
Abstract
Transitioning to a circular plastics economy will require use of renewable feedstocks, energy-efficient processes, and closed-loop recyclable polymers, such as polyesters. A key challenge lies in sustainably sourcing monomers used to make recyclable polyesters. This work presents a catalytic platform utilizing earth-abundant Cu(x)Ca(1-x)O mixed metal oxides for the oxidative dehydrocyclization of bio-based diols to lactones, which are advantaged for energy-efficient ring-opening polymerization. Operating below 200 °C, at ambient pressure, and without solvent, the process uses air as the sole oxidant, achieving high yields of lactones across a broad substrate scope of C4-8 diols in the liquid phase. The oxidative dehydrocyclization reaction is thermodynamically downhill due to water formation and energy-efficient compared to incumbent, non-redox pathways utilized in fossil carbon-based industrial processes for lactone production. Mechanistic studies reveal facile redox cycling of Cu2+-O(Ca2+)-Cu2+ interfacial sites unique to the developed catalyst. Techno-economic analysis and life cycle assessment estimate 40% lower energy demand and 15% lower GHG intensity per mass of butyrolactone produced compared to the fossil carbon-based route. Liquid-phase oxidative dehydrocyclization offers a promising approach for scalable lactone production from renewable, bio-based diols to enable circular polyesters.
Similar content being viewed by others
Introduction
Global production of primary plastics generated ~2.24 gigatonnes of carbon dioxide equivalent in 2019, representing 5.3% of total global greenhouse gas (GHG) emissions. Importantly, 40% of the total energy use and 75% of the total GHG emissions from primary plastic production occur from the steps prior to polymerization e.g., monomer production1. Moving toward a circular economy necessitates adoption of renewable carbon feedstocks in lieu of fossil-carbon sources, a drastic reduction in energy demands and GHG emissions of processes, especially for monomer production, and development and deployment of closed-loop recyclable polymers (e.g., polyesters)2. Various formulations of circular polyesters capable of facile depolymerization back to polymerizable monomers have been reported, with polyester architecture, composition, and co-monomer identity offering promising levers to tune polymer properties3,4,5,6,7,8,9,10,11. A common strategy to synthesize circular polyesters is the ring-opening polymerization of lactones, which in turn raises the challenge of sourcing lactone monomers sustainably.
In this regard, a promising pathway to access lactones is from bio-derived diols like propanediol, butanediol, and pentanediol, which are readily accessible from waste and renewable carbon sources12,13,14,15,16,17,18,19. Today, bio-based diols are industrially produced at tens-to-hundreds of kiloton/year scale, typically from sugars20,21,22,23. Diols can be catalytically dehydrocyclized to lactones via, as summarized in Fig. 1: (a) non-oxidative (acceptor-less) pathways that co-produce H2, or through (b) oxidative (acceptor-mediated) pathways using oxidants such as O2 or H2O2 that co-produce H2O or hydrogen acceptors like carbonyl compounds that co-produce alcohols24. In the non-oxidative route, one hydroxyl group oxidizes to a carbonyl, forming a hydroxy aldehyde intermediate, which undergoes intramolecular nucleophilic attack to form a cyclo-ester alcohol intermediate. Cyclization is followed by a second dehydrogenation step to yield the final lactone25,26,27,28. The oxidative mechanism proceeds analogously, besides the key difference that H-transfer occurs to an oxidant, e.g., O2, to form H2O instead of evolving H2. Using O2 in the oxidative route makes the overall transformation exergonic and exothermic under standard conditions, eliminating the need for high temperatures if a suitable catalyst is employed. In contrast, the non-oxidative route remains endergonic and endothermic regardless of kinetics, necessitating elevated temperatures18. For instance, the industrial production of γ-butyrolactone from 1,4-BDO (ΔH: +61.6 kJ mol−1) occurs at 300–500 °C29,30, with a co-flow of H2 to keep the metal catalysts reduced. Therefore, redox pathways of dehydrocyclization of diols to lactones require lower intrinsic energy demand and offer improved sustainability for polymerizable monomer production.

A Acceptor-less/non-oxidative and B acceptor-mediated/oxidative dehydrocyclization of diols to lactones. The non-oxidative route produces H2 as the co-product, while the oxidative route produces H2O when O2 is used as the oxidant.
Oxidative dehydrocyclization reactions are often conducted using supported precious metal such as Pt31, Au26,27, or Pd32 supported on oxide supports like SnO231, TiO226, AlOOH27, in the presence of solvents like tri-butyl phosphate, mesitylene, or α,α,α-trifluorotoluene at ambient or high pressures of air. The use of precious elements like Au, Pd, and Pt instead of earth-abundant ones not only adds to the economic cost burden of the process, but also results in significantly higher GHG emissions and carbon footprint of the catalyst. For instance, in comparison to Cu production (mining and concentration), Pd, Pt, and Au production results in three orders of magnitude higher CO2eq emissions per kg of metal33,34. Furthermore, the use of organic solvents presents two major challenges: first, these solvents impose economic and environmental burdens associated with their use and recovery, which are exacerbated under the oxidative reaction conditions, where the co-presence of oxygen/air, organic solvents, and redox-active catalysts may lead to undesirable side reactions or safety risks. Second, solvent-catalyst interactions can adversely affect catalyst stability and performance, potentially inducing structural modification of metal-containing solid catalysts through chelation and leaching, block active sites via competitive adsorption, and interact with the support in ways that promote physical restructuring. Thus, an optimal oxidative dehydrocyclization process would ideally avoid organic solvents and precious-metal catalysts to improve economic feasibility and exhibit low environmental impacts. In this context, the water generated in situ during the oxidative pathway can serve as a benign reaction medium, offering advantages in process safety and environmental impacts. Moreover, the large differences in boiling points between water, diol substrates, and the lactone products enable straightforward product recovery.
To this end, here we develop an earth-abundant mixed metal oxide (MMO) catalyst containing oxides of Ca and Cu that oxidatively dehydrocyclizes neat (solventless), liquid-phase diols, at temperatures below 200 °C, at ambient pressures, using air (O2) as the oxidant. The catalyst developed here achieves quantitative lactone yields, minimizing units and energy-intensive separations downstream of the process. Further, we show that this catalyst can ring-close linear, cyclic, and aromatic C4-8 diols in the liquid-phase to the corresponding lactones. Using differential kinetics and in situ characterization of the catalyst, we show that the Cu-O-Ca interfacial sites readily redox-cycle between +2, +1, 0 oxidation states during reduction and oxidation half cycles, while crystalline CuO and other Cu- and Ca-containing supported catalysts do not under these reaction conditions. Lastly, we develop a process model and conduct techno-economic analysis (TEA) and life cycle assessment (LCA) of this process using 1,4-BDO conversion to γ-butyrolactone as an example reaction, showing that this process is cost competitive and energy-advantaged, leading to lower GHG emissions compared to incumbent fossil carbon-based processes for lactone production.
Results
Catalyst screening via probe reaction and substrate scope
Since literature precedence is the richest for 1,4-BDO to γ-butyrolactone conversion, we decided to use this as a probe reaction to screen catalysts. Typically, redox dehydrocyclization of diols to lactones are catalyzed by redox-active catalysts such as single-atom transition metal catalysts (M-N-C type), and bulk or supported basic oxides, hydroxides, and bifunctional (basic+redox) MMOs35,36,37,38,39. Conversely, non-oxidative/acceptor-less dehydrocyclization reactions are catalyzed by supported transition metals in their metallic state31,40. Taking inspiration from the broader literature on catalytic alcohol dehydrogenation, we recently hypothesized18 that the oxidative cleavage of C–H bonds during oxidative dehydrocyclization of diols to lactones should be achievable over group 10–11 transition metal oxide sites with highly occupied d orbitals that do not exhibit strong Lewis acidity unlike group 5–7 transition metal oxides with fully vacant d orbitals. Moreover, we hypothesized that basic oxide supports or promoters primarily suppress acid-catalyzed dehydration of the diols, which typically occurs on common oxidic supports that contain acidic hydroxyls. Based on this hypothesis, we designed and synthesized a series of Cu(x)Ca(1-x)O MMO catalysts by varying Cu/Ca nominal ratio in the final catalyst.
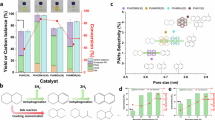
The conditions for our screens were semi-batch, liquid-phase, solventless reactions, at 180 °C to minimize vapor-phase evaporative losses, ambient pressure air (O2) (see Supplementary Fig. S1 for setup details). Typical dehydrocyclization catalysts, including various compositions containing precious metals, single-atom transition metal catalysts, bulk or supported basic oxides, hydroxides, etc, were all found to be inactive (Fig. 2A). Notably, this does not mean these catalysts would be inactive for the probe reaction under all conditions, however, under the conditions targeted here, they do not exhibit measurable reactivity. The Cu(x)Ca(1-x)O MMO catalysts exhibited tunable activity towards lactone production from a wide range of C4-8 diols (Fig. 2B). These diols were screened to demonstrate the catalyst’s activity and the impact of diol size and substituent effects on the catalytic rate. Note that lactone yields in Fig. 2B should not be interpreted as “upper limits”—each lactone can be produced in 100% yield when optimized reaction conditions are used i.e., longer reaction time, higher temperature, lower diol/catalyst ratio, etc. Considering the probe reaction screening results in Fig. 2, in the next sections, we focus on understanding the origin of this unique activity over these Cu(x)Ca(1-x)O MMO catalysts for liquid-phase dehydrocyclization of diols to lactones.

(A) Catalyst screening results of liquid-phase, solventless, ambient-pressure oxidative dehydrocyclization of 1,4-BDO to γ-butyrolactone probe reaction for various families of solid catalysts inspired by literature reports. Reaction setup details are provided in Supplementary Fig. S1. Reaction temperature and time for the screens were held constant at 180 °C and 24 h, respectively. The boiling point of 1,4-BDO and γ-butyrolactone are 235 °C and 204 °C, respectively, and the screening temperature was chosen to minimize evaporative losses. (B) Substrate screening results at an arbitrary but constant condition of 180 °C, diol/catalyst mass ratio: 20, reaction time: 24 h, using Cu0.18Ca0.82O MMO catalyst. NMR spectra are shown in Supplementary Fig. S2. Prior literature precedence has already established the direct and indirect polymerization and depolymerization routes of the polymers formed using the lactones shown in (B). We list the representative studies for various (lactone ↔ polymer) systems for the reader’s convenience: γ-butyrolactone ↔ poly(butyrolactone)3, γ-valerolactone and derivatives → corresponding thiolactones ↔ poly(thioesters)87,88, δ-valerolactone ↔ poly(valerolactone)89, methyl-valerolactone ↔ poly(methyl-valerolactone)90,91, (7) hexahydrophthalide ↔ poly(hexahydrophthalide)5. All yields are NMR based. Figure 2A was created in BioRender (https://BioRender.com/e09t023), and is licensed under CC BY 4.0.
