
Abstract
Efficient CO2 electroreduction to multi-carbon (C2+) oxygenates in acidic electrolytes remains a great challenge, especially under high current density conditions. In this study, we prepare an ionic liquid (IL)-modified Cu electrode (IL@Cu), which achieve a Faradaic efficiency (FE) of 82.7% toward C2+ products at a current density of 2.0 A cm−2 in 0.5 M K2SO4 (pH = 1, adjusted with H2SO4), with a single-pass carbon efficiency reaching 78.5%. Under the same conditions, the partial current density for C2+ oxygenates and ethanol exceed 1.2 A cm−2 and 1.0 A cm−2, respectively, over IL@Cu. Mechanism study has shown that K+ cations are repelled by the IL cations during the reaction, allowing water molecules to access the electrode surface. The displacement of K+ enhances C–C coupling, while the proximity of water to the electrode surface facilitates the incorporation of oxygen-containing intermediates into the hydrogen bond network, thereby promoting the formation of C2+ oxygenates.
Similar content being viewed by others


Introduction
Electrochemical CO2 reduction reaction (CO2RR) into chemicals and fuels is a promising method for storing intermittent renewable energy in chemical bonds1,2,3. Among the various products derived from CO2RR, multi-carbon (C2+) oxygenates, such as acetic acid, ethanol, and propanol, hold significant industrial value4,5,6. Notable progress has been made in converting CO2 into C2+ oxygenates over Cu-based electrodes under alkaline electrolyte conditions7,8,9, but the alkaline environment can cause CO2 loss and salt precipitation, which hinder CO2RR performance. Acidic electrolytes can mitigate these issues, but in environments with higher proton concentrations at the electrode surface, hydrogenation at the oxygen atom is favored10,11,12, leading to C–O bond cleavage of the key C2+ oxygenate intermediate, *CH2CHO, to form C2H4.
Integrating the *CH2CHO intermediate into the interfacial hydrogen bond network could prolong their lifetime, thereby promoting the production of C2+ oxygenates13,14,15. Reconstructing the interfacial hydrogen bond network is a promising approach to stabilize *CH2CHO16,17,18, thereby enhancing selectivity toward oxygenates over C2H4. Alkali metal cations play a significant role in acidic electrolytes by increasing the local pH, which suppresses the hydrogen evolution reaction (HER) and promotes C–C coupling19,20,21. However, the distribution and hydration of alkali metal cations at the electrode surface influence the interfacial hydrogen bond network. Therefore, the optimal configuration of alkali metal cations at the electrode surface is crucial for simultaneously favoring C–C coupling and reconstructing the interfacial hydrogen bond network, thereby enhancing the production of C2+ oxygenates.
Ionic liquids (ILs), composed of tunable organic cations and paired anions, can generate complex interactions that modulate the surface microenvironment22,23. They have been widely employed as supporting electrolytes to enhance solution conductivity and CO2 interaction24,25,26, or as modifiers to tailor the electronic structure of catalysts27,28. The state of the IL, the electrolyte, and the reactor type each lead to distinct reaction pathways and mechanisms. In particular, IL cations can electrostatically repel alkali metal cations while forming hydrogen bonds with water molecules, which is likely to induce restructuring of the cation configuration and the interfacial hydrogen bond network. In this study, IL@Cu electrode was prepared by the electroreduction of CuO in 0.46 M 1-butyl-3-methylimidazolium perchlorate (BmimClO4, Supplementary Fig. 1) aqueous solution, with Bmim+ anchored onto the IL@Cu surface. The resulting IL@Cu electrode delivered outstanding catalytic performance, ranking among the top-reported systems for CO2RR to C2+ oxygenates. In situ characterization and theoretical calculations collectively elucidated the mechanism underlying the efficient CO2RR to C2+ oxygenates over the IL@Cu catalyst.
Results
Electrode preparation and characterization
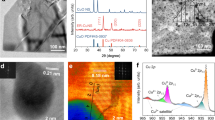
In partially miscible systems, the nanodomain size in solution increased dramatically near the phase boundary upon concentration changed29,30. BmimClO4 exhibited partial miscibility with water, undergoing phase separation at a concentration of 11.0 wt%. At 0.46 M BmimClO4 (10.9 wt%), a concentration considered to be in the pre-phase separation region near the phase boundary, the domain size reached approximately 3.8 nm, as confirmed by dynamic light scattering (DLS) and small-angle X-ray scattering (SAXS) (Supplementary Figs. 2–4). The IL@Cu was prepared by the electro-reduction of commercial CuO at −0.5 V vs. RHE in 0.46 M BmimClO4 aqueous solution (Supplementary Fig. 5). X-ray diffraction (XRD) confirmed that Cu(111) was the predominant surface facet (Supplementary Fig. 6). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images revealed that IL@Cu consisted of nanoparticles (Supplementary Fig. 7 and Fig. 1a). High-resolution TEM further showed a primary interplanar spacing of 0.21 nm (Fig. 1b and Supplementary Fig. 8), matching the theoretical d-spacing of Cu(111). Elemental mapping from TEM revealed a uniform distribution of Cu and N (Fig. 1c), suggesting that Bmim+ was evenly distributed on the surface of IL@Cu. X-ray absorption near-edge structure (XANES) and Cu 2p X-ray photoelectron spectroscopy (XPS) demonstrated Cu(0) valence state (Fig. 1d and Supplementary Figs. 9–10). The N 1 s XPS spectrum of IL@Cu was deconvoluted into three peaks, corresponding to Cu–N, C–N, and C = N species (Fig. 1e)31,32, confirming the coordination of nitrogen Bmim+ with Cu. The red shift of characteristic peaks, attributed to CH3(N) and CH2(N) bond variations of Bmim+ from attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), further confirmed the nitrogen atom of Bmim+ interacts directly with the Cu surface (Fig. 1f)33,34,35. Also, the zeta potential of IL@Cu was 20.4 mV (Supplementary Fig. 11), verifying that its surface was positively charged. The process for the electroreduction of CuO to IL@Cu was monitored using the in situ SAXS-XAS technique (Supplementary Fig. 12). As the electroreduction progressed, the steadily increasing SAXS signals indicated that more Bmim+ aggregates accumulated on the electrode surface over time (Fig. 1g). In situ XANES analysis revealed a gradual shift in the Cu K-edge energy from 8995 eV [Cu(II)] to 8979 eV [Cu(0)] during the electroreduction of CuO (Fig. 1h and Supplementary Fig. 13). Correspondingly, in the FT-EXAFS spectra, the Cu–O peak at 1.5 Å diminished while the Cu–Cu peak at 2.2 Å intensified (Fig. 1i), indicating the progressive reduction of CuO to metallic Cu(0).

