM-out orientation, the flaps can then anneal between each other to produce in situ duplications63 (Fig. 2c. v). This provides a clever approach for manipulating repeat sequences in mammalian cells.
Currently, both E. coli and S. cerevisiae have become highly efficient assembly platforms, which enabled the assembly of Mb-scale human genomic regions47,49,50. Regarding repetitive DNA assembly, assembly techniques have progressed from relying on random methods to utilizing precise assembly strategies. AE represents a significant breakthrough, enabling Mb-scale in situ duplication in the mammalian genome. However, the customized assembly of large-scale repetitive DNA remains a challenge. The optimized repetitive DNA assembly technology in the future should focus on the customized and reprogrammed assembly of different types and lengths of repetitive DNA. Large DNA constructs assembled in E. coli or S. cerevisiae can be delivered into mammalian cells using either direct cell-to-cell transfer or in vitro transfer. The details of the transfer technologies will be elaborated in the following section. Furthermore, assembly techniques developed in S. cerevisiae provide a reference for rewriting the mammalian genome15,71,72,73,74, as exemplified by the mSwAP-In method that directly enables genome modifications in mammalian cells59.
Technologies for the transfer of large DNA to mammalian cells
Efficient transfer methods for large DNA are essential prerequisites for its functionality in target cells. Conventional DNA transfer methods primarily include lipid transfection92,93, electroporation (Fig. 3a. i)94, microinjection95, and viral vectors96,97,98,99 (Table 2). However, delivering large DNA to mammalian cells remains a significant challenge. Compared to the delivery of small-sized DNA, large DNA purified in vitro is more susceptible to fragmentation caused by shear forces, and in vivo transfer methods are often limited by efficiency. Recent advancements have provided desirable solutions to address these issues (Fig. 3).

a Physical and chemical techniques. The red curve represents transferred DNA. i. The schematic of electroporation. ii. The schematic of lipid transfection. iii. The schematic of microinjection. b Biological techniques. i. The schematic of microcell-mediated chromosome transfer. The purple ring and the blue circle respectively represent the cell surface proteins and the envelope proteins. ii. The schematic of yeast spheroplast-mammalian cell fusion. iii. The schematic of bacteria-mediated transfer methods. iv. The schematic of viral vectors.
Physical and chemical techniques
Employing some materials for compacting large DNA, as well as cleverly utilizing the natural protection of the cell nucleus, can effectively shield large DNA from shear stress during isolation and purification. When combined with physical and chemical transfer methods, these approaches can significantly improve the integrity of large DNA during transfer.
Polycations are commonly used materials for compacting large DNA, which are positively charged materials that tightly bind to the negatively charged DNA (Fig. 3a. ii). They help to highly condense the DNA, preventing large DNA fragments from mechanical forces during in vitro isolation100. The Globin-HAC was isolated using polyamine buffer and 25% sucrose cushion centrifugation and transferred from K562 cells to target mammalian recipient cells via lipid transfection101. Besides, 1.7 and 2.3 Mb yeast artificial chromosomes (YACs) were purified through high-quality pulsed field gel and compacted against degradation by poly-L-lysine (PLL) or polyethyleneimine (PEI), successfully delivered into HT1080 cells via lipid transfection100.
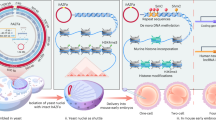
Nucleus extraction is a clever strategy to protect large DNA, not only to avoid breakage caused by the direct extraction of large DNA, but also to remove excess cellular components and material. By employing the nucleus isolation for chromosomes extraction (NICE) method, which isolates yeast nuclei and preserves chromosome structure, in conjunction with nuclear microinjection (Fig. 3a. iii), a massive 1.14 Mb synthetic human DNA was successfully delivered into mouse metaphase II (MII) oocytes51.
Biological techniques
Natural membrane fusion, spheroplast fusion, conjugation, and viral infection phenomena provide new insights for developing mammalian cell delivery methods. These methods primarily include microcell-mediated chromosome transfer (MMCT), yeast spheroplast-mammalian cell fusion, bacterial-to-cell transfer techniques, and viral vectors.
MMCT enables the transfer of chromosome-scale DNA between mammalian cells (Fig. 3b. i). The core steps of MMCT include: (1) Induce micronucleation: Donor cells are treated with mitotic spindle inhibitors, such as colcemid to induce micronuclei formation. (2) Enucleation of micronucleate cells: Cytochalasin B can promote micronuclei extrusion and centrifugation to collect microcells. (3) Fusion and selection: Microcells are fused with recipient cells, and clones carrying transferred chromosomes are selected via selection102. MMCT efficiency depends on the percentage of micronucleate cells and the number of micronuclei per cell during mitotic arrest (step 1), the degree of actin cytoskeleton disarrangement during micronuclei extrusion (step 2), and the fusion efficiency between microcell and recipient cell membranes (step 3). Traditional MMCT exhibits low efficiency, typically yielding approximately one clone per 10⁵–10⁶ recipient cells. This procedure conventionally employs CHO and A9 cells as standard donor cells, owing to their high efficiency in generating micronuclei. Recent advancements have targeted the optimization of each core step. For Steps 1-2, replacement of colcemid with TN-16+Griseofulvin, substitution of Cytochalasin B with Latrunculin B, and use of Collagen/Laminin surface coating collectively increased MMCT efficiency ~6 times103. For Step 3, employing the hemagglutinating virus of Japan envelope (HVJ-E) system instead of polyethylene glycol (PEG) improved artificial chromosome transfer efficiency from CHO cells to HT1080 and hiMSC by ~3 and 8 times104. Using measles virus (MV) fusogenic envelope proteins (H/F), which specifically bind to human CD46 or SLAM cell surface protein and trigger membrane fusion, enhanced chromosome transfer efficiency by 50 and 100 times in HT1080 and hiMSC cells105. Engineering the MV-H protein fused with single-chain antibodies further expanded the range of targeted human cells, which barely express native MV receptors106. Modified murine leukemia viruses (MLVs) envelope proteins enabled cross-species chromosome transfer (human, monkey, mouse, rat, and rabbit)107. Cryopreservation of microcells at −80 °C has also enhanced experimental flexibility108.
Preparing yeast spheroplasts via enzymatic degradation of the cell wall followed by fusion with mammalian cells represents a promising approach for transferring large DNA from yeast to mammalian systems (Fig. 3b. ii). Current methods have demonstrated the capacity to deliver Mb-scale DNA into diverse cell lines109, including goat fetal fibroblasts110, LA-9 mouse cells111, RCC-1 cells112, mouse embryonic stem (ES) cells113, and mouse L cells114. Notably, this approach exhibits significantly higher fidelity in delivering intact DNA compared to conventional methods, such as lipid transfection, albeit with efficiencies remaining modest (10−5–10−6). Recent optimizations involving mitotic arrest and refined reaction conditions have elevated delivery efficiency to 1/840 in HEK293 cells while expanding applicability to other cells115. Combining the mature DNA assembly techniques from yeast, this method may provide a simplified workflow for synthetic rewriting in mammalian cells, from DNA assembly to transfer. Transgenic mice harboring human TCRαβ loci (1.1 Mb and 0.7 Mb) have been successfully generated through yeast spheroplast-mammalian cell fusion113,116. However, current optimization strategies predominantly focus on PEG-mediated fusion protocols. Future developments could employ other membrane fusion mechanisms to improve throughput and broaden the range of ideal cell types.
Conjugation facilitates the transfer of DNA across bacteria and promotes the transfer of plasmids to be assembled within the bacterial host49,50,117. Similar processes can also occur between distantly related species118, primarily via broad-host-range and F-factor plasmids. Using the RK2 broad-host range conjugative plasmid system with modified E. coli as donor cells enables the transfer of DNA to mammalian cells119. Agrobacterium tumefaciens facilitates T-DNA transfer from Ti-plasmids to human cells120 via VIRD2 endonuclease-mediated single-strand DNA excision and genomic integration. Highly attenuated pathogenic strains have also been utilized as delivery vehicles for transferring DNA into mammalian cells121,122,123,124, which involves bacterial invasion of host cells, followed by their lysis and subsequent plasmid release. Invasive genes can be introduced into other bacteria for DNA delivery. Modified E.coli using the invasion gene of Yersinia pseudotuberculosis allows it to invade HeLa cells and deliver DNA, which can transfer a ~ 200 kb BAC to HeLa cells with an efficiency of 2.8%125. A similar technique can also enhance DNA transfer to COS-1 and CHO cells126. This approach will greatly improve the utility of existing BAC libraries from humans and other mammalian genomes in functional research (Fig. 3b. iii).