Kinetics and apparent energetics of dehydrocyclization of diols to lactones over Cu(x)Ca(1-x)O mixed metal oxide catalysts
Figure 3A–E summarizes the performance screening of the Cu(x)Ca(1-x)O MMO catalyst series for the probe reaction of 1,4-BDO conversion to γ-butyrolactone. Figure 3A compares O2-rich and O2-limited conditions for catalysts with 1, 5, 10, and 18% Cu. Across all compositions, higher lactone yields are observed under O2-rich (semi-batch) conditions compared to O2-limited (batch), indicating a positive oxygen order. However, we refrain from quantifying the O2 reaction order since pressurizing with air is not feasible from a safety standpoint. These results corroborate the involvement of O2 and suggest a redox mechanism (Fig. 1B) as opposed to a completely non-redox pathway catalyzed by supported metallic Cu catalysts25,28,30,41.

A Probe reaction yield (1,4-BDO conversion to γ-butyrolactone) dependence on open-system (i.e., semi-batch) vs. closed-system (i.e., batch) experiments. B Lactone yields at 180 °C, 24 h, diol/catalyst mass ratio 20 g/g. The shades of burgundy represent Cu(x)Ca(1-x)O MMO catalysts where darker shades imply higher Cu-content, while the green datapoints represent the reference compounds like CuO and Cu powder. The pink data points (bulk compounds CaO and Ca(OH)2) are not shown due to the log-scale x-axis and are indicated with an arrow instead. Error bars are standard deviations of n = 3–15 replicates, and when not visible are hidden behind the data point. C The lactone yield data are used to calculate a nominal STY by normalizing by moles of Cu. D STY as a function of lactone yield. E Catalyst mass-normalized rates used for Koros-Nowak /Madon–Boudart test, showing that as Cu-content increases, the reaction rate increases non-linearly (i.e rate ∝ [Cu]0.5). F Arrhenius plot estimating apparent activation barriers for 1,4-BDO, 1,4-PDO, and 1,5-PDO converting to corresponding lactones over Cu(0.05)Ca(0.95)O MMO catalyst, 500 rpm stirring rate, diol/catalyst mass ratio: 20 g/g. All numerical data shown in this figure are provided in supplementary data tables.
To probe the influence of Cu-content on the catalytic performance, Cu(x)Ca(1–x)O MMO catalysts with x = 0.01–0.9, i.e., Cu: 1–90%, were tested for γ-butyrolactone production, and results summarized in the semi-log plots of Fig. 3B, C, where each catalyst formulation was run at least 3 times; Cu(0.05)Ca(0.95)O was run 15 times, to account for catalyst batch to batch variability and random experimental errors. Reference materials (CuO, Ca(OH)₂,CaO) were found to be inactive. In contrast, lactone yield increased substantially with Cu-content in MMO catalysts (Fig. 3B), though the trend is non-linear, likely due to mass-transfer limitations at high conversions and due to diminishing Cu returns (i.e., not all added Cu atoms form active surface sites). Site-time yield (STY) analysis (Fig. 3C) reveals a volcano trend with highest STY at ~5% Cu-content; higher or lower Cu-contents leads to lower STY values. We note that although convenient, analyzing data based on the nominal STY framework overlooks the actual number of active surface sites since the STY parameter simply lumps surface and bulk copper into a single quantity via the assumption that all added Cu is active, underestimating the true reaction rate per active Cu site42,43, Furthermore, catalyst reuse up to three times did not evince any change in performance at partial conversions. In agreement, elemental analysis via ICP-OES (Supplementary Fig. S3) of a wide range of representative samples from reactions at 180–200 °C, representing lactone yields in 5–90% range, using Cu-content in the 5–18% range also show that Cu remains below detection limit of 2 ppm in the filtrate (residual diol + product lactone + product H2O). At very high Cu-content, e.g., 90%, minor Cu-leaching occurred, presumably via chelation44. Therefore, we focus on 5–18% Cu-content catalysts for further analysis.
Correlating nominal STY with lactone yield or extent of reaction, shown in Fig. 3D, reveals that initial STY values are nearly an order of magnitude higher than STY values estimated at higher reaction extents. Initial STYs plateau around ~0.055 molelactone per moleCu per min. The observed decline in STY with higher conversion extents is attributed to possible mass-transfer limitations due to diol depletion at higher conversions and possible product inhibition. Product inhibition was confirmed by spiking reactions with lactone (50% by volume lactone), which leads to ~88% reduction in STY values due to the lactone competitively binding to the active sites. Conversely, spiking the diol with H2O does not affect the STY up to 10% by volume H2O, while further H2O addition up to 50% leads to ~60% decrease in the STY values compared to the neat diol case (Supplementary Fig. S4). Accordingly, all kinetic analyses were performed using initial STYs at ≤15% lactone yield to minimize transport and product inhibition artifacts, though these non-idealities cannot be completely eliminated in the liquid-phase, semi-batch operation.
In Fig. 3E, a log-log plot of initial STY for Cu(x)Ca(1-x)O MMOs catalysts as a function of Cu-content (5–18%) reveals a slope of −0.5, indicating that increasing Cu leads to a lower STY due to the formation of inactive crystalline CuO phase (vide infra). The same panel also includes a Madon–Boudart plot (ln(rate) as a function of ln(Cu content)), which yields a slope of +0.5 – suggestive of internal mass-transfer limitations even in the initial STY data45,46,47. However, this test is best suited to supported catalysts where the active phase is on the external surface and accessible. In MMOs, a significant fraction of Cu resides in the bulk and is inaccessible to the substrate, meaning that even in the absence of mass-transfer limitations, a slope of +1 is unlikely43,45,46.
Figure 3F summarizes the apparent activation barriers for lactone formation as a function of substrate structure by comparing: (i) 1,4-BDO with 1,4-pentanediol (1,4-PDO) to assess the effect of methyl substitution, and (ii) 1,4-BDO with 1,5-pentanediol (1,5-PDO) to evaluate chain length and lactone ring size effects. Under identical conditions (fixed stirring rate, temperature range, and Cu0.05Ca0.95O catalyst), initial rates yield apparent barriers of 147, 117, and 89 kJ mol⁻¹ for γ-butyrolactone, γ-valerolactone, and δ-valerolactone formation, respectively. While derived from initial rates, these activation barriers reflect combined effects of intrinsic factors e.g., ring strain and alkyl substituents (Thorpe-Ingold effect)18, as well as extrinsic factors e.g., substrate size and active-site accessibility, diol viscosity, O2 solubility in each diol at a given temperature, and product inhibition from lactone and water binding during reaction. Experimental error ≤10% in Ea estimates can be reasonably assumed, as repeats at 180 °C for 1,4-BDO and 1,5-PDO indicate the average STY values to be 0.092 ± 0.002 and 0.136 ± 0.010 mollactonemolcu−1 min−1, respectively. The error in activation barrier estimates stems from factors like temperature variations during the holding time, errors in mass measurements, etc.
Molecular structure of Cu–O–Ca active sites in Cu(x)Ca(1-x)O MMO catalysts
Next, we probed the origin of the activity observed in Figs. 2 and 3 over the Cu(x)Ca(1-x)O MMO catalysts by studying their molecular structure before and during reaction with in situ Raman spectroscopy. Figure 4A shows Raman spectra of bulk references (Ca(OH)2 and CuO) and the MMO catalyst. The band assignments are summarized in Table 1 for the reviewer’s convenience. Curious readers are directed to Supplementary Fig. S5 for extended comparisons. Since the Raman spectra of Ca(OH)2 and CuO are well-known in the broader literature cited in Table 1, we do not discuss specific band assignments for bulk compounds for brevity.

A Ex situ Raman spectra (785 nm laser) of bulk reference compounds including Ca(OH)2 and CuO and Cu0.05Ca0.95O MMO catalyst. B Ex situ Raman spectra (785 nm laser) of Cu(x)Ca(1-x)O catalysts as a function of Cu-content from 5 to 18% Cu. Bands marked with (*) represent vibrational modes of CuO and Ca(OH)2 related domains present in the CuCaO MMO catalysts. C In situ Raman spectra evolution of Cu0.05Ca0.95O catalyst during reaction with 1,4-BDO at 25°C, 150 °C under inert (i.e., reduction half cycle) and under air (i.e., oxidation half cycle). The gray shaded spectrum in each panel is the reference Raman spectrum of as-synthesized Cu0.05Ca0.95O. The as-synthesized spectrum is presented in each panel to draw the eye to the Raman bands assigned to Cu-O-Ca interfacial bonds (500–710 cm−1) that diminish in intensity upon reduction of the catalyst and then reappear upon reoxidation of the catalyst.
On the other hand, the Cu0.05Ca0.95O catalyst, synthesized by adding aqueous Cu2+ to Ca(OH)2 and annealing at 500 °C under N2, shows distinct Raman bands, not present in the reference compounds including Ca(OH)2 and CuO. Unique Raman bands observed in the MMO catalysts include lattice breathing modes at 122 and 140 cm−1, Cu–O vibrations at 253 cm−1, and overlapping contributions from CuO and Ca(OH)2 domains at 274–290 and 355 cm−1, respectively. Bands at 518, 550, and 615 cm−1 – absent in both Ca(OH)2 and CuO/Cu2O – are attributed to interfacial Cu–O(Ca2+)–Cu bonds unique to the catalyst. The Cu–O(Ca2+)–Cu vibrations are blue-shifted relative to Cu–O–Cu in CuO due to shorter Cu–O bond lengths and increased bond polarity from the higher Pauling electronegativity of Cu2+ (~1.5) than Ca2+(~1.1)48. The higher polarity of the Cu–O(Ca2+)–Cu bonds is expected to make the corresponding vibrational modes more Raman-active that Cu–O–Cu vibrations in CuO. We note that our band assignments align with prior reports of Ca–Cu mixed oxides such as CaCuO249,50,51. Lastly, as with other Ca-containing materials, surface CO32− bands at 1086–1400 cm⁻¹ stem from adsorbed CO2 on residual basic Ca(OH)2 domains.
