
Abstract
Protecting crops from diseases is vital for the sustainable agricultural systems that are needed for food security. Introducing functional resistance genes to enhance the plant immune system is highly effective for disease resistance, but identifying new immune receptors is resource intensive. We observed that functional immune receptors of the nucleotide-binding domain leucine-rich repeat (NLR) class show a signature of high expression in uninfected plants across both monocot and dicot species. Here, by exploiting this signature combined with high-throughput transformation, we generated a wheat transgenic array of 995 NLRs from diverse grass species to identify new resistance genes for wheat. Confirming this proof of concept, we identified new resistance genes against the stem rust pathogen Puccinia graminis f. sp. tritici and the leaf rust pathogen Puccinia triticina, both major threats to wheat production. This pipeline facilitates the rapid identification of candidate NLRs and provides in planta gene validation of resistance. The accelerated discovery of new NLRs from a large gene pool of diverse and non-domesticated plant species will enhance the development of disease-resistant crops.
Similar content being viewed by others


Main
Protecting plant health is vital for building the sustainable food systems needed to end hunger and poverty, as plant diseases and pests cause major losses to crop yields worldwide1,2. New pathogen species and virulent strains can appear suddenly and spread rapidly, aided by globalization and climate change3,4,5,6,7, so there is an urgent need to accelerate methods for disease control. Using the plant immune system for defence is an effective method of crop protection. Plants contain immune receptors that recognize pathogen invasion, and a major class of intracellular plant disease resistance genes encode nucleotide-binding domain leucine-rich repeat (NLR) proteins8. Transferring NLRs within and between plant species has proved successful for disease resistance breeding; however, pathogens are constantly evolving and can overcome and evade existing NLRs used in the field. Introducing multiple NLRs in gene stacks can provide strong defence and limit this breakdown9, yet few NLRs are available in modern crop cultivars10,11,12. Wild relatives of crop species are a valuable source for new NLRs for disease resistance as they are often resistant to major agricultural pathogens13,14. Accessing the causal genes can be difficult due to limited availability of genetic resources; therefore, large-scale projects to characterize NLRs are required to find new resistances useful against a wide range of pathogens11,12,15,16.
NLRs recognize pathogen infection by directly interacting with pathogen molecules or via recognizing pathogen-induced modifications to plant host proteins17,18. Successful recognition results in defence responses to prevent the spread of infection, often including localized cell death19. The presence or altered regulation of some NLRs has been shown to cause deleterious effects: the presence of Arabidopsis thaliana RPM1 reduced silique and seed production20, overexpression of RPW8 (ref. 21) and LAZ5 (ref. 22) can cause spontaneous cell death, and the lack of PigmR suppression in Oryza sativa causes a decrease in grain weight23. These observations, combined with the cell death function, resulted in the pervasive idea that NLRs require strict regulation to control defence responses24,25,26,27,28,29,30,31,32,33. NLRs were thought to be transcriptionally repressed across plants, but recent work has shown the cross-species transfer of NLRs without penalty9,34, and new knowledge of NLR function challenges these assumptions.
Here we found that multiple copies of the barley NLR Mla7 are required for full complementation of resistance. This supports similar findings for Mla3 (ref. 35), challenging the prevailing view that NLR expression must be maintained at a low level. We observed that an unexpectedly large number of NLRs are expressed in uninfected plants and that known functional NLRs are present among highly expressed NLR transcripts. We used this expression signature to predict functional NLR candidates at scale to find new resistance against two major diseases of wheat: stem rust caused by Puccinia graminis f. sp. tritici (Pgt) and leaf rust caused by Puccinia triticina (Pt). To date, 13 NLRs with efficacy against Pgt have been cloned36,37,38,39,40,41,42,43,44,45,46 and 7 against Pt47,48,49,50,51,52,53,54,55. We generated a transgenic array of 995 NLRs, using high-efficiency wheat transformation56, and identified 31 new resistant NLRs: 19 to stem rust and 12 to leaf rust. This proof-of-concept pipeline is applicable across plant species to rapidly identify new NLRs against various pathogens, enabling the development of disease-resistant crops.
Results
The NLR Mla7 requires multiple copies to confer resistance to barley powdery mildew and wheat stripe rust
NLR resistance genes are thought to be maintained at low expression levels in uninfected plants to control defence responses. In barley (Hordeum vulgare), alleles of the NLR Mla are known to confer resistance to the barley powdery mildew pathogen Blumeria hordei (Bh). However, through transgene complementation, we observed that single insertions of Mla7 driven by the Mla6 promoter were insufficient to complement the resistance phenotype, whereas multicopy insertion lines expressed resistance to Bh isolate CC148 carrying the recognized effector AVRa7 (Fig. 1a). To mitigate the challenges associated with multisite insertion and transgene silencing, we developed single-copy transgenic lines expressing Mla7 under its native promoter. We crossed two T1 families to develop an F2 population segregating for zero to four copies of Mla7. Higher-order copies were required for resistance to Bh, as only transgenic lines carrying two or more copies showed resistance to Bh isolate CC148 (AVRa7) (Fig. 1a), with full recapitulation of native Mla7-mediated resistance in lines with four copies (Fig. 1b). Mla7 multicopy insert lines retained race specificity, as resistance was observed to only Bh isolates carrying AVRa7 (Fig. 1c). Although considerable variation was observed within and between transgenic families, increased copy number of Mla7 does not cause auto-activity of resistance.

a–c, Powdery-mildew-susceptible barley cv. SxGP DH-47 was transformed with Mla7 driven by the Mla6 promoter–5′UTR and the Mla6 3′UTR–terminator. Three single-copy insert lines (T1-1, T1-2 and T1-3) and three multiple-copy insert lines (T1-4, T1-8 and T1-12) were identified for Mla7. Resistance to Bh isolates carrying AVRa7 was observed in transgenic barley lines carrying multiple copies of Mla7. Specific recognition of the effector AVRa7 was retained across transgenic lines. Panel a shows infection phenotypes for the presence or absence of transfer DNA (T-DNA) for Mla7 T1 families and controls inoculated with Bh isolate CC148. The presence and absence of T-DNA are shown in orange and blue, respectively. All phenotypes are on a scale from 0 to 4. Transparency and jittering were used to visualize multiple overlapping data points. As shown in b, multiple copies, not single copies, of Mla7 under its native promoter/terminator are required to confer full resistance to Bh isolate CC148. Individual phenotypes from an F2 population with varying numbers of Mla7 transgene copies derived from a cross of two single-insertion transgenic lines (T1-117 and T1-121) are plotted. As shown in c, multicopy lines carrying Mla7 driven by the Mla6 promoter/terminator confers race-specific resistance to barley powdery mildew (Bh). Controls include SxGP DH-47 and near-isogenic lines in the Pallas (Mla8) genetic background: P04B (Mla7) and P06 (Mla7). Bh isolates are ordered according to the presence (AVRa7) or absence (avra7) of the effector recognized by Mla7. Isolates include 3-33 (A), Race I (B), X-4 (C), I-167 (D), K-200 (E), M-236 (F), Z-6 (G), C-132 (H), 120 (I), R86/1 (J), K-3 (K), KM18 (L) and MN-B (M). All experiments were performed twice with similar results; the data shown are the average of these experiments.
Mla alleles recognize multiple pathogens35,57 and in previous work, Mla7 was shown to be in complete genetic coupling with Rps7, which confers resistance to Puccinia striiformis f. sp. tritici (Pst), indicating that they are probably the same gene57. Using the Mla7 transgenic lines, we confirmed that Mla7 also confers resistance to Pst (Supplementary Fig. 1a). In addition, as observed with Bh, multiple insertions of the Mla7 transgene were required for Pst resistance (Supplementary Fig. 1b). Progeny of these multicopy lines showed unstable resistance, probably due to transgene silencing (Supplementary Fig. 2). Due to the correlation of copy number and phenotype, increased NLR expression is hypothesized to increase with higher copy number. Mla7 natively exists with three identical copies in the haploid genome of barley cv. CI 16147 (Supplementary Fig. 3), supporting the hypothesis that a specific threshold of expression is required for function.
Functional NLRs exhibit high steady-state expression levels
To investigate NLR expression, we assessed the expression levels of known characterized NLRs across six plant species of both monocots and dicots using sequencing data from uninfected leaf tissue. In monocots, the barley resistance genes Rps7/Mla8 and Rps7/Mla7 against Bh and Pst are present in highly expressed transcripts (Fig. 2a and Supplementary Data 1). The Aegilops tauschii-derived Pgt resistance genes Sr46, SrTA1662 and Sr45 are also present in highly expressed NLR transcripts across accessions (Fig. 2b). As a model species, the dicot A. thaliana contains a large complement of characterized functional NLRs, and highly expressed NLR transcripts are enriched with known genes (Fig. 2c). The most highly expressed NLR in ecotype Col-0 is ZAR1, and collectively across ecotypes, highly expressed NLRs provide resistance to diverse pathogen species (Fig. 2c and Supplementary Data 2). Using the de novo assembled transcriptome of accession Col-0, we found that known NLRs are significantly enriched in the top 15% of expressed NLR transcripts compared with the lower 85% (χ2 (1, n = 616) = 4.2979, P = 0.038). Using a non-redundant set of the highest-expressed transcript for each NLR, we found that the top 14% of expressed NLR transcripts are enriched for known NLRs (χ2 (1, n = 141) = 4.5767, P = 0.032). NLRs in the top 15% are in NLR classes containing coiled-coil, nucleotide-binding-site, leucine-rich-repeat and Toll/interleukin1 receptor domains without additional non-canonical domains (CNL, NL, TN, TNL and TNLT; Supplementary Figs. 4 and 5). Overall, the expression level of the most highly expressed NLR is above the median and mean expression levels for all genes in A. thaliana accession Col-0, confirming that NLRs are not transcriptionally repressed in uninfected plants (Supplementary Figs. 4 and 5).

