
Abstract
Senescent cells, characterized by a state of irreversible proliferative arrest and inflammatory profile, have emerged as drivers of age-related decline. Growing evidence suggests that alterations in mitochondrial function and morphology play a key role in the induction and maintenance of senescence, as well as in promotion of the proinflammatory senescence-associated secretory phenotype (SASP). In this review, we seek to survey the relationship between mitochondrial dysfunction and senescence, focusing on the consequences of changes in oxidative phosphorylation efficiency, calcium handling, mitochondrial metabolites, mitochondrial dynamics and quality control, and release of damage-associated molecular patterns. We first describe these changes before illustrating the pathways and mechanisms through which mitochondrial dysfunction results in cell cycle arrest and the SASP. Lastly, we showcase evidence relating cellular senescence to neurodegenerative disease and propose that mitochondrial dysfunction may act as a bridge between the two.
Similar content being viewed by others


Introduction
The nature and cause of aging have long remained a topic of considerable debate, despite its widespread appearance across species. One of the most prevalent and influential theories to explain aging was proposed by Denham Harman in 1955 as the free radical theory of aging1. In it, Harman postulates that aerobic respiration could produce reactive oxygen species (ROS) that contain an unpaired electron, which can damage molecules within cells and impair their function. The accumulation of oxidative damage over time leads to a decline in tissue and organ function, ultimately resulting in the aging phenotype. Nearly two decades later, Harman revised his theory to include the role of mitochondria, the organelles responsible for cellular respiration, proposing the mitochondrial free radical theory of aging (MFRTA)2. The MFRTA states that, by virtue of their role in respiration, mitochondria are responsible for producing damaging ROS. Later refinement of the theory proposed a positive feedback loop or “vicious cycle” in which damage to mitochondria and mitochondrial DNA (mtDNA) by ROS would promote the production of further ROS, causing a decline in mitochondrial function and a cascade of damage to cells, tissues, organs, and the organism3.
Efforts to test the MFRTA using various methods have yielded mixed and sometimes conflicting results. An assertion arising from the MFRTA is that the mitochondria of longer-lived species should produce fewer ROS than those of shorter-lived species. In general, this association appears to hold true, at least in regards to superoxide and hydrogen peroxide (H2O2) production4,5,6. However, species that do not hold to this trend, such as the naked mole rat and questions regarding the validity of the methods used to measure H2O2 production have called the accuracy of these findings into question5,7. A more direct way of testing the MFRTA is through the manipulation of the cell’s ability to combat the effects of ROS via free radical-quenching antioxidant molecules or enzymes, which should presumably reduce oxidative damage and thus extend lifespan, while decreasing these defenses should decrease lifespan. Antioxidant supplements have been tested in several different model organisms, including Caenorhabditis elegans, Drosophila melanogaster, and Mus musculus, with mixed results. In invertebrates, many antioxidants tested do exert a positive influence on lifespan6,8; again, with exceptions. For example, chronic administration of dietary thiols, important cellular antioxidants, decreases lifespan in C. elegans9. In addition, exposure of C. elegans to ROS-generating compounds can, in fact, increase lifespan6,10, likely via activation of the unfolded protein response of the mitochondria11. Results of antioxidant supplementation in mice are even more heterogenous8,12,13. Notably, no extension of lifespan occurs with administration of coenzyme Q10, a mitochondrial antioxidant14 and no consistent effect on lifespan is observed either with overexpression or knockouts of several antioxidant enzymes6,12,15,16,17. Likely, these inconsistent results are driven by the complex roles of ROS as a signaling molecule in diverse pathways, some of which can have beneficial effects18.
The ambiguous results produced from efforts to validate the MFRTA have now shifted the field to characterize oxidative damage from ROS as being one of many sources of damage19 that contribute to a diverse array of interconnected aging hallmarks20, rather than being a sole, primary driver of aging. However, progress in mitochondrial research made since the inception of the MFRTA has revealed mitochondria as dynamic players in many cellular processes. Mitochondria undergo a variety of changes that can have both beneficial and detrimental effects on cellular processes, influencing metabolism, signaling, and cell fate decisions in complex ways. The regulation of mitochondrial function is thus central to cellular function, organismal health, aging, and disease. Given the breadth of this topic, we focus here on a specific area of interest to the aging field: how mitochondrial alterations contribute to cellular senescence, another key hallmark and driver of aging. Here, we will review the changes that mitochondria undergo in senescent cells and describe the role this plays in driving senescence-related pathology, ultimately explaining how these changes manifest as a driver of age-related neurodegenerative disease.
Cellular senescence and aging
Cellular senescence, first described in the early 1960s21, is a state of irreversible cell cycle arrest that results in widespread changes to gene expression and cell morphology22,23. Senescent cells play important physiological roles in development, wound healing, and cancer suppression, but contribute to age-related pathology in later life24. Senescence can be induced by a wide variety of means, including telomere shortening (replicative senescence (RS)), activation of oncogenes (oncogene-induced senescence (OIS)), and exposure to genotoxic agents, such as radiation (stress-induced senescence (SIS))23. These cause DNA damage, leading to the recruitment of protein kinases, such as ataxia-telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related (ATR) and initiation of the DNA damage response (DDR)25. This ultimately induces a stable cell-cycle arrest through the cyclin-dependent kinase inhibitors p16 and p21 via the tumor-suppressive transcriptional regulators pRB and p53, respectively23. In addition to cell cycle arrest, senescent cells undergo a change in morphology to become large and flat, increase lysosomal β-D-galactosidase activity termed senescence-associated β-galactosidase (SA-β-gal)26,27, and display resistance to apoptosis28. Increases in markers of DNA damage, including γH2AX and 53BP1, changes in heterochromatin, loss of Lamin B1, relocation of high mobility group box 1 (HMGB1) from the nucleus to cytoplasm, and fragments of chromatin present in the cytoplasm29,30 are also observed in senescence. A combination of these and other phenotypic changes is used to identify senescent cells in culture and tissues.