a, b TEM images. c Corresponding elemental mappings. d Cu K-edge FT–EXAFS spectrum. e N 1 s XPS spectrum. f ATR-SEIRAS spectra. g–i Variations of SAXS curve g, Cu K-edge XANES spectrum h, and contour map of Cu K-edge FT–EXAFS spectrum i, over time during the reduction of CuO to IL@Cu at −0.5 V vs. RHE in 0.46 M BmimClO4. Source data for the figure are provided as a Source data file.
For comparison, pristine Cu electrode was prepared by the electroreduction of CuO at −0.5 V vs. RHE in 0.46 M KHCO3. The as-prepared Cu electrode exhibited identical structure, morphology and valence state to that of IL@Cu (Supplementary Figs. 14–16). Compared with IL@Cu, Cu showed lower zeta potential and conductivity due to the absence of Bmim+ (Supplementary Figs. 11 and 17). In contrast to IL@Cu formation, during the CuO electroreduction in KHCO3 to form the Cu electrode, there was negligible variation in the SAXS pattern (Supplementary Fig. 18), and Cu–Cu bonds emerged very quickly, indicating that CuO was reduced rapidly in the absence of Bmim+ (Supplementary Figs. 19–20).
CO2RR in acidic electrolyte
Both Cu and IL@Cu exhibited a metallic Cu structure with a Cu(0) valence state under negative potentials, which is efficient for CO2RR to various products36,37. All CO2RR experiments were performed in 0.5 M K2SO4 (pH = 1, adjusted with H2SO4) in a flow cell (Supplementary Fig. 21). Linear sweep voltammetry (LSV) revealed that while both Cu and IL@Cu showed CO2RR activity, the IL@Cu electrode displayed markedly improved catalytic performance (Supplementary Fig. 22). Constant-current electrolysis of CO2RR was conducted at a current density range of 1.2–2.4 A cm−2. Upon completion of the reaction, all products in the gas and liquid phases were collected and quantified by chromatography (GC) and hydrogen nuclear magnetic resonance spectroscopy (1H NMR), respectively. As the current density increased, the Faradaic efficiency toward C2+ (FEC2+) exhibited a volcano-like trend, reaching its maximum value at a current density of 2.0 A cm−2 over both Cu and IL@Cu (Fig. 2a–b). FEC2+ reached 82.7% at 2.0 A cm−2 over IL@Cu, while it was only 59.3% over Cu. The high FEC2+ over IL@Cu primarily arose from the low CO production, suggesting that IL@Cu promoted the coupling of *CO rather than its desorption. At 2.0 A cm−2, FEC2+oxygenates values over IL@Cu and Cu were determined to be 60.3% and 26.8%, respectively (Fig. 2c and Supplementary Fig. 23), while the partial current density for C2+ oxygenates (jC2+-oxygenates) exceeded 1.2 A cm−2 over IL@Cu (Fig. 2d). Particularly, the FEethanol reached 50.1%, with a partial current density of higher than 1.0 A cm−2 over IL@Cu. Cyclic voltammetry (CV) at different scan rates was used to determine the double-layer capacitance (Cdl), from which the electrochemical surface area (ECSA) was calculated. The ECSA values for Cu and IL@Cu were 41.72 and 55.86, respectively (Supplementary Figs. 24–26 and Supplementary Table 1). The larger ECSA of IL@Cu highlighted its enhanced density of electrochemically active sites. The ECSA-normalized jC2+-oxygenates over IL@Cu remained much larger than that over Cu (Supplementary Fig. 27). This suggested that the increased ECSA of IL@Cu was not the main reason for enhanced C2+ oxygenate production. Moreover, IL@Cu achieved a very high single-pass carbon efficiency (SPCE) of 78.5% (Fig. 2e). IL@Cu exhibited superior catalytic performance, surpassing most state-of-the-art catalysts documented in the literature (Supplementary Table 2). Additionally, after 100 h of CO2RR at 2.0 A cm−2 over IL@Cu, the FEC2+-oxygenates remained 60% (Fig. 2f), indicative of the excellent stability and effectiveness. XRD, TEM, XPS, and ATR-SEIRAS analyses confirmed that IL@Cu retained its original structure, morphology, and valence states after the reaction (Supplementary Figs. 28–31), indicative of the stability of IL@Cu. Furthermore, in situ XPS and ATR-SEIRAS provided complementary evidence for the stability of the Bmim+ on Cu, revealing its distribution, binding configuration, and dynamic evolution during CO2RR (Supplementary Figs. 32–33).