In Fig. 4B, the Raman spectra of Cu(x)Ca(1-x)O MMO catalysts are summarized for different Cu loadings from 5 to 18%, demonstrating that the molecular structure of the catalysts does not drastically change, and that the interfacial Cu–O–Ca sites, signified by Raman bands in the 500–700 cm−1 region, are present in these MMO catalysts at all studied Cu loadings. At higher copper content, bulk-like CuO domains form, which would convolute the Raman signatures, preventing clean analysis possible in the case of lower Cu-content samples. Summarily, the Raman analysis shows that Cu–O(Ca2+)–Cu interfacial sites are present in the MMO, and also that the Cu migrates to some extent into the lattice of the Ca(OH)2 matrix, as seen from the presence of unique lattice breathing modes below 200 cm−1. Cu2+ ions are smaller than Ca2+ ions (73 pm vs. 99 pm radius, respectively)48, which increases the likelihood that at least some of the added copper diffuses into the bulk matrix during catalyst synthesis and annealing steps, thus forming an MMO instead of a supported metal oxide.
To further examine the formation of MMO, in situ DRIFTS was used to study the hydroxyls in the Ca(OH)2 with and without Cu addition (Supplementary Fig. S6A). The spectra of dehydrated samples clearly show that the parent Ca(OH)2 exhibits IR peaks at 3639 and 3710 cm−1 from O–H vibrations of the bulk and surface hydroxyls. Upon adding 5 wt% Cu to Ca(OH)2, the 3639 and 3710 cm−1 peaks entirely disappear, instead of experiencing a small decrease from stoichiometric anchoring of copper species on the surface hydroxyl expected when a supported metal oxide forms, as opposed to a MMO52,53. The disappearance of all peaks from bulk and surface hydroxyls implies, in agreement with Raman spectroscopy results, that the Ca(OH)2 matrix undergoes significant restructuring to form a Cu(x)Ca(1-x)O MMO instead of forming an overlayer of CuOx species anchored on the hydroxyls of calcium oxide/hydroxide support.
Figure 4C shows in situ Raman spectra of a representative catalyst, Cu0.05Ca0.95O before, during, and after reaction with 1,4-BDO. The bottom panel shows the spectrum at room temperature of catalyst with a small amount of liquid 1,4-BDO added to the catalyst under inert gas flow. Even at room temperature, the Raman bands from interfacial Cu–O(Ca2+)–Cu sites at 500–700 cm−1 significantly diminish in intensity due to the diol adsorbing on these sites. The diminishing of Cu–O(Ca2+)–Cu Raman bands also confirms that these bonds are at the catalyst surface where the diol adsorbs. Conversely, the sharp band at 344–354 cm−1 attributed to Ca(OH)2 remains unaffected indicating that those domains are not interacting with the diol or in the bulk. As the temperature increases to ~150 °C under an inert atmosphere, the diol dehydrogenates, and the catalyst reduces i.e., the reduction half-cycle (RHC) occurs. The Cu–O(Ca2+)–Cu vibrations completely diminish, indicating that the interfacial sites are consumed during the reduction half cycle, yielding oxygen vacancies. In a later step, as the gas feed is switched to air to initiate the oxidation half-cycle (OHC), rapid reoxidation of the interfacial sites leads to full recovery of the corresponding Raman bands in the 500–700 cm−1 region. In fact, a comparison of the Raman spectrum of as-synthesized/fresh catalyst (black shaded spectrum) shows that the reoxidizied MMO catalyst after a full sequence of reduction and oxidation quantitatively matches the fresh catalyst and no indication of restructuring is observed, proving the facile, reversible redox cycling of the Cu–O(Ca2+)–Cu sites during liquid-phase dehydrocyclization of diols to lactones.
In an analogous experiment to Fig. 4C, the surface intermediates were tracked via in situ DRIFTS at identical gas flows and temperature over this representative catalyst, shown in Supplementary Fig. S6B. As a function of time, the diagnostic IR peak at 1760 cm−1 corresponding to the C=O stretching evolves, indicating lactone formation, confirming that as Cu–O–Ca sites redox cycle (Raman experiment), the forward reaction occurs to yield the lactone on the MMO surface (DRIFTS experiment).
Electronic structure & redox dynamics of Cu–O–Ca active sites in Cu(x)Ca(1-x)O MMO catalysts
Next, the electronic structure and redox dynamics of the Cu0.05Ca0.95O catalyst were further investigated using a combination of in situ UV–Vis diffuse reflectance spectroscopy (UV-VisDRS) and X-ray absorption spectroscopy (XAS), shown in Fig. 5A–C. Figure 5A displays the UV–Vis DRS spectra of the fresh MMO catalyst, which shows minimal absorption in the 400–800 nm range—where d–d transitions typically appear in bulk CuO as well as reported UV–Vis spectra of catalysts containing copper like copper-loaded zeolites, copper-containing mixed metal oxides, supported copper oxide catalysts54,55,56,57. Typically, d–d transitions in Cu2+Ox bulk compounds like CuO and copper-loaded catalysts exhibit substantial absorption bands in 400–800 nm region, especially in the 600–800 nm region. The suppressed absorption suggests a symmetric octahedral environment around Cu2+ (where d–d transitions are Laporte-forbidden) or increased electron density on Cu2+, shifting its character toward Cu+ (d10, with no possible d–d transitions). The 200–400 nm ligand-to-metal charge transfer (LMCT) region also shows distinct features. By analogy to literature on Cu-zeolites, the 216 nm band is assigned to isolated CuOx species, and the 340 nm band to oligomeric CuxOy species with Cu2+–O–Cu2+ bridging bonds54,55,56,58. A unique 268 nm band is observed and tentatively attributed to LMCT from interfacial Cu2+–O(Ca2+)–Cu2+ sites. The blue-shifted absorption of the LMCT compared to Cu2+–O–Cu2+ is consistent with higher electron density in the Cu–O–Ca, requiring more energy for LMCT due to reduced d-orbital vacancy.

A In situ UV-VisDRS of fresh Cu0.05Ca0.95O MMO catalyst and during reaction with 1,4-BDO at 150 °C under inert (RHC) and under air (OHC). B In situ XANES at 150 °C of Cu0.05Ca0.95O MMO catalyst during reaction with 1,4-BDO. As the reaction proceeds (0–75 min), the catalyst reduces. C The relative population of Cu2+, Cu+, and Cu0 extracted from panel (B) as a function of reaction time at 150 °C. H2-TPR profiles of D crystalline CuO, crystalline Cu2O, and E Cu(x)Ca(1-x)O MMO catalysts with 5–36% Cu-content. F Results of TPR peak deconvolution quantifying crystalline CuO and CuCaO domains in the MMO catalysts.
Once the diol is added to the catalyst and temperature increased to 150 °C to initiate the reaction, all LMCT bands diminish rapidly and the highest energy LMCT at 216 nm shifts to even higher energy, which coincides with the catalyst color darkening. If the catalyst and diol are kept under an inert gas flow at 150 °C, the reoxidation cannot occur and the copper species stay reduced (reduction half-cycle), as indicated by the band intensities diminishing. In the next step, when air is turned on while the temperature is held constant at 150 °C, the LMCT bands rapidly appear again and within a few minutes, the intensity of the absorption bands at 216, 268, and 340 nm increases, indicating reoxidation (oxidation half-cycle). The redox cycling experiment in Fig. 5A confirms that the MMO catalysts contain unique Cu2+–O(Ca2+)–Cu2+sites distinct from crystalline reference compounds like CuO or Cu-loaded catalysts. Upon reaction with 1,4-BDO without air present, the Cu in the MMO catalyst reduces, which causes the LMCT bands to diminish. Upon reoxidation with air, the LMCT bands reappear, indicating reformation of the original sites. These redox cycling experiments are in good agreement with the redox cycling experiments studied via Raman spectroscopy in Fig. 4.
Figure 5B summarizes XAS results, focusing on X-ray near-edge absorption spectra (XANES) analysis. Compared to reference materials (CuO, Cu₂O, and Cu foil representing Cu2+, Cu+, and Cu0, respectively), the fresh Cu0.05Ca0.95O catalyst shows pre-edge and white line features consistent with octahedral Cu2+. We note that XAS probes the bulk structure, and minority sites e.g., Cu1+–O–Ca surface sites may be obscured by higher population bulk species unless advanced analysis is applied. Anyhow, upon heating the catalyst with 1,4-BDO to 150 °C under oxygen-limited conditions using a capillary tube reactor, a rapid shift of the pre-edge and white line to lower energies signifies reduction of Cu2+ species as the diol dehydrocyclizes.
Through linear combination fitting of the normalized XANES data, the fractional population of copper species in various oxidation states is estimated, shown in Fig. 5C. As the reaction progresses, Cu1+ and Cu0 both co-evolve. The formation of Cu0 from the onset of reaction indicates a 2-electron redox cycle (Cu2+ ⇌ Cu0). Conversely, formation of the Cu1+ species also at the onset of reaction indicates another possibility, i.e., two consecutive 1-electron reduction steps occurring on the same copper site (Cu2+ ⇌ Cu+ ⇌ Cu0). If two consecutive steps occur and no re-oxidation occurs in between, then Cu1+ should form transiently as it eventually will be consumed to form Cu0, which is inactive for the reaction. However, we observe a steady persistence of Cu1+ species (15-20% of total Cu), suggesting that some reoxidation occurred during the reaction. All three oxidation states of copper, Cu2+, Cu1+, and Cu0, were found to co-exist, where the relative population balance will be a function of redox dynamics and O2 availability. Taken together, the in situ XAS data quantitatively verify the redox cycling insights from Raman and UV–Vis and indicate that the dehydrocyclization of diols to lactones over the Cu0.05Ca0.95O MMO catalyst occurs via a redox mechanism as opposed to a non-redox pathway, where the redox cycle seems to operate via both 1- and 2-electron pathways.
Next, the reduction of copper species was quantified with H2-temperature-programmed reduction experiments (H2-TPR), summarized in Fig. 5D–F. The reduction peaks for reference CuO and Cu2O are shown in Fig. 5D, indicating that CuO reduces to Cu0 via a single-step 2e− process at 217 °C59. Likewise, Cu2O reduced to Cu0 via a single-step 1e− process at 306 °C. While the exact peak reduction temperature (Tp) values are a function of H2 flow rate, temperature ramp rate, amount of catalyst loaded, hydrodynamics of the TPR reactor, and domain size (i.e., micro vs. nano vs. isolated CuOx surface species), the trends observed here match the literature, where CuO is found to be more reducible than Cu2O60,61,62,63.