The lack of a definitive senescence marker has led to some controversy over what constitutes a senescent cell. For example, in response to immune stimulation, macrophages express p16 and SA-β-gal, which reflect an activated state and not senescence31. In addition, single-cell and single-nucleus transcriptomic approaches have revealed the heterogeneous nature of senescent cells32. Single-cell RNA-seq of RS, irradiation-induced senescence, and etoposide-induced senescence revealed similarities in the transcriptome between both SIS models, but significant variation from RS cells33. Furthermore, several distinct clusters emerged from this analysis, including growth-arrested cells, which did not exhibit other senescent phenotypes, a group displaying the traditional senescence markers, and another senescent group associated with changes in ECM proteins, anti-apoptotic pathways, and long non-coding RNAs. These studies demonstrate that cellular senescence cannot be viewed as a monolith but instead manifests in diverse ways, highlighting the necessity of testing for multiple markers of senescence.
One significant pathological aspect of senescent cells is the secretion of signaling factors, including cytokines, chemokines, interleukins, growth factors, proteases, insoluble proteins, and extracellular matrix (ECM) components34. The central regulator of these inflammatory factors is nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which is activated as a result of DNA damage35. Together, these factors make up what is known as the senescence-associated secretory phenotype (SASP) and are believed to be one of the primary ways in which senescent cells contribute to tissue dysfunction and aging.
Current interest in cellular senescence largely focuses on the role senescent cells play in the aging process. Senescent cells accumulate with age in a number of species across a wide variety of tissues36,37,38,39, although the exact proportion of senescent cells in aged tissues remains controversial due to a lack of a single biomarker for senescence. Senescent cell accumulation is believed to be due to a decline in immune surveillance with age, as impairment to immune cell function accelerates senescent cell burden40,41. In tissue, senescent cells contribute to a proinflammatory microenvironment via the SASP42, which can act in both an autocrine and paracrine manner43,44, reinforcing senescence as well as inducing senescence in neighboring cells45,46. Thus, recent work has focused on demonstrating that elimination of senescent cells—either through genetic means or the use of senolytic drugs that selectively induce apoptosis in senescent cells—can extend healthspan and lifespan47,48,49, and a multitude of clinical trials targeting diverse conditions using senolytics are underway50,51. Like any other therapeutic, it will be important to consider possible side effects when administering senolytic treatments, particularly in light of the role senescent cells play in wound healing52 and liver regeneration53. Nevertheless, by targeting the largest risk factor for the diseases of late life, aging itself, senotherapeutics hold promise for the future of medicine, spurring the need to further understand the mechanisms that underlie the senescent phenotype. As investigations into these mechanisms have progressed, mitochondria have emerged as central players in both senescence induction and the SASP.
Mitochondrial dysfunction in cellular senescence
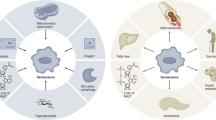
Mitochondria of senescent cells exhibit a myriad of changes in activity and morphology (Fig. 1), many of them similar to those that drive aging pathology20. Here, we describe these changes, which are consistent with mitochondrial dysfunction. Classically, the definition of mitochondrial dysfunction is a reduction in mitochondrial membrane potential and a corresponding loss in respiratory capacity per mitochondrion when at a steady state. We expand this definition beyond bioenergetics to encompass impairment in the maintenance of mitochondrial form and any of its multifaceted functions within the cell. Further, we explore studies which demonstrate that mitochondrial dysfunction is causative in senescence induction and that targeting mitochondria has therapeutic potential. Mitochondrial-based interventions from studies that demonstrate anti-senescence effects are summarized in Table 1.

Mitochondrial membrane potential (ΔΨm) decreases during senescence as a result of accumulating calcium from the endoplasmic reticulum (ER) through mitochondria-ER contact sites (MERCS) and increased proton leakage. These and other changes decrease electron transport chain (ETC) efficiency, increasing reactive oxygen species (ROS) production and decreasing ATP production. An imbalance in the mitochondrial metabolites NAD+/NADH exacerbates declining ATP levels, promoting activation of AMPK. In addition to supporting cell cycle arrest, activated AMPK promotes mitochondrial biogenesis. Mitochondrial biogenesis and additional changes to morphological regulation, including increased mitochondrial fusion and decreased mitophagy, further aid ROS production. DNA damage caused by ROS enforces cell cycle arrest and promotes the SASP. In addition, ROS activates JNK, which promotes the formation of cytosolic chromatin fragments (CCFs), activating the cytosolic DNA sensor cGAS, which, through STING activation, enhances SASP production. Minority mitochondrial outer membrane permeabilization (miMOMP) releases inflammatory DAMPs, including mtDNA and cardiolipin, which further enhances the SASP.