By utilizing the ability of viruses to infect cells, viral vectors can also be used for DNA transfer. The commonly used DNA viral vectors are lentiviral, adenoviral, and adeno-associated (AAV) viral vectors96. However, widely used high-efficiency viral vectors have a small carrying capacity, typically within 10 kb127. There are also viral vectors capable of carrying larger DNA, such as the Epstein-Barr virus (EBV) amplicon vector and herpes simplex virus type 1 (HSV-1) amplicon vector97,98,99,128, which can accommodate foreign DNA larger than 100 kb into mammalian cells (Fig. 3b. iv). EBV amplicon delivered a 123 kb insert (total size 152 kb) to the Loukes B-cell line, and achieved an efficiency 2000 times higher than conventional transfection in B cells99. HSV-1 amplicon delivered 100 kb of 17α DNA into HUES-2 and HT1080 cells with efficiencies of 16% and 25%, respectively128.
Currently, technologies at the forefront of the field have enabled the delivery of Mb-scale DNA into mammalian cells. These approaches encompass a variety of strategies, ranging from optimized MMCT103,104,105,106,107,108 and yeast spheroplast-mammalian cell fusion115 to novel strategies that ingeniously exploit the native protective mechanisms of the nucleus51. However, the current low transfer efficiency and significant manipulation difficulty remain the key limitations for the pipeline for synthetic rewriting in mammalian cells. Future technical optimization will focus on further protecting DNA integrity and improving the transfer efficiency into diverse cell types.
Methods for maintenance of exogenous large DNA in mammalian cells
Key strategies for the maintenance of large-scale DNA in mammalian cells are integration methods and HACs/ mammalian artificial chromosomes (MACs). Recent studies have made significant advancements in large DNA integration technologies, particularly in terms of scale, precision, and iterability. The successful establishment of single-copy HACs has also provided new possibilities for the application of artificial chromosomes (Fig. 4).

a Large DNA integration into the genome. i. Site-specific recombination-mediated precise integration. The schematic of the Big-in technique. The triangles represent lox sites, the pink rectangle represents the Cre recombinase, and the blue rectangle represents the payload DNA. ii. Homologous recombination-mediated precise iterative integration. The schematic of the mSwAP-in technique. The pink rectangle represents payload A, the blue rectangle represents payload B, and the deep blue rectangle and the orange rectangle represent selection markers. iii. PE-mediated precise integration. A schematic of PASTE technique. The blue line represents Bxb1 attB site, and the pink line represents integrated gene. iv. Transposable elements-mediated precise integration. The left section displays the CAST technique, where the yellow line represents the target site and the blue line represents the edit sequence. The middle section displays the STITCHR technique, where the pink line represents the target site and the blue line represents the edit sequence. The right section displays the PRINT technique, where the red line and the yellow line represent the target sites, and the blue line represents the edit sequence. b Large DNA loading into HACs/MACs. i. The schematic of large DNA loading into alphoidtetO HAC. ii. The schematic of large DNA loading into 4q21LacO HAC. The blue circle represents the M. mycoides genomic DNA.
Techniques for the integration of large DNA into the genome
After the transfer of large DNA into mammalian cells, it can be integrated into the genome to persistently maintain through (1) random integration, (2) site-specific recombinase-mediated precise integration, (3) homologous recombination-mediated precise integration, (4) prime editing (PE)-mediated precise integration, or (5) transposable elements-mediated precise integration.
Large DNA can be stably integrated into cells through random integration113,116,129, but the insertion site, copy number, and orientation are uncertain. The site-specific recombination system-mediated integration of large DNA130,131,132,133 can achieve hundreds of kilobases at a time without relying on homologous recombination. For example, using sequential recombinase-mediated cassette exchange (S-RMCE) in ES cells, the entire human immunoglobulin variable-gene repertoire (about 2.7 Mb) was precisely inserted into the mouse genome94. A systematic pipeline for the precise integration of large DNA, called Big-in (Fig. 4a. i), was established through the introduction of a landing pad (LP) that permits payload integration via recombinase-mediated cassette exchange (RMCE). This platform incorporates efficient positive/negative selection, with integration events validated by hybridization capture sequencing (Capture-seq) and bamintersect134.
Homologous recombination-mediated methods can be used for large DNA replacement in mammalian cells. A strategy utilizing customized, large-scale BAC with homology arms achieved the in situ replacement of 6 Mb of mouse immunoglobulin genes with the corresponding human genes135. This technology also enabled the replacement of the murine TCRαβ variable regions, along with the ectodomains of MHC-I and MHC-II and co-receptors CD4 and CD8α/CD8β, with corresponding human sequences136. Researchers have developed a large-scale, efficient, scarless, iterative, and biallelic integration approach in mouse ES cells, called mammalian switching antibiotic resistance markers progressively for integration (mSwAP-In)59. This method is derived from the yeast genome rewriting method (SwAP-In)15,137 and utilizes CRISPR/Cas9-assisted homologous recombination combined with efficient positive and negative selection strategies (Fig. 4a. ii). Using this method, they achieved iterative genome rewriting of up to 115 kb of a customized Trp53 locus, and humanization of mice by 116 and 180 kb human ACE2 loci. This approach theoretically enables the infinite iterative integration of large DNA in mammalian cells, which have a similar homologous recombination ability to mouse ES cells.
With the continuous advancement of gene editing technologies, significant progress has been made in improving the scale and efficiency of gene integration. Programmable PE can be combined with site-specific integrases to achieve gene integration at precisely placed LPs without inducing DNA double-strand breaks (DSBs)53,54,56. The developed technique, called programmable addition via site-specific targeting elements (PASTE), enables the more efficient and precise insertion of DNA fragments (up to 36 kb) into mammalian and human cells (Fig. 4a. iii)53. Compared to PASTE, PrimeRoot utilizes many optimized components to enhance integration efficiency, offering an alternative strategy for optimizing large-scale DNA integration in mammalian cells138.
The large DNA targeted integration technology based on transposable elements achieves precise genomic insertion that is DSB-free, fundamentally avoiding the risk of random mutagenesis and cellular toxicity associated with the CRISPR/Cas9 system (Fig. 4a. iv). The CRISPR-associated transposases (CAST) utilize Tn7-like transposons for precise large DNA integration. Researchers applied phage-assisted continuous evolution (PACE) to perform hundreds of rounds of mutagenesis on the transposase module of type I-F CAST, resulting in the development of an optimized version, evoCAST, which achieved targeted integration efficiencies of 10%-30% for exogenous genes in human HEK293T cells58. Another system relies on R2 retrotransposons for precise large DNA integration. By harnessing the targeting flexibility of the CRISPR/nCas9 system in combination with the large DNA integration capacity of R2 retrotransposons, site-specific target-primed insertion through targeted CRISPR homing of retroelements (STITCHER) enables scarless integration of DNA up to 12.7 kb57. The precise RNA-mediated insertion of transgenes (PRINT) method marks a breakthrough by utilizing the eukaryotic R2 retrotransposon protein to achieve gene integration. By delivering only two in vitro transcribed RNAs (the message RNA encoding the R2 protein and the template RNA carrying the transgene), the technology achieved stable insertion up to 4 kb139. Further advancements were made through systematic analysis and rational engineering design, which significantly optimized the integration efficiency of the R2 retrotransposon system in mammalian cells. For example, the engineered system successfully achieved the gene integration efficiency exceeding 60% in mouse embryos and 25% in human liver cells. The engineered R2 system (en-R2Tg) demonstrated remarkable on-target integration specificity, reaching up to 99% at the intended locus. This near-elimination of random insertion events underscores its high safety profile for potential gene therapy applications140.
Design and construction of HACs/MACs
HACs/MACs offer key advantages, such as avoiding host genome perturbations and ensuring high mitotic stability. The construction of HACs/MACs is mainly divided into two approaches: top-down and bottom-up. Top-down HACs/MACs are typically formed by directional chromosome truncation mediated by artificial telomeres or the Cre-loxP system. By reducing the natural chromosomes to HACs/MACs that contain functional elements, and then adding payloads into the HACs/MACs. Artificial chromosomes for the human chromosomes X, Y, 14, and 21 have already been constructed using this method, with scales in the Mb-range141,142,143,144. Bottom-up HACs/MACs are typically achieved by introducing 30-200 kb of natural or assembled α-satellite sequences into mammalian cells89,90,91,145,146,147,148. Recently, the bottom-up HACs have made significant progress, providing new strategies for the functional applications of HAC.