a, b Plot of FEs of various products vs. current density over Cu a and IL@Cu b. c Plot of FEC2+ and FEC2+-oxygenates. d Plot of jC2+-oxygenates vs. total current density. e Plot of FEs of various products and SPCE vs. CO2 flow rate over IL@Cu electrode at 2.0 A cm–2. f Plot of potential and FEC2+-oxygenates vs. time over IL@Cu at 2.0 A cm–2. The potential is iR corrected. Source data for the figure are provided as a Source data file.
Detection of surface microenvironment
In situ surface-enhanced Raman spectroscopy (SERS) and in situ ATR-SEIRAS were conducted over Cu and IL@Cu at various potentials38,39. In situ SERS (Fig. 3a, b, Supplementary Fig. 34 and Supplementary Table 3) revealed a more intense of Cu–CO peak at 310 cm−1 over IL@Cu compared with Cu. After normalization, the larger peak area indicated a higher *CO coverage over IL@Cu (Fig. 3c), which is likely related to the enhanced local electric field induced by IL40. A larger Stark slope of Cu–CO was observed on IL@Cu than on Cu (Supplementary Fig. 35), suggesting that the IL enhanced the local electric field at the electrode surface39. This enhancement led to a greater sensitivity of the *CO vibrational frequency to the applied potential, reflecting stronger electronic coupling between *CO intermediate and the IL@Cu surface. The in situ ATR-SEIRAS spectra showed a peak at 1570 cm−1 corresponding to the *OCCOH intermediate, with IL@Cu exhibiting weaker intensity (Fig. 3d, e, Supplementary Fig. 36 and Supplementary Table 4), indicating its promotion of *OCCOH transformation into deeply reduced products. The key ethanol intermediate, *OC2H5, identified at 1040 cm−1, showed that the area peaks over IL@Cu was 2.5 times higher than that over Cu at −1.3 V vs. RHE (Fig. 3f), indicating that IL@Cu facilitated C–C coupling toward C2+ oxygenates production.

a, b In situ SERS spectra over Cu a and IL@Cu b during CO2RR. c Plot of normalized *CO peak area vs. potential. d, e In situ ATR-SEIRAS over Cu d and IL@Cu e. f Plot of normalized *OC2H5 peak area as a function of the applied potential over IL@Cu and Cu. g Plot of the population of 2H-H2O vs. potential. h Plot of the simulated density of K+ vs. distance from electrode surface. i Plot of probability of interfacial water vs. angle over Cu and IL@Cu electrodes. Source data for the figure are provided as a Source data file.
The SERS spectra in the range of 3000–3800 cm−1 correspond to the OH stretching vibrations of water molecules, with Gaussian deconvolution revealing three distinct categories of interfacial water: hydrated cation water (K+-H2O), two-coordinated water (2H-H2O) and four-coordinated water (4H-H2O) with reliable fitting quality (R2 ≈ 0.99) (Supplementary Fig. 37)41,42. The population of 2H-H2O on the surface of IL@Cu was significantly higher than that on Cu (Fig. 3g), indicating that water dissociation was favored over IL@Cu, as 2H-H2O served as a proton source to produce *H. The adsorption pseudo-capacitance (Cϕ), representing the *H adsorption charge, further supported this observation; IL@Cu exhibited higher *H coverage compared with Cu, as evidenced by its higher Cϕ at the same potential (Supplementary Fig. 38)43,44. Besides, the water dissociation kinetics at the electrode surface were further probed using the kinetic isotope effect (KIE). The significantly lower KIEH2O/D2O value observed on IL@Cu (1.28) compared to Cu (2.07) provided direct experimental evidence that the IL enhanced water dissociation (Supplementary Fig. 39). The Stark slope for all types of water over IL@Cu was higher than that over Cu (Supplementary Fig. 40), suggesting that water molecules were positioned closer to the IL@Cu surface. Lower K+ concentration can be found on IL@Cu than that of Cu using K+ retention experiment45 and ATR-SEIRAS analysis46 (Supplementary Figs. 41–42), which elucidated that some water molecules occupied the positions of a portion of K+ cations, thereby reconstructing the hydrogen bond network on the electrode surface. Molecular dynamics (MD) simulations confirmed that K+ was far away from to the electrode on IL@Cu than Cu (Fig. 3h). The distributions of K+ and water revealed by AIMD simulations were in agreement with the experimental results. Moreover, the presence of IL can also influence the water orientation at the electrode surface. AIMD simulations revealed that the introduction of Bmim+ suppressed the population of chemisorbed water (∼60°) and shifted the interfacial water dipole distribution toward ∼140°, indicating a pronounced reorganization of the hydrogen bond network (Fig. 3i and Supplementary Fig. 43).
DFT calculations
From the discussion above, K+ cations moved further away from the IL@Cu surface. As we know, K+ is crucial for C–C coupling. The dependence of the K+ distance to the Cu(111) surface on the C–C coupling performance was investigated through density functional theory (DFT) calculations. As K+ moved further away from the Cu(111) surface, the *CO adsorption energy decreased, while the water adsorption energy increased (Supplementary Figs. 44–45). This optimized *CO adsorption strength on IL@Cu is crucial, as it directly influences the energetics of C–C coupling. DFT calculations confirmed that the IL lowered the free energy for all major coupling pathways (*CO–*CO, *CO–*COH, and *CO–*CHO), with the *CO–*CO route being the most favorable (Supplementary Fig. 46). Furthermore, enhanced sampling Ab Initio molecular dynamics (SG-AIMD) including explicit water molecules showed that Bmim+ reduced the C–C coupling barrier from 1.28 to 0.84 eV (Supplementary Figs. 47–48), highlighting the pivotal role of the IL in promoting C–C coupling (Fig. 4a, b). As ethanol is the primary C2+ oxygenate, to gain deeper insights into the enhanced CO2RR to C2+ oxygenates over IL@Cu compared with Cu, we calculated the reaction pathways for the reduction of *CH2CHO intermediate (Supplementary Figs. 49–50)47,48,49, which can proceed via two potential routes: C–O bond cleavage leading to *C2H4 in the ethylene pathway or retention of the C–O bond yielding *CH3CHO, which subsequently forms ethanol (Fig. 4c, d). From the Gibbs free energy profiles, Cu favors ethylene production, whereas IL@Cu facilitates ethanol formation (Supplementary Fig. 51).