OXPHOS dysfunction and ROS generation
The generation of ATP through oxidative phosphorylation (OXPHOS) is dependent upon a negative mitochondrial membrane potential (ΔΨm) between the mitochondrial intermembrane space and matrix established by the electron transport chain (ETC). The ETC is made up of four protein complexes (complex I–IV), which pump protons as they are reduced by electrons from NADH and FADH2 generated in the tricarboxylic acid (TCA) cycle. This proton gradient provides the energetic basis needed to catalyze the formation of ATP from ADP mediated by ATP synthase. The loss of ΔΨm emerges as a unifying theme across both cellular senescence and aging. Cells induced to senesce by a wide variety of stimuli undergo a decrease in ΔΨm54,55,56,57, which coincides with an increase in proton leakage across the inner mitochondrial membrane57. These changes are caused in part by an increase in mitochondrial uncoupling via upregulation of mitochondrial uncoupling protein 255,57,58,59, which dissipates the proton gradient and reduces OXPHOS60. Mitochondrial uncoupling may initially be in response to an increase in ROS production60; however, chronic uncoupling compromises respiratory chain function and causes ROS accumulation, which can drive senescent phenotypes55,57,59,61,62,63,64. This leads to damage of mitochondrial components, such as mtDNA55, mitochondrial proteins65, and lipids66, which can further exacerbate damage in a feed-forward loop. For example, senescent cells exhibit increased ROS-mediated protein modification, including to enzymes involved in mitochondrial protein quality control and the repair of oxidized proteins, such as methionine sulfoxide reductase A and B65. Additionally, peroxidation of lipids by ROS leads to the formation of 4-hydroxy-2-nonenal (HNE), which can react with proteins to form adducts67; an increase in HNE-modified proteins is found in senescent cells, with many of the modified proteins localized to the mitochondria65. Misfolded or oxidized mitochondrial proteins—especially those involved in protein quality control—can result in further impairment of ETC complexes, which disrupt electron flow and increase electron leakage to generate ROS, which could abet the formation and persistence of further oxidative damage. In particular, ROS-mediated DNA damage has been implicated in senescence (see Box 1).
Multiple studies have shown that inhibition of ETC enzymes leads to senescence. Genetic or pharmacological disruption of complex I56,61, complex II68, and complex III56,69 induces senescence, and complex IV deficiency is a feature of senescent cells70,71. These interventions are accompanied by increases in ROS and decreases in ATP production and ΔΨm. Similarly, disruption of other mitochondrial enzymes that regulate TCA activity also induces senescence, even in the absence of DNA damage72,73,74. Interestingly, while chronic ETC inhibition promotes senescence, transient inhibition of complex III may paradoxically improve mitochondrial function75, perhaps through hormesis. OXPHOS dysfunction further promotes senescence by activating the SASP. Reduced OXPHOS gene expression and loss of ΔΨm increase ROS, which activate c-Jun N-terminal kinase (JNK) and promote formation of cytoplasmic chromatin fragments (CCFs)76. CCFs trigger cyclic GMP-AMP synthase - stimulator of interferon genes (cGAS-STING) signaling, amplifying inflammatory SASP factors and reinforcing senescence in neighboring cells77,78,79,80.
Interventions that act to either increase mitochondrial ROS or decrease ROS-protective mechanisms can induce senescence. For example, depletion of key antioxidant enzymes, including superoxide dismutase (SOD) 181, SOD282, catalase83, and glutathione84,85 promotes senescence. Administration of the cytokine and SASP factor transforming growth factor-β (TGF-β) in vitro to corneal endothelium cells decreases SOD2 expression, which increases ROS and senescence markers, including p16, p21, and SA-β-gal86. Conversely, other studies have demonstrated that increasing ROS defenses delays senescence. Mitoquinone mesylate (MitoQ) is a mitochondrial antioxidant, which can prevent induction of senescent markers in doxorubicin-induced human endothelial cells87. Overexpression of SOD3 in fibroblasts also increases replicative lifespan under both normoxic and hyperoxic conditions88. Finally, pretreatment of MRC-5 fibroblasts with the free radical scavenger α-phenyl-t-butyl-nitrone extends replicative lifespan89. Other antioxidants have also been shown to extend replicative lifespan, including methylene blue (MB)90, carnosine91, aminoguanidine92,93, and the NAD+ precursor nicotinamide94, although not every antioxidant does so95. This suggests that a reduction in ROS per se may not be solely responsible for extension in replicative lifespan but is likely due to additional mechanisms of action beyond antioxidant defense.
Mitochondrial elongation and impaired mitophagy
Senescent cells may compensate for functional defects in mitochondria by increasing mitochondrial biogenesis. Increased mitochondrial mass in senescent cells is observed in vitro55,96,97 as well as in vivo98. This is the result of increased expression of a number of proteins that are responsible for mitochondrial biogenesis, including peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), PGC-1β, mitochondrial transcription factor A (TFAM), and nuclear factor erythroid 2-related factor 2 (NRF2)56,96. A reduction in mitochondrial turnover further contributes to increases in mitochondrial mass and dysfunction99. For example, basal levels of mitophagy – a lysosome-mediated degradation of dysfunctional mitochondria100 – driven by the PTEN-induced kinase 1 (PINK1)/Parkin/p62 pathway are reduced within two hours of SIS induction, suggesting impairment in mitophagy occurs early in senescence101. Several mechanisms likely drive this impairment. First, separating a dysfunctional mitochondrion from the mitochondrial network through fission is a prerequisite for mitophagy102,103. In senescent cells, mitochondria display an elongated and interconnected morphology that coincides with a downregulation in expression of the fission machinery dynamin-related protein 1 (DRP1) and mitochondrial fission 1 protein (FIS1)104. p53 also plays a large role in altering mitochondrial dynamics and impairing mitophagy in senescence (see Box 2). In addition, a decrease in S-nitrosoglutathione reductase activity in senescent cells causes excessive S-nitrosylation of Parkin, inhibiting its activity and thus hindering mitophagy105. Finally, elevated mTORC1 activity results in reduced capacity for lysosomal degradation106,107. Altogether, these changes reduce mitophagy, which results in accumulation of lower quality mitochondria, ultimately increasing ROS production56,98.