a–f, Known functional NLRs from each species are among the most highly expressed NLR transcripts from unchallenged plant tissue (Supplementary Data 2). Transcript abundance was estimated from self-aligned RNA-seq data and measured in transcripts per million (TPM). Panel a shows transcript abundance of NLRs from the de novo assembled leaf transcriptomes of Hordeum vulgare accessions Golden Promise, CI16153 and CI16147. The expression of the functional resistance gene Rps7 against Bh and Pst is indicated. Panel b shows transcript abundance of NLRs from the de novo assembled leaf transcriptomes of Aegilops tauschii accessions KU2025, KU2075, KU2078, KU2093, KU2124 and PI499262. The expression of the functional resistance genes Sr45, Sr46 and SrTA1662 against Pgt is indicated. Panel c shows transcript abundance of NLRs from the leaf transcriptomes of Arabidopsis thaliana accessions Col-0, Ler-0, Sf-2 and Ws-0. The expression of the functional resistance genes RPP4, RPP5, RPP8 and RPP13 to downy mildew (caused by Hyaloperonospora arabidopsidis); WRR4 against white rust (Albugo candida); ZAR1 against Pseudomonas syringae and Xanthomonas campestris pv. campestris; RPS2 and RPM1 against Pseudomonas syringae; RCY1 against cucumber mosaic virus; and alleles of RLM3 against grey mould (Botrytis cinerea), dark leaf spot of cabbage (Alternaria brassicicola) and dark spot of crucifers (Alternaria brassicae) is indicated. Panel d shows transcript abundance of NLRs from the Cajanus cajan accession G119-99 leaf transcriptome. The expression level of CcRpp1, which confers resistance to Asian soybean rust (Phakopsora pachyrhizi)58, is indicated. Panel e shows transcript abundance of NLRs from the de novo assembled leaf transcriptome of Solanum americanum accession SP2273. The expression of the functional resistance genes Rpi-amr1 and Rpi-amr3 to late blight (Phytophthora infestans) is indicated. An allele of Ptr1, which recognizes Pseudomonas syringae pv. tomato and Ralstonia pseudosolanacearum in Solanum lycopersicoides, is also shown. Helper NLRs are annotated as NRC1, NRC2, NRC3, NRC4 and NRC0 as defined by Kourelis et al.140. NRC6 is not present in the dataset. Panel f shows transcript abundance of NLRs from Solanum lycopersicum cultivars VFNT Cherry and Motelle carrying Mi-1 with resistance to root-knot nematodes (Meloidogyne spp.), the potato aphid (Macrosiphum euphorbiae) and the sweet potato whitefly (Bemisia tabaci). Mi-1 is highly expressed in both cultivars in both leaf and root tissue. The expression levels of additional known functional NLRs Tm-2 for resistance to tobamoviruses including tomato mosaic virus and tobacco mosaic virus, Prf for resistance to Pseudomonas syringae pv. tomato, Sw5 for resistance to a broad range of viruses across paralogues and Ph-3 for resistance to P. infestans are indicated. Helper NLRs are annotated as NRC1, NRC2, NRC3, NRC4, NRC6 and NRC0 as defined by Kourelis et al.140.
NLRs previously identified via traditional methods and bioinformatic approaches, such as CcRpp1 from Cajanus cajan58 and Rpi-amr1 from Solanum americanum59, were also found to be present in highly expressed NLRs in the respective species (Fig. 2d,e). The tomato NLR Mi-1 provides resistance to potato aphid and whitefly in foliar tissue and the root-knot nematode in the roots. We found that Mi-1 is highly expressed in both the leaves and roots of the resistant cultivars Motelle and VFNT Cherry, alongside the additional characterized NLRs (Fig. 2f and Supplementary Data 2). Rpi-amr1 and Mi-1 are dependent on additional NLRs for function and are present in a wider network described in Solanaceae species59,60. NLRs that recognize pathogen products or host modifications directly are described as ‘sensor’ NLRs, and these partner with ‘helper’ NLRs that facilitate immune signalling61. Known helper NLRs, designated with the prefix NRC in the Solanaceae, are also highly expressed (Fig. 2e,f and Supplementary Data 2). In addition, many helper NLRs display tissue specificity; NRC6 is highly expressed in the roots but not the leaves of tomato cvs. VFNT Cherry and Motelle, and NRC0 is highly expressed in the roots of cv. VFNT Cherry but lowly expressed in the leaves of both cultivars and in the roots of cv. Motelle (Fig. 2f). These results therefore show the importance of investigating the appropriate plant tissue relevant for the pathogen and indicate the tissue specificity of resistance.
The most highly expressed isoform of Rpi-amr1 is the functional NLR
Multiple isoforms of each NLR are present in transcriptomes, and while the function of alternative splicing and isoform variation across NLRs is broadly uncharacterized, alternatively spliced variants of a few NLRs have been shown to modulate defence62,63,64,65. Different isoforms of Rpi-amr1 are present in the assembled transcriptome of S. americanum accession SP2273 at varied expression levels. Rpi-amr1 isoforms show sequence variation, including the presence/absence of the final exon (Supplementary Fig. 6). Expressing the different isoforms under the same NRC4 promoter in transient assays in Nicotiana benthamiana, we found that the most abundant isoform, i3, provides resistance to Phytophthora infestans (Fig. 3). The isoform i3 contains all exons as compared to the published sequence. Isoform i1 also confers resistance to P. infestans as it contains all exons and is 99.3% similar to i3. Other isoforms present at lower expression levels in the transcriptome confer reduced levels of resistance to P. infestans. Transcript variants may be due to alternative splicing, the presence of paralogues or transcript assembly. As the most highly expressed isoform of Rpi-amr1 is the functional transcript, this supports the idea that selecting the highest-expressed transcript is a feasible approach to select functional variants for other NLRs.

a, Point plot and box plots of the lesion area of P. infestans infection in transient assays in N. benthamiana using different isoforms of Rpi-amr1. Individual biological replicates in each replicate are indicated with different coloured shapes (red circles for 1, green triangles for 2 and blue squares for 3). b, Representative photographs of lesions across replicates. Controls of resistant Rpi-amr3 from accession SP1102, susceptible Rpi-amr1 from accession SP2271 and resistant Rpi-amr1 from accession SP2273 were included. Rpi-amr1 isoforms from accession SP2273 are present in descending order of expression level: i3 at 6.53 TPM, i8 at 1.64 TPM, i1 at 1.39 TPM, i4 at 0.523343 TPM, i7 at 0.30 TPM and i6 at 0.07 TPM.
Building an NLR array for Pgt resistance
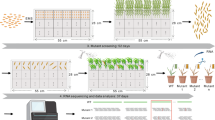
We hypothesized that we could use the signature of higher expression levels in unchallenged tissue to mine functional NLRs from diverse plant germplasm. As a proof of concept, we sought to identify new NLRs against Pgt and Pt, which are major threats to global wheat production. NLRs providing resistance to rust have been previously characterized from close relatives of wheat36,43,66. We therefore investigated a total of 30 accessions of Aegilops bicornis, A. longissima, A. searsii and A. sharonensis to capture the genetic diversity present in this genus (Fig. 4a and Supplementary Table 1). To represent the breadth of diversity across the grasses, we included several species across the Triticeae and Poeae through to the distant wheat relatives spanning the Pooideae—many of which have not previously been assessed for rust resistance.

a, Phylogenetic tree of grass species in the PACMAD and BOP clades of the Poaceae. Species used for NLR discovery are indicated with pink arrows. b, The pipeline for the identification of highly expressed NLRs from RNA-seq data. The expression of transcripts containing a nucleotide-binding (NB) domain was used to identify the top 25% of expressed NLRs. Primers were designed on the open reading frame (ORF) of the NLRs, and the coding sequences were amplified via PCR and assembled into the transformation vector under the maize ubiquitin promoter.
We identified highly expressed candidate functional NLRs from a total of 68 accessions of 18 plant species (Supplementary Table 1). We excluded NLRs from the MIC1 clade, as this clade is enriched with NLRs with integrated domains that require an additional NLR to function together as a pair67,68,69. The corresponding clade of paired helper NLRs was retained for the possibility of multi-sensor or interspecific NLR pairings. We chose the top 25% of NLR transcripts as this threshold encompasses known NLRs across plant species, particularly monocots (Fig. 2 and Supplementary Data 9). We selected the most highly expressed isoform for each NLR under the hypothesis that it is the functional variant. A total of 6,260 transformation events using high-efficiency Agrobacterium-mediated transformation of the wheat cultivar Fielder56 generated 5,177 independent T1 families for 995 NLR constructs driven by the maize ubiquitin promoter to create an array of transgenic wheat (Fig. 4b and Supplementary Data 3, 9 and 10). This array also includes the controls of known NLRs conferring stem rust resistance: Sr33 (ref. 39), Sr35 (ref. 40) and Sr50 (ref. 38). Lr21 was included as a control for leaf rust55.
Identification of 19 NLRs conferring resistance to Pgt
We interrogated the transgenic wheat array for resistance to Pgt in greenhouse seedling assays using Pgt race QTHJC, a highly virulent isolate on wild-type Fielder (Fig. 5a,b, Supplementary Data 4 and Supplementary Table 2). A total of 19 NLRs provided resistance to Pgt race QTHJC when expressed in Fielder. Of these, resistance was observed in two or more independent T1 families for six NLRs and in one independent T1 family for 13 of the NLRs. Control lines carrying the known resistance gene Sr50 were resistant to Pgt race QTHJC9 (Fig. 5a, Supplementary Data 4 and Supplementary Table 2). The resistant phenotypes observed were comparable to the differential wheat lines carrying known Sr resistance genes, which display phenotypes of 0; to 22+ (weighted averages 0 to 5.33) (Supplementary Data 6). All other remaining NLRs showed a susceptible phenotype (Fig. 5a, Supplementary Fig. 7 and Supplementary Data 4), including those carrying controls Sr33 and Sr35. Sr35 is ineffective against QTHJC as previously reported40, whereas the susceptible phenotype observed in Sr33 transgenics may represent insufficient complementation by the transgene70.