A type of strategy uses a synthetic alphoid DNA array carrying multiple tet operator (tetO) sequences that enable the attachment of functional proteins to the array as tet repressor (tetR) fusions. By linking natural α-satellite monomers with synthetic monomers that replace the CENP-B binding motif with a 42 bp tetO sequence, a 50 kb alphoid tetO dimeric repeat sequence can be constructed, enabling de novo HAC formation. This designed HAC can be controlled by a tTA transcriptional activator or tTS transcriptional silencer fused to TetR, thereby allowing for the inactivation of centromere activity when not required149. Using a similar method, the histone acetyltransferase (HAT) domains were attached to the array, breaking the HAC barrier in HeLa cells150.
Furthermore, structural analysis151 and further functional exploration of the formed alphoid tetO HAC have also been conducted. These structurally confirmed HACs/MACs were transferred into homologous recombination-efficient DT40 cells and CHO cells that are amenable to MMCT operations for editing and then transferred into target cells (Fig. 4b. i). Through multiple rounds of MMCT and gene editing, the structure of the HAC remains largely unchanged. The retrofitting of a loxP cassette into DT40 cells by homologous recombination for alphoid tetO HAC gene loading, followed by Cre-loxP recombination in CHO cells to insert EGFP, allows stable expression for at least 12 weeks152. Using similar methods, the 90 kb BRCA1 gene153, ~55 kb NBS1, and ~25 kb VHL genes154 were loaded into HACs/MACs in CHO cells through Cre-loxP recombination and delivered into target cells via MMCT to correct genetic defects and perform a series of functional tests. Chromatin insulators can be used for protection to prevent gene expression from being affected by heterochromatin domains and to avoid potential interference with the function of the centromere155. Recently, the maintenance of alphoid tetO HAC in mouse ES cells and their differentiated progeny in mice, as well as germline transmission in mice, have been evaluated156,157. They found that alphoid tetO HAC is well-tolerated in mouse ES cells, preserving pluripotency in vitro and their differentiated derivatives, and exhibits robust maintenance and expression during mouse ontogeny. Alphoid tetO HAC could be maintained as extrachromosomal elements without selection and could be transmitted through both ova and sperm. These have done preliminary characterization work for subsequent applications in these systems.
Another strategy tries to induce centromere formation by inducing CENP-A nucleosome assembly through targeted recruitment. Seeding CENP-A nucleosome assembly by a pulse expression of targeted LacI-HJURP can induce centromere formation on chr7α-sat BAC LacO and chr11α-sat BAC LacO, which come from CENP-A-poor regions and are deemed incapable of forming de novo HACs. And what’s even more exciting is that 4q21 BAC LacO can form HACs that bypass centromeric DNA, and targeted LacI-HJURP could induce HAC formation without the need for genomic sequences158. Using the 4q21 BAC LacO to load 550 kb M.mycoides DNA in S. cerevisiae has efficiently formed a single-copy HAC in human cells52, offering a potential approach for functional sequence loading (Fig. 4b. ii).
In mammalian cells, large DNA can be maintained via two primary strategies, including integration into the genome and loading into HACs/MACs. Integration strategies have evolved from random integration to precise, targeted integration. Key technologies include the mSwAP-In iterative rewriting technique59 and large DNA integration mediated by DSB-free gene editing tools53,54,56,57,58,139,140. Regarding HACs/MACs construction, de novo centromere formation independent of canonical repetitive DNA can be induced by seeding CENP-A nucleosome assembly158. Future research will focus on increasing the payload size and achieving efficient, multiplexed-target, and precise genomic integration. Moreover, a key objective is to design and construct convenient and efficient HACs/MACs (analogous to BACs and YACs) that can ensure the stable expression of large-scale DNA in mammalian cells. These tools should feature compact structures and high sequence stability, thereby further enriching the synthetic rewriting toolbox for mammalian cells.
Genome rearrangement techniques in mammalian cells
Genome rearrangement is closely associated with driving biological evolution and plays a pivotal role in the development of various complex diseases159,160. The Sc2.0 project enables large-scale rearrangements by incorporating recombination sites during the construction of synthetic chromosomes15,137. However, this process remains highly challenging in mammals. Recent advancements have provided examples of systematically introducing recombination sites into the genome or directly inducing seamless rearrangements (Fig. 5).

a Site-specific recombination systems-based genome rearrangement techniques. i. PE-mediated insertion of recombination sites. ii. PiggyBac transposon-mediated insertion of recombination cassette library. The red rectangle represents repeat sequences, and the diamond represents recombination sites. The rectangles of different colors represent the barcodes. b RNA-guided editing systems-based genome rearrangement techniques. i. CRISPR/Cas9 systems-mediated rearrangements. The top section displays that sgRNAs target specific genomic sequences for precise rearrangements. The gray ovals represent telomeres and black ovals represent centromeres. The bottom section displays that sgRNAs target genomic repeat sequences for random rearrangements. The red rectangle represents repeat sequences. ii. Programmable bridge recombinases-mediated rearrangements. The top section displays bridge recombinases-mediated large DNA inversion, and the bottom section displays bridge recombinases-mediated large DNA excision.
Site-specific recombination systems-based genome rearrangement techniques
The site-specific recombination system, such as the Cre-loxP system, can be used to induce recombination between two specific DNA sites, resulting in deletions, inversions, or other structural variations161. The symmetric loxP (loxPsym) site lacks the directional characteristic of the classical loxP site, and the DNA between two loxPsym sites undergoes inversion and deletion with equal probability. This method has been widely used in the Sc2.0 project to achieve a highly adaptable and multifunctional synthetic yeast genome29,162,163,164,165,166,167,168,169,170,171. Due to the large size of mammalian cell genomes compared to S. cerevisiae, it is currently challenging to introduce site-specific recombination sites at the whole-genome level, as done in yeast through de novo synthesis. Recent research has primarily focused on systematically inserting recombinase recognition sites into the genome through engineered approaches to rearrange the human genome using recombinase systems.
PE was applied to insert loxPsym sites into the targeted enhancer region172. Through Cre recombinase-induced rearrangements, this method was used on the distal super-enhancer of the OTX2 gene to generate a diverse cell pool with a randomized regulatory landscape. Using PE targeting of high-copy-number long interspersed nuclear element-1 (LINE-1) retrotransposons, thousands of recombinase recognition sites were inserted into the human genome (Fig. 5a. i)173. Recombinase-mediated recombination events subsequently generated variants covering three times the human genome in a single experiment. These events included a variety of rearrangements, such as deletions, inversions, extrachromosomal DNAs, fold-backs, and translocations. The resulting cell pool contained cells with an average of over a hundred SVs. Clones with multiple large-scale rearrangements were further characterized, revealing that when variant copy numbers were altered, they significantly impacted gene expression, although they did not affect the expression of adjacent genes. To further improve the efficiency and scale of DNA rearrangements, Sun et al. developed a programmable chromosome engineering (PCE) system, which enabled inversions of up to 12 Mb in mammalian cells174.
Another study developed a method called Genome-Shuffle-seq, which utilizes transposons to integrate loxPsym sites and distinct barcodes into various genomic locations (Fig. 5a. ii). Changes in barcode pairing during the recombination process represent the occurrence of different SVs. By coupling T7 in situ transcription with single-cell RNA-seq, precise localization of SVs and their impact on gene expression can be achieved without the need for whole-genome sequencing. In each experiment with mouse ES cells and human cancer cells, thousands of SVs were generated and localized. The functional effects of large-scale SVs on cellular adaptability, gene expression, and other aspects were further analyzed175. These two technologies provide potential technical approaches for in-depth exploration of the relationship between genotype and phenotype, as well as for the construction of the minimal mammalian genome.
In addition to the Cre-lox system, researchers have discovered a multiplexed site-specific inversion system that can be used in mammalian cells176. When multiple sites coexist, it preferentially promotes inversions between reverse sites rather than deletions between same-oriented sites, allowing for complex DNA rearrangements through random inversions between multiple sites and providing a diverse tool for rearrangements in mammalian cells.
RNA-guided editing systems-based genome rearrangement techniques
RNA-guided editing systems enable genome rearrangements by binding to the target sites on the genome through RNA navigation and functional enzymes to act at the targeted sites. These include CRISPR/Cas9-mediated rearrangements (Fig. 5b. i) as well as bridge recombinases-mediated rearrangements (Fig. 5b. ii).