a Illustration of the specific adsorption configuration of intermediate during the *CO–*CO coupling. H, C, O, K, and Cu atoms are shown in pink, gray, red, purple, and blue, respectively. b The Gibbs free energy profile of *CO–*CO coupling over Cu and IL@Cu. c, d Gibbs free energy profiles for the reduction of *CH2CHO intermediate to ethylene or ethanol over Cu c and IL@Cu d. e Schematic illustration for the surface microenvironment on Cu and IL@Cu. Source data for the figure are provided as a Source data file.
Mechanism discussion
MD simulations confirmed that Bmim+ in IL@Cu significantly enriches CO2 concentration at the electrode surface (Supplementary Fig. 52), thereby increasing the probability of CO2 transformation. Bmim+ also exhibited strong electrostatic interactions with surface K+ cations, repelling them further away from the electrode surface (Fig. 4e). Additionally, the displacement of K+ allowed water molecules to be positioned closer to the electrode surface, and the *CH2CHO intermediate can participate in the hydrogen bond network of the interfacial water (Fig. 4e), preventing its cleavage to form C2H4 and promoting its transformation into C2+ oxygenates. Bmim+ on IL@Cu exerted strong electrostatic repulsion against K+, driving K+ to migrate away from the electrode surface. This suppressed the salting-out effect generated by high K⁺ concentrations under high current density, thus contributing to the long-term stability of IL@Cu.
Discussion
In this work, we prepared a IL@Cu electrode, incorporating Bmim+ on the electrode surface. At 2.0 A cm−2 in 0.5 M K2SO4 (pH = 1, adjusted with H2SO4), IL@Cu electrode achieved FEC2+ and FEC2+-oxygenates of 82.7% and 60.3%, respectively, significantly higher than the 59.3% and 26.8% observed over Cu. The partial current density for C2+ oxygenates and ethanol exceeded 1.2 A cm−2 and 1.0 A cm−2 over IL@Cu. Additionally, the SPCE at 2.0 A cm−2 in the acidic electrolyte reached 78.5% over IL@Cu. Compared with pristine Cu, the surface microenvironment on IL@Cu was reconstructed by altering the distribution of K+ through the repulsion of Bmim+, leading to three key effects: (i) promotion of C–C coupling to C2+ products, (ii) positioning of water closer to the electrode surface, reconstructing the hydrogen bond network, preventing C–O cleavage of *CH2CHO to enhance formation of C2+ oxygenates, and (iii) suppression of the salting-out effect, ensuring long-term stability. This approach offers valuable insights into surface microenvironment reconstruction, which can be used to control reaction pathways for a wide range of electrocatalytic reactions.
Methods
Chemicals
CuO (>99%), K2SO4 ( >99%), D2O ( >99%), Pt foil, Nafion D-521 dispersion (5 wt%), H2SO4 ( >99%), KHCO3 ( >99%), isopropanol (>99%), DMSO ( >99%), proton exchange membrane (N117) and gas diffusion layer (CP, YLS-30) were purchased from Alfa Aesar China Co., Ltd. CO2 (99.999%) and CO (99.999%) were provided by Beijing Analytical Instrument Company. BmimClO4, EmimClO4, and BmimBF4 were supplied by Lanzhou Yulu Fine Chemical Co. All chemicals were used as received without further purification.
Preparation of Cu-based electrodes
10 mg of CuO, along with 30 μL of Nafion solution, was dispersed into 1 mL of isopropanol and sonicated for 30 min to achieve a homogeneous ink. The catalyst ink was then coated onto hydrophobic CP (2 cm × 1.5 cm) and dried under an infrared lamp for 5 min to obtain the gas diffusion electrode. The catalyst loading on the electrode surface was approximately 1.0 mg cm−2. Finally, the as-prepared electrode was subjected to electroreduction in different electrolytes of 0.46 M at −0.5 V vs. RHE for 120 min.
Characterizations
The morphology was characterized by TEM JEM-2100F. XRD patterns were obtained on an X-ray diffractometer (Model D/MAX2500, Rigaka) with Cu-Kα radiation at a scan speed of 10o/min. XPS analysis was performed on the Thermo Scientific ESCA Lab 250Xi with a 200 W monochromated Al Kα radiation. The aggregation behavior of BmimClO4 solutions were characterized by DLS and SAXS. DLS and zeta potential experiments were performed on a Zetasizer Nano ZS ZEN3600 (Malvern, UK) at 25 °C. SAXS was conducted at 1W2A beamline station of Beijing Synchrotron Radiation Facility (BSRF). The liquid sample was loaded in a sample cell and characterized at 25 °C. Conductivity of electrode materials were measured using a digital multimeter (Vicior VC890D).