In contrast, as shown in Fig. 5E, the Cu(x)Ca(1-x)O catalysts exhibit a Tp significantly higher than CuO but lower than Cu2O. Since the domain size is expected to be lower for Cu(x)Ca(1-x)O than CuO, it is expected that the Cu2+→Cu0 transformation should have occurred at temperatures lower than that of CuO, unless Cu2+ species are strongly interacting with the Ca matrix via Cu–O–Ca bonds, which in turn leads to the increase in Tp value, in agreement with the unique Raman bands in Fig. 4, and blue-shifted LMCT in Fig. 5A. The shift in the Tp to higher temperatures upon Ca2+ interaction with Cu-O sites agrees with broader literature on binary and ternary cuprates being less reducible than CuO64. It may suggest higher electron density in the Cu centers in cuprates, yielding Cu(2-δ)+ type sites, which would shift the reduction temperature to higher values, noting Cu1+ in Cu2O reduces less readily than Cu2+ in CuO.
Figure 5F shows the results of TPR peak deconvolution, where two Gaussian peaks centered at 217 and 243 °C, corresponding to bulk-like CuO and Cu(x)Ca(1-x)O MMO phase, respectively, were fit to the TPR profiles (Supplementary Fig. S7). The deconvoluted envelopes were used to estimate the amount of reducible Cu in Cu(x)Ca(1-x)O MMO catalysts, assuming Cu:H2 ratio of 1:160,61,62,63. Deconvolution results, in agreement with STY values reported in Fig. 3, show that as the overall Cu content increases in the Cu(x)Ca(1-x)O MMOs, a significant portion of the Cu is tied up in a bulk CuO-like phase (~30% Cu for Cu0.36Ca0.64O), which explains the decrease in STY values with increasing Cu content, noting that bulk CuO is inactive for this reaction and may physically block the Cu–O(Ca2+)–Cu active sites in the MMO domains. The TPR results also corroborate the Raman findings where these materials exhibit a mixture of phases including CuO, CuCaO, Ca(OH)2 and are not homogeneously one phase e.g., CuCaO2.
Techno-economic analysis and life cycle assessment of bio-lactone production
A process model for catalytic dehydrocyclization of diols to corresponding lactones was developed in Aspen Plus using bio-derived 1,4-BDO to γ-butyrolactone as the representative case, using experimental insights and kinetic data from the experimental work. Figure 6A shows a simplified process flow diagram. The process model is based on a continuous supply of bio-derived 1,4-BDO from an upstream facility with a production capacity of 65 kilotonnes per annum (ktpa), yielding 63 ktpa of γ-butyrolactone. The conversion of 1,4-BDO to γ-butyrolactone is conducted in batch-operated bubble column reactors at ambient pressure and 195 °C, with a 1,4-BDO-to-catalyst mass ratio of 20. Complete conversion is achieved within 24 h and reactor scheduling ensures semi-continuous operation (Supplementary Fig. S8). Compressed air is introduced at a flow rate of 0.62 vessel volumes per minute to provide the required oxygen for the reaction. Water formed as a byproduct of the reaction is continuously removed from the reactor in the exhaust gas stream, preventing its accumulation in the liquid phase. Complete assumptions and a detailed process flow diagram are provided in Supplementary Table S1 and Supplementary Figs. S8–11.

A Simplified process flow diagram for ambient-pressure, solventless, liquid-phase oxidative dehydrocyclization of 1,4-BDO to γ-butyrolactone (abbreviated in figure labels as GBL for clarity) using the Cu(x)Ca(1-x)O MMO catalyst. B Breakdown of direct installed cost in millions of dollars ($MM). All costs and prices shown are on a 2022 USD basis. C Feedstock contribution to total annual operating cost in millions of dollars per year ($MM/yr). D Breakdown of other cost contributions in $MM/yr for the annual operating cost. E Breakdown of minimum selling price in $/kg GBL by cost category. The five-year average price for GBL is shown for reference; since industrial market prices for GBL are not publicly available, import data from the U.S. Trade Census (Supplementary Fig. S12) was used to estimate its cost. F Breakdown of greenhouse gas (GHG) emissions. G Normalized results comparing fossil resource use, land use and water use for bio-based and fossil-based GBL. Error bars represent standard deviation determined from Monte Carlo analysis with n = 1000 runs. H Univariate sensitivity analysis for 1,4-BDO conversion to GBL. I Illustration of dependence of saturation oxygen solubility on conversion. Impact of second generation (gen. 2) sugars for 1,4-BDO production and renewable utility usage on J the MSP ($/kg GBL) and K the GHG emissions (kg CO2/kg GBL). Renewable electricity is modeled using the Regional Energy Development System ReEDS mid-case scenario for 95% decarbonization by 203592, while renewable heat is derived from biomethane. Second-generation sugars are derived from corn stover and are modeled using the life cycle inventory reported in ref. 79. Model inputs and detailed diagrams are provided in Supplementary Figs. S8–12 and Supplementary Tables S1–3. All numerical data shown in this figure are provided in supplementary data tables.
The resulting mass and energy balances from the process model were used to inform the TEA. Key assumptions and parameters for the TEA are summarized in Supplementary Tables S2, 3. The total direct installed capital cost was estimated at $15 MM and the annual operating cost at $165 MM. The reactor represents the largest capital investment, accounting for 79% of the total installed cost (Fig. 6B). For operational expenses, feedstock represents the dominant expense, contributing 96% of the total annual operating cost while other costs for raw materials and utilities contribute less than 1% each (Fig. 6C, D). Discounted cash flow analysis estimated a minimum selling price of $2.89/kg for γ-butyrolactone, with feedstock costs ($2.43/kg65, Supplementary Fig. S12) contributing 88% overall (Fig. 6E). Compared to the average 5-year market price of $3.53/kg γ-butyrolactone66, this represents an 18% reduction.
The mass and energy balances derived from the process models were also used to develop a foreground process inventory to conduct a cradle-to-gate LCA for the process, with a functional unit of 1 kg GBL. γ-Butyrolactone produced from bio-based 1,4-BDO65 resulted in a 15% reduction in GHG emissions and 40% reduction in energy use compared to traditional fossil-based industrial route (Fig. 6F). This bio-based γ-butyrolactone production route also resulted in a 5–40% reduction in other environmental impacts including water use, particulate matter emissions, and ionizing radiation compared to fossil-based γ-butyrolactone (Fig. 6G). The majority (95–100%) of all environmental impacts for bio-based γ-butyrolactone come from the 1,4-BDO feedstock, while steam, electricity, and cooling water, combined, are responsible for less than 2% of overall climate change impacts. The 1,4-BDO feedstock is derived from glucose from corn maize. The agricultural impacts of corn production, driven by land occupation, fertilizer inputs and runoff, and use of agricultural machinery, therefore result in a >15% increase in land-use, acidification, ecotoxicity, and marine eutrophication compared to fossil-based γ-butyrolactone production.
A univariate sensitivity analysis, shown in Fig. 6H, suggests that the MSP is respondent to changes in feedstock price, plant capacity, and reaction rate. A 66% increase and 50% decrease in feedstock price results in a 60% increase and 47% decrease in MSP, respectively. When the reaction rate is reduced by an order of magnitude, the MSP increases by 18%; however, a tenfold increase in the reaction rate yields only a 1% decrease in MSP. Among all parameters analyzed, feedstock price is the only factor capable of raising the MSP of bio-derived γ-butyrolactone above the prevailing average market price of $3.53/kg. The solubility of oxygen in the liquid phase is a critical parameter in ensuring complete conversion, and its variation with the extent of reaction is presented in Fig. 6I. Given that all the environmental impacts are driven by 1,4-BDO production, the sensitivity analysis did not result in notable changes. For example, a change in air flow rates resulted in minimal changes (less than 1%) to environmental impacts. A decarbonization scenario was also considered wherein the electricity and heat used to produce the 1,4-BDO as well as γ-butyrolactone are assumed to be renewable (Fig. 6J, K). By utilizing renewable energy, a 22% and 43% reduction in GHG emissions is achieved for bio-based and fossil-based γ-butyrolactone, respectively, making the fossil-based route slightly lower in terms of GHG emissions (3.12 kg CO2e/kg fossil-γ-butyrolactone vs. 3.68 kg CO2e/kg bio-based γ-butyrolactone). Bio-based 1,4-BDO could also be produced from second-generation sugars rather than glucose (i.e., first-generation sugars), further lowering the impacts of bio-based γ-butyrolactone by 45%. Therefore, when combining the impacts of utilizing second-generation sugars for 1,4-BDO, along with renewable energy, the GHG emissions for bio-based butyrolactone have the potential to be roughly 45% lower than fossil-based.
Discussion
We found no literature-reported catalyst capable of converting neat diols to lactones in the liquid phase at ambient pressure using air (O2) as the sole oxidant. Guided by mechanistic site requirements detailed in our prior work18, we designed an earth-abundant Cu(x)Ca(1–x)O MMO catalyst featuring both basic and redox-active sites that enable diol dehydrocyclization to lactones at mild conditions. In situ spectroscopies (Raman, DRIFTS, UV–Vis DRS, XAS; Figs. 3–5) reveal the presence of unique Cu–O(Ca)–Cu interfacial sites not found in crystalline reference oxides. These sites are identified by characteristic Raman bands (500–700 cm⁻¹), a distinct 268 nm LMCT in UV–Vis DRS, weak basicity in CO2-DRIFTS, intermediate H₂-TPR reduction temperature to Cu2+ and Cu+, and rapid redox cycling observed in XAS. Collectively, the data indicate that Cu–O(Ca)–Cu sites drive liquid-phase C–H bond activation at <200 °C, which crystalline oxides and hydroxides like Ca(OH)₂, CaO, CuO, Cu2O, and physical mixtures of these compounds cannot accomplish.
Based on the characterization data, a mechanism is proposed (Fig. 7) that involves stepwise C–H activation in the diol on Cu2+–O(Ca)–Cu sites, forming oxygen vacancies and reducing the Cu species. Since UV–Vis and XAS cannot resolve between 1- and 2-electron redox pathways due to coexisting Cu2+, Cu+, and Cu0 species, we refrain from assigning a specific active oxidation state. In future studies, we will further probe the redox dynamics to delineate if a singular oxidation state constitutes the active site or if different oxidation states contribute partially to the overall activity. The first dehydrogenation of the diol results in the production of H2O, which is expected to leave the catalyst surface faster than the reaction time scales under these conditions of 150–200 °C. The hydroxyaldehyde intermediate produced is expected to cyclize rapidly via a non-surface-catalyzed step, owing to nucleophilic attack of the hydroxyl oxygen on the electron-deficient carbonyl carbon, resulting in a lactol. The lactol then undergoes a second dehydrogenation step to form lactone and a second equivalent of H2O. During the reduction half-cycle, if the second dehydrogenation occurs on a different Cu2+–O(Ca)–Cu site than the first dehydrogenation step, two Cu+1 sites would be formed after the reduction half-cycle is complete (i.e., 2 Cu2+→2 Cu1+). Conversely, if the second dehydrogenation occurs on the same site, which would now be present as Cu+–O(Ca)–Cu, Cu0 is expected to form (i.e., Cu2+→Cu0). Once Cu0 forms, it may reoxidize back to Cu1+/2+ depending on the nature and concentration of the dominant oxidant species at the catalyst surface, for example, hydroxyl ions OH−, atomic oxygen O2−, super oxide O2−, etc. Conversely, some Cu0 may persist under reaction at steady state and contribute by serving as adsorption sites for dissociative adsorption of dissolved O2 or as active sites for C–H cleavage non-oxidatively (Fig. 1) if the oxidant coverage is low67,68,69,70.