CRISPR/Cas9-mediated rearrangements primarily occur under the guidance of sgRNA, where the Cas9 nuclease specifically cleaves the DNA double-strand and then religates the DNA through non-homologous end joining (NHEJ) or homologous-directed repair (HDR) pathways, stimulating rearrangements. By designing precise targeting sites, large fragment deletions, inversions, translocations, and copy number variations can be achieved in mammalian cells177,178,179,180. This method can also induce genome-wide rearrangements by targeting repetitive DNA in the mammalian genome, but this method is typically challenging because the extensive DSBs lead to the death of most cells181.
The emergence of bridge recombinase offers a new perspective for precise genome-wide rearrangement182. Bridge RNA (bRNA), binding with IS622 recombinase, specifically recognizes the donor and recipient DNA, ultimately enabling precise rearrangements, including large DNA integration, inversion, and excision. Using this technology, 0.92 Mb inversion and 0.13 Mb deletion were achieved in human cells183 (Table 3).
Currently, both site-specific recombination systems and RNA-guided editing systems have enabled customized Mb-scale inversion and whole-genome random rearrangements in mammalian cells173,174,175,181,183. Moreover, integration tools, such as PE and transposons have facilitated the pre-installation of recombination sites173,175. These technological advancements enable the generation of thousands of SVs at the whole-genome level, allowing for rapid functional analysis. Separately, CRISPR/Cas9 stimulates rearrangement by inducing DSBs. However, its primary limitation in large-scale rearrangement is the high cellular toxicity associated with extensive DSBs181. A transformative breakthrough is the DSB-free RNA-guided bridge recombinase system, which can perform precise, Mb-scale DNA rearrangements183. Nevertheless, large-scale rearrangement remains constrained by operationally complex and low efficiency. Future efforts in genome rearrangement technology should focus on expanding the types and enhancing the efficiency of rearrangements via DSB-free RNA-guided enzyme systems. The ultimate goal is to create a scalable platform to enable in-depth exploration of genotype-phenotype relationships and investigation of the genomic regulatory landscape.
Applications of synthetic rewriting techniques to mammalian systems
Synthetic rewriting technologies, such as assembly, transfer, maintenance, and rearrangement, facilitate the introduction of diverse modifications into mammalian genomes and enable the customization of genetic material. These advancements have significantly expanded both the scale and diversity of the customizable genome range. Such progress allows for the construction of better cell models to understand diseases and test new therapies, while also offering insights into the long-standing complexities of genome regulation.
Constructing mammalian cell models and animal models
Synthetic rewriting technologies can support the construction of models that are difficult to achieve through traditional methods, including simulating cell models caused by abnormal gene amplification and complex loss or mutations in genes, as well as developing humanized mouse models and aneuploid mouse models.
The in situ abnormal amplification of genes, as well as the extensive deletion or mutation of gene sequences, are difficult to simulate or treat using traditional methods. The AE technology facilitates the study of the functions and underlying mechanisms of genomic SVs by simulating gene amplification and generating repetitive DNA at different scales. The HbH model was established in the erythroid-related K562 cell line for the study of α-thalassemia, and the chromosome microduplication genetic model was established in mouse and human ES cells to explore the functional impact of SVs on chromosome scales63. Duchenne muscular dystrophy (DMD) is caused by multiple deletions or mutations in the dystrophin gene. Using a HAC with the 2.4 Mb complete human genome sequence of the dystrophin gene, the genetic defects in iPS cells of human DMD patients were successfully corrected184. Missense p53 mutations are commonly observed in cancer, owing to frequent deamination of 5-methylcytosine, resulting in C to T conversion. Using the mSwAP-in technology, leading to the generation of the 115 kb synTrp53 in mouse ES cells, which demonstrates enhanced resistance to spontaneous mutagenesis59.
Mice are commonly employed as preclinical models. However, due to evolutionary divergence, human disease physiology or immune responses are not fully recapitulated. Humanized mouse models constructed via mSwAP-in technology enable better modeling of human diseases. Using the mSwAP-in technology, humanized mice can be efficiently constructed by replacing the mouse Ace2 gene with human ACE2 loci of 116 kb and 180 kb, creating a more human-like infection model59. The introduction of Mb-scale human immunoglobulin genes or human TCR repertoire sequences into mice establishes models for studying human immune responses and developing monoclonal antibodies. Based on the assembly-yeast spheroplast-ES cell fusion-integration pathway, mice with human TCRαβ gene loci113,116 have been successfully established. Mice with human immunoglobulin genes135,185 were established through precise customized BACs and in situ replacements. These mice provide a platform for the discovery of therapeutic antibodies and the identification of pathogenic and therapeutic human TCRs. Constructing aneuploid mouse models is crucial for understanding chromosomal disorders and cancer186. To date, trisomic mouse models, such as Tc1 and TcMAC21 have been established using technologies like MMCT and MAC, successfully recapitulating many phenotypes of Down syndrome (DS) with broad potential to support basic and clinical research187,188. Furthermore, the induction of chromosome loss by simultaneously generating multiple DSBs at target chromosomal locations using CRISPR/Cas9 has successfully deleted sex chromosomes or autosomes in various cell lines, mouse embryos, and in vivo tissues189,190. Prospectively, leveraging insights from chromosomal rearrangement in S. cerevisiae29,162,163,164,165,166,167,168,169,170, synthetic rewriting technologies could be used to introduce programmable recombination sites into aneuploid cells via strategies, such as de novo design and assembly or multiplex editing. These approaches would facilitate the establishment of a flexible and reconfigurable mouse trisomy model. Such systems enable the systematic induction of chromosomal rearrangements, thereby elucidating the key genomic regions and intergenic interactions underlying aneuploidy-associated pathologies.
Revealing the complex regulatory mechanisms of the genome
Synthetic rewriting technologies can be used to study the complex regulatory mechanisms of the genome, including the basic principles of epigenetic establishment, the complex regulatory roles of non-coding elements, and the complex biological effects caused by chromosome fusion.
Due to the heritable and stable characteristics of epigenetic modifications on natural chromosomes, it is difficult to learn the basic principles of their establishment by erasing epigenetic marks. The SynNICE method provides a unique platform for exploring de novo epigenomic regulatory mechanisms. The de novo assembled 1.14 Mb human AZFa (hAZFa) locus was delivered into early mouse embryos, where spontaneous incorporation of mouse histones and the establishment of DNA methylation at the single-cell stage were observed51.
Non-coding ‘dark matter’ constitutes approximately 98% of the human genome, a space where multiple distant regulatory variants interact in unknown ways, and it is increasingly recognized as a significant factor in the genetics of numerous diseases and traits4,191,192,193. Traditional methods struggle to comprehensively analyze the complex regulatory mechanisms of the genome by merely removing, disrupting individual regulatory components, or overlooking their context. The design-assembly-transfer-maintenance approach enables complete design freedom for synthetic DNA sequences, helping us address long-standing challenges in regulatory genomics. By de novo designing and assembling an 86 kb α-globin super-enhancer, a new class of regulatory elements called ‘facilitator’ was revealed. Facilitators lack intrinsic enhancer activity but potentiate the activity of classical enhancers in a position-dependent manner194. Using Big-IN to rewrite the mouse Sox2 locus to construct deletions, rearrangements, and inversions of single or multiple distal clusters of DNase I hypersensitive sites (DHSs), as well as surgical alterations to transcription factor (TF) recognition sequences, the expression of the endogenous Sox2 locus was analyzed, demonstrating the role of context in regulating the function of genomic regulatory elements195. By introducing a synthetic ~100 kb human HPRT1 locus reverse sequence (HPRT1R) into S. cerevisiae and mouse ES cells, a new perspective on the default transcriptional state of eukaryotic genomes was explored. In S. cerevisiae, both the reverse and natural HPRT1 loci exhibit widespread activity, whereas the inverted locus shows no activity at all in mouse ES cells, instead displaying repressive chromatin characteristics196. By constructing different rat HoxA variants ranging from 130-170 kb in yeast and integrating them into ectopic loci in the genome, the independent ability of these variants to reconstruct different aspects of HoxA regulation at ectopic clusters was tested197.
There are no effective methods to explore the complex biological effects caused by chromosome fusion. Employing CRISPR/Cas9-mediated genome rearrangement in mouse cells, centromere-centromere chromosome fusions and centromere-telomere chromosome fusions have been achieved, yielding mice with fused chromosomes177,179,198. These models have been utilized to dissect the multifaceted impact of chromosomal fusion phenomena on gene expression, stem cell differentiation, chromatin organization, mouse phenotypes, and mouse development.