In situ SAXS-XAS
In situ SAXS-XAS was conducted at 1W2B beamline station of BSRF. The beamline was equipped with a combined setup (Supplementary Fig. 12), incorporating an area detector for SAXS measurements and a point detector for XAS signal acquisition. The experiments were performed in a custom flow cell using a three-electrode system, with CuO as the working electrode, Ag/AgCl as the reference electrode, and Ni foam as the counter electrode. The electrochemical cell was positioned at the center of the SAXS beam path, with both the beam path and the cell window carefully aligned. The electroreduction of CuO was carried out at −0.5 V vs. RHE in either 0.46 M KHCO3 or BmimClO4, with in situ SAXS measurements taken at 10-minute intervals. The 2D SAXS images were obtained from the detector and then transformed into the profiles of intensity (I) vs. wavevector (q) using the software SAXS Data Preprocess V2.0.050. In situ small-angle X-ray scattering (SAXS) was utilized to track the dynamic evolution of nanoscale electrolyte domains during electroreduction of CuO. The SAXS pattern of the electrode collected before electroreduction was used for background subtraction. The XAS spectrum was recorded every 10 min. The Cu K-edge data were analysed by the ATHENA and ARTEMIS modules of the Demeter software package51. All X-ray absorption spectra were processed by normalising the edge-jump step to obtain an EXAFS spectrum and correcting it for energy. Cu K-edge k3-weighted χ(k) data from k-space was converted to R-space by Fourier transform. The obtained curves were converted into heatmaps using Origin software for enhanced visualization.
CO2RR experiments
Electrochemical experiments were conducted in a flow cell on the CHI 660E electrochemical workstation (Shanghai CH Instruments Co., China) equipped with a high current amplifier CHI680c at 25 °C. The flow-cell configuration consisted of a three-electrode system, and the as-prepared electrode, Pt foil and Ag/AgCl electrode were used as the working electrode, counter electrode and reference electrode, respectively. A sealing gasket with a 0.5 cm × 1 cm window was employed to prevent electrolyte leakage, which defined the actual geometric working area of the electrode to 0.5 cm2. All potentials were converted to the RHE reference scale using the relation:
The catholyte and anolyte used were 0.5 M K2SO4 (pH = 1, adjusted with H2SO4). The catholyte and anolyte, each with a volume of 30 mL, were circulated at a flow rate of 30 mL min−1 using a peristaltic pump and a gas-liquid circulation pump, respectively. The CO2 flow rate was controlled to be 30 mL min−1 using a digital gas flow controller. Upon completion of CO2RR, the gaseous product of electrochemical experiments was collected using a gas bag and analyzed by gas chromatography (GC, HP 4890D), which was equipped with FID and TCD detectors using Ar as the carrier gas. From the GC peak areas and calibration curves for FID and TCD detector, we can obtain the A % of gaseous products. Since the flow rate of the outlet was monitored to be constant, the moles of gaseous products can be calculated. The liquid products were analyzed by 1H NMR Bruker Ascend 400 MHz using DMSO as an internal standard. To ensure reproducibility, all reactions were carried out with three independently prepared electrode batches, with each batch tested at least three times, and the resulting error bars indicate variations within ±5%. The FEs of different products were calculated based on the following equation.
where nproduct is the amount of product (mol) from GC (for gaseous products) or 1H NMR spectroscopy (for liquid products), nelectron is electron transfer number (varying by product)52, F is the Faraday constant (96485 C mol−1), and Q is the total charge passed during CO2RR.
The SPCE was calculated as follows:53
where jtotal is the total current density, FE is the Faradaic efficiency of different products VM is the molar volume of CO2 under 1 atm and 25 °C, vinCO2 is the CO2 flow rate.
The long-term stability test was performed at 2.0 A cm–2. After a period of time, the catholyte was removed, and a certain amount of deionized water was injected into the gas and cathode chambers to remove the accumulated salts on the electrode surface54,55. Subsequently, fresh catholyte was circulated to maintain a stable pH.
Electrochemical characterizations
Electrochemical impedance spectra (EIS) experiments were conducted at various potentials, with the alternating current frequency ranging from 0.1 to 106 Hz. The Cϕ data obtained from the EIS measurement was fitted using ZView software (version 2.4, Scribner Associates). For the KIE study, D2O was used to replace H2O in the electrolyte.
The K+ retention experiment was conducted in 0.5 M K2SO4 (pH = 1, adjusted with H2SO4), employing a three-electrode configuration within a single cell. The experimental setup comprised a Pt foil electrode, an Ag/AgCl reference electrode, and Cu-based catalyst as the working electrode. The rear surface of the as-prepared electrode was entirely sealed with Kapton tape. A constant potential of −1.8 V vs. Ag/AgCl was applied immediately after immersing the electrode into the electrolyte. After 120 s, the electrode was withdrawn from the solution, rinsed with 10 mL of deionized water, and the applied potential was terminated. The K⁺ ions adsorbed on the electrode were subsequently eluted into the deionized water. The concentration of K⁺ in the water was quantified using an ICP-OES spectrometer capable of covering the full wavelength range, and the K⁺ concentration was normalized to the geometric area of the catalyst. The K+ adsorption ATR-SEIRAS experiment was conducted on a BRUKER VERTEX 70 v spectrometer, featuring an MCT detector cooled with liquid nitrogen. The catalyst ink was deposited onto a germanium ATR crystal, which was pre-coated with a thin gold film. The electrolyte was 30 mM KHCO3 with 100 mM methyl4N(HCO3) using a three-electrode setup over different electrodes, A platinum electrode was employed as the counter electrode, while an Ag/AgCl electrode served as the reference electrode. A constant potential of −0.45 V vs. RHE was applied for 180 s.