a, A total of 19 NLRs conferred resistance against Pgt race QTHJC (Supplementary Data 4 and 5). Phenotypic scores from individuals in T1 families from each construct inoculated with Pgt race QTHJC are plotted on a weighted and transformed Stakman scale from completely resistant (0) to susceptible (9). Phenotypes of known Sr genes are indicated to the right of the plot (Supplementary Data 6). Circle size indicates the number of individuals with each phenotypic score. Individuals from the other 971 susceptible NLRs shown on the right were susceptible to Pgt and exhibited phenotypic scores similar to wild-type Fielder. b, Seedling leaves of T2 individuals from T1 families infected with Pgt race QTHJC under greenhouse conditions. An additional inoculation experiment was performed to obtain the photographs. From top to bottom: the wild-type (WT) Fielder susceptible control and four resistant individuals from resistant NLRs with the NLR construct and independent T1 family shown. The Stakman phenotype and corresponding weighted average (wa) per individual are also shown. S, susceptible; HR, highly resistant. c, Stem sections of selected individuals infected with Pgt race QTHJC under field conditions. From top to bottom: the wild-type Fielder susceptible control, a susceptible wheat control of cv. Morocco, a susceptible individual from a segregating family from NLR NLR_01_48 T1 family 3, a resistant individual from NLR NLR_01_48 T1 family 2 and a resistant individual from NLR NLR_04_40 T1 family 3. The NLR construct, the independent T1 family, and the phenotype of per cent severity and infection response are shown. S, susceptible; HR, highly resistant. d, Nine NLRs conferred resistance against Pgt race TTKSK. Individuals with resistance against Pgt race QTHJC were screened with Pgt race TTKSK. Phenotypes were scored on a weighted Stakman scale from highly resistant (0) to susceptible (9). Circle size indicates the number of individuals with each phenotypic score. Individuals from T1 families of NLRs NLR_01_21, NLR_01_48, NLR_04_40, NLR_04_67, NLR_06_10, NLR_06_36, NLR_06_52, NLR_08_79 and NLR_09_55 showed resistant phenotypes to Pgt race TTKSK. e, Seedling leaves of individuals from T1 families inoculated with Pgt race TTKSK under greenhouse conditions. From top to bottom: a susceptible individual of NLR NLR_06_02 independent T1 family 5 and four resistant individuals from resistant NLRs. The NLR construct, the independent T1 family, the Stakman phenotype and the corresponding weighted average per individual are shown. f, A total of 12 NLRs conferred resistance against Pt race THBJ (Supplementary Table 3 and Supplementary Data 8). Phenotypic scores from individuals in T1 families from each construct inoculated with Pt race THBJ are plotted on a weighted and transformed phenotypic scale from highly resistant (0) to susceptible (9). Circle size indicates the number of individuals with each phenotypic score. Individuals from the other 980 susceptible NLRs shown on the right were susceptible to Pt and exhibited phenotypic scores similar to that of wild-type Fielder.
Seed from individual resistant transgenic lines was bulked following the first round of inoculations, and T2 families were then tested to confirm efficacy in the field. A total of 17 of the 19 resistant NLRs were tested under field conditions, and four NLRs showed resistance to Pgt race QTHJC in the field (Fig. 5c, Supplementary Fig. 8 and Supplementary Data 5). To further validate resistance, we performed a secondary phenotypic screen using Pgt race QTHJC on all independent T1 transgenic lines per resistant NLR construct, as a random sub-sampling of independent lines was used in the primary screen. Resistance was observed in at least one T1 family for 16 of the resistant NLRs and in at least two T1 families for 10 of the NLRs (Supplementary Figs. 9 and 10, Supplementary Table 3 and Supplementary Data 7). Collectively across screens, resistance was observed in two or more T1 families for 14 of the resistant NLRs and in at least one T1 family for 19 of the NLRs. As all bulked T2 plants from the primary screen were used in the field, T2 lines of the collection that were bulked separately were screened to assess the heritability of resistance. Resistance was observed in two or more T2 families for 9 of the NLRs and in at least one T2 family for 13 NLRs. Reduced resistance at the T2 generation was observed for the controls, with one family of Sr50 showing resistance and Sr33 showing susceptibility (Supplementary Figs. 9 and 10, Supplementary Table 3 and Supplementary Data 7). Reduced resistance could be due to transgene silencing or insufficient expression for complementation of the phenotype, as these lines were bulked without phenotypic selection, indicating the importance of promoter optimization for sufficient NLR expression.
To assess the breadth of pathogen recognition, the 19 NLRs resistant to Pgt race QTHJC were further tested against the widely virulent Pgt race TTKSK. Nine NLRs also conferred resistance to Pgt race TTKSK. Ten NLRs exhibited race specificity, conferring resistance to Pgt race QTHJC and susceptibility to Pgt race TTKSK. The controls Sr35 and Sr50 showed resistance to TTKSK, and Sr33 showed susceptibility (Fig. 5d,e, Supplementary Data 4 and Supplementary Table 2).
Identification of 12 NLRs conferring resistance to Pt
The value of the transgenic array is the ability to repeatedly screen it against diverse pathogens. We further evaluated the array against Pt, the fungal wheat leaf rust pathogen. A total of 12 new NLRs provided resistance to Pt race THBJ in seedling glasshouse assays (Fig. 5f, Supplementary Fig. 7, Supplementary Data 8 and Supplementary Table 4). Of these, resistance was observed in two or more independent T1 families for eight NLRs and in one independent T1 family for four of the NLRs. The cloned leaf rust resistance gene, Lr21 (ref. 55), was included as a resistant control and showed resistance to THBJ (Fig. 5f and Supplementary Fig. 10). Two NLRs identified in this screen matched known resistance genes: NLR_01_19, which matches Yr87/Lr85 (Aegilops sharonensis), and NLR_10_01 which is Mla37-1 (Hordeum vulgare)52,71. All transgenic lines carrying the NLRs resistant to Pgt were susceptible to Pt. NLRs within the transgenic array therefore retain pathogen recognition specificity, and the resistant phenotypes observed are not due to constitutive defence activation by the transgenes.
NLR hits are present across diverse phylogenetic clades
We investigated the distribution of the functional NLR hits against Pgt and Pt using a previously annotated phylogenetic tree of NLRs from grass species67. We found that the NLR hits are present across different phylogenetic clades (Supplementary Tables 2 and 4 and Supplementary Fig. 11). Six Pgt hits are present in clade 17, which contains the known rust resistance genes Lr10, Sr33 and Sr35 (ref. 67). Two Pgt hits are present in clade 24, which contains the known leaf rust resistance genes Lr1 and Lr21. Five Pgt hits are derived from clade 7 NLRs enriched with known paired NLRs with a helper function from the MIC1 clade67. For Pt hits, two NLRs are present in each of clades 7, 17, 19, 20, 22 and 24 (Supplementary Table 4). Several putative orthologous groups of NLRs in clade 7 confer rust resistance, such as NLR_03_49 (H. lanatus), NLR_06_52 (Aegilops sharonensis) and NLR_09_55 (B. media). Aegilops-derived orthologous NLRs include clade 7 NLR_04_67 (Aegilops bicornis) and NLR_06_36 (Aegilops searsii); one NLR group in clade 20 consisting of NLR_03_68 (Aegilops sharonensis) and NLR_06_10 (Aegilops bicornis); and different orthologues conferring resistance to wheat stem rust and wheat leaf rust in clade 22, NLR_01_74 (H. lanatus) and NLR_08_16 (Aegilops sharonensis).
Discussion
Here we demonstrated that selecting highly expressed NLRs in uninfected plants facilitates rapid prediction of new NLR candidates from diverse germplasm. In our proof-of-concept study, the cross-species transfer of single NLRs—including NLRs from species never genetically investigated for resistance to wheat pathogens—provided 19 new resistance genes with efficacy against Pgt, including nine NLRs with resistance to the widely virulent Pgt race TTKSK (Ug99 lineage), and a further 12 against Pt. This method improves on previous cloning timelines, as 13 NLRs with efficacy against Pgt36,37,38,39,40,41,42,43,44,45 and 7 against Pt47,48,49,50,51,52,53,54,55 have been cloned to date. Variation in resistance was observed for the 19 NLRs across heritability, field efficacy and isolate specificity. Additional experimentation to investigate biological and/or technical mechanisms for this variation would further validate resistance. Among these new NLRs identified is the barley powdery mildew resistance gene Mla37-1 (ref. 71), which we showed also confers resistance to wheat leaf rust, and the recently cloned Yr87/Lr85 from Aegilops sharonensis independently identified through traditional genetic mapping and mutagenesis methods52. The majority of genes conferring resistance to wheat leaf rust in Aegilops sharonensis and the closely related species Aegilops longissima can be explained by Yr87/Lr85, as orthologues are present across accessions and confer the underlying resistance observed. This broad approach to curating NLR collections across species therefore minimizes redundancy and maximizes value to collect diverse NLRs. This NLR array is a valuable resource that can be repeatedly interrogated against other pests and diseases to validate new effective resistance genes in planta.
Further investigation is required to elucidate the mechanism of recognition of these new NLRs. While the association of NLR functionality and high expression occurs broadly across plant species, this may be non-uniform across different classes of NLRs and dependent on additional mechanisms. Several of the new Pgt- and Pt-resistant NLRs fall into phylogenetic clades with known rust resistance genes. Clade 17 contains the NLRs Sr35 and Mla, which directly bind pathogen effectors72,73, and we hypothesize that clade 17 hits also function as single NLRs via direct recognition. Members of clade 7 are known to function as pairs, often with members of the MIC1 (C16) clade67,68,69. NLRs in pairs have distinct roles: NLRs in clade 16 are sensors that recognize pathogen effectors, and they require the clade 7 NLR helpers to activate immune signalling following recognition18. Here, the transfer of single clade 7 NLRs from divergent grass species provided resistance in wheat. These hits may represent a newly described function for helper NLRs in direct pathogen recognition, or they may be functioning with a new partner in the Fielder genetic background. If so, introducing new sensor NLRs or components of NLR pairs can expand or revive pathogen recognition with endogenous genes74.
In this study, no obvious macroscopic detrimental growth traits were observed across transgenic wheat lines in the greenhouse or field experiments. Moreover, transgenic lines were obtained for 99.6% of all transformed NLRs. Further testing is required to detect small differences in agro-morphological traits, but the absence of negative pleiotropic phenotypes is consistent with the cross-species transfer of NLRs into barley34 and a multi-transgene cassette transformed into wheat9. Further studies are required to elucidate the biological mechanisms of NLR expression, and subsequent work may be required for optimization; for example, NLRs in the array may need additional regulatory, promoter, terminator or intronic elements to function, so it is not possible to estimate the false-negative rate. Alternatively, these NLRs may provide resistance to other pathogens.