A growing body of evidence has demonstrated that direct impairment of mitophagy induces senescence108,109,110, while activation of mitophagy can mitigate senescence111,112,113,114, revealing mitophagy as a potential therapeutic target for senescence. In lung fibroblasts, treatment with a mitophagy inhibitor causes mitochondrial elongation, increased mitochondrial mass, and increased senescence markers106. Conversely, activation of mitophagy using small molecules reduces senescence markers in SIS human dermal fibroblasts, although this does not affect the SASP factors IL-6 and IL-8101. Similarly, insulin-like growth factor 1 (IGF-1) suppresses RS in smooth muscle cells, an effect dependent on the activity of PINK1115. In mice, PINK1 knockout leads to mitochondrial dysfunction, increased cellular senescence, and elevated SASP in aging renal cells116. Impaired mitochondrial dynamics and quality control may also promote inflammation, as mitochondrial elongation facilitates NLR family pyrin domain-containing 3 (NLRP3) inflammasome assembly117. NLRP3 inflammasome activation also occurs via the generation of ROS through complex I or III inhibition or impaired mitophagy118,119. Additionally, mitophagy impairment increases ROS production, which activates JNK signaling to promote inflammation and senescence120. In summary, impaired mitochondrial quality control produces dysfunctional mitochondria which induces senescence via ROS-mediated DNA damage and supports inflammation through NLRP3 inflammasome and JNK activation.
Changes in mitochondrial morphology also directly influence senescence. Overexpression of PGC-1 in human fibroblasts increases mitochondrial mass causing increased SA-β-gal activity and slowed cellular proliferation121. This apparent pro-senescent effect is likely due to elevated ROS production rather than greater mitochondrial mass per se, as a separate study found that treatment with resveratrol and salidroside suppress senescence by increasing mitochondrial biogenesis without increasing ROS122. In addition to changes in mitochondrial mass, inducing an elongated mitochondrial morphology by knockdown of FIS1 also leads to cells with a senescent morphology and increased SA-β-gal activity; conversely, reintroducing FIS1 reduces SA–β-gal activity123. Furthermore, knockdown of OPA1 causes mitochondrial fragmentation and reduces senescent phenotypes in FIS1-knockdown cells123. Promotion of mitochondrial elongation by targeting other fission or fusion regulators, such as downregulation of DRP1124,125 or overactivation of mitofusin 1 (MFN1)126, can also induce senescence. Altogether, these findings support a causal role for disrupted mitochondrial dynamics and impaired mitophagy in the induction of senescence.
Emerging evidence suggests that interactions between mitochondria and the endoplasmic reticulum (ER) play a role in determining mitochondrial dynamics in senescence. Mitochondria-ER contact sites (MERCS) help mediate a wide variety of processes, including calcium handling, lipid synthesis and trafficking, autophagy, and inflammation127. It is known that the number of MERCS generally increases in senescence, and artificial linkage of the mitochondria to ER promotes senescence128,129. MERCS also act as regulators of mitochondrial dynamics by localizing fusion and fission machinery130,131. A recent study found that in senescent cardiomyocytes, the senolytic quercetin inhibits cellular senescence by preserving MERCS, which improves mitochondrial morphology and function132, suggestive of a link between MERCS, mitochondrial dynamics, and senescence.
Mitochondrial calcium accumulation
Altered calcium handling is another characteristic of mitochondrial dysfunction in senescence in which MERCS play a role. Inositol 1,4,5-trisphosphate receptor, type 2 (ITPR2) is a calcium channel present in the ER and at MERCS responsible for the movement of calcium into the cytosol. At MERCS, ITPR2-mediated calcium release allows for uptake into the mitochondria via the mitochondrial calcium uniporter (MCU). During OIS and RS, increased calcium release via ITPR2, combined with MCU-mediated mitochondrial uptake, causes calcium accumulation in the mitochondrial matrix, reducing ΔΨm and elevating ROS production133. Notably, treating cells under oxidative stress with a calcium chelator prevents senescence134. Similarly, knockdown of ITPR2 prevents the establishment of senescence in response to oncogenic stress, and activation of ITPR2 induces expression of senescence markers133. Forcing the formation of MERCS via expression of mitochondria-ER linkers can also promote senescence through the accumulation of calcium in the ER135. Conversely, knockout of Itpr2 in mouse embryonic fibroblasts reduces the number of MERCS, thereby decreasing calcium flux and mitochondrial calcium accumulation and ultimately limiting senescence induction135.
Calcium accumulation activates the mitochondrial permeability transition pore (mPTP), a nonselective, voltage-gated channel located in the inner mitochondrial membrane that opens in response to elevated calcium in the matrix136. The consequences of pore opening depend upon its duration, where sustained activation leads to the collapse of ΔΨm, loss of ATP production, and ultimately cell death137. Activation of the mPTP increases with age across various cell types138, is triggered by ROS, and is capable of promoting further ROS production139. Senescent cells exhibit increased mPTP activity140,141, although current evidence suggests this may function as a compensatory mechanism to support cell survival rather than a driver of senescence. Cyclophilin D (CypD), the primary positive regulator of mPTP opening, was recently identified as a senolytic target141. Inhibiting CypD in senescent cells leads to mitochondrial calcium accumulation and cell death. Notably, senescent cells display frequent transient opening of the mPTP, a phenomenon termed mPTP “flickering”, which serves as a mechanism to offload excessive calcium into the cytosol, thereby preventing apoptosis. Conversely, sustained mPTP activation has also been shown to function as a senolytic142. Together, these findings suggest that senescent cells regulate mPTP dynamics in an attempt to balance calcium homeostasis and ensure survival, making the mPTP an important target for future development of senolytics.