The ECSA was determined from Cdl measurements, derived from scan-rate-dependent CV. CV was conducted within a potential window of −0.20 to −0.30 V vs. Ag/AgCl, using scan rates of 20, 50, 80, 100, 150, and 200 mV s−1 in a potential window where no Faradaic process occurs. Cdl was estimated by plotting the capacitive current difference (Δj = ja − jc) at −0.25 V vs. Ag/AgCl against the scan rate. The ECSA was calculated using the formula:
where Rf is the roughness factor, and S represents the geometric area of the electrode (1 cm2). Polycrystalline Cu was used as the reference standard, assuming a roughness factor of 1 and a double-layer capacitance of 29 μF cm−2. The roughness factor was calculated as:
The ECSA-normalized jC2+-oxygenates was obtained as follows:
In situ spectra during CO2RR
In situ SERS experiments were conducted using a HORIBA LabRAM HR Evolution Raman spectrometer equipped with a 785 nm solid-state laser as the excitation source. Measurements were performed in a custom-designed flow cell, configured similarly to the flow cell reactor. The OH stretching band of interfacial water was deconvoluted into three distinct components corresponding to strongly hydrogen-bonded, weakly hydrogen-bonded, and isolated water molecules. To minimize subjectivity in the fitting, Gaussian functions were employed with initial peak positions fixed at 3200, 3400, and 3600 cm−1, respectively, consistent with literature assignments for these aqueous environments. The robustness of the fitting procedure was confirmed by the low reduced Chi-squared (~10−9) and high R2 ( ~ 0.99), ensuring the reliability of the derived spectral parameters.
In situ ATR-SEIRAS experiments were carried out on a BRUKER VERTEX 70 v spectrometer, featuring an MCT detector cooled with liquid nitrogen. The catalyst ink was deposited onto a germanium ATR crystal, which was pre-coated with a thin gold film. A platinum electrode was employed as the counter electrode, while an Ag/AgCl electrode served as the reference electrode. Before measurement, the initial state was scanned under open-circuit conditions to establish a baseline.
Classical molecular dynamics simulations
MD simulations were carried out using the GROMACS software package56. The model system consisted of a four-layer Cu(111) slab (24 × 24 supercell, 2304 atoms) in contact with a 0.5 M K₂SO₄ aqueous electrolyte containing 100 K+ ions, 50 SO42− ions, 200 CO2 molecules, and 20 Bmim+ cations immobilized on the Cu surface, solvated by ~6000 water molecules. The simulation box dimensions were 5.21 × 6.02 × 6.00 nm3. Force field parameters for Cu were taken from Naik et al.57, water was described by the SPC/E model58, and all other species were modeled using the OPLS-AA force field59. Simulations were performed in the NVT ensemble at 300 K using a Nosé–Hoover thermostat, the Verlet cutoff scheme, and particle mesh Ewald (PME) for long-range electrostatics60. The positions of Cu and Bmim⁺ were fixed during the runs, while initial velocities were assigned according to a Maxwell–Boltzmann distribution. Each trajectory was propagated for 40 ns, with the final 20 ns extracted for analysis to ensure equilibration.
AIMD simulations
The benchmark model comprised a 4 × 4 × 4 Cu(111) supercell with 62 water molecules and two CO molecules adsorbed at the interface, enclosed in a periodic box of 8.854 × 10.224 × 30.0 Å3. Experimental variants were generated by introducing either one K+ ion or both K+ and Bmim+ ions, denoted as CO–CO–K+, and CO–CO–Bmim+–K+, respectively. Geometry optimization was performed using the CP2K package61 with the LBFGS algorithm, applying convergence thresholds of 0.001 Å for atomic displacements and 0.001 eV Å−1 for forces. A maximum of 200 optimization steps was allowed.
AIMD simulations were carried out in CP2K at 300 K using the Nosé–Hoover thermostat62, a timestep of 0.5 fs, and a total simulation length of 10 ps. Periodic boundary conditions were applied in all three dimensions. Interatomic forces were evaluated within the GPW framework using the PBE functional63 with DFT-D3 dispersion corrections64. To mimic the bulk region, the first Cu atoms were fixed, while trajectories were recorded at every step for subsequent analysis.
From the final snapshot of the equilibrated 10 ps AIMD trajectories, enhanced sampling simulations were performed using the SG-AIMD method implemented in Vienna Ab-initio Simulation Package (VASP)65. The PBE-D3(BJ) functional with a plane-wave cutoff of 400 eV was employed under NVT conditions at 300 K. The C–C coupling process was modeled by gradually reducing the C–C bond distance at a rate of −0.001 Å step−1 to drive the reaction coordinate. Each trajectory was propagated for 10 ps with tight electronic (10−5 eV) and ionic (<0.03 eV Å−1) convergence criteria. Three independent simulations were performed for each system, and the averaged free-energy profiles were used in the analysis.
DFT calculations
DFT calculations were executed employing the VASP65. The van der Waals correction was considered by applying the DFT-D3 approach64,66. The utilization of projector-augmented plane wave pseudopotentials was employed, coupled with the application of the generalized gradient approximation using the Perdew-Burke-Ernzerhof functional to effectively describe the exchange-correlation functional63,67. The Brillouin zone was sampled with Gamma-centered Monkhorst-Pack mesh sampling for geometry relaxation, using the K-mesh of 0.03 Å−1. A cutoff energy of 500 eV and relaxation using a conjugate gradient algorithm until the total force on each atom was less than 0.03 eV/Å were set throughout this study. The Cu(111) was chosen as the reaction interface and 6 layers of atoms (96 Cu atoms) were considered and the bottom 3 layers were fixed, of which, the supercell with 2*2*2 was constructed. For the influence of K+ caions on the catalyst surface, we constructed three H2O-coordinated K+ models (K-3H2O) and placed them on the Cu(111) surface. The structure-activity relationship was then further elucidated by placing the K-3H2O structure at different distances of 2, 5, and 8 Å from the surface.