The concept that highly expressed NLRs may be detrimental was based on early observations of deleterious phenotypes20,23,24,25,26,27,28,29,30,31,32,33,75,76,77,78,79 and the induction of NLR expression following pathogen infection29,80. New understanding of NLR function can alleviate detrimental effects. For example, cell death or yield penalties caused by some NLRs can be suppressed via co-expression with their required partner or suppressor18,23,68,81. Other deleterious effects may be caused by mutations in the sequence or regulatory requirements of NLRs rather than expression levels. In A. thaliana, overexpression of the wild-type SSI4 TIR-NLR sequence did not cause the stunting and cell death observed in the mutant variant, indicating that the phenotypes were caused by sequence mutation and not expression level82. Similarly, overexpression of RPW8 alleles under the 35S promoter showed no spontaneous cell death, whereas cell death was correlated with increased transgene copies of the genomic fragment carrying RPW8 (ref. 21), indicating the disruption of gene regulation as causal83. The reduced height observed from induced mutations in Rht13b in wheat was independent of transgene copy number or gene expression84. Described examples of deleterious NLRs also involve guards or NLRs with additional integrated domains20,85. RPM1 is lowly expressed in transcripts in A. thaliana Col-0, as are other known NLRs that guard host proteins such as LOV1 (refs. 20,86). Different expression levels are observed across ecotypes for alleles, so effects may also be genotype dependent. In comparison, the expression of NLRs that directly recognize pathogen products may have less influence from host processes and proteins. Here we observed the requirement of high expression for Mla7 function, and as other Mla alleles directly recognize effectors from Bh73, increased gene expression could compensate for any reduction in binding affinity towards their recognized effector under a positive dosage model87. Sufficient expression and protein abundance may also be required for NLR oligomerization and resistosome formation72,88,89. These results, alongside further advances in our understanding of NLR function, may explain the observation of deleterious phenotypes via mechanisms that are independent of NLR expression.
We have produced a high-throughput pipeline, designated NLRseek, which facilitates large-scale mining of resistance genes and provides access to genetic resistance in diverse plant species previously inaccessible to traditional methods. High-throughput phenotypic screening of plant transgenic arrays has been used successfully for identifying drought tolerance in rice90 and is a powerful tool that enables the study of pathogens less amenable to in vitro culture, such as shown here for Pgt and Pt. The value of this array is that it can be repeatedly screened with diverse pathogen species, isolates and races to validate NLRs directly in planta. The many diverse NLRs found using this approach would support the breadth of variation to deploy tailored gene stacks against major pathogens. This capability, combined with advances in gene cloning and genotype-independent transformation technology, ignites an exciting potential for the future of biotechnology to protect plant health and improve food security.
Methods
Native copy number variation of Mla7
Genomic DNA was extracted using a CTAB extraction approach from the Manchuria near-isogenic lines CI 16147 and CI 16153, which carry Mla7 from two different donors (Multan and Long Glumes)91. Illumina sequencing was performed at Novogene (Cambridge, UK) using 150-bp paired-end reads. The reads were trimmed using Trimmomatic (v.0.39)92 with adapter clipping using TruSeq3-PE adapters and parameters 2:30:10, trimming of low-quality leading and trailing sequence with parameter 5, sliding window trimming with parameters 4:10 and a final minimum length of 36 bp. The copy numbers of Mla7 and the control gene Bpm were determined using the k-mer analysis toolkit (KAT; v.2.4.1)93. The module sect was used with the default parameters to determine k-mer coverage over the gene sequences of Mla7 and Bpm. R (v.4.1.2)94 and ggplot2 (v.3.3.6)95 were used to estimate copy number variation of Mla7.
Transgenic complementation of Mla7
Promoter regions, UTRs and terminator regions of Mla6 were amplified from barley CI 16151 (Mla6) and native genomic fragments of Mla7 from CI 16153 (Mla7)96 using GoTaq Long PCR Master Mix (Promega). Constructs were developed as described in Bettgenhaeuser et al.57. Briefly, PCR fragments were assembled into the pBract202 binary vector (BRACT) via the Gibson reaction97. PCR fragments for assembly were produced with Phusion High-Fidelity DNA Polymerase (NEB) with 40-mer chimeric primers. Barley transformation was performed using the technique described in ref. 98 using the hygromycin resistance gene (hyg) as a selectable marker. Transformation was performed with the barley powdery mildew (Bh) and wheat stripe rust (Pst) susceptible line SusPtrit × Golden Promise DH-47 (SxGP DH-47)99. Copy number variation in transgenic plants was determined by quantitative real-time PCR using the selectable marker gene hyg (AttoDNA100). A population segregating for two T-DNA inserts was generated by crossing transgenic lines T1-117 and T1-121 (SxGP DH-47 transformed with pMla7::Mla7::tMla7) with the Bh- and Pst-susceptible accession Manchuria. T-DNA was mapped in segregating F2 populations to chromosomes 3H and 5H using SNP markers derived from the OPA markers 1_0702 and 2_1012 (ref. 101). F2 lines heterozygous for the T-DNA and homozygous at the Mla locus for the Manchuria allele (the non-functional mla allele) were crossed and validated in the resulting progeny for their allelic state at both the T-DNA and Mla loci. Selfed seed of this line was used for pathogen assays with Bh and Pst.
Pathogen assays for Mla7
Pathogen assays with Bh and Pst were performed as described in Bettgenhaeuser et al.57. A collection of Bh isolates (n = 13) were selected from a collection containing 59 reference isolates collected in 12 countries in all non-polar continents over a period of 66 years (1953–2019) and maintained at the Agricultural Research Institute Kroměříž Ltd. Virulence patterns to 35 differential barley genotypes are shown in ref. 102. Prior to inoculation, purity was verified on standard barley lines103. Bh isolates were multiplied on leaf segments of the susceptible cultivar Bowman. Bh isolate CC148 was propagated on barley cv. Manchuria (CI 2330) prior to inoculation. Seedlings were placed horizontally, inoculated, rotated 180 °C after a resting period of 2 min for conidia to settle and then inoculated again. Susceptible controls in every experiment included Manchuria (CIho 2330), Pallas (CIho 11313), Siri (CIho 14846) and the barley cv. Siri-derived set of near-isogenic lines, each carrying a single mildew resistance gene104. Bh isolate CC148 assays were carried out in a negative-pressure containment greenhouse with supplemental lighting and temperature set at 18 °C day and 12 °C night. For pathogen assays using the diverse collection of Bh isolates, inoculation and evaluation protocols are described in detail in ref. 105. Briefly, seed was grown in a mildew-proof greenhouse under natural daylight. Central leaf segments of 15 mm were cut from fully expanded primary leaves after 14 days for each transgenic family and controls. The leaves were placed on water agar (0.8%) containing benzimidazole (40 mg l−1) in petri dishes with adaxial surfaces facing upwards. Leaf segments were placed at the bottom of a settling tower, and conidia from a fresh leaf segment of the susceptible cultivar were blown into the settling tower at a concentration of approximately 8 conidia per mm2. The petri dishes of leaf segments were incubated at 20 ± 2 °C under artificial light (cool-white fluorescent lamps providing 12 h light at 30 ± 5 μmol m−2 s−1). Infection responses were scored seven days after inoculation using a 0–4 scale where 0 indicates no visible mycelium or sporulation, and 4 indicates strong mycelial growth and sporulation106. Scoring was repeated a day later. Two replications were performed.
Pst inoculations were performed using a suspension of urediniospores in talcum powder at a weighted ratio of spores:talcum powder of 1:16 and applied to leaves using a spinning table. Plants were sealed and stored at 8 °C for 48 h immediately after inoculation. The plants were grown in a controlled-environment room under 16 h light/8 h dark. The phenotypes of the first leaves were scored 14 days post-inoculation using an incremental scale of 0 to 4 representing the surface area displaying an infection phenotype where 0 represented no chlorosis or no pustules of Pst, and 4 indicated infection across 100% of the surface area.
Plant materials and growth conditions for RNA-seq analysis
Seeds of the grass species Achnatherum hymenoides, Aegilops bicornis, Aegilops longissima, Aegilops searsii, Aegilops sharonensis, Agropyron cristatum, Avena abyssinica, Brachypodium distachyon, Briza media, Cynosurus cristatus, Echinaria capitata, Holcus lanatus, Hordeum vulgare, Koeleria macrantha, Lolium perenne, Melica ciliata, Phalaris coerulescens and Poa trivialis were used for candidate NLR gene discovery.
Seeds were germinated on damp filter paper on petri dishes and placed at 4 °C for six to seven days to break seed dormancy. Germinated seeds were transferred into an in-house custom soil mix prepared by the horticultural services department at the John Innes Centre (JIC cereal mix: 65% peat, 25% loam, 10% grit, 3 kg m−3 dolomitic limestone, 1.3 kg m−3 PG mix and 3 kg m−3 Osmocote Exact). The seedlings were grown in a pest- and disease-free controlled-environment chamber under 16 h light at 20 °C/8 h dark at 16 °C. For RNA isolation, leaves of plants 12 to 35 days post germination were used depending on the species. The first and second leaves of plants were harvested for most plant species; however, the first to sixth leaves were used from species with smaller leaf sizes.
Seeds of Solanum lycopersicum cultivars VFNT Cherry (LA1221) and Motelle (LA2823) were obtained from the C.M. Rick Tomato Genetics Resource Center (https://tgrc.ucdavis.edu/). The seeds were germinated in an in-house custom soil mix prepared by the horticultural services department at the John Innes Centre (JIC multipurpose + grit: 90% peat, 10% grit, 4 kg m−3 dolomitic limestone, 0.75 kg m−3 PG mix and 1.5 kg m−3 Osmocote Bloom). Seedlings were grown in a pest- and disease-free controlled-environment chamber under 16 h light/8 h dark at 18 °C. Tissue was sampled for the RNA isolation after one month. Fully expanded leaves were used for leaf tissue, and the entire root system was used after washing in distilled water. For each tissue type, samples were pooled from three seedlings per cultivar.