Metabolic changes
As key regulators of cellular metabolism, mitochondria are both affected by and contribute to the diverse array of metabolic changes that occur during senescence143. Mitochondria play important roles in regulating the levels of nicotinamide adenine dinucleotide (NAD), generating its reduced form (NADH) during the citric acid cycle and subsequently its oxidized form (NAD+) during OXPHOS. NAD serves as a cofactor in numerous metabolic reactions; for example, malate dehydrogenase utilizes the reduction of NAD+ to NADH to catalyze the reversible conversion of oxaloacetate to malate. In senescent cells, the activity and protein level of cytosolic malate dehydrogenase 1 (MDH1) is reduced, which leads to decreased levels of NAD+144. During senescence, reduced activity of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in NAD+ salvage, further decreases NAD+ levels145,146. Compounding this, opening of the mPTP facilitates the loss of NAD+ from the matrix, where it is subsequently hydrolyzed by the glycohydrolase CD38 in the intermembrane space138,147.
Altogether, these processes result in a decreased NAD+/NADH ratio. NAD+ is necessary for glycolysis, where it is utilized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the conversion of glyceraldehyde 3-phosphate to 1,3-bisphosphoglyceric acid. Thus, reduced NAD+/NADH inhibits glycolysis and ATP production148, leading to an increased AMP + ADP/ATP ratio149,150. Decreases in the efficiency of the ETC in senescent cells57,59 also decrease production of ATP, further increasing the ratio. An increased AMP + ADP/ATP ratio then activates the energy sensor and regulator AMP-activated protein kinase (AMPK)151. AMPK activation in turn induces mitochondrial biogenesis through direct phosphorylation of PGC-1α152, as well as inducing epigenetic changes that increase transcription of genes involved in mitochondrial biogenesis153,154.
The reduction in the NAD+/NADH ratio and increase in the AMP + ADP/ATP ratio act to reinforce and promote senescent phenotypes. NAD+ is used in a number of DNA repair systems, including poly-ADP ribose polymerases (PARPs) and sirtuins155. PARP1 inhibition in irradiated cancer cells facilitates the persistence of DNA damage foci and thereby promotes senescence156. Similarly, inhibition of sirtuins can promote senescence as these NAD+-dependent histone deacetylases mediate access of DNA repair machinery to sites of DNA damage157. In addition, sirtuin activity limits the SASP by deacetylation of the promoter regions of IL-6 and IL-8 and repressing NF-κB activation158,159. The mitochondrial sirtuin SIRT3 also plays a role in inhibiting mPTP opening by deacetylating CypD160. Direct modulation of enzymes that regulate the NAD+/NADH ratio can impact senescence. For example, reduction of the cytosolic NAD+/NADH ratio via knockdown of MDH1 induces senescence144. Interestingly, knockdown of the mitochondrial malate dehydrogenase 2 (MDH2), which affects mitochondrial–but not cytosolic—NAD+ levels, does not affect cellular senescence in young human dermal fibroblasts144. Similarly, inhibiting the shuttling of NADH produced during glycolysis to mitochondria by the malate-aspartate shuttle with aminooxyacetate, which lowers cytosolic but not mitochondrial NAD+/NADH ratios, causes senescence149. These data suggest that cytosolic, but not mitochondrial, NAD+ levels impact senescence induction.
These findings are not limited to NAD+; increasing the AMP + ADP/ATP ratio of a cell by the addition of exogenous AMP also induces senescence161. A consequence of an increased AMP + ADP/ATP ratio is the activation of AMPK, which directly phosphorylates p53, promoting p21 transcription and cell cycle arrest162. Activated AMPK also prevents the translocation of the mRNA-stabilizing protein human antigen R (HuR) from the nucleus to the cytosol163. HuR binds to and stabilizes mRNA of proliferative genes c-Fos, cyclin A, and cyclin B; thus, its reduced presence in the cytosol reduces translation of these genes and aids proliferative arrest164. Interestingly, in OIS, but not RS, an increase in NAMPT expression increases the NAD+/NADH ratio, which suppresses AMPK activity150. This reduces p53 signaling and leads to enhanced activation of NF-κB through increased p38 mitogen-activated protein kinase (MAPK) signaling, promoting the SASP150. These findings suggest that the role of AMPK in senescence is context-dependent and influenced by the mode of senescence induction. Lastly, multiple studies have demonstrated that activation of AMPK through small molecules or by genetic means induces senescence162,163,165,166; however, this is context dependent, as AMPK activation can also suppress senescence, particularly in SIS models167,168,169,170. Altogether, these studies highlight the significance of metabolic changes in driving senescence, although it is important to recognize that this is not solely limited to the changes in NAD+/NADH or AMP + ADP/ATP ratios described here. Fatty acid oxidation (FAO), for example, has been implicated in senescence, discussed in Box 3.
Release of damage-associated molecular patterns (DAMPs)
Certain mitochondrial components can act as damage-associated molecular patterns (DAMPs), which elicit an inflammatory response when present in the cytoplasm or extracellular space. This is likely due to the bacterial origin of mitochondria, as many DAMPs contain elements that are characteristic of bacteria171. DAMPs are recognized by immune cells via pattern recognition receptors, inducing an immune response. Some examples of DAMPs are mtDNA172,173; cardiolipin, an inner mitochondrial membrane lipid174; and TFAM175,176. Aspects of mitochondrial dysfunction observed in senescent cells, such as ΔΨm depolarization and impairment in mitophagy, likely result in increased release of DAMPs176,177.