[] represents an oxygen vacancy. 2e− redox cycle involves a single -O(Ca)-Cu-O(Ca)- site for the entire catalytic cycle involving two dehydrogenation steps, while 1e− redox cycle involves two different -O(Ca)-Cu-O(Ca)- sites, with each site catalyzing one dehydrogenation step. The pink annotations highlight the oxidation state of the Cu site involved.
Importantly, at low conversions, e.g., <5% 1,4-BDO conversion at short reaction times, the putative hydroxy aldehyde and lactol intermediates were not detected by NMR, with the only species detected in the samples being diol and the corresponding lactone. NMR quantification suggests that the first dehydrogenation may be the rate-limiting step and once that occurs, the cyclization and the second dehydrogenation steps are energetically downhill and occur rapidly, leading to the lactone product. Had the second dehydrogenation been as rate-limiting as the first dehydrogenation, the reaction intermediate would presumably be observed, which is not the case in our experiments, although we acknowledge the limitations of NMR quantifications at low concentrations.
Notably, the current data do not offer quantitative insights into the kinetics and rate-limitations stemming from transport, dissolution, and diffusion of gaseous O2 in air into the reaction media (diol primarily), followed by O2 activation to replenish the oxygen vacancies on the catalyst surface during the oxidation half cycle. Since the apparent activation barriers estimated are 147 kJ mol−1, 117 kJ mol−1, and 89 kJ mol−1 for the formation of γ-butyrolactone, γ-valerolactone, and δ-valerolactone, respectively, it can be reasonably argued that at least the estimated barriers are not entirely dominated by O2 activation kinetics as O2 activation during the oxidation half cycle is expected to be similar owing to identical reaction conditions across the experiments. Under the tested conditions, the catalyst is clearly able to complete the redox cycle and since an open-system configuration was used for these tests instead of a completely sealed system, we expect that especially at low diol conversions, the headspace of the reactors have sufficient air to prevent O2 limitations.
TEA and LCA for the conversion of 1,4-BDO to γ-butyrolactone highlighted the high impact of the feedstock contributions across both economic and environmental metrics. For the MSP, feedstock cost accounted for 88% of the total with process operations contributing only 12%. The estimated MSP of γ-butyrolactone is $2.89/kg, representing an 18% reduction compared to the five-year average market price ($3.53/kg)66, highlighting the economic viability of the proposed process. Across environmental impacts, similar findings were observed with feedstock accounting for 95–100% across most metrics. Although we only evaluated conversion of 1,4-BDO, the process design is representative of other diols that result in high conversion to lactone product or do not require final purification. While differences in utility demands, reactor sizing, etc. would be expected for conversion other diols, the dominance of the feedstock contribution would still yield costs and impacts that closely resemble those of the diol feedstock.
Beyond feedstock contributions, oxygen solubility emerged as a critical parameter, as complete conversion of 1,4-BDO to γ-butyrolactone within 24 h depends on sustained oxygen dissolution in the liquid phase. Under constant reaction rate and airflow, and in a mass transfer-limited regime, oxygen is immediately and stoichiometrically consumed with 1,4-BDO upon dissolution, keeping the required oxygen mass transfer rate constant. As the reaction proceeds, both 1,4-BDO concentration and oxygen solubility decline (Fig. 6B and Supplementary Fig. S10). This lowers the concentration driving force for oxygen transfer. Hence, maintaining a constant oxygen mass transfer rate requires an increasing the gas-liquid mass transfer coefficient (kLa) as conversion progresses. This is achieved through a system design that combines mechanical agitation with intensive aeration. A tenfold increase in reaction rate shortens reaction time, reduces reactor count, and lowers capital costs. However, it also demands a tenfold increase in oxygen mass transfer rate, necessitating higher kLa, more intensive aeration and mixing, and greater energy input, resulting in only 1% reduction in the MSP. The combination of mixing, aeration, and changing liquid-phase composition during reaction results in liquid entrainment. To minimize losses of reactants and products, the evolved gas stream is condensed, and liquid is recycled back to the reactor, while water is vented with exhaust gas71. The exhaust gas stream retains less than 0.03 mol% of 1,4-BDO and γ-butyrolactone (Supplementary Fig. S11).
In the LCA, the biogenic carbon associated with bio-based γ-butyrolactone is not accounted for. This is because the end-of-life impacts of γ-butyrolactone are not considered, and therefore, the fate of the biogenic carbon is unknown. For example, if γ-butyrolactone is polymerized to poly(γ-butyrolactone) (PGBL) there is the possibility of biogenic carbon storage if the polymer is landfilled or recycled at end-of-life. Recent studies have shown the complete recyclability potential of PGBL5,72,73, which would enable the biogenic carbon to remain stored in the material as it is continuously depolymerized and repolymerized via circular methods. Based on the carbon content of γ-butyrolactone, this biogenic carbon would result in 2 kg CO2e/kg polymer of carbon storage, resulting in a 51% reduction in GHG emissions compared to fossil-based γ-butyrolactone. Given the significant contribution of the 1,4-BDO feedstock to environmental impacts, it is important to consider additional 1,4-BDO production routes. In this study, we examine second-generation sugars from corn stover as an alternative feedstock to glucose from corn maize, which further lowered the environmental impacts of bio-based γ-butyrolactone by 45%, and also improved other environmental impacts such as ecotoxicity, water use, particulate matter emissions and human toxicity. Future work should consider additional 1,4-BDO production routes and expand the scope of LCA to include end-of-life and account for biogenic carbon to draw further comparisons to fossil-based γ-butyrolactone.
Catalytic dehydrocyclization of scalable bio-derived diols offers a direct route to lactone monomers for sustainable polyesters. We report a low-temperature (<200 °C), ambient-pressure process using a Cu(x)Ca(1–x)O MMO catalyst that converts neat liquid diols to lactones with air as the oxidant. Our results identify Cu–O–Ca interfacial sites unique to this catalyst as the active sites for low-temperature C–H activation in liquid diols, as confirmed by in situ Raman spectroscopy (peaks at 500–610 cm−1 that diminish during RHC and reappear during OHC), DRIFTS (C=O formation), UV–Vis DRS and XAS (Cu2+ reduction to Cu1+ and Cu0), and H2-TPR (higher Tp of Cu(x)Ca(1-x)O than CuO). Taken together, we posit that this catalyst operates through a redox mechanism involving both 1e− and 2e− pathways, with Cu cycling among Cu2+, Cu1+, and Cu0. NMR quantification suggests that initial dehydrogenation is likely rate-limiting; cyclization and the second dehydrogenation proceed rapidly. Apparent activation barriers are 147, 117, and 89 kJ mol−1 for γ-butyrolactone, γ-valerolactone, and δ-valerolactone formation from corresponding diols, respectively. It suffices to note that detailed mechanistic analysis including the reactive intermediates, rate-determining steps, and active-site dynamics will be addressed separately in future studies. TEA suggests that bio-derived 1,4-BDO feedstock price dominates economics (~88%), yielding an MSP of $2.89/kg, below the market price of fossil-derived lactone ($3.53/kg). LCA suggests that the bio-1,4-BDO feedstock also accounts for ~95–100% of environmental impacts; other process operations including the catalytic steps contribute little to environmental footprint of this process owing to the lack of solvent use, minimal unit op requirements, earth-abundant catalyst composition, and mild temperatures.
Overall, Cu(x)Ca(1–x)O MMO catalysts provide a viable platform for converting bio- and waste-derived diols to lactones for circular polyester production. Remaining challenges include long residence times and limited efficiency of these catalysts towards four- and seven-membered lactone production, motivating further compositional and process optimization. The mechanistic insights developed here may extend to upgrading other oxygen- and nitrogen-containing feedstocks, including amino-alcohol dehydrocyclization to lactams. Moreover, future work should also explore additional BDO production routes and include end-of-life impacts in LCA for more accurate comparisons
Methods
Catalyst synthesis
The catalysts that were inactive were not analyzed further for physicochemical properties and instead we focused on Cu(x)Ca(1-x)O series. Sigma Aldrich Ca(OH)2 catalog # 239232 (BET 32 m2g−1) was used as a support where the hydroxide yielded a more robust anchoring of copper species than calcium oxide due to the presence of surface hydroxyls. 0.5 mL g−1 of copper nitrate solution was impregnated sequentially with drying at room temperature in between. For example, 1.0 g of Cu(0.05)Ca(0.95)O was synthesized as follows: 0.95 g of Ca(OH)2 was impregnated with 0.5 mL of aq. solution containing 0.19 g of Cu(NO3)2.3H2O. 0.19 g Cu(NO3)2.3H2O contains 7.86 × 10−4 moles Cu, which would give 0.05 g Cu (metal basis) in 0.95 g Ca(OH)2 support. Once the impregnation process was complete, the resulting dark blue powder was dried in the fumehood overnight. The dried powder was then further dried in a muffle furnace under N2 flow at 120 °C for 4 h, followed by annealing at 500 °C for 4 h. The annealed catalysts looked white to light gray in color. CuO (Sigma Aldrich #241741), Cu2O (Sigma Alrich #566284), Cu metallic powder (Sigma Aldrich #203122) were all sourced commercially and used as reference materials.
All supported catalysts e.g., Cu-Ca/SiO2, Cu-Ca-Na/Al2O3, etc. were prepared via incipient-wetness-impregnation (IWI) method. Appropriate amounts of copper(II)nitrate trihydrate (Sigma Aldrich 61194-100G), calcium nitrate tetrahydrate (Sigma Aldrich C4955), sodium hydroxide (Supelco 1064980500) were dissolved in deionized water via sonication, and the resulting solution impregnated sequentially onto various supports via a micropipette. The following supports were purchased and used without further treatment/purification besides grinding and sieving to <200 µm particle size when needed: Sasol Puralox SBA-200 γ-Al2O3 (BET: 217 m2 g−1), Sigma Davisil amorphous SiO2 catalog # 243981 (BET: 300 m2 g−1), ACS Materials mesoporous SiO2 catalog # MSB15011SBA-15 (BET 550 m2 g−1). All IWI samples were thermally dried and annealed as described above. All bulk bases were sources from Sigma Aldrich as well.