For Arabidopsis thaliana, seeds of the lines Col-0, Ler-0, Mt-0 and Ws-0 were obtained from the Nottingham Arabidopsis Stock Centre (https://arabidopsis.info/). The seeds were surface sterilized in a sterilization chamber using chlorine gas for 5 h and sown on Murashige and Skoog media with 1% sucrose supplemented with 0.8% agar. The seeds were kept at 4 °C for two to three days and then grown at 22 °C under 16 h light. Seedlings were transferred to liquid Murashige and Skoog media with 1% sucrose in a 24-well tissue culture plate with seedlings in each well. The controlled-environment chamber used was a clean germination room free of plant pests and pathogens. The seedlings were sampled for RNA isolation nine to ten days post-germination.
RNA extraction and sequencing
Total RNA was extracted from leaves of grass species, seedlings of A. thaliana, and leaf and root tissue of S. lycopersicum using a Trizol-phenol-based protocol according to the manufacturer’s instructions (Sigma-Aldrich; T9424). Barcoded Illumina TruSeq RNA HT libraries were constructed and pooled with four samples per lane on a single HiSeq 2500 lane run in Rapid Run mode using 150-bp paired-end reads. Reads were assessed for quality using FastQC (v.0.11.7)107 and trimmed before assembly using Trimmomatic (v.0.39) with the parameters set at ILLUMINACLIP, 2:30:10; LEADING, 5; TRAILING, 5; SLIDINGWINDOW, 4:15; and MINLEN, 36. De novo transcriptome assemblies were generated using Trinity108 with the default parameters (v.2013-11-10). kallisto (v.0.43.1)109 was used to estimate expression levels for all transcripts using the default parameters and 100 bootstraps.
Rpi-amr1 isoform characterization and Phytophthora infestans assays
Isoforms were identified from the transcriptome of Solanum americanum accession SP2273 using BLAST+ (v.2.2.31)110 of Rpi-amr1 (GenBank: MW348763, NCBI). Sequence analysis was performed using Geneious Prime (v.2024.0.3) (https://www.geneious.com/features/prime). Gene isoforms were synthesized via Gene Universal (Supplementary Data 11). Coding sequences were expressed under the NRC4 promoter and terminator, and constructs were assembled using Golden Gate into the Level 1 acceptor pICH47732. Constructs of Rpi-amr1 from the resistant accession SP2273, the susceptible accession SP2271 and Rpi-amr3 from SP1102 (ref. 59) were used as controls. Transient complementation assays and P. infestans inoculation were performed as described previously111,112. Briefly, Agrobacterium liquid cultures were resuspended in MES buffer (10 mM MES, 10 mM MgCl2 and 150 mM acetosyringone) and adjusted to 0.3 OD600. A total of three to four leaves of different N. benthamiana plants were infiltrated with each construct per replicate with a total of three replicates. P. infestans 88069 was grown on rye media, and sporangia were harvested after ten days. Leaves were inoculated with two 10-μl droplets of a zoospore suspension (50,000 zoospores per ml). The inoculated leaves were incubated for 7 to 12 days on damp paper towels in a Sanyo cabinet at 16 °C under 16 h light and 8 h dark before the phenotypes were scored. The lesion area was measured from images using Fiji (ImageJ2, v.2.14.0/1.5f)113 and analysed using RStudio (v.2023.12.1+402)114.
Identification of highly expressed NLRs
TransDecoder (v.4.1.0) LongOrfs115 was used to predict all open reading frames in de novo assembled transcriptomes. Transcript abundance was quantified using kallisto109. InterProScan (v.5.27-66.0)116 was used to annotate domains using Coils and the Pfam, Superfamily and ProSite databases. Any protein that contained both a nucleotide-binding domain and a leucine-rich repeat domain was retained. Histograms were generated using RStudio. The transcripts of known and characterized NLRs were identified from the transcriptome using a BLAST+ (v.2.2.31) search using the publicly available nucleotide sequence. Sequence similarity of the coding sequences was assessed using MUSCLE (v.5.1)117 using the default parameters.
Building the NLR array and molecular cloning
Sequencing, de novo RNA-seq assembly, NLR identification and PCR primer development were completed for 81 accessions of 18 grass species. Of the 81 accessions sequenced, 68 accessions were progressed to molecular cloning including species in the genera Achnatherum, Aegilops, Agropyron, Avena, Brachypodium, Briza, Cynosurus, Echinaria, Holcus, Hordeum, Koeleria, Lolium, Melica, Phalaris and Poa.
Highly expressed NLRs were identified according to the pipeline described above, with the addition of Fast Approximate Tree Classification (FAT-CAT; https://github.com/shailen/FAT-CAT; ref. 118), which was used to classify nucleotide-binding domains on the basis of a phylogenetic tree developed from rice, Brachypodium distachyon and barley nucleotide-binding domains derived from NLRs67. NLR-encoding genes were advanced on the basis of the following requirements: the transcript must contain either a complete or 5′ partial open reading frame, the gene must be among the top 25% expressed NLRs and the gene must not belong to NLR families (C15/16) known to require an additional NLR. Among the candidate NLRs, redundancy was removed using CD-HIT (v.4.7)119 requiring 100% identity (c = 1.0).
For molecular cloning, PCR primers were developed using Gateway adapters attB1 and attB2 fused to the first 20 nucleotides of the start or end of the coding sequence, respectively. The proportion of cloned NLRs is variable according to species, guided by the available diversity of accessions in each species and the prevalence of resistance to target pathogens. PCR primers were developed for a total of 1,909 NLRs. In total, 1,019 NLRs were cloned into the Gateway pDONR entry vector. This set includes known resistance genes: Sr33 (wheat stem rust39), Sr35 (wheat stem rust40), Sr50 (wheat stem rust38), Lr21 (wheat leaf rust, Huang et al.120), Yr10 (wheat stripe rust121), Pm3b (wheat powdery mildew122,123), Mla3 (barley powdery mildew and rice blast35), Mla7 (barley powdery mildew) and Mla8/Rps7 (barley powdery mildew and wheat stripe rust57,124).
Plant transformation
The NLRs in the entry clones were transferred to the destination binary vector pDEST2BL by the LR reaction of the Gateway system. NLRs were expressed under the maize ubiquitin promoter (Supplementary Data 10). The destination vector pDEST2BL includes the DsRed2 fluorescent protein for a visual selectable marker in the seed125. The resultant transformation vectors were introduced into Agrobacterium tumefaciens strain EHA105 by electroporation. Agrobacterium strains carrying the transformation vectors were used to transform wheat cv. Fielder56 with the modification that an immature embryo was cut into three pieces when transferred to the second selection medium. Fielder seeds were obtained from Kihara Institute for Biological Research, Yokohama City University. Briefly, 15 immature embryos were infected with each of the Agrobacterium strains, and up to seven independent events per NLR were grown to maturity. A total of 6,260 transformation events were achieved for 999 NLRs, and seed was obtained from T1 plants from 995 NLRs. Fifty or more seeds were obtained from 96.4% of the harvested events. A total of 5,177 T1 families were generated; the families were further subdivided on the basis of the fluorescence of the seed to a total of 10,646 DsRed2 groups.
Inoculation and phenotyping with the wheat stem rust pathogen (Pgt) and the wheat leaf rust pathogen (Pt) at the seedling stage
Independent T1 families for each NLR construct were subsampled, and three seedlings from each T1 family were used for resistance phenotyping. Rust inoculations were performed according to the standard protocols used at the USDA-ARS Cereal Disease Laboratory and the University of Minnesota126. Briefly, on the day before inoculation, urediniospores of the rust pathogens were removed from a −80 °C freezer, heat-shocked in a 45 °C water bath for 15 min and then rehydrated in an 80% relative humidity chamber overnight. After germination rates were assessed127, 10 mg of urediniospores were placed into a gelatin capsule (size 00), and 700 ml of the light mineral oil (Soltrol 170, Chevron Phillips Chemical Company) carrier was added. The inoculum suspension was applied to 12-day-old plants (second leaf fully expanded) using custom atomizers (Tallgrass Solutions) pressured by a pump set at 25–30 kPa. Approximately 0.15 mg of urediniospores were applied per plant. Immediately after inoculation, the plants were placed in front of a small electric fan for 3–5 min to hasten the evaporation of the oil carrier from the leaf surfaces. The plants were off-gassed for an additional 90 min before being placed inside mist chambers. In the mist chambers, ultrasonic humidifiers (Vick’s model V5100NSJUV; Proctor & Gamble) were run continuously for 30 min to provide sufficient initial moisture for the germination of urediniospores. For the next 16–20 h, the plants were kept in the dark, and the humidifiers were run for 2 min every 15 min to maintain moisture on the plants. Light (400-W high-pressure sodium lamps emitting 300 mmol photons per s per m2) was provided for 2 to 4 h after the dark period. The chamber doors were opened halfway to allow the leaf surfaces to dry completely before the plants were returned to the greenhouse under the same conditions as described above126.
All rust phenotyping experiments were conducted in a completely randomized design. Accessions exhibiting variable reactions across experiments were repeated in an additional experiment if sufficient seeds were available. Stem rust infection types on the accessions were scored 12 days after inoculation using a 0 to 4 Stakman scale (0, 1−, 1, 1+, 2−, 2, 2+, 3−, 3, 3+, and 4)128,129. The semicolon symbol ‘;’ indicates a hypersensitive fleck. Raw seedling infection type data were converted to a numerical 0–9 linear scale as detailed in ref. 130. The initial phenotyping of transgenic lines was performed with the Minnesota, USA, Pgt race QTHJC (isolate 69MN399, originally collected in Minnesota, USA, and provided by Y. Jin, USDA-ARS Cereal Disease Laboratory, St. Paul, MN, USA). Lines found resistant to Pgt race QTHJC were further phenotyped against the more broadly virulent Pgt race TTKSK (isolate 04KEN156/04 provided by Y. Jin) from Kenya. Both races are virulent on the wheat cultivar Fielder. T2 plants were used for photographs of Pgt race QTHJC infection and for NLRs NLR_05_75, NLR_05_92, NLR_08_16, NLR_08_79 and NLR_09_55 for Pgt race TTKSK screening due to seed availability.
Wheat leaf rust screening was carried out using a protocol similar to that used for wheat stem rust screening but without the light period being provided at the end of the infection period126. Pt race THBJ (isolate 99ND588DLL, provided by J. Kolmer, USDA-ARS Cereal Disease Laboratory, St. Paul, MN, USA) was used for the assays. Leaf rust infection types were scored after inoculation using a 0 to 4 scale131, and the data were converted to a numerical 0–9 linear scale as described above.