mtDNA has received the most attention in its capacity as a DAMP to drive the SASP. Like bacterial DNA, mtDNA contains unmethylated CpG islands, which are recognized by toll-like receptor 9 (TLR9) in endosomes178, initiating a signaling cascade that causes inflammation by activation of p38 MAPK and increased expression of NF-κB172,173,179,180. Cytoplasmic mtDNA is also detected by cGAS, which catalyzes the formation of the second messenger cyclic GMP-AMP dinucleotide (cGAMP) and activates STING, culminating in the expression of type I interferons181. Circulating mtDNA in serum is known to both increase with age and correlate with levels of inflammatory cytokines182, and an inducer of senescence, cigarette smoke, causes release of mtDNA106. A recent study elucidated the mechanism by which mtDNA is released in senescence183. Mitochondria in senescent cells were found to undergo low-grade apoptotic stress, which, although insufficient to induce apoptosis, does cause limited mitochondrial outer membrane permeabilization (MOMP) termed minority MOMP (miMOMP). This facilitates the release of mtDNA into the cytosol through BAX/BAK channel pores, activating the cGAS-STING pathway and driving the SASP, but not affecting other senescence markers. Blocking mtDNA release by treating mice with a BAX inhibitor reduced SASP factor expression in the bone and improved measures of healthspan, with no impact on lifespan. Interestingly, miMOMP only occurs at fragmented mitochondria, suggesting mitochondrial elongation during senescence may be a means of decreasing miMOMP. Accordingly, promoting mitochondrial fusion does indeed reduce miMOMP183.
Aside from cGAS-STING, mtDNA also activates additional inflammatory pathways. Oxidized mtDNA can bind to and activate the NLRP3 inflammasome184, promoting IL-1β secretion185 and downstream NF-κB activation186. Interestingly, activation of NLRP3 requires mtDNA synthesis118,187, supporting a role for mitochondrial dysfunction in inflammasome engagement. Other mitochondrial DAMPs may also contribute to SASP release. Cardiolipin accumulates in senescent cells and can induce senescence188, in addition to binding to and activating the NLRP3 inflammasome174. Extracellular TFAM is also proinflammatory189,190. When bound to mtDNA, TFAM interacts with the membrane-bound receptor for advanced glycation end-products (RAGE) of plasmacytoid dendritic cells191. This facilitates the internalization of mtDNA and subsequent recognition by TLR9, resulting in expression of inflammatory factors. While suggestive, more work is needed to fully elucidate the extent different DAMPs contribute to the senescent phenotype.
Direct mitochondrial manipulations reveal causality in cellular senescence
Certain interventions tested predominately in mouse and in vitro models broadly enhance mitochondrial function to delay the onset of senescence. Methylene blue (MB), a tricyclic phenothiazine drug historically used to treat malaria and methemoglobinemia192, acts as a catalytic redox cycler. MB accepts electrons from complex I, forming LeucoMB, which donates electrons to complex IV, regenerating MB193. This redox cycling enhances complex IV activity and may protect against oxidative damage by outcompeting oxygen for electrons193,194. MB also increases complex IV expression, elevates the NAD+/NADH ratio194,195, improves ΔΨm, reduces lipid peroxidation, and increases ATP production in rodent models of cholestatic disease and multiple sclerosis196,197. Notably, MB extends the replicative lifespan of normal human fibroblasts194,195 and reduces SA-β-gal activity and p16 levels in aged primary human fibroblasts90, suggesting that improved mitochondrial function may be delaying the onset of senescence.
Klotho is a protein present in both transmembrane and secreted forms that modulates oxidative stress, growth factor signaling, and ion transport198. Reduced Klotho levels have been implicated in senescence199,200. In a mouse model of kidney disease, ectopic Klotho expression lowers ROS, increases mitochondrial mass, and improves mitochondrial morphology201. These mitochondrial improvements were accompanied by a reduction in senescence markers, including SA–β-gal, p16 mRNA and protein levels, and γH2AX foci in renal tubular epithelial cells. In human cortical brain organoids, extended culture causes downregulation of Klotho and coincides with increases in p16 and p21 mRNA levels and SA-β-gal activity202. Upregulation of Klotho reduces these markers of senescence and expression of several SASP factors.
Caloric restriction (CR), one of the most well-established methods of increasing lifespan in a diverse variety of organisms203, reduces senescent cell markers in mice204, as well as humans205. Evidence suggests that this occurs at least in part through alteration in mitochondrial function. In aged mouse skeletal muscle, CR decreases ROS production, increases antioxidant scavenging, and reduces oxidative damage206. Similarly, CR reduces mitochondrial activity in mouse subcutaneous adipose-derived stem cells, which correlates with a decrease in SA-β-gal, suggesting a link between mitochondrial function and senescence207.
Due to the multifaceted mechanisms by which these interventions enhance mitochondrial function and the potential for other affects on the cell, it is difficult to definitively prove causality between improved mitochondrial function and reduced senescence in these instances. A direct test of causality comes from a study in which healthy mitochondria were transplanted into senescent cells208. The mitochondria of RS human retinal pigment epithelial cells produce more ROS, have impaired mitophagy, and altered DRP1 and MFN1 levels consistent with elongation compared to proliferating cells, indicative of mitochondrial dysfunction. Transplantation of mitochondria from human umbilical cord-derived mesenchymal stem cells into these senescent cells reversed mitochondrial dysfunction and reduced p16 and p21 levels as well as the SASP factors TNF-α and IL-8, providing strong evidence that mitochondrial dysfunction is a significant driver of cellular senescence.