Discussion
Synthetic rewriting technologies can be used either independently or within the assembly-transfer-maintenance-rearrangement pipeline and have unique value for constructing mammalian disease models and studying complex regulatory mechanisms of genomes. Despite the substantial progress made in mammalian synthetic rewriting technologies, several limitations remain. (1) Using existing technologies, Mb-scale DNA assembly can be completed efficiently in E. coli and S. cerevisiae. However, challenges remain for assembling mammalian genomic DNA. Key limitations include the absence of methods for larger-scale assembly (such as at the 10 Mb-100 Mb scale) and difficulties in the fidelity and stability of repetitive DNA. Base insertions and deletions have been observed within mononucleotide and TA-rich repeats in some E. coli assembly strains49,50, while deleted DNA fragments have been reported in interspersed repeats at the IGH locus in some S. cerevisiae assembly strains47. To advance mammalian genome rewriting, future efforts should focus on scaling up assembly capacity, developing or modifying assembly platforms to improve the accurate assembly and stabilization of highly repetitive regions, and establishing automated workflows for large DNA construction. Additionally, it is essential to implement convenient detection technologies to identify and correct unintended errors introduced during genome manipulation. (2) Emerging technologies related to the transfer of large DNA into mammalian cells offer new solutions to overcome the limitations of in vitro operations on large DNA. However, they are still constrained by challenges, such as low efficiency, limited recipient cell types, and potential contamination from the host genome. Therefore, developing more efficient, universal, and safe transfer methods is the future trend of large DNA transfer technologies. (3) Recent advancements in single-copy artificial chromosomes, combined with the construction of a ~ 12 Mb yeast chromosome199,200, imply that yeast centromere vectors can serve as hosts for synthetic mammalian chromosomes. These studies may enable the exploration of larger artificial chromosomes constructed in yeast before transferring them into mammalian cells, offering a new possibility for rewriting mammalian chromosomes. (4) Recently, the genome rearrangement technologies in mammalian cells have enabled large-scale rearrangements, facilitating the study of the potential mechanisms of SVs in mammalian cells. In terms of rearrangement strategies, enhancing the efficiency and developing multi-site rearrangements of bridge recombinases is expected to achieve more extensive and precise rearrangements without the need to introduce recombination sites. Additionally, combined with technologies, such as DNA assembly, it is possible to synthesize re-arrangeable sequences and induce rearrangements in specific regions. In terms of functionality, like the Sc2.0 project, genome-scale rearrangements with high-throughput analysis and detection methods not only support basic genomic research in mammalian cells but also have the potential to be used to explore phenotypic evolution.
Given that mammals are multicellular organisms, the genetic stability of their germline and tissue function is crucial, making stem cells an important cornerstone in the synthetic rewriting technology-mediated modification of mammalian cells. Stem cells, particularly pluripotent and embryonic stem cells, have expandability and potent differentiation capacity, which provides abundant platforms for subsequent disease modeling and therapy201. Currently, strategies based on DNA assembly, transfer, and maintenance have enabled the construction of animal models and the large-scale analysis of complex regulatory mechanisms in stem cells59,194,195,196,197. Notably, haploid embryonic stem cells (haESCs) are a type of stem cell containing a single set of chromosomes202,203,204, which can be combined with various synthetic rewriting technologies, providing a powerful platform for the construction of complex models and enabling large-scale functional gene screening.
The rapid development of artificial intelligence (AI) in multiple fields provides technical support for the synthetic rewriting of mammalian genomes. Generative AI has demonstrated its potential in designing and predicting functional elements205,206,207,208. For example, AlphaGenome can interpret the function of large-scale non-coding regions in the mammalian genome209, and Evo2 can predict the functional impact of DNA sequence variations, generate genomic-scale sequences, and control epigenetic structures210,211. Cello provides logical gate structures and mathematical models to represent the dynamic behavior of gene circuits212. The combination of AI-assisted genome design and synthetic rewriting technologies will further advance the process of mammalian genome rewriting, significantly enhance the understanding of mammalian synthetic biology, and contribute to the development of human medicine and health.
References
Venter, J. C., Glass, J. I., Hutchison, C. A. III & Vashee, S. Synthetic chromosomes, genomes, viruses, and cells. Cell 185, 2708–2724 (2022).
James, J. S., Dai, J., Chew, W. L. & Cai, Y. The design and engineering of synthetic genomes. Nat. Rev. Genet. 26, 298–319 (2025).
Chari, R. & Church, G. M. Beyond editing to writing large genomes. Nat. Rev. Genet. 18, 749–760 (2017).
Zhang, W., Mitchell, L. A., Bader, J. S. & Boeke, J. D. Synthetic Genomes. Annu. Rev. Biochem. 89, 77–101 (2020).
Schindler, D., Dai, J. & Cai, Y. Synthetic genomics: a new venture to dissect genome fundamentals and engineer new functions. Curr. Opin. Chem. Biol. 46, 56–62 (2018).
Fredens, J. et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019).
Ostrov, N. et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016).
Lajoie, M. J. et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013).
Wang, K. et al. Defining synonymous codon compression schemes by genome recoding. Nature 539, 59–64 (2016).
Hutchison, C. A. et al. Design and synthesis of a minimal bacterial genome. Science 351, aad6253 (2016).
Pelletier, J. F. et al. Genetic requirements for cell division in a genomically minimal cell. Cell 184, 2430–2440 (2021).
Thornburg, Z. R. et al. Fundamental behaviors emerge from simulations of a living minimal cell. Cell 185, 345–360 (2022).
Luo, Z. et al. Compacting a synthetic yeast chromosome arm. Genome Biol. 22, 5 (2021).
Jiang, S. et al. Building a eukaryotic chromosome arm by de novo design and synthesis. Nat. Commun. 14, 7886 (2023).
Richardson, S. M. et al. Design of a synthetic yeast genome. Science 355, 1040–1044 (2017).
Dai, J., Boeke, J. D., Luo, Z., Jiang, S. & Cai, Y. Sc3.0: revamping and minimizing the yeast genome. Genome Biol. 21, 205 (2020).
Ma, Y. et al. Convenient synthesis and delivery of a megabase-scale designer accessory chromosome empower biosynthetic capacity. Cell Res 34, 309–322 (2024).
Dormitzer, P. R. et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 5, 185ra168 (2013).
Thi Nhu Thao, T. et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 582, 561–565 (2020).
Blight, K. J., Kolykhalov, A. A. & Rice, C. M. Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 (2000).
Gibson, D. G. et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56 (2010).
Gibson, D. G. et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220 (2008).
Robertson, W. E. et al. Escherichia coli with a 57-codon genetic code. Science 390, eady4368 (2025).
Zhou, S. et al. Dynamics of synthetic yeast chromosome evolution shaped by hierarchical chromatin organization. Natl. Sci. Rev. 10, nwad073 (2023).
Zhao, Y. et al. Debugging and consolidating multiple synthetic chromosomes reveals combinatorial genetic interactions. Cell 186, 5220–5236 (2023).
Zhang, W. et al. Manipulating the 3D organization of the largest synthetic yeast chromosome. Mol. Cell 83, 4424–4437 (2023).
Luo, J. et al. Synthetic chromosome fusion: Effects on mitotic and meiotic genome structure and function. Cell Genom. 3, 100439 (2023).
Foo, J. L. et al. Establishing chromosomal design-build-test-learn through a synthetic chromosome and its combinatorial reconfiguration. Cell Genom. 3, 100435 (2023).
Shen, Y. et al. Dissecting aneuploidy phenotypes by constructing Sc2.0 chromosome VII and SCRaMbLEing synthetic disomic yeast. Cell Genom. 3, 100364 (2023).
Williams, T. C. et al. Parallel laboratory evolution and rational debugging reveal genomic plasticity to S. cerevisiae synthetic chromosome XIV defects. Cell Genom. 3, 100379 (2023).
McCulloch, L. H. et al. Consequences of a telomerase-related fitness defect and chromosome substitution technology in yeast synIX strains. Cell Genom. 3, 100419 (2023).
Blount, B. A. et al. Synthetic yeast chromosome XI design provides a testbed for the study of extrachromosomal circular DNA dynamics. Cell Genom. 3, 100418 (2023).
Lauer, S. et al. Context-dependent neocentromere activity in synthetic yeast chromosome VIII. Cell Genom. 3, 100437 (2023).
Schindler, D. et al. Design, construction, and functional characterization of a tRNA neochromosome in yeast. Cell 186, 5237–5253 (2023).