Inoculation and phenotyping with the wheat stem rust pathogen (Pgt) at the adult stage
Individual lines found resistant in the seedling assays with Pgt race QTHJC were bulked, and T2 families from multi-transgene lines and control lines were planted at the University of Minnesota Rosemount Research and Outreach Center in Rosemount, MN, in 2021 and 2022. The wheat cultivars Fielder, Morocco and LMPG-6 were used as susceptible controls. Field inoculations were performed with Pgt race QTHJC and as previously described in Luo et al.9. Several of the lines found resistant in the seedling stage were not available for testing in the field due to low seed production, and therefore partially overlapping field trials were used. Approximately 25 seeds per line were planted in each plot. When the first nodes of plants were detectable (31 Zadoks scale132), they were inoculated with a suspension of Pgt urediniospores (1 g urediniospores per 1 l Soltrol 170 mineral oil) using an ultra-low-volume sprayer (Mini-ULVA, Micron Group). Three additional inoculations were made in successive weeks to ensure high infection levels during the later stages of crop development. Severity was recorded as the visual percentage (0–100%) of stem and leaf sheath tissue covered by uredinia. Ratings were assessed using the modified Cobb scale133. The infection responses were recorded as highly resistant (clear hypersensitive infection sites but with no pathogen sporulation), resistant (minute to small uredinia surrounded by chlorosis or necrosis), moderately resistant (medium-sized uredinia often surrounded by chlorosis), moderately susceptible (medium to large erumpent uredinia with little or no chlorosis) or susceptible (very large erumpent uredinia with little or no chlorosis).
Phylogenetic analyses
Phylogenetic analysis of the nucleotide-binding domains of NLRs was carried out as described by Bailey et al.67 using updated NLR gene annotations for barley134, B. distachyon (v.3.1; NCBI PRJNA32607 and PRJNA74771) and wheat135. Nucleotide-binding domains from NLRs were identified using HMMer (v.3.3.2) (http://hmmer.org/) hmmalign with the hidden Markov model encompassing the NB-ARC1-ARC2 domains67. The alignment was converted to an aligned FASTA file using esl-reformat and processed using the QKphylogeny script QKphylogeny_alignment_analysis.py with the parameters d = 0.3 (non-redundant), b = 0.5 (breadth coverage of alignment greater than or equal to 50%) and d = 0.3 (depth of coverage at each residue of greater than or equal to 30%). The phylogenetic tree was constructed using RAxML (v.8.2.12)136 using the PROTGAMMAJTT model and 1,000 bootstraps and visualized using iTOL (https://itol.embl.de/).
The Pooideae species phylogenetic tree was generated using the QKbusco pipeline (https://github.com/matthewmoscou/QKbusco). BUSCO (v.3.0.2)137 with the default parameters and the embryophyte_odb9 library was used to identify genes using annotated coding sequences (sequenced genomes) or open reading frames predicted using TransDecoder (v.4.1.0) from de novo assemblies (transcriptomes). QKbusco_merge.py was used to parse the BUSCO output and prepare FASTA files for multiple sequence alignment. The parameter status was set to fragmented to allow fragmented coding sequences to be included in the analysis. Codon-based multiple sequence alignment of individual genes was performed using PRANK (v.170427)138. Individual gene multiple sequence alignments were merged using QKbusco_phylogeny.py using a coverage depth of 40% at individual sites for inclusion in the alignment. The maximum likelihood phylogenetic tree was generated using RAxML (v.8.2.12) with the GTRGAMMA model.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The whole-genome sequencing data from barley accessions CI 16147 and CI 16153 have been deposited in NCBI under BioProject PRJNA952654. The RNA-seq data for Arabidopsis thaliana, tomato and diverse Pooideae species have been deposited in NCBI under BioProjects PRJNA928100, PRJNA927036 and PRJNA913397, respectively. The GenBank identifiers for the transformation construct sequence for Mla7 under the Mla6 promoter/terminator and the native sequence are MZ555770 and OQ859100, respectively. The databases used for protein domain analysis include Pfam, Superfamily and ProSite. The raw data and uncropped images are available via figshare at https://doi.org/10.6084/m9.figshare.28680800.v1 (ref. 139).
Code availability
The scripts used for data analysis and figure preparation are available via figshare at https://doi.org/10.6084/m9.figshare.28680800.v1 (ref. 139).
Change history
06 February 2026
In the version of the article initially published, affiliation 7 was incomplete and has now been amended to “Department of Integrated Plant Protection, Agrotest Fyto Ltd, Kroměříž, Czech Republic” in the HTML and PDF versions of the article.
References
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439 (2019).
Ristaino, J. B. et al. The persistent threat of emerging plant disease pandemics to global food security. Proc. Natl Acad. Sci. USA 118, e2022239118 (2021).
Bebber, D. P. Range-expanding pests and pathogens in a warming world. Annu. Rev. Phytopathol. 53, 335–356 (2015).
Fisher, M. C. et al. Emerging fungal threats to animal, plant and ecosystem health. Nature 484, 186–194 (2012).
Shaw, M. W. & Osborne, T. M. Geographic distribution of plant pathogens in response to climate change. Plant Pathol. 60, 31–43 (2011).
Kettles, G. J. & Luna, E. Food security in 2044: how do we control the fungal threat? Fungal Biol. 123, 558–564 (2019).
Hovmøller, M. S., Thach, T. & Justesen, A. F. Global dispersal and diversity of rust fungi in the context of plant health. Curr. Opin. Microbiol. 71, 102243 (2022).
Dodds, P. N. & Rathjen, J. P. Plant immunity: towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548 (2010).
Luo, M. et al. A five-transgene cassette confers broad-spectrum resistance to a fungal rust pathogen in wheat. Nat. Biotechnol. 39, 561–566 (2021).
van Esse, H. P., Reuber, T. L. & van der Does, D. Genetic modification to improve disease resistance in crops. N. Phytol. 225, 70–86 (2020).
Hafeez, A. N. et al. Creation and judicious application of a wheat resistance gene atlas. Mol. Plant 14, 1053–1070 (2021).
Wulff, B. B. & Krattinger, S. G. The long road to engineering durable disease resistance in wheat. Curr. Opin. Biotechnol. 73, 270–275 (2022).
Schulze-Lefert, P. & Panstruga, R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 16, 117–125 (2011).
Panstruga, R. & Moscou, M. J. What is the molecular basis of nonhost resistance? Mol. Plant Microbe Interact. 33, 1253–1264 (2020).
Bevan, M. W. et al. Genomic innovation for crop improvement. Nature 543, 346–354 (2017).
Mascher, M. et al. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 51, 1076–1081 (2019).
Van Der Biezen, E. A. & Jones, J. D. Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem. Sci. 23, 454–456 (1998).
Cesari, S., Bernoux, M., Moncuquet, P., Kroj, T. & Dodds, P. N. A novel conserved mechanism for plant NLR protein pairs: the ‘integrated decoy’ hypothesis. Front. Plant Sci. 5, 606 (2014).
Saur, I. M., Panstruga, R. & Schulze-Lefert, P. NOD-like receptor-mediated plant immunity: from structure to cell death. Nat. Rev. Immunol. 21, 305–318 (2021).
Tian, D., Traw, M. B., Chen, J. Q., Kreitman, M. & Bergelson, J. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423, 74–77 (2003).
Xiao, S., Brown, S., Patrick, E., Brearley, C. & Turner, J. G. Enhanced transcription of the Arabidopsis disease resistance genes RPW8.1 and RPW8.2 via a salicylic acid–dependent amplification circuit is required for hypersensitive cell death. Plant Cell 15, 33–45 (2003).
Palma, K. et al. Autoimmunity in Arabidopsis acd11 is mediated by epigenetic regulation of an immune receptor. PLoS Pathog. 6, e1001137 (2010).
Deng, Y. et al. Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355, 962–965 (2017).
Lai, Y. & Eulgem, T. Transcript-level expression control of plant NLR genes. Mol. Plant Pathol. 19, 1267–1281 (2018).
Brown, J. K. M. Yield penalties of disease resistance in crops. Curr. Opin. Plant Biol. 5, 339–344 (2002).
Brown, J. K. M. & Rant, J. C. Fitness costs and trade-offs of disease resistance and their consequences for breeding arable crops. Plant Pathol. 62, 83–95 (2013).
Richard, M. M. S., Gratias, A., Meyers, B. C. & Geffroy, V. Molecular mechanisms that limit the costs of NLR-mediated resistance in plants. Mol. Plant Pathol. 19, 2516–2523 (2018).
Bergelson, J. & Purrington, C. B. Surveying patterns in the cost of resistance in plants. Am. Nat. 148, 536–558 (1996).
Tan, X. et al. Global expression analysis of nucleotide binding site-leucine rich repeat-encoding and related genes in Arabidopsis. BMC Plant Biol. 7, 56 (2007).
Balint‐Kurti, P. The plant hypersensitive response: concepts, control and consequences. Mol. Plant Pathol. 20, 1163–1178 (2019).
Karasov, T. L., Chae, E., Herman, J. J. & Bergelson, J. Mechanisms to mitigate the trade-off between growth and defense. Plant Cell 29, 666–680 (2017).
Stokes, T. L., Kunkel, B. N. & Richards, E. J. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 16, 171–182 (2002).
Howles, P. et al. Autoactive alleles of the flax L6 rust resistance gene induce non-race-specific rust resistance associated with the hypersensitive response. Mol. Plant Microbe Interact. 18, 570–582 (2005).
Hatta, M. A. M. et al. The wheat Sr22, Sr33, Sr35 and Sr45 genes confer resistance against stem rust in barley. Plant Biotechnol. J. 19, 273–284 (2021).
Brabham, H. J. et al. Barley MLA3 recognizes the host-specificity effector Pwl2 from Magnaporthe oryzae. Plant Cell 36, 447–470 (2024).
Arora, S. et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 37, 139–143 (2019).
Chen, S., Zhang, W., Bolus, S., Rouse, M. N. & Dubcovsky, J. Identification and characterization of wheat stem rust resistance gene Sr21 effective against the Ug99 race group at high temperature. PLoS Genet. 14, e1007287 (2018).