Additionally, we constructed 3 layers and 210 Cu atoms (for Cu catalyst) and 3 layers and 210 Cu atoms Cu with Bmim+ cluster attached to the surface (for IL@Cu) to investigate the C–C coupling and reduction of *CH2CHO reaction pathways.
References
He, M., Sun, Y. & Han, B. Green carbon science: efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew. Chem. Int. Ed. 61, e202112835 (2022).
Yang, Y. et al. Operando studies reveal active Cu nanograins for CO2 electroreduction. Nature 614, 262–269 (2023).
Jia, S. et al. Electrochemical conversion of CO2 via C−X bond formation: recent progress and perspective. Chem. Synth. 4, 60 (2024).
Zhang, Y. et al. Low-coordinated copper facilitates the *CH2CO affinity at enhanced rectifying interface of Cu/Cu2O for efficient CO2-to-multicarbon alcohols conversion. Nat. Commun. 15, 5172 (2024).
Gao, X. et al. Intermediate-regulated dynamic restructuring at Ag-Cu biphasic interface enables selective CO2 electroreduction to C2+ fuels. Nat. Commun. 15, 10331 (2024).
Yin, Y. et al. Modulating CO2 electroreduction pathways through controlled ionomer arrangement on catalyst surfaces via solvent dispersion. Innovation 6, 100882 (2025).
Yang, P. & Gao, M. Enrichment of reactants and intermediates for electrocatalytic CO2 reduction. Chem. Soc. Rev. 52, 4343–4380 (2023).
Li, Z. et al. Mesostructure-specific configuration of *CO adsorption for selective CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. 64, e202413832 (2025).
Bi, J. et al. Construction of 3D copper-chitosan-gas diffusion layer electrode for highly efficient CO2 electrolysis to C2+ alcohols. Nat. Commun. 14, 2823 (2023).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Cao, Y. et al. Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions. Nat. Commun. 14, 2387 (2023).
Cao, X., Cha, S. & Gong, M. Interfacial electrical double layer in electrocatalytic reactions: fundamentals, characterizations and applications. Acta Phys. -Chim. Sin 41, 2410018 (2025).
Shayesteh Zeraati, A. et al. Carbon- and energy-efficient ethanol electrosynthesis via interfacial cation enrichment. Nat. Synth. 4, 75–83 (2025).
Liu, Z. et al. Switching CO2 electroreduction toward ethanol by delocalization state-tuned bond cleavage. J. Am. Chem. Soc. 146, 14260–14266 (2024).
Li, J. et al. Twin heterostructure engineering and facet effect boosts efficient reduction CO2-to-ethanol at low potential on Cu2O@Cu2S catalysts. ACS Catal. 14, 266–3277 (2024).
Fu, W. et al. Preserving molecular tuning for enhanced electrocatalytic CO2-to-ethanol conversion. Angew. Chem. Int. Ed. 63, e202407992 (2024).
Zhang, H. et al. Dynamic ionization equilibrium-induced “oxygen exchange” in CO electroreduction. ACS Catal. 14, 10737–10745 (2024).
Zhou, J. et al. Regulating interfacial hydrogen-bonding networks by implanting Cu sites with perfluorooctane to accelerate CO2 electroreduction to ethanol. Angew. Chem. Int. Ed. 64, e202418459 (2024).
Li, J. et al. MOFs-based single/dual-atom catalysts: atomically active sites engineering for efficient CO2 reduction. Chem. Synth. 5, 69 (2025).
Monteiro, M. C. O. et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654–662 (2021).
Zhang, Z. et al. Molecular understanding of the critical role of alkali metal cations in initiating CO2 electroreduction on Cu(100) surface. Nat. Commun. 15, 612 (2024).
Tan, Z. et al. Alkaline ionic liquid microphase promotes deep reduction of CO2 on copper. J. Am. Chem. Soc. 145, 21983–21990 (2024).
Ahmed, Y. et al. Interface modification by ionic liquid for efficient and stable FAPbI3 perovskite solar cells. Acta Phys. Chim. Sin 40, 2303057 (2024).
Zhang, M. et al. Mechanism of efficient electroreduction of CO2 to CO at Ag electrode in imidazolium-based ionic liquids/acetonitrile solution. Appl. Catal. B 359, 124508 (2024).
Dongare, S. et al. A bifunctional ionic liquid for capture and electrochemical conversion of CO2 to CO over silver. ACS Catal 13, 7812–7821 (2023).
Coskun, O. K. et al. Tailoring electrochemical CO2 reduction on copper by reactive ionic liquid and native hydrogen bond donors. Angew. Chem. Int. Ed. 136, e202312163 (2024).
Sha, Y. et al. Anchoring ionic liquid in copper electrocatalyst for improving CO2 conversion to ethylene. Angew. Chem. Int. Ed. 61, e202200039 (2024).
Cai, H. et al. Ionic liquid-induced product switching in CO2 electroreduction on copper reaction interface. Adv. Funct. Mater. 34, 2404102 (2024).
Kang, X. et al. Synthesis of hierarchical porous metals using ionic-liquid-based media as solvent and template. Angew. Chem. Int. Ed. 56, 12683–12686 (2017).
Kang, X. et al. Formation of large nanodomains in liquid solutions near the phase boundary. Chem. Commun. 52, 14286–14289 (2016).