Robertson, W. E. et al. Sense codon reassignment enables viral resistance and encoded polymer synthesis. Science 372, 1057–1062 (2021).
Nyerges, A. et al. A swapped genetic code prevents viral infections and gene transfer. Nature 615, 720–727 (2023).
Zürcher, J. F. et al. Refactored genetic codes enable bidirectional genetic isolation. Science 378, 516–523 (2022).
Grome, M. W. et al. Engineering a genomically recoded organism with one stop codon. Nature 639, 512–521 (2025).
Boeke, J. D. et al. The genome project-write. Science 353, 126–127 (2016).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Vollger, M. R. et al. Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 (2022).
Aganezov, S. et al. A complete reference genome improves analysis of human genetic variation. Science 376, eabl3533 (2022).
Altemose, N. et al. Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022).
Gershman, A. et al. Epigenetic patterns in a complete human genome. Science 376, eabj5089 (2022).
Church, D. M. A next-generation human genome sequence. Science 376, 34–35 (2022).
Hoyt, S. J. et al. From telomere to telomere: the transcriptional and epigenetic state of human repeat elements. Science 376, eabk3112 (2022).
He, B. et al. YLC-assembly: large DNA assembly via yeast life cycle. Nucleic Acids Res. 51, 8283–8292 (2023).
Ma, Y. et al. Assembly and delivery of large DNA via chromosome elimination in yeast. Nat. Protoc. 20, 3755–3782 (2025).
Zürcher, J. F. et al. Continuous synthesis of E. coli genome sections and Mb-scale human DNA assembly. Nature 619, 555–562 (2023).
Zhong, L. et al. The conjugation-associated linear-BAC iterative assembling (CALBIA) method for cloning 2.1-Mb human chromosomal DNAs in bacteria. Cell Res. 35, 309–312 (2025).
Liu, Y. et al. De novo assembly and delivery of synthetic megabase-scale human DNA into mouse early embryos. Nat. Methods 22, 1686–1697 (2025).
Gambogi, C. W. et al. Efficient formation of single-copy human artificial chromosomes. Science 383, 1344–1349 (2024).
Yarnall, M. T. N. et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol. 41, 500–512 (2023).
Pandey, S. et al. Efficient site-specific integration of large genes in mammalian cells via continuously evolved recombinases and prime editing. Nat. Biomed. Eng. 9, 22–39 (2025).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Fell, C. W. et al. Reprogramming site-specific retrotransposon activity to new DNA sites. Nature 642, 1080–1089 (2025).
Witte, I. P. et al. Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science 388, eadt5199 (2025).
Zhang, W. et al. Mouse genome rewriting and tailoring of three important disease loci. Nature 623, 423–431 (2023).
Engler, C., Kandzia, R. & Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647 (2008).
Shetty, R. P., Endy, D. & Knight, T. F. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5 (2008).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Zhang, R. et al. Amplification editing enables efficient and precise duplication of DNA from short sequence to megabase and chromosomal scale. Cell 187, 3936–3952 (2024).
Cui, Y., Wu, Y. & Yuan, Y. Amplification editing empowers in situ large-scale DNA duplication. Innovation 5, 100716 (2024).
O’Connor, M., Peifer, M. & Bender, W. Construction of large DNA segments in Escherichia coli. Science 244, 1307–1312 (1989).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000).
Wannier, T. M. et al. Improved bacterial recombineering by parallelized protein discovery. Proc. Natl. Acad. Sci. USA 117, 13689–13698 (2020).
Zhang, Y., Buchholz, F., Muyrers, J. P. P. & Stewart, A. F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20, 123–128 (1998).
Robertson, W. E. et al. Creating custom synthetic genomes in Escherichia coli with REXER and GENESIS. Nat. Protoc. 16, 2345–2380 (2021).
Wang, T. et al. Reconstruction of a robust bacterial replication module. Nucleic Acids Res. 52, 11394–11407 (2024).
Mitchell, L. A. et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355, eaaf4831 (2017).
Shen, Y. et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science 355, eaaf4791 (2017).
Xie, Z.-X. et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science 355, eaaf4704 (2017).
Wu, Y. et al. Bug mapping and fitness testing of chemically synthesized chromosome X. Science 355, eaaf4706 (2017).
Zhang, W. et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science 355, eaaf3981 (2017).
Zhou, J., Wu, R., Xue, X. & Qin, Z. CasHRA (Cas9-facilitated Homologous Recombination Assembly) method of constructing megabase-sized DNA. Nucleic Acids Res. 44, e124 (2016).
de Koning, A. P., Gu, W., Castoe, T. A., Batzer, M. A. & Pollock, D. D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7, e1002384 (2011).
Jurka, J., Kapitonov, V. V., Kohany, O. & Jurka, M. V. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics Hum. Genet. 8, 241–259 (2007).
Martens, U. M. et al. Short telomeres on human chromosome 17p. Nat. Genet. 18, 76–80 (1998).
Logsdon, G. A. et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Henikoff, S., Ahmad, K. & Malik, H. S. The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293, 1098–1102 (2001).
Blackburn, E. H. Structure and function of telomeres. Nature 350, 569–573 (1991).
Payer, L. M. & Burns, K. H. Transposable elements in human genetic disease. Nat. Rev. Genet. 20, 760–772 (2019).
Ebersole, T. et al. Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Res. 33, e130 (2005).
Jiang, S. et al. High plasticity of ribosomal DNA organization in budding yeast. Cell Rep. 43, 113742 (2024).
Chen, Y. et al. Multiplex base editing to convert TAG into TAA codons in the human genome. Nat. Commun. 13, 4482 (2022).
Yuan, Q. & Gao, X. Multiplex base- and prime-editing with drive-and-process CRISPR arrays. Nat. Commun. 13, 2771 (2022).
Basu, J., Stromberg, G., Compitello, G., Willard, H. F. & van Bokkelen, G. Rapid creation of BAC-based human artificial chromosome vectors by transposition with synthetic alpha-satellite arrays. Nucleic Acids Res. 33, 587–596 (2005).
Harrington, J. J., Bokkelen, G. V., Mays, R. W., Gustashaw, K. & Willard, H. F. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 15, 345–355 (1997).
Ohzeki, J., Nakano, M., Okada, T. & Masumoto, H. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 159, 765–775 (2002).
Lamb, B. T. et al. Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [corrected]. Nat. Genet. 5, 22–30 (1993).
Mejía, J. E., Willmott, A., Levy, E., Earnshaw, W. C. & Larin, Z. Functional complementation of a genetic deficiency with human artificial chromosomes. Am. J. Hum. Genet. 69, 315–326 (2001).
Lee, E. C. et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 32, 356–363 (2014).
Gnirke, A., Huxley, C., Peterson, K. & Olson, M. V. Microinjection of intact 200- to 500-kb fragments of YAC DNA into mammalian cells. Genomics 15, 659–667 (1993).
Schiedner, G. et al. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 18, 180–183 (1998).
Wade-Martins, R., Smith, E. R., Tyminski, E., Chiocca, E. A. & Saeki, Y. An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat. Biotechnol. 19, 1067–1070 (2001).
Moralli, D. & Monaco, Z. L. Developing de novo human artificial chromosomes in embryonic stem cells using HSV-1 amplicon technology. Chromosome Res 23, 105–110 (2015).
White, R. E., Wade-Martins, R. & James, M. R. Infectious delivery of 120-kilobase genomic DNA by an epstein-barr virus amplicon vector. Mol. Ther. 5, 427–435 (2002).
Marschall, P., Malik, N. & Larin, Z. Transfer of YACs up to 2.3Mb intact into human cells with polyethylenimine. Gene Ther. 6, 1634–1637 (1999).
Suzuki, N., Itou, T., Hasegawa, Y., Okazaki, T. & Ikeno, M. Cell to cell transfer of the chromatin-packaged human β-globin gene cluster. Nucleic Acids Res. 38, e33 (2010).
Fournier, R. E. & Ruddle, F. H. Microcell-mediated transfer of murine chromosomes into mouse, Chinese hamster, and human somatic cells. Proc. Natl. Acad. Sci. USA 74, 319–323 (1977).
Liskovykh, M., Lee, N. C., Larionov, V. & Kouprina, N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 3, 16043 (2016).
Yamaguchi, S. et al. A new method of microcell-mediated transfer of human artificial chromosomes using a hemagglutinating virus of Japan envelope. Chromosome Sci. 9, 65–73 (2006).
Katoh, M. et al. Exploitation of the interaction of measles virus fusogenic envelope proteins with the surface receptor CD46 on human cells for microcell-mediated chromosome transfer. BMC Biotechnol. 10, 37 (2010).