Mago, R. et al. The wheat Sr50 gene reveals rich diversity at a cereal disease resistance locus. Nat. Plants 1, 15186 (2015).
Periyannan, S. et al. The gene Sr33, an ortholog of barley Mla genes, encodes resistance to wheat stem rust race Ug99. Science 341, 786–788 (2013).
Saintenac, C. et al. Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 341, 783–786 (2013).
Zhang, J. et al. A recombined Sr26 and Sr61 disease resistance gene stack in wheat encodes unrelated NLR genes. Nat. Commun. 12, 3378 (2021).
Zhang, W. et al. Identification and characterization of Sr13, a tetraploid wheat gene that confers resistance to the Ug99 stem rust race group. Proc. Natl Acad. Sci. USA 114, E9483–E9492 (2017).
Steuernagel, B. et al. Rapid cloning of disease-resistance genes in plants using mutagenesis and sequence capture. Nat. Biotechnol. 34, 652–655 (2016).
Luo, J. et al. Identification and characterization of Sr22b, a new allele of the wheat stem rust resistance gene Sr22 effective against the Ug99 race group. Plant Biotechnol. J. 20, 554–563 (2022).
Zhang, J. et al. Single amino acid change alters specificity of the multi-allelic wheat stem rust resistance locus SR9. Nat. Commun. 14, 7354 (2023).
Upadhyaya, N. M. et al. Genomics accelerated isolation of a new stem rust avirulence gene–wheat resistance gene pair. Nat. Plants 7, 1220–1228 (2021).
Cloutier, S. et al. Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol. Biol. 65, 93–106 (2007).
Feuillet, C. et al. Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc. Natl Acad. Sci. USA 100, 15253–15258 (2003).
Hewitt, T. et al. Wheat leaf rust resistance gene Lr13 is a specific Ne2 allele for hybrid necrosis. Mol. Plant 14, 1025–1028 (2021).
Lin, G. et al. Cloning of the broadly effective wheat leaf rust resistance gene Lr42 transferred from Aegilops tauschii. Nat. Commun. 13, 3044 (2022).
Mapuranga, J. et al. Harnessing genetic resistance to rusts in wheat and integrated rust management methods to develop more durable resistant cultivars. Front. Plant Sci. https://doi.org/10.3389/fpls.2022.951095 (2022).
Sharma, D. et al. A single NLR gene confers resistance to leaf and stripe rust in wheat. Nat. Commun. 15, 9925 (2024).
Thind, A. K. et al. Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat. Biotechnol. 35, 793–796 (2017).
Yan, X. et al. High-temperature wheat leaf rust resistance gene Lr13 exhibits pleiotropic effects on hybrid necrosis. Mol. Plant 14, 1029–1032 (2021).
Huang, L. et al. Evolution of new disease specificity at a simple resistance locus in a crop–weed complex: reconstitution of the Lr21 gene in wheat. Genetics 182, 595–602 (2009).
Ishida, Y., Tsunashima, M., Hiei, Y. & Komari, T. Wheat (Triticum aestivum L.) transformation using immature embryos. Methods Mol. Biol. 1223, 189–198 (2015).
Bettgenhaeuser, J. et al. The barley immune receptor Mla recognizes multiple pathogens and contributes to host range dynamics. Nat. Commun. 12, 6915 (2021).
Kawashima, C. G. et al. A pigeonpea gene confers resistance to Asian soybean rust in soybean. Nat. Biotechnol. 34, 661–665 (2016).
Witek, K. et al. A complex resistance locus in Solanum americanum recognizes a conserved Phytophthora effector. Nat. Plants 7, 198–208 (2021).
Wu, C. H. et al. NLR network mediates immunity to diverse plant pathogens. Proc. Natl Acad. Sci. USA 114, 8113–8118 (2017).
Adachi, H. & Kamoun, S. NLR receptor networks in plants. Essays Biochem. 66, 541–549 (2022).
Zhang, X.-C. & Gassmann, W. RPS4-mediated disease resistance requires the combined presence of RPS4 transcripts with full-length and truncated open reading frames. Plant Cell 15, 2333–2342 (2003).
Dinesh-Kumar, S. P. & Baker, B. J. Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proc. Natl Acad. Sci. USA 97, 1908–1913 (2000).
Tang, F., Yang, S., Gao, M. & Zhu, H. Alternative splicing is required for RCT1-mediated disease resistance in Medicago truncatula. Plant Mol. Biol. 82, 367–374 (2013).
Cesari, S. et al. The rice resistance protein pair RGA4/RGA5 recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell 25, 1463–1481 (2013).
Periyannan, S. et al. Identification of a robust molecular marker for the detection of the stem rust resistance gene Sr45 in common wheat. Theor. Appl. Genet. 127, 947–955 (2014).
Bailey, P. C. et al. Dominant integration locus drives continuous diversification of plant immune receptors with exogenous domain fusions. Genome Biol. 19, 23 (2018).
Cesari, S. et al. The NB-LRR proteins RGA4 and RGA5 interact functionally and physically to confer disease resistance. EMBO J. 33, 1941–1959 (2014).
Wang, X. et al. The rpg4-mediated resistance to wheat stem rust (Puccinia graminis) in barley (Hordeum vulgare) requires Rpg5, a second NBS-LRR gene, and an actin depolymerization factor. Mol. Plant Microbe Interact. 26, 407–418 (2013).
Rouse, M. N., Olson, E. L., Gill, B. S., Pumphrey, M. O. & Jin, Y. Stem rust resistance in Aegilops tauschii germplasm. Crop Sci. 51, 2074–2078 (2011).
Maekawa, T. et al. Subfamily-specific specialization of RGH1/MLA immune receptors in wild barley. Mol. Plant Microbe Interact. 32, 107–119 (2018).
Förderer, A. et al. A wheat resistosome defines common principles of immune receptor channels. Nature 610, 532–539 (2022).
Saur, I. M. et al. Multiple pairs of allelic MLA immune receptor–powdery mildew AVRA effectors argue for a direct recognition mechanism. eLife https://doi.org/10.7554/eLife.44471 (2019).
Contreras, M. P. et al. Resurrection of plant disease resistance proteins via helper NLR bioengineering. Sci Adv. 9, eadg3861 (2023).
Li, X., Clarke, J. D., Zhang, Y. & Dong, X. Activation of an EDS1-mediated R-gene pathway in the snc1 mutant leads to constitutive, NPR1-independent pathogen resistance. Mol. Plant Microbe Interact. 14, 1131–1139 (2001).
Oldroyd, G. E. D. & Staskawicz, B. J. Genetically engineered broad-spectrum disease resistance in tomato. Proc. Natl Acad. Sci. USA 95, 10300–10305 (1998).
Yang, S. et al. Rapidly evolving R genes in diverse grass species confer resistance to rice blast disease. Proc. Natl Acad. Sci. USA 110, 18572–18577 (2013).
Yi, H. & Richards, E. J. A cluster of disease resistance genes in Arabidopsis is coordinately regulated by transcriptional activation and RNA silencing. Plant Cell 19, 2929–2939 (2007).
Yi, H. & Richards, E. J. Phenotypic instability of Arabidopsis alleles affecting a disease resistance gene cluster. BMC Plant Biol. 8, 36 (2008).
Halterman, D. A., Wei, F. & Wise, R. P. Powdery mildew-induced Mla mRNAs are alternatively spliced and contain multiple upstream open reading frames. Plant Physiol. 131, 558–567 (2003).
Guo, H., Wang, S. & Jones, J. D. Autoactive Arabidopsis RPS4 alleles require partner protein RRS1-R. Plant Physiol. 185, 761–764 (2021).
Shirano, Y., Kachroo, P., Shah, J. & Klessig, D. F. A gain-of-function mutation in an Arabidopsis Toll Interleukin1 Receptor–Nucleotide Binding Site–Leucine-Rich Repeat type R gene triggers defense responses and results in enhanced disease resistance. Plant Cell 14, 3149–3162 (2002).
Yang, X.-M. et al. Broad-spectrum resistance gene RPW8.1 balances immunity and growth via feedback regulation of WRKYs. Plant Biotechnol. J. 22, 116–130 (2024).
Borrill, P. et al. An autoactive NB-LRR gene causes Rht13 dwarfism in wheat. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2209875119 (2022).
Tong, M. et al. E3 ligase SAUL1 serves as a positive regulator of PAMP-triggered immunity and its homeostasis is monitored by immune receptor SOC3. N. Phytol. 215, 1516–1532 (2017).
Lorang, J. et al. Tricking the guard: exploiting plant defense for disease susceptibility. Science 338, 659–662 (2012).
Innan, H. & Kondrashov, F. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108 (2010).
Ma, S. et al. Oligomerization-mediated autoinhibition and cofactor binding of a plant NLR. Nature 632, 869–876 (2024).
Wang, J. et al. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 364, eaav5870 (2019).
Komori, T. et al. High-throughput phenotypic screening of random genomic fragments in transgenic rice identified novel drought tolerance genes. Theor. Appl. Genet. 133, 1291–1301 (2020).
Moseman, J. G. Isogenic barley lines for reaction to Erysiphe graminis f. sp. hordei. Crop Sci. 12, 681–682 (1972).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics https://doi.org/10.1093/bioinformatics/btu170 (2014).
Mapleson, D. et al. KAT: a K-mer Analysis Toolkit to quality control NGS datasets and genome assemblies. Bioinformatics https://doi.org/10.1093/bioinformatics/btw663 (2016).
R Core Team R: A Language and Environment for Statistical Computing (R Core Team, 2025).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Halterman, D., Zhou, F., Wei, F., Wise, R. P. & Schulze-Lefert, P. The MLA6 coiled-coil, NBS-LRR protein confers AvrMla6-dependent resistance specificity to Blumeria graminis f. sp. hordei in barley and wheat. Plant J. 25, 335–348 (2001).
Gibson, D. G. Programming biological operating systems: genome design, assembly and activation. Nat. Methods 11, 521–526 (2014).
Hensel, G., Kastner, C., Oleszczuk, S., Riechen, J. & Kumlehn, J. Agrobacterium-mediated gene transfer to cereal crop plants: current protocols for barley, wheat, triticale, and maize. Int. J. Plant Genom. https://doi.org/10.1155/2009/835608 (2009).