Another direct test of the role of mitochondria in senescence involves complete mitochondrial depletion. This can be achieved through administration of the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), which targets mitochondria for degradation via Parkin-mediated mitophagy98. In irradiation-induced senescent human fibroblasts, CCCP treatment effectively depletes mitochondria and causes a significant reduction in the senescent markers p16, p21, SA-β-gal, and senescence-associated heterochromatin foci, as well as a reduction in cell size and ROS production98. SASP factor secretion, specifically IL-6, IL-8, GRO, and MCP-1, was nearly abolished in these cells. Similar effects were found in RS, OIS, and oxidative-damaged induced senescent fibroblasts. Mitochondria-depleted cells continued to proliferate, albeit at a greatly reduced rate, until twenty days post-irradiation. Once arrested, senescence was not accompanied by the SASP. Thus, mitochondria appear to be central to the process of cellular senescence, particularly its inflammatory qualities.
Mitochondrial dysfunction links neurodegenerative disease to cellular senescence
Aging is the greatest risk factor for neurodegenerative disease209, suggesting that fundamental biological hallmarks of aging, including mitochondrial dysfunction and cellular senescence, may also be drivers of neurodegenerative disease. Mitochondrial dysfunction is a hallmark of both inherited and sporadic neurodegenerative diseases210, and cellular senescence is similarly implicated in their progression211,212. These findings support a potential mechanistic link in which mitochondrial dysfunction promotes senescence, which in turn contributes to neurodegeneration. Notably, many of the same defects in mitochondria that occur in senescence are also observed in neurodegenerative disorders (Fig. 2).
Neurodegeneration is characterized by the progressive loss of neurons in the brain, which leads to impaired cognitive and physiological function213. Many common neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) involve the accumulation of misfolded and aggregated proteins that disrupt mitochondrial function214. Given the brain’s exceptionally high metabolic demand215, neuronal health is particularly sensitive to these changes. In the following sections, we examine mitochondrial dysfunction and cellular senescence in AD, PD, and HD, focusing on shared pathology between aging, senescence, and neurodegenerative disease.

The mitochondria of senescent cells and neural cells in the context of neurodegeneration exhibit a wide array of commonalities, including OXPHOS dysfunction leading to a loss of mitochondrial membrane potential (ΔΨm), increased ROS production, and altered energy metabolism characterized by reduced NAD+ and ATP levels. Mitochondrial calcium accumulation further exacerbates these changes. Mitophagy is also impaired which reduces mitochondrial quality. Lastly, damage-associated molecular pattern (DAMP) release promotes inflammation. These many shared features suggest mitochondrial dysfunction in neurodegenerative disease may be inducing senescence to further drive neurodegeneration.
Mitochondrial dysfunction and cellular senescence in Alzheimer’s disease
Mitochondrial dysfunction is a well-described feature of AD216,217,218,219 and has even been proposed as one of its primary drivers217,220. Many of the same features of mitochondrial dysfunction that occur in senescent cells are observed in AD contexts, suggesting overlapping mechanisms. Some of the hallmark pathologies of AD—including β-amyloid (Aβ) plaques and intercellular phosphorylated tau tangles—directly contribute to mitochondrial impairment and neurodegeneration221. For example, Aβ222 and certain forms of phosphorylated tau223 are taken up by mitochondria, where they negatively interact with ETC complexes. Exposure to Aβ significantly reduces complex I and IV activity in neurons224,225, likely due to ROS-induced mitochondrial membrane peroxidation225, while tau impairs complex IV activity in the P301L transgenic tau mouse model226. These alterations decrease ΔΨm and increase ROS production214,225,226,227,228, driving oxidative damage to both nuclear and mtDNA229.
Aβ also impairs calcium regulation214,230,231, which further amplifies mitochondrial dysfunction. In transgenic mice overexpressing human Aβ, mitochondrial calcium accumulation overload coincides with increased CypD expression and mPTP formation232,233. Aβ may promote this accumulation by increasing plasma membrane permeability and activating N-methyl-D-aspartate (NMDA) receptors234. Similar to what is found in senescent cells, MERCS are also altered in AD. AD brains exhibit a higher number of MERCS and shorter ER-mitochondria distances, which promote calcium accumulation and mPTP activation235,236. Importantly, reduction of MERCS in the 3xTg-AD mouse model, which develops both Aβ plaques and tau tangles, prevents mitochondrial calcium accumulation and ΔΨm depolarization in hippocampal neurons and reduces Aβ levels237, positioning MERCS as potential targets for future AD therapeutics.
As in senescence, OXPHOS dysfunction in AD disrupts cellular energetics. Decreased levels of ATP are found in cell and animal models of AD224,226,238,239, and dysregulation of AMPK activity has also been reported, although its precise role in AD progression, whether protective or detrimental, remains uncertain240,241. NAD+ depletion, a hallmark of brain aging and AD, further contributes to energetic failure; restoring NAD+ ameliorates pathology and improves cognition in several AD mouse models242. Increased CD38 activity may be partially responsible for this loss of NAD+, as its knockout or inhibition in the APP/PS1 amyloidogenic mouse model reduces Aβ levels in the brain and improves cognition243,244.
Alterations in mitochondrial morphology and quality control accompany these bioenergetic defects. For example, reduced NAD+ impairs sirtuin activity, compromising mitophagy in AD242,245,246. Mitophagy is also impaired in other ways by AD pathology247,248, including reduced recruitment of Parkin to PINK1-labeled mitochondria249. Unlike senescent cells, mitochondrial morphology in AD is typically characterized by fragmentation rather than elongation, as both Aβ250 and tau251 interact with DRP1 to promote mitochondrial fission. In addition, changes in expression of DRP1, FIS1, MFNs, and OPA1 in favor of fission occur in the AD brain225,252. However, some evidence suggests that mitochondrial elongation may occur, as tau disrupts the localization of DRP1 to mitochondria in a Drosophila model of AD253 and primary fibroblasts from AD patients exhibit mitochondrial elongation254. Unlike cellular senescence, mitochondrial biogenesis declines in AD, via decreased expression of PGC-1α255,256.