Hiratsuka, M. et al. Retargeting of microcell fusion towards recipient cell-oriented transfer of human artificial chromosome. BMC Biotechnol. 15, 58 (2015).
Suzuki, T., Kazuki, Y., Oshimura, M. & Hara, T. Highly efficient transfer of chromosomes to a broad range of target cells using chinese hamster ovary cells expressing murine leukemia virus-derived envelope proteins. PLoS One 11, e0157187 (2016).
Uno, N. et al. The transfer of human artificial chromosomes via cryopreserved microcells. Cytotechnology 65, 803–809 (2013).
Allshire, R. C. et al. A fission yeast chromosome can replicate autonomously in mouse cells. Cell 50, 391–403 (1987).
Zhang, X. F. et al. Transfer of an expression YAC into goat fetal fibroblasts by cell fusion for mammary gland bioreactor. Biochem. Biophys. Res. Commun. 333, 58–63 (2005).
Huxley, C., Hagino, Y., Schlessinger, D. & Olson, M. V. The human HPRT gene on a yeast artificial chromosome is functional when transferred to mouse cells by cell fusion. Genomics 9, 742–750 (1991).
Jülicher, K. et al. Yeast artificial chromosome transfer into human renal carcinoma cells by spheroplast fusion. Genomics 43, 95–98 (1997).
Li, L. & Blankenstein, T. Generation of transgenic mice with megabase-sized human yeast artificial chromosomes by yeast spheroplast-embryonic stem cell fusion. Nat. Protoc. 8, 1567–1582 (2013).
Pachnis, V., Pevny, L., Rothstein, R. & Costantini, F. Transfer of a yeast artificial chromosome carrying human DNA from Saccharomyces cerevisiae into mammalian cells. Proc. Natl. Acad. Sci. USA 87, 5109–5113 (1990).
Brown, D. M. et al. Efficient size-independent chromosome delivery from yeast to cultured cell lines. Nucleic Acids Res. 45, e50 (2017).
Li, L. P. et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat. Med. 16, 1029–1034 (2010).
Itaya, M. et al. Far rapid synthesis of giant DNA in the Bacillus subtilis genome by a conjugation transfer system. Sci. Rep. 8, 8792 (2018).
Lacroix, B. & Citovsky, V. Transfer of DNA from bacteria to eukaryotes. mBio 7, e00863–16 (2016).
Waters, V. L. Conjugation between bacterial and mammalian cells. Nat. Genet. 29, 375–376 (2001).
Kunik, T. et al. Genetic transformation of HeLa cells by Agrobacterium. Proc. Natl. Acad. Sci. USA 98, 1871–1876 (2001).
Sizemore, D. R., Branstrom, A. A. & Sadoff, J. C. Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization. Science 270, 299–302 (1995).
Krusch, S. et al. Listeria monocytogenes mediated CFTR transgene transfer to mammalian cells. J. Gene Med. 4, 655–667 (2002).
Weiss, S. & Krusch, S. Bacteria-mediated transfer of eukaryotic expression plasmids into mammalian host cells. Biol. Chem. 382, 533–541 (2001).
Dietrich, G. et al. Delivery of antigen-encoding plasmid DNA into the cytosol of macrophages by attenuated suicide Listeria monocytogenes. Nat. Biotechnol. 16, 181–185 (1998).
Narayanan, K. & Warburton, P. E. DNA modification and functional delivery into human cells using Escherichia coli DH10B. Nucleic Acids Res 31, e51 (2003).
Grillot-Courvalin, C., Goussard, S., Huetz, F., Ojcius, D. M. & Courvalin, P. Functional gene transfer from intracellular bacteria to mammalian cells. Nat. Biotechnol. 16, 862–866 (1998).
Jacobs, R., Singh, P., Smith, T., Arbuthnot, P. & Maepa, M. B. Prospects of viral vector-mediated delivery of sequences encoding anti-HBV designer endonucleases. Gene Ther. 32, 8–15 (2025).
Mandegar, M. A. et al. Functional human artificial chromosomes are generated and stably maintained in human embryonic stem cells. Hum. Mol. Genet. 20, 2905–2913 (2011).
Schedl, A., Montoliu, L., Kelsey, G. & Schütz, G. A yeast artificial chromosome covering the tyrosinase gene confers copy number-dependent expression in transgenic mice. Nature 362, 258–261 (1993).
Iacovino, M. et al. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells 29, 1580–1588 (2011).
Zhu, F. et al. DICE, an efficient system for iterative genomic editing in human pluripotent stem cells. Nucleic Acids Res. 42, e34 (2014).
Bouhassira, E. E., Westerman, K. & Leboulch, P. Transcriptional behavior of LCR enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood 90, 3332–3344 (1997).
Wallace, H. A. et al. Manipulating the mouse genome to engineer precise functional syntenic replacements with human sequence. Cell 128, 197–209 (2007).
Brosh, R. et al. A versatile platform for locus-scale genome rewriting and verification. Proc. Natl. Acad. Sci. USA 118, e2023952118 (2021).
Macdonald, L. E. et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl. Acad. Sci. USA 111, 5147–5152 (2014).
Moore, M. J. et al. Humanization of T cell-mediated immunity in mice. Sci. Immunol. 6, eabj4026 (2021).
Dymond, J. S. et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 477, 471–476 (2011).
Sun, C. et al. Precise integration of large DNA sequences in plant genomes using PrimeRoot editors. Nat. Biotechnol. 42, 316–327 (2024).
Zhang, X. et al. Harnessing eukaryotic retroelement proteins for transgene insertion into human safe-harbor loci. Nat. Biotechnol. 43, 42–51 (2025).
Chen, Y. et al. All-RNA-mediated targeted gene integration in mammalian cells with rationally engineered R2 retrotransposons. Cell 187, 4674–4689 (2024).
Katoh, M. et al. Construction of a novel human artificial chromosome vector for gene delivery. Biochem. Biophys. Res. Commun. 321, 280–290 (2004).
Kakeda, M. et al. A new chromosome 14-based human artificial chromosome (HAC) vector system for efficient transgene expression in human primary cells. Biochem. Biophys. Res. Commun. 415, 439–444 (2011).
Mills, W., Critcher, R., Lee, C. & Farr, C. J. Generation of an ∼2.4Mb human X centromere-based minichromosome by targeted telomere-associated chromosome fragmentation in DT40. Hum. Mol. Genet. 8, 751–761 (1999).
Heller, R., Brown, K. E., Burgtorf, C. & Brown, W. R. Mini-chromosomes derived from the human Y chromosome by telomere directed chromosome breakage. Proc. Natl. Acad. Sci. USA 93, 7125–7130 (1996).
Laner, A., Schwarz, T., Christan, S. & Schindelhauer, D. Suitability of a CMV/EGFP cassette to monitor stable expression from human artificial chromosomes but not transient transfer in the cells forming viable clones. Cytogenet. Genome Res. 107, 9–13 (2004).
Ikeno, M. et al. Construction of YAC–based mammalian artificial chromosomes. Nat. Biotechnol. 16, 431–439 (1998).
Kouprina, N. et al. Cloning of human centromeres by transformation-associated recombination in yeast and generation of functional human artificial chromosomes. Nucleic Acids Res. 31, 922–934 (2003).
Okamoto, Y., Nakano, M., Ohzeki, J., Larionov, V. & Masumoto, H. A minimal CENP-A core is required for nucleation and maintenance of a functional human centromere. Embo J. 26, 1279–1291 (2007).
Nakano, M. et al. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell 14, 507–522 (2008).
Ohzeki, J. et al. Breaking the HAC Barrier: histone H3K9 acetyl/methyl balance regulates CENP-A assembly. Embo J. 31, 2391–2402 (2012).
Kouprina, N. et al. Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synth. Biol. 1, 590–601 (2012).
Iida, Y. et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. 17, 293–301 (2010).
Kononenko, A. V. et al. A portable BRCA1-HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Res. 42, e164 (2014).
Kim, J.-H. et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc. Natl. Acad. Sci. USA 108, 20048–20053 (2011).
Lee, N. C. O. et al. Protecting a transgene expression from the HAC-based vector by different chromatin insulators. Cell. Mol. Life Sci. 70, 3723–3737 (2013).
Liskovykh, M. et al. Stable maintenance of de novo assembled human artificial chromosomes in embryonic stem cells and their differentiated progeny in mice. Cell Cycle 14, 1268–1273 (2015).