Yeo, F. K. et al. Golden SusPtrit: a genetically well transformable barley line for studies on the resistance to rust fungi. Theor. Appl. Genet. 127, 325–337 (2014).
Bartlett, J. G., Alves, S. C., Smedley, M., Snape, J. W. & Harwood, W. A. High-throughput Agrobacterium-mediated barley transformation. Plant Methods 4, 22 (2008).
Close, T. J. et al. Development and implementation of high-throughput SNP genotyping in barley. BMC Genom. 10, 582 (2009).
Dreiseitl, A. A novel resistance against powdery mildew found in winter barley cultivars. Plant Breed. 138, 840–845 (2019).
Kølster, P., Munk, L., Stølen, O. & Løhde, J. Near-isogenic barley lines with genes for resistance to powdery mildew. Crop Sci. 26, 903–907 (1986).
Kølster, P. & Stølen, O. Barley isolines with genes for resistance to Erysiphe graminis f. sp. hordei in the recurrent parent ‘Siri’. Plant Breed. 98, 79–82 (1987).
Dreiseitl, A. Postulation of specific disease resistance genes in cereals: a widely used method and its detailed description. Pathogens 11, 284 (2022).
Torp, J., Jensen, H. P. & Jørgensen, J. H. Powdery mildew resistance genes in 106 northwest European spring barley cultivars. Kongelige Veterinaer- Og Landbohoejskole, 75–102 (1978).
Andrews, S. FastQC. Babraham Informatics http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. https://doi.org/10.1038/nbt.1883 (2011).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. https://doi.org/10.1186/1471-2105-10-421 (2009).
Witek, K. et al. Accelerated cloning of a potato late blight-resistance gene using RenSeq and SMRT sequencing. Nat. Biotechnol. 34, 656–660 (2016).
Foster, S. J. et al. Rpi-vnt1.1, a Tm-22 homolog from Solanum venturii, confers resistance to potato late blight. Mol. Plant Microbe Interact. 22, 589–600 (2009).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods https://doi.org/10.1038/nmeth.2019 (2012).
Posit Team RStudio: Integrated Development Environment for R (Posit Software, 2025).
Haas, B. J. TransDecoder. GitHub https://github.com/TransDecoder/TransDecoder (2024).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. https://doi.org/10.1093/nar/gkh340 (2004).
Afrasiabi, C., Samad, B., Dineen, D., Meacham, C. & Sjölander, K. The PhyloFacts FAT-CAT web server: ortholog identification and function prediction using fast approximate tree classification. Nucleic Acids Res. 41, W242–W248 (2013).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics https://doi.org/10.1093/bioinformatics/btl158 (2006).
Huang, L. et al. Map-based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics https://doi.org/10.1093/genetics/164.2.655 (2003).
Liu, W. et al. The stripe rust resistance gene Yr10 encodes an evolutionary-conserved and unique CC-NBS-LRR sequence in wheat. Mol. Plant 7, 1740–1755 (2014).
Srichumpa, P., Brunner, S., Keller, B. & Yahiaoui, N. Allelic series of four powdery mildew resistance genes at the Pm3 locus in hexaploid bread wheat. Plant Physiol. 139, 885–895 (2005).
Yahiaoui, N., Srichumpa, P., Dudler, R. & Keller, B. Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J. 37, 528–538 (2004).
Halterman, D. A. & Wise, R. P. A single-amino acid substitution in the sixth leucine-rich repeat of barley MLA6 and MLA13 alleviates dependence on RAR1 for disease resistance signaling. Plant J. 38, 215–226 (2004).
Yarbrough, D., Wachter, R. M., Kallio, K., Matz, M. V. & Remington, S. J. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-Å resolution. Proc. Natl Acad. Sci. USA 98, 462–467 (2001).
Huang, S., Steffenson, B. J., Sela, H. & Stinebaugh, K. Resistance of Aegilops longissima to the rusts of wheat. Plant Dis. 102, 1124–1135 (2018).
Scott, J. C., Manisterski, J., Sela, H., Ben-Yehuda, P. & Steffenson, B. J. Resistance of Aegilops species from Israel to widely virulent African and Israeli races of the wheat stem rust pathogen. Plant Dis. 98, 1309–1320 (2014).
Roelfs, A. P. & Martens, J. An international system of nomenclature for Puccinia graminis f. sp. tritici. Phytopathology 78, 526–533 (1988).
Stakman, E. C., Stewart, D. & Loegering, W. Q. Identification of Physiologic Races of Puccinia graminis var. tritici (USDA, 1962).
Zhang, D., Bowden, R. L., Yu, J., Carver, B. F. & Bai, G. Association analysis of stem rust resistance in U.S. winter wheat. PLoS ONE 9, e103747 (2014).
Kolmer, J. Leaf rust of wheat: pathogen biology, variation and host resistance. Forests 4, 70–84 (2013).
Zadoks, J. C., Chang, T. T. & Konzak, C. F. A decimal code for the growth stages of cereals. Weed Res. 14, 415–421 (1974).
Peterson, R., Campbell, A. & Hannah, A. A diagrammatic scale for estimating rust severity on leaves and stem of cereals. Can. J. Res. 26, 490–500 (1948).
Mascher, M. et al. Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33, 1888–1906 (2021).
Zhu, T. et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 107, 303–314 (2021).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics https://doi.org/10.1093/bioinformatics/btu033 (2014).
Tegenfeldt F. et al. OrthoDB and BUSCO update: annotation of orthologs with wider sampling of genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae987 (2025).
Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. https://doi.org/10.1007/978-1-62703-646-7_10 (2014).
Moscou, M. & Brabham, H. Source data for Brabham et al. 2024 bioRxiv. figshare https://doi.org/10.6084/m9.figshare.28680800.v1 (2025).
Kourelis, J., Sakai, T., Adachi, H. & Kamoun, S. RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biol. 19, e3001124 (2021).
Acknowledgements
We thank V. Sham, M. Neequaye, H. Jennings, S. Sharpe, I. Amarnath, S. Perkins and K.-P. Nguyen for their technical help. We also thank P. Nicholson, Y. Gupta, J. Jones, N. Talbot, J. Rhodes and R. Heal for scientific discussion. In addition, we thank D. Horvath for scientific discussion and editing of the manuscript. Funding for this research came from the 2Blades Foundation, the Lieberman-Okinow Endowment at the University of Minnesota, Japan Tobacco Inc., Kaneka Corporation, the United Kingdom Research and Innovation-Biotechnology and Biological Sciences Research Council Institute Strategic Programme (grant no. BBS/E/J/000PR9795 to M.J.M.), the Gatsby Charitable Foundation (M.J.M.), and United States Department of Agriculture Agricultural Research Service CRIS no. 5062-21220-025-000D (M.J.M.). This research used resources provided by the SCINet project and the AI Center of Excellence of the USDA Agricultural Research Service, ARS project number 0500-00093-001-00-D.
Author information
Authors and Affiliations
Contributions
M.J.M., H.P.v.E., N.T. and R.P.F. conceptualized the project. M.J.M., H.J.B., B.S., O.N.M., I.H.-P. and T. Komori curated the data. H.J.B., M.J.M., O.N.M. and B.S. conducted the formal analysis. H.P.v.E., N.T. and R.P.F. acquired the funding. H.J.B., M.J.M., K.W., I.H.-P. and A.D. conducted the investigation. M.J.M., H.J.B., I.H.-P., C.Y., N.I., N.T., T. Komari, T. Komori, H.N., P.G., A.D., A.H., B.S., O.N.M. and A.F. developed the methodology. M.J.M., H.J.B. and T. Komori were responsible for project administration. M.J.M., A.H., N.T. and B.S. provided the resources for the project. M.J.M. implemented the software. M.J.M., H.J.B., H.P.v.E., B.S. and T. Komori supervised the project. M.J.M., H.J.B. and K.W. validated the data. M.J.M. and H.J.B. visualized the results. H.J.B. wrote the original draft of the paper. M.J.M., H.J.B., H.P.v.E., K.W., B.S., O.N.M., T. Komori, T. Komari and A.D. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.J.M. and H.P.v.E. are inventors on a US provisional patent application no. 63/186,986 filed by 2Blades and relating to the use of preparing a library of plant disease resistance genes for functional testing for disease resistance. R.P.F. is a principal advisor to the Gatsby Foundation, and at the time of this work he was executive chairman of the 2Blades Foundation. H.P.v.E. serves on the 2Blades board. The other authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks Zhiyong Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11 and Tables 1–4.
Supplementary Data 1
Additional references for known NLR genes.
Supplementary Data 2
Expression of NLRs from the transcriptomes of Arabidopsis and Solanum species.
Supplementary Data 3
Details of the NLRs present in the NLRseek transgenic wheat collection.
Supplementary Data 4
Details on the NLRseek transgenic lines and their raw infection types, linearized infection type scores and weighted averages to races QTHJC and TTKSK of the stem rust pathogen (Pgt) at the seedling stage in the greenhouse.
Supplementary Data 5
Details on the NLRseek transgenic lines and their rust severity and infection responses to race QTHJC of the stem rust pathogen (Pgt) in field trials conducted in Rosemount, Minnesota, in 2021 and 2022.
Supplementary Data 6
Detailed infection type and weighted average phenotypes of known stem-rust-resistant Sr genes.
Supplementary Data 7
Details on the secondary NLRseek transgenic lines and their raw infection types including previous screening tests for comparison, linearized infection type scores and weighted averages to races QTHJC and TTKSK of the stem rust pathogen (Pgt) at the seedling stage in the greenhouse across T1 and T2.
Supplementary Data 8
Details on the NLRseek transgenic lines and their raw infection types, linearized infection type scores and weighted averages to race THBJ of the leaf rust pathogen (Pt) at the seedling stage in the greenhouse.
Supplementary Data 9
Details of the NLRs identified from donor species and transformed as part of the NLRseek collection.
Supplementary Data 10
Sequence of the maize ubiquitin promoter used in construct development.
Supplementary Data 11
Sequences of the synthesized Rpi-amr1 isoforms.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brabham, H.J., Hernández-Pinzón, I., Yanagihara, C. et al. Discovery of functional NLRs using expression level, high-throughput transformation and large-scale phenotyping. Nat. Plants 11, 2100–2114 (2025). https://doi.org/10.1038/s41477-025-02110-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41477-025-02110-w