Reaction tests and product analysis
All catalytic tests were conducted in custom-built 12-well microwave vial setup, (pictured in Supplementary Fig. S1). Each vial was 20 mL volume, 23 ×75 mm (Agilent 5183-4474), sealed with a crimp top cap with PTFE lining and silicone septa (Biotage 352298). In a typical trial, 2.00 mL of 1,4-BDO (2.04 g diol; 0.027 mmol diol) was pipetted into each vial using a calibrated micropipette, followed by the addition of 100 mg of catalyst powder sieved to <140 mesh, and a 10 mm rare-earth elliptical stir bar. Typically, the diol/catalyst mass ratio was maintained in the range of 20–80 for various experiments to control the extent of diol conversion. Each vial was stirred at 500 RPM throughout reaction. The vials are housed in a thick-walled aluminum block (60 mm thick sides) to ensure isothermal operation and heat retention over prolonged reaction times. Additionally, the aluminum block was further insulated by a 3D-printed polyetherimide thermoplastic jacket (ULTEM™ 9085 resin from Stratasys) to ensure isothermal operation. A 3 ml plastic syringe, with the plunger removed, fitted with a narrow bore needle (0.5 ×25 mm) is fixed to each vial through the crimp-top septum to allow for the escape of volatile vapors or gases that are expected to form during reaction and prevent pressure build up. The narrow-bore needle is intentionally used to leverage Venturi effect to promote condensation of the vapor in the syringe as the vapors exit into the syringe and cool down to enhance mass-balance closure. For all tests, a mass balance of >90% was ensured.
After reaction, the vials were cooled down to room temperature. 2.00 ml of methanol was added into the vials and vigorously shaken to help mix the solvent with the diol, lactone mixture. The resulting solution was then filtered using a 0.2 micron syringe filter to remove catalyst powder from the solution. 100 μL of the diol, lactone, methanol solution was pipetted into an NMR tube and diluted with 0.3 ml of DMSO solvent for all NMR analysis. NMR was conducted on a 400 MHz Bruker instrument at ambient temperature. Chemical shifts were referenced to internal solvent resonances and reported as parts per million (ppm) relative to tetramethylsilane. The following abbreviations (and their combinations) are used to label the multiplicities: s (singlet), d (doublet), t (triplet), m (multiplet) and br (broad). Representative samples covering 10-90% lactone yield from NMR quantification were cross-verified with quantitative GC-MS were the diol + lactone + methanol mixtures (without DMSO) were directly analyzed. GC-MS and NMR quantifications of lactone yields resulted in excellent agreement, but NMR quantifications are preferred as GC-MS can lead to ring-opening of strained lactones, and dehydration of some diols in the stationary phase due to high temperatures used in the GC-MS.
Safety consideration
During oxidative conditions, care must be taken to prevent an uncontrolled exotherm and ensure that explosive mixtures of diol and air are not formed by exercising heat management, O2 concentration management, and product separation strategies. During oxygen-lean conditions, if Cu0 forms due to lack of reoxidation and initiates H2 production (as opposed to H2O) via the non-redox route, further care must be taken to prevent H2-related hazards.
In situ Raman spectroscopy
The Raman spectra were obtained with a Renishaw InVia Raman spectrometer equipped with 785, 514, and 325 nm (excitation) lasers. For this study, the ex situ measurements were conducted with the 785 nm (1200 l/mm grating) and 514 nm lasers (2400 l/mm grating) and Leica N PLAN EPI objective with 20× magnification, using a spectral range of 100–1400 cm−1. Only CuO and Cu2O required the 514 nm laser. All Raman spectra were calibrated to an internal Si (111) standard at 521 cm−1. These measurements were recorded in extended scan mode with 60 s exposure time with three accumulations. The 785 nm laser was used at 10% power and the 514 nm laser at 20% power.
The in situ measurements were taken with a Linkam CCR1000 cell attached to the MS 20 Encoded Stage and Olympus LMPlanFLN objective with 50× magnification utilizing a 60 s exposure time with three accumulations in extended scan mode. The cell was packed with quartz wool, a quartz filter, then a flattened layer of catalyst on top. The cell temperature was controlled by a Linkam T95-HT system. Under 8.0 ml min−1 N2 (Airgas, Industrial grade) flow, spectra of the reference samples were taken at room temperature and during heat treatment to obtain dried material spectra. For the catalyst samples, room temperature in situ spectra were recorded under 8 mL/min N2. After room temperature spectra were obtained, the catalysts were doped with 1,4-BDO then ramped at 1 °C min−1 to 150 °C under 8.0 ml min−1 N2 and held at 150 °C for ~1 h while measurements were taken. After the spectra were recorded at 150 °C under N2, the gas was switched to air (Airgas, Medical grade) and held at 150 °C under air for ~1 h while taking spectra during oxidation.
H2 temperature programmed reduction
H2-TPR were carried out using a homebuilt setup utilizing an SRI 8690 as a TCD. Approximately 50 mg of each sample was packed in a quartz reactor tube in between quartz wool and quartz chips. The samples were reduced in 50 sccm 5% H2/N2 (Airgas, Industrial grade) and heated to 400 °C with a ramp rate of 2 °C/min. After the TPR the flow gas was switched to He (Airgas, UHP grade). The temperature was then lowered to 150 °C for N2O pulse chemisorption experiments. A 250-μL sample loop was filled with 10% N2O/He (Airgas, Industrial grade) and flown to the samples using He as the carrier gas. A total of 100 pulses were performed on each sample. A cold trap filled with liquid nitrogen was utilized to condense unreacted N2O.
In situ UV-VisDR spectroscopy
UV–Vis DR spectra were collected using an Agilent Cary 5000 UV–vis-NIR spectrophotometer equipped with a Harrick Praying Mantis diffuse reflectance attachment and reaction chamber. In situ spectra reported herein were taken at room temperature post dehydration and during heat treatment to obtain dried material spectra. For the catalyst samples, room temperature in situ spectra were recorded under 50 mL/min N2. After room temperature spectra were obtained, the cell was opened and catalyst was doped with 1-2 drops of liquid 1,4-BDO. The temperature was ramped at 5 °C min−1 to 150 °C under 50 ml min−1 N2 and held at 150 °C for ~1 h while measurements were taken (constituting the reduction half cycle). After the spectra were recorded at 150 °C under N2, the gas was switched to 10% air (Airgas, Medical grade) and held at 150 °C under air for ~30 min while taking spectra during oxidation half cycle.
X-ray absorption spectroscopy
XAS data were collected at beamline 9-3 of Stanford Synchrotron Radiation Lightsource (SSRL). The beamline was configured to Cu K-edge using a double crystal monochromator with Si (220) phi 0 crystals. Data were collected as continuous scan mode (~2 min/scan) using WebXAS/CXAS software. The energy was calibrated to a reference foil of Cu (7.5 μm thickness), which was set to 8979.0 eV. Beam size for ex situ and in situ measurements were 1.0 mm × 3 mm and 1.0 mm and 0.5 mm, respectively. The ex situ samples were fresh Cu0.05Ca0.95O MMO powder, post-reaction Cu0.05Ca0.95O MMO powder (on a filter paper). Powder samples were pressed into 7 mm pellets (of about 20 mg, diluted with cellulose if necessary) and X-ray absorption coefficient retrieved in transmission mode (µ(E)≈ln(I0/I1)), while data for the filter paper sample was collected in fluorescence mode (µ(E) ≈ IF/I0).
The in situ XAS data for Cu0.05Ca0.95O MMO catalyst with 1,4 BDO was also recorded in fluorescence mode. The Co-ACCESS/SSRL experimental cell was modified to hold a vertical one-end-closed glass capillary of 8 cm length, 0.9 mm diameter and 10 micrometer wall thickness. About 5 mg catalyst powder was added at the bottom of the capillary, followed by 15 mg of 1,4 BDO (about 1:3 dilution) resulting in a slurry of catalyst-reactant between 0.5 and 2.0 cm from the bottom of the capillary. The top of the capillary was open to atmosphere. Radiative-convective heating was provided using resistant coils with PID controller pre auto-tuned to 150 °C. The X-ray measurement position was about 1 cm from the bottom with the active catalyst presence. A thermocouple was inserted into the capillary (touching the slurry e.g., from 1.5 cm from the bottom). The sample was heated from 25 °C with 3 °C/min ramp rate to 150 °C, kept for 75 min at this temperature (e.g., 30 in situ scans), and then cooled at 10 °C /min to 25 °C, while collecting continuous XAS scans at all steps. Calibration, normalization and linear combination fitting of XAS data was performed using Athena software. The three standard spectra used in linear combination fitting range of −20 + 30 eV were Cu foil (as Cu0), Cu2O (as Cu+1), and the room temperature scan for the Cu0.05Ca0.95O in 1,4 BDO (as Cu2+) therefore enabling the estimation of the fraction of copper species during the reaction.
Process modeling, TEA, and LCA
Process simulations for the catalytic conversion of 1,4-BDO to GBL were performed using Aspen Plus V14. The feedstock, bio-derived 1,4-BDO, was assumed to be sourced from a facility with 65 ktpa capacity, aligned with the planned scale of Qore’s commercial plant74. The NRTL activity coefficient model was employed to represent non-ideal behavior in the liquid phase, while the vapor phase was assumed to be ideal under the ambient pressure conditions considered. A detailed process description and corresponding stream compositions are provided in Supplementary Figs. S8–12 and Supplementary Table S1, respectively. The material and energy balances derived from the simulations served as the basis for estimating CAPEX, and OPEX associated with the process. These financial inputs were subsequently used to construct discounted cash flow (DCF) models to evaluate the economic viability of the system. The minimum selling price (MSP) of γ-butyrolactone—defined as the price required to achieve a net present value (NPV) of zero over a 30-year plant lifetime—was determined through this analysis. Further details regarding the process simulation method and TEA can be found in Supplementary Tables S2, S3.