Xu, B. et al. Highly efficient electrocatalytic CO2 reduction to C2+ products on a poly(ionic liquid)-based Cu(0)-Cu(I) tandem catalyst. Angew. Chem. Int. Ed. 61, e20211065 (2022).
Qin, D. et al. Enhanced electrochemical nitrate-to-ammonia performance of cobalt oxide by protic ionic liquid modification. Angew. Chem. Int. Ed. 62, e202304935 (2023).
Kiefer, J., Fries, J. & Leipertz, A. Experimental vibrational study of imidazolium-based ionic liquids: raman and infrared spectra of 1-ethyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide and 1-ethyl-3-methylimidazolium ethyl sulfate. Appl. Spectrosc. 61, 1306–1311 (2007).
Dhumal, N., Noack, K., Kiefer, J. & Kim, H. Molecular structure and interactions in the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide. J. Phys. Chem. A 118, 2547–2557 (2014).
Cao, Y. et al. A transparent, self-healing, highly stretchable ionic conductor. Adv. Mater. 29, 1–9 (2017).
Lian, Z., Dattila, F. & López, N. Stability and lifetime of diffusion-trapped oxygen in oxide-derived copper CO2 reduction electrocatalysts. Nat. Catal. 7, 401 (2024).
Zhang, P. et al. Tuning the surface field by embedding cations into metals to direct the reaction pathway of CO2 electroreduction. CCS Chem 6, 631–640 (2024).
Liu, S. et al. Temperature-dependent pathways in carbon dioxide electroreduction. Sci. Bull. 70, 889–896 (2025).
Gunathunge, C. M. et al. Surface-adsorbed CO as an infrared probe of electrocatalytic interfaces. ACS Catal 10, 11700–11711 (2020).
Wang, P. et al. Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Wang, Y. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Wang, Y. et al. Characterizing surface-confined interfacial water at graphene surface by in situ Raman spectroscopy. Joule 7, 1–11 (2023).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe−N−C single-atom catalyst for enhanced CO2 electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Li, J. et al. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover. Energy Environ. Sci. 12, 2298–2304 (2019).
Zhu, Z. et al. Covalent organic framework ionomer steering the CO2 electroreduction pathway on Cu at industrial-grade current density. J. Am. Chem. Soc. 146, 1572–1579 (2024).
Ovalle, V. J. et al. Correlating hydration free energy and specific adsorption of alkali metal cations during CO2 electroreduction on Au. Nat. Catal. 5, 624–632 (2022).
Wang, S. et al. Manipulating C-C coupling pathway in electrochemical CO2 reduction for selective ethylene and ethanol production over single-atom alloy catalyst. Nat. Commun. 15, 10247 (2024).
Zhao, Z. et al. Highly efficient electroreduction of CO2 to ethanol via asymmetric C-C coupling by a metal-organic framework with heterodimetal dual sites. J. Am. Chem. Soc. 145, 26783–26790 (2023).
Zhan, C. et al. Key intermediates and Cu active sites for CO2 electroreduction to ethylene and ethanol. Nat. Energy 9, 1485–1496 (2024).
Li, Z. et al. A Small-angle X-ray scattering station at Beijing synchrotron radiation facility. Instrum. Sci. Technol. 42, 128–141 (2014).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Sun, Z. et al. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chem 3, 560–587 (2017).
Li, S. et al. Ampere-level CO2 electroreduction with single-pass conversion exceeding 85% in acid over silver penetration electrodes. Nat. Commun. 15, 6101 (2024).
Li, Z. et al. Directing CO2 electroreduction pathways for selective C2 product formation using single-site doped copper catalysts. Nat. Chem. Eng. 1, 159–169 (2024).
Jiao, J. et al. Lattice strain engineering boosts CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. 63, e202409563 (2024).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Heinz, H., Vaia, R. A., Farmer, B. L. & Naik, R. R. Accurate simulation of surfaces and interfaces of face-centered cubic metals using 12−6 and 9−6 lennard-jones potentials. J. Phys. Chem. C 112, 17281–17290 (2008).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Melander, M. M. Frozen or dynamic? — An atomistic simulation perspective on the timescales of electrochemical reactions. Electrochim. Acta. 446, 142095 (2023).
Kuhne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Martyna, G. J., Klein, M. L. & Tuckerman, M. Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643 (1992).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Acknowledgements
We thank the National Natural Science Foundation of China (22273108, 22033009, 22293015, and 22121002), the Youth Innovation Promotion Association CAS (Y2022017), the CAS Project for Young Scientists in Basic Research (YSBR-050), the ICCAS Carbon Neutral Chemistry Program (CCNC-202403) and the National Key Research and Development Program of China (2023YFA1507400) for their financial support of this research. In situ SAXS-XAS measurements were performed on the 1W2B beamline of BSRF.
Author information
Authors and Affiliations
Contributions
Y.Y. and Z.L.: syntheses and characterizations of catalysts. S.L., S.Z., H.W., and L.J.: CO2RR experiments. W.L. and H.Q.: MD and DFT simulations. R.F. and X.S.: collection and analysis of in situ SERS and in situ ATR-SEIRAS spectra data. X.X., Y.X., and Q.Z.: collection and analysis of in situ XAS/SAXS combined technology. Q.Q. and J.Z.: mechanism analysis. X.K. and B.H.: overall design and direction of the project. Y.Y., X.K., and B.H.: preparation of the manuscript with help from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yi Xiao and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, Y., Ling, Z., Liu, S. et al. Ampere-level CO2 electroreduction to multi-carbon oxygenates in acidic electrolyte through surface microenvironment reconstruction. Nat Commun 17, 2353 (2026). https://doi.org/10.1038/s41467-026-68739-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68739-z