Wudzinska, A. et al. Germline Transmission of a Circular Human Artificial Chromosome in the Mouse. Preprint at https://doi.org/10.1101/2022.06.22.496420 (2022).
Logsdon, G. A. et al. Human artificial chromosomes that bypass centromeric DNA. Cell 178, 624–639.e619 (2019).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138 (2013).
Kilby, N. J., Snaith, M. R. & Murray, J. A. Site-specific recombinases: tools for genome engineering. Trends Genet. 9, 413–421 (1993).
Ma, L. et al. SCRaMbLE generates evolved yeasts with increased alkali tolerance. Microb. Cell Fact. 18, 52 (2019).
Liu, W. et al. Rapid pathway prototyping and engineering using in vitro and in vivo synthetic genome SCRaMbLE-in methods. Nat. Commun. 9, 1936 (2018).
Jia, B. et al. Precise control of SCRaMbLE in synthetic haploid and diploid yeast. Nat. Commun. 9, 1933 (2018).
Blount, B. A. et al. Rapid host strain improvement by in vivo rearrangement of a synthetic yeast chromosome. Nat. Commun. 9, 1932 (2018).
Wu, Y. et al. In vitro DNA SCRaMbLE. Nat. Commun. 9, 1935 (2018).
Wang, J. et al. Ring synthetic chromosome V SCRaMbLE. Nat. Commun. 9, 3783 (2018).
Gowers, G. O. F. et al. Improved betulinic acid biosynthesis using synthetic yeast chromosome recombination and semi-automated rapid LC-MS screening. Nat. Commun. 11, 868 (2020).
Cheng, L. et al. Large-scale genomic rearrangements boost SCRaMbLE in Saccharomyces cerevisiae. Nat. Commun. 15, 770 (2024).
Luo, Z. et al. Identifying and characterizing SCRaMbLEd synthetic yeast using ReSCuES. Nat. Commun. 9, 1930 (2018).
Yin, H. et al. Inducible chromosomal rearrangement reveals nonlinear polygenic dosage effects in driving aneuploid yeast traits. Nat. Commun. 16, 10884 (2025).
Koeppel, J. et al. Resolution of a human super-enhancer by targeted genome randomisation. Preprint at https://doi.org/10.1101/2025.01.14.632548 (2025).
Koeppel, J. et al. Randomizing the human genome by engineering recombination between repeat elements. Science 387, eado3979 (2025).
Sun, C. et al. Iterative recombinase technologies for efficient and precise genome engineering across kilobase to megabase scales. Cell 188, 4693–4710 (2025).
Pinglay, S. et al. Multiplex generation and single-cell analysis of structural variants in mammalian genomes. Science 387, eado5978 (2025).
Li, J. et al. Creation of a eukaryotic multiplexed site-specific inversion system and its application for metabolic engineering. Nat. Commun. 16, 1918 (2025).
Wang, L.-B. et al. A sustainable mouse karyotype created by programmed chromosome fusion. Science 377, 967–975 (2022).
Maddalo, D. et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014).
Zhang, X. M. et al. Creation of artificial karyotypes in mice reveals robustness of genome organization. Cell Res 32, 1026–1029 (2022).
Murillo-Pineda, M. et al. Induction of spontaneous human neocentromere formation and long-term maturation. J. Cell Biol. 220, e202007210 (2021).
Liu, Y. et al. Global chromosome rearrangement induced by CRISPR-Cas9 reshapes the genome and transcriptome of human cells. Nucleic Acids Res. 50, 3456–3474 (2022).
Durrant, M. G. et al. Bridge RNAs direct programmable recombination of target and donor DNA. Nature 630, 984–993 (2024).
Perry, N. T. et al. Megabase-scale human genome rearrangement with programmable bridge recombinases. Science 387, adz0276 (2025).
Kazuki, Y. et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol. Ther. 18, 386–393 (2010).
Murphy, A. J. et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl. Acad. Sci. USA 111, 5153–5158 (2014).
Tosh, J., Tybulewicz, V. & Fisher, E. M. C. Mouse models of aneuploidy to understand chromosome disorders. Mamm. Genome 33, 157–168 (2022).
O’Doherty, A. et al. An aneuploid mouse strain carrying human chromosome 21 with down syndrome phenotypes. Science 309, 2033–2037 (2005).
Kazuki, Y. et al. A non-mosaic transchromosomic mouse model of down syndrome carrying the long arm of human chromosome 21. eLife 9, e56223 (2020).
Adikusuma, F., Williams, N., Grutzner, F., Hughes, J. & Thomas, P. Targeted deletion of an entire chromosome using CRISPR/Cas9. Mol. Ther. 25, 1736–1738 (2017).
Zuo, E. et al. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. 18, 224 (2017).
Ling, H., Girnita, L., Buda, O. & Calin, G. A. Non-coding RNAs: the cancer genome dark matter that matters!. Clin. Chem. Lab. Med. 55, 705–714 (2017).
Ruffo, P., Traynor, B. J. & Conforti, F. L. Unveiling the regulatory potential of the non-coding genome: insights from the human genome project to precision medicine. Genes Dis. 12, 101652 (2025).
Fulco, C. P. et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773 (2016).
Blayney, J. W. et al. Super-enhancers include classical enhancers and facilitators to fully activate gene expression. Cell 186, 5826–5839 (2023).
Brosh, R. et al. Synthetic regulatory genomics uncovers enhancer context dependence at the Sox2 locus. Mol. Cell 83, 1140–1152 (2023).
Camellato, B. R., Brosh, R., Ashe, H. J., Maurano, M. T. & Boeke, J. D. Synthetic reversed sequences reveal default genomic states. Nature 628, 373–380 (2024).
Pinglay, S. et al. Synthetic regulatory reconstitution reveals principles of mammalian Hox cluster regulation. Science 377, eabk2820 (2022).
Wang, Y. et al. Chromosome territory reorganization through artificial chromosome fusion is compatible with cell fate determination and mouse development. Cell Discov. 9, 11 (2023).
Shao, Y. et al. Creating a functional single-chromosome yeast. Nature 560, 331–335 (2018).
Shao, Y. et al. A single circular chromosome yeast. Cell Res 29, 87–89 (2019).
Shi, Y., Inoue, H., Wu, J. C. & Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130 (2017).
Yang, H. et al. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell 149, 605–617 (2012).
Li, Z. K. et al. Generation of bimaternal and bipaternal mice from hypomethylated haploid ESCs with imprinting region deletions. Cell Stem Cell 23, 665–676 (2018).
Li, Z. K. et al. Adult bi-paternal offspring generated through direct modification of imprinted genes in mammals. Cell Stem Cell 32, 361–374 (2025).
Wang, J. et al. Scaffolding protein functional sites using deep learning. Science 377, 387–394 (2022).
Dauparas, J. et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
de Boer, C. G. et al. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 38, 56–65 (2020).
Fu, Z. H., He, S. Z., Wu, Y. & Zhao, G. R. Design and deep learning of synthetic B-cell-specific promoters. Nucleic Acids Res. 51, 11967–11979 (2023).
Callaway, E. DeepMind’s new AlphaGenome AI tackles the ‘dark matter’ in our DNA. Nature 643, 17–18 (2025).
Brixi, G. et al. Genome modeling and design across all domains of life with Evo 2. Preprint at https://doi.org/10.1101/2025.02.18.638918 (2025).
Merchant, A. T., King, S. H., Nguyen, E. & Hie, B. L. Semantic design of functional de novo genes from a genomic language model. Nature (2025).
Jones, T. S., Oliveira, S. M. D., Myers, C. J., Voigt, C. A. & Densmore, D. Genetic circuit design automation with Cello 2.0. Nat. Protoc. 17, 1097–1113 (2022).
Acknowledgements
This manuscript was supported by the National Key R&D Program of China [2024YFA0917400 for Y.W.], the National Natural Science Foundation of China [32471483 for Y.W.], and the Natural Science Foundation of Tianjin [23JCYBJC00220 for Y.W.].
Author information
Authors and Affiliations
Contributions
Y.W. conceived the original idea of this review. Y.W., Y.C., G.R.Z., Y.W., and Y.-J.Y. contributed to organizing the structure of the manuscript. Y.W., Y.C., G.-R.Z., Y.W., and Y.-J.Y. contributed to the literature review and wrote sections of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The Authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Weimin Zhang, Wei Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Cui, Y., Zhao, GR. et al. Synthetic rewriting technologies in mammalian cells. Nat Commun 17, 1308 (2026). https://doi.org/10.1038/s41467-025-68066-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68066-9