LCA was conducted using Brightway75, and version 2.11 of the Activity Browser76 to evaluate the production of 1,4-BDO from biologically derived 1,4-BDO and compare against conventional fossil-based production. The LCA follows a cradle-to-gate system boundary and uses a functional unit of 1 kg of GBL. Life cycle stages analyzed include the cultivation of bio-based feedstocks (corn/corn stover), production of BDO, and final conversion to bio-based γ-butyrolactone. The ReCiPe 2016 v1.03 midpoint H impact assessment method77 was used for all impact categories except for climate change, which was analyzed using the Intergovernmental Panel on Climate Change (IPCC) 2021 methodology. Foreground data for γ-butyrolactone production from bio-based BDO are obtained from the process models developed herein using Aspen plus. Background data for bio-based 1,4-butanediol from glucose were developed based on the work of Satam et al.65, and background data for bio-based γ-butyrolactone production as well as life cycle inventories for traditional fossil-based γ-butyrolactone production were derived from ecoinvent v3.9.178. Glucose production for the base-case scenario for bio-based BDO production is also derived from ecoinvent, while the second-generation sugars scenario is derived from literature79. For the decarbonization scenarios, the renewable electricity grid is based on the mid-case scenario modeled in the Regional Energy Development System (ReEDS) for 95% decarbonization by 2035. The renewable heat scenario leverages an ecoinvent life cycle inventory for decarbonized heat from biomethane sources, For the sensitivity analysis, the standard deviations for each environmental impact category were derived from a Monte Carlo assessment using 10,000 iterations. The uncertainty assessment was conducted using a semi-quantitative pedigree matrix approach outlined in our previous work80.
Data availability
All data are available from the corresponding author, Gregg T. Beckham (gregg.beckham@nlr.gov), upon request. Additionally, supplementary experimental details regarding the reaction setup, additional experimental results including NMR spectra, product inhibition studies, IR spectra, ICP-OES results, process-flow diagram and TEA/LCA details. Supplementary data tables also contain numerical data from Fig. 3 and Fig. 6 of main text.
References
Karali, N., Khanna, N. & Shah, N. Climate Impact of Primary Plastic Production (Lawrence Berkeley National Laboratory, 2024).
Aarsen, C. V., Liguori, A., Mattsson, R., Sipponen, M. H. & Hakkarainen, M. Designed to degrade: tailoring polyesters for circularity. Chem. Rev. 124, 8473–8515 (2024).
Hong, M. & Chen, E. Y. X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of γ-butyrolactone. Nat. Chem. 8, 42–49 (2016).
Tang, X. & Chen, E. Y. X. Chemical synthesis of perfectly isotactic and high melting bacterial poly(3-hydroxybutyrate) from bio-sourced racemic cyclic diolide. Nat. Commun. 9, 2345 (2018).
Zhu, J.-B., Watson, E. M., Tang, J. & Chen, E. Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science 360, 398–403 (2018).
Shi, C. et al. Design principles for intrinsically circular polymers with tunable properties. Chem 7, 2896–2912 (2021).
Li, X.-L. et al. Dual recycling of depolymerization catalyst and biodegradable polyester that markedly outperforms polyolefins. Angew. Chem. Int. Ed. 62, e202303791 (2023).
Zhang, Z., Gowda, R. R. & Chen, E. Y. X. Chemosynthetic P4HB: a ten-year journey from a “non-polymerizable” monomer to a high-performance biomaterial. Acc. Mater. Res. 5, 1340–1352 (2024).
Zhou, L. et al. Chain-end controlled depolymerization selectivity in α,α-disubstituted propionate PHAs with dual closed-loop recycling and record-high melting temperature. J. Am. Chem. Soc. 146, 29895–29904 (2024).
Kim, M. S. et al. A review of biodegradable plastics: chemistry, applications, properties, and future research needs. Chem. Rev. 123, 9915–9939 (2023).
Lowe, J. R., Martello, M. T., Tolman, W. B. & Hillmyer, M. A. Functional biorenewable polyesters from carvone-derived lactones. Polym. Chem. 2, 702–708 (2011).
Cortright, R. D., Davda, R. R. & Dumesic, J. A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 418, 964–967 (2002).
He, J. et al. New catalytic strategies for α,ω-diols production from lignocellulosic biomass. Faraday Discuss. 202, 247–267 (2017).
Burt, S. P. et al. Production of 1,6-hexanediol from tetrahydropyran-2-methanol by dehydration–hydration and hydrogenation. Green. Chem. 19, 1390–1398 (2017).
Huang, K. et al. Conversion of furfural to 1,5-pentanediol: process synthesis and analysis. ACS Sustain. Chem. Eng. 5, 4699–4706 (2017).
Eagan, N. M., Kumbhalkar, M. D., Buchanan, J. S., Dumesic, J. A. & Huber, G. W. Chemistries and processes for the conversion of ethanol into middle-distillate fuels. Nat. Rev. Chem. 3, 223–249 (2019).
Stadler, B. M., Wulf, C., Werner, T., Tin, S. & de Vries, J. G. Catalytic approaches to monomers for polymers based on renewables. ACS Catal. 9, 8012–8067 (2019).
Kiani, D. et al. Production of bio-based lactones as monomers for circular polymers. Nat. Rev. Chem. 9, 749–765 (2025).
Krishna, S. H. et al. Oxygenated commodity chemicals from chemo-catalytic conversion of biomass derived heterocycles. AIChE J. 64, 1910–1922 (2018).
Cargill. Bio-based coatings, https://www.cargill.com/bioindustrial/coatings (2025).
ACS. Cargill to build biobased 1,4-butanediol plant, https://pubs.acs.org/doi/10.1021/cen-09922-buscon2 (2021).
Gantrade. Bio-Based 1,4-Butanediol (BDO), https://www.gantrade.com/products/bio-based-14-butanediol-bdo (2025).
Geno. Bio-based BDO, https://www.genomatica.com/products/ (2025).
Tan, X. et al. Lactonization of diols over highly efficient metal-based catalysts. ChemSusChem n/a, e202400909 (2024).
Ichikawa, N., Sato, S., Takahashi, R., Sodesawa, T. & Inui, K. Dehydrogenative cyclization of 1,4-butanediol over copper-based catalyst. J. Mol. Catal. A Chem. 212, 197–203 (2004).
Huang, J., Dai, W.-L., Li, H. & Fan, K. Au/TiO2 as high efficient catalyst for the selective oxidative cyclization of 1,4-butanediol to γ-butyrolactone. J. Catal. 252, 69–76 (2007).
Huang, J., Wang, Y., Zheng, J., Dai, W.-L. & Fan, K. Influence of support surface basicity and gold particle size on catalytic activity of Au/γ-AlOOH and Au/γ-Al2O3 catalyst in aerobic oxidation of α,ω-diols to lactones. Appl. Catal. B Environ. 103, 343–350 (2011).
Huang, Z. et al. Hydrogenation of γ-butyrolactone to 1,4-butanediol over CuCo/TiO2 bimetallic catalysts. ACS Catal. 7, 8429–8440 (2017).
Schwarz, W., Schossig, J., Rossbacher, R., Pinkos, R. & Höke, H. In Ullmann’s Encyclopedia of Industrial Chemistry 1–7 (Wiley, 2019).
Jochen, H. et al. Dehydrogenation of 1,4-butanediol to γ-butyrolactone. USA patent (1999).
Touchy, A. S. & Shimizu, K. -i Acceptorless dehydrogenative lactonization of diols by Pt-loaded SnO2 catalysts. RSC Adv. 5, 29072–29075 (2015).
Tana, T. et al. Non-plasmonic metal nanoparticles as visible light photocatalysts for the selective oxidation of aliphatic alcohols with molecular oxygen at near ambient conditions. Chem. Commun. 52, 11567–11570 (2016).
Torrubia, J., Valero, A. & Valero, A. Energy and carbon footprint of metals through physical allocation. Implications for energy transition. Resour. Conserv. Recycling 199, 107281 (2023).
Han, L. et al. Multifunctional high-entropy materials. Nat. Rev. Mater. 9, 846–865 (2024).
Zhong, W., Liu, H., Bai, C., Liao, S. & Li, Y. Base-free oxidation of alcohols to esters at room temperature and atmospheric conditions using nanoscale Co-based catalysts. ACS Catal. 5, 1850–1856 (2015).
Tang, D. et al. Aerobic oxidative lactonization of diols at room temperature over defective titanium-based oxides in water. J. Catal. 418, 237–246 (2023).
Bagley, M. C., Lin, Z., Phillips, D. J. & Graham, A. E. Barium manganate in microwave-assisted oxidation reactions: synthesis of lactones by oxidative cyclization of diols. Tetrahedron Lett. 50, 6823–6825 (2009).
Bhanushali, J. T. et al. Tailoring the catalytic activity of basic mesoporous Cu/CeO2 catalyst by Al2O3 for selective lactonization and dehydrogenation of 1,4-butanediol to γ-butyrolactone. Catal. Commun. 143, 106049 (2020).
Mitran, G., Nguyen, T. L. P. & Seo, D.-K. Effect of solvent, in the sol–gel synthesis of CoAl2O4, on the structure and catalytic properties in 1,4-butanediol dehydrocyclization. React. Chem. Eng. 8, 1901–1913 (2023).
Bhatia, A., Kannan, M. & Muthaiah, S. Ruthenium-promoted acceptorless and oxidant-free lactone synthesis in aqueous medium. Synlett 30, 721–725 (2019).
Liu, H., Jiang, Y., Zhao, H. & Hou, Z. Preparation of highly dispersed Cu catalysts from hydrotalcite precursor for the dehydrogenation of 1,4-butanediol. J. Ind. Eng. Chem. 102, 251–259 (2021).
Schweitzer, N. M., Gounder, R. & Rioux, R. M. Addressing Rigor and Reproducibility in Thermal, Heterogeneous Catalysis (1.00). Zenodo https://doi.org/10.5281/zenodo.8029159 (2023).
Kiani, D. & Wachs, I. E. Practical considerations for understanding surface reaction mechanisms involved in heterogeneous catalysis. ACS Catal. 14, 16770–16784 (2024).
Danks, A. E., Hall, S. R. & Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 3, 91–112 (2016).
Kiani, D. & Wachs, I. E. The conundrum of “Pair Sites” in Langmuir–Hinshelwood reaction kinetics in heterogeneous catalysis. ACS Catal. 14, 10260–10270 (2024).
Kiani, D., Ibrahim, F., Hayden, S., Hermans, I. & Beckham, G. T. Understanding the origin of negative temperature dependence and activity of N-coordinated cobalt sites during ethylene dimerization. Appl. Catal. B Environ. Energy 365, 124952 (2025).
Madon, R. J. & Boudart, M. Experimental criterion for the absence of artifacts in the measurement of rates of heterogeneous catalytic reactions. Ind. Eng. Chem. Fundam. 21, 438–447 (1982).
Li, K. & Xue, D. Estimation of electronegativity values of elements in different valence states. J. Phys. Chem. A 110, 11332–11337 (2006).
Di Castro, D. et al. Raman spectroscopy study of the interface structure in (CaCuO2)n/(SrTiO3)m superlattices. Appl. Phys. Lett. 103, 191903 (2013).
Kan, D., Yamanaka, A., Terashima, T. & Takano, M. Preparation and optical properties of single-crystalline CaCuO2 thin films with infinite layer structure. Phys. C Superconductivity 412-414, 298–302 (2004).
Fouad, O. A. et al. Synthesis, characterization and application of some nanosized mixed metal oxides as high heat resistant pigments: Ca2CuO3, Ca3Co2O6, and NiSb2O6. J. Alloy. Compd. 537, 165–170 (2012).
Chakrabarti, A. & Wachs, I. E. Molecular structure–reactivity relationships for olefin metathesis by Al2O3-supported surface MoOx sites. ACS Catal. 8, 949–959 (2018).
Kiani, D. & Baltrusaitis, J. A spectroscopic study of supported-phosphate-catalysts (SPCs): evidence of surface-mediated hydrogen-transfer. ChemCatChem 13, 2064–2073 (2021).
Bravo-Suárez, J. J., Subramaniam, B. & Chaudhari, R. V. Ultraviolet–visible spectroscopy and temperature-programmed techniques as tools for structural characterization of Cu in CuMgAlOx mixed metal oxides. J. Phys. Chem. C. 116, 18207–18221 (2012).
Angelici, C., Velthoen, M. E. Z., Weckhuysen, B. M. & Bruijnincx, P. C. A. Effect of preparation method and CuO promotion in the conversion of ethanol into 1,3-butadiene over SiO2–MgO catalysts. ChemSusChem 7, 2505–2515 (2014).
Smeets, P. J., Groothaert, M. H. & Schoonheydt, R. A. Cu based zeolites: a UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 110, 303–309 (2005).
Giordanino, F. et al. Characterization of Cu-exchanged SSZ-13: a comparative FTIR, UV-Vis, and EPR study with Cu-ZSM-5 and Cu-β with similar Si/Al and Cu/Al ratios. Dalton Trans. 42, 12741–12761 (2013).
Lezcano-Gonzalez, I. et al. Determining the storage, availability and reactivity of NH3 within Cu-Chabazite-based Ammonia Selective Catalytic Reduction systems. Phys. Chem. Chem. Phys. 16, 1639–1650 (2014).
Gu, M. et al. Structure–activity relationships of copper- and potassium-modified iron oxide catalysts during reverse water–gas shift reaction. ACS Catal. 11, 12609–12619 (2021).
Wang, B., Jin, M., An, H., Guo, Z. & Lv, Z. Hydrogenation performance of acetophenone to 1-phenylethanol on highly active nano Cu/SiO2 catalyst. Catal. Lett. 150, 56–64 (2020).
Fierro, G. et al. A study of anomalous temperature-programmed reduction profiles of Cu2O, CuO, and CuO-ZnO catalysts. J. Catal. 148, 709–721 (1994).
Kim, J. Y., Rodriguez, J. A., Hanson, J. C., Frenkel, A. I. & Lee, P. L. Reduction of CuO and Cu2O with H2: H embedding and kinetic effects in the formation of suboxides. J. Am. Chem. Soc. 125, 10684–10692 (2003).
Villarroel-Rocha, J. & Gil, A. Modeling the temperature-programmed reduction of metal oxide catalysts by considering the particle-size distribution effect. Chem. Eng. J. 487, 150722 (2024).
Klissurski, D. & Rives, V. High-temperature superconductors in catalysis. Appl. Catal. A Gen. 109, 1–44 (1994).
Satam, C. C., Daub, M. & Realff, M. J. Techno-economic analysis of 1,4-butanediol production by a single-step bioconversion process. Biofuels Bioprod. Bioref. 13, 1261–1273 (2019).
United States Census Bureau USA Trade Import and Export Data, https://usatrade.census.gov/ (2025).
van Bree, R. A. B. & Kroes, G. J. O2 dissociation on Cu(111) dynamics on a novel screened hybrid van der Waals DFT potential energy surface. J. Phys. Chem. C. 128, 19182–19196 (2024).
Junell, P., Ahonen, M., HirsimÄKi, M. & Valden, M. Influence of surface modification on the adsorption dynamics of O2 on Cu{100}. Surf. Rev. Lett. 11, 457–461 (2004).
Hodgson, A., Lewin, A. K. & Nesbitt, A. Dissociative chemisorption of O2 on Cu(110). Surf. Sci. 293, 211–226 (1993).
Orozco, I. et al. In situ studies of methanol decomposition over Cu(111) and Cu2O/Cu(111): effects of reactant pressure, surface morphology, and hot spots of active sites. J. Phys. Chem. C. 125, 558–571 (2021).
Seo, W.-W., Yim, J.-H., Kang, J. W. & Lim, J. S. Isobaric vapor–liquid equilibrium data of binary mixtures of [water + 2,3-butanediol] and [water + 1,4-butanediol] at 40, 50, 60, 66.7, 80, and 101 kPa. Korean J. Chem. Eng. 41, 1457–1466 (2024).
Xu, G. & Wang, Q. Chemically recyclable polymer materials: polymerization and depolymerization cycles. Green. Chem. 24, 2321–2346 (2022).
Liu, Y., Wu, J., Hu, X., Zhu, N. & Guo, K. Advances, Challenges, and Opportunities of Poly(γ-butyrolactone)-Based Recyclable Polymers. ACS Macro Lett. 10, 284–296 (2021).
Cargill to build biobased 1,4-butanediol plant. CEN Glob. Enterp. 99, 10–10 (2021).
Mutel, C. Brightway: an open source framework for life cycle assessment. J. Open Source Softw. 2, 236 (2017).
Steubing, B., De Koning, D., Haas, A. & Mutel, C. L. The activity browser—an open source LCA software building on top of the brightway framework. Softw. Impacts 3, 100012 (2020).
Huijbregts, M. A. et al. ReCiPe2016: a harmonised life cycle impact assessment method at midpoint and endpoint level. Int. J. Life cycle Assess. 22, 138–147 (2017).
Wernet, G. et al. The ecoinvent database version 3 (part I): overview and methodology. Int. J. Life Cycle Assess. 21, 1218–1230 (2016).
Mokwatlo, S. C. et al. Bioprocess development and scale-up for cis,cis-muconic acid production from glucose and xylose by Pseudomonas putida. Green. Chem. 26, 10152–10167 (2024).
Uekert, T. et al. Life cycle assessment of enzymatic poly(ethylene terephthalate) recycling. Green. Chem. 24, 6531–6543 (2022).
Padanyi, Z. V. The Raman spectrum of Ca(OH)2. Solid State Commun. 8, 541–543 (1970).
Balachandran, U. & Eror, N. G. Laser-induced Raman scattering in calcium titanate. Solid State Commun. 44, 815–818 (1982).
Lu, B. et al. Spatially resolved product speciation during struvite synthesis from magnesite (MgCO3) particles in ammonium (NH4+) and phosphate (PO43–) aqueous solutions. J. Phys. Chem. C. 123, 8908–8922 (2019).
Xu, J. F. et al. Raman spectra of CuO nanocrystals. J. Raman Spectrosc. 30, 413–415 (1999).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of cuiii oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Luo, M.-F., Fang, P., He, M. & Xie, Y.-L. In situ XRD, Raman, and TPR studies of CuO/Al2O3 catalysts for CO oxidation. J. Mol. Catal. A Chem. 239, 243–248 (2005).
Xia, Y., Yuan, P., Zhang, Y., Sun, Y. & Hong, M. Converting non-strained γ-valerolactone and derivatives into sustainable polythioesters via isomerization-driven cationic ring-opening polymerization of thionolactone intermediate. Angew. Chem. Int. Ed. 62, e202217812 (2023).
Yuan, P. et al. Library of stereoregular polythioesters for stereocomplex formation enabled by isomerization-driven cationic ring-opening polymerization. Angew. Chem. Int. Ed. e202501485, https://doi.org/10.1002/anie.202501485.
Li, X.-L., Clarke, R. W., Jiang, J.-Y., Xu, T.-Q. & Chen, E. Y. X. A circular polyester platform based on simple gem-disubstituted valerolactones. Nat. Chem. 15, 278–285 (2023).
Schneiderman, D. K. et al. Chemically recyclable biobased polyurethanes. ACS Macro Lett. 5, 515–518 (2016).
Brutman, J. P., De Hoe, G. X., Schneiderman, D. K., Le, T. N. & Hillmyer, M. A. Renewable, degradable, and chemically recyclable cross-linked elastomers. Ind. Eng. Chem. Res. 55, 11097–11106 (2016).
Ho, J. et al. Regional Energy Deployment System (reeds) Model Documentation (version 2020) (National Renewable Energy Lab.(NREL), Golden, CO (United States), 2021).
Acknowledgements
Funding was provided by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Materials and Manufacturing Technologies Office (AMMTO), and Bioenergy Technologies Office (BETO). This work was performed as part of the BioOptimized Technologies to keep Thermoplastics out of Landfills and the Environment (BOTTLE) Consortium and was supported by AMMTO and BETO at the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. The BOTTLE Consortium includes members from SLAC. D.K. was in part supported by the Director’s Fellowship - Laboratory Directed Research and Development (LDRD) Program at NREL. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. We thank Professor Eugene Chen (CSU) and Dr. Tobias Hull (NREL) for their insightful comments and discussion.
Author information
Authors and Affiliations
Contributions
Conceptualization, D.K., G.R., G.T.B.; methodology, D.K., G.R., F.I., O.B., J.S.D., A.P., E.V.R.; investigation, D.K., G.R., O.B., F.I., J.S.D., A.P. E.V.R.; writing—original draft, D.K., G.R., O.B., F.I., J.S.D., A.P., E.V.R.; writing—review & editing, D.K., G.R., O.B., F.I., J.S.D., A.P., E.V.R., G.T.B., I.H., S.R.B.; funding acquisition, G.T.B., I.H., S.R.B.; resources G.T.B., I.H., S.R.B.; supervision, G.T.B., I.H., S.R.B.
Corresponding authors
Ethics declarations
Competing interests
D.K., G.R., and G.T.B. have filed a provisional patent application related to this work, No.63/790,923. Other authors have no competing interests to declare.
Peer review
Peer review information
Nature Communications thanks Saikat Dutta, Bhari Mallanna Nagaraja, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kiani, D., Rosetto, G., Ibrahim, F. et al. Solventless, ambient-pressure production of bio-based lactones over earth-abundant, mixed metal oxide catalysts for circular polyesters. Nat Commun 17, 2804 (2026). https://doi.org/10.1038/s41467-026-69362-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69362-8