Mitochondrial-derived inflammatory signaling also contributes to AD pathology. Neuroinflammation is recognized as a prominent feature of AD257,258, and recent studies implicate mitochondrial DAMPs and the cGAS-STING pathway in this process. Cytosolic mtDNA levels are elevated in the brains of both the amyloidogenic 5×FAD mouse and naturally aged mice, and pharmacological inhibition of the cGAS-STING pathway reduces Aβ pathology in the 5×FAD model259,260. In the amyloidogenic APP/PS1 model, increased cytosolic mtDNA also triggers cGAS-STING activation, and a reduction in cytosolic mtDNA release by administration of melatonin or the NAD+ precursor nicotinamide riboside reduces inflammation261. Even in naturally aged mice, release of mtDNA into the cytosol of microglia similarly promotes cGAS-STING pathway signaling to drive microglia activation, inflammation, and neurodegeneration262, which can impair neurocognitive function in mice263. Overall, through their diverse means of inducing inflammation, mitochondrially-derived DAMPs are increasingly being recognized as players in AD and neurodegenerative disease more broadly264,265,266,267.
There exists substantial evidence linking cellular senescence to AD268,269,270, although much of it remains correlative or derived from aggressive transgenic mouse models. In the inferior parietal cortex of AD patients, up to 80% of large Aβ plaques are associated with p21-positive oligodendrocyte precursor cells271. In addition, neurons containing hyperphosphorylated tau neurofibrillary tangles exhibit a transcriptional profile characteristic of senescence, including upregulation of genes related to cell survival, stress response, and inflammation272,273. Consistently, neurons from AD prefrontal cortex display elevated senescence markers relative to age-matched controls274, and direct conversion of AD patient fibroblasts to neurons yields cells with senescence-like transcriptomic and epigenomic signatures and an inflammatory SASP capable of activating astrocytes274.
Single-nuclei transcriptomics and imaging mass cytometry further reveal that microglia are particularly susceptible to senesce275. Microglia Aβ load, proximity to Aβ plaques, and severity of AD all correlate with upregulation of genes associated with senescence. Significantly, microglia with Aβ load display a gene signature indicative of mitochondrial dysfunction, particularly loss of ΔΨm, suggesting Aβ may be inducing mitochondrial dysfunction and senescence in microglia275. Tau also mediates senescence in microglia. Co-culture of primary murine microglia with human tau monomers induces senescence and accompanying SASP276. Importantly, senescent microglia are functionally impaired, having a slower migration rate and reduced ability to clear tau.
Growing preclinical evidence supports senescent cells as a potential therapeutic target for AD. The senolytic cocktail dasatinib and quercetin (D + Q) is the most widely tested senolytic in both preclinical and clinical AD trials. D is a tyrosine kinase inhibitor FDA-approved to treat cancer, and Q is a flavonoid that, when combined with D, suppresses anti-apoptotic pathways in senescence to drive senolysis277. D is known to cross the blood-brain barrier (BBB) in humans278 while the pharmacokinetics of Q are less characterized in humans, although evidence in rats suggests it is also BBB permeable279. In a mouse model of Aβ accumulation, intermittent administration of D + Q for 11 weeks significantly reduced Aβ load and improved measures of memory271. In this model, p16 mRNA-positive cells were almost exclusively oligodendrocyte precursor cells localized to areas of Aβ deposition, suggesting their clearance mediated these positive effects. In the rTg(tauP301L)4510 mouse model of tauopathy, which develops aggressive tau pathology in the forebrain and consequently neurodegeneration, the forebrain exhibits increased DNA damage, p16 and p21 expression, and NF-κB activation272. Interestingly, decreased respiratory capacity was also described in the cortex and hippocampus, although no changes in mitochondrial mass were present. Intermittent treatment of these mice with D + Q decreased the expression of senescence genes and the number of tau neurofibrillary tangle-bearing cortical neurons as compared to vehicle-treated animals272. Similar findings were reported in the PS19 tauopathy model, which expresses mutant tau specifically in neurons, where p16-positive astrocytes and microglia accumulate in the hippocampus and cortex280. Genetic clearance of these cells prevented neuronal loss in the dentate gyrus and improved short-term memory. A more recent study which administered D + Q in PS19 mice at month 3 of age to month 9 validated these findings281. D + Q administration maintained BBB, attenuated hippocampal and neocortex brain atrophy, reduced hyperphosphorylated tau levels, and improved performance on a memory task. In addition, D + Q treatment shifted microglia from a disease-associated state to a homeostatic state281.
With growing preclinical evidence supporting a causal role for cellular senescence in AD, several Phase I clinical trials using D + Q in small patient populations with mild-cognitive impairment or AD have been completed282,283,284. D + Q was found to be safe with only a small incidence of mild adverse effects reported, and although the studies were not sufficiently powered to assess efficacy, results suggest possible decreases in inflammatory SASP factors, paving the way for future studies.
Mitochondrial dysfunction and cellular senescence in Parkinson’s disease
Mitochondrial dysfunction has been strongly implicated in PD285,286,287,288, perhaps more than any other neurodegenerative disease, and shares many of the features observed in AD. PD is characterized by the accumulation of the protein α-synuclein (α-syn), encoded by SNCA, into Lewy body aggregates and the selective loss of dopaminergic neurons in the substantia nigra289. α-syn preferentially binds mitochondrial membranes through interactions with cardiolipin290,291, which may underlie the association between mitochondrial dysfunction and PD. ETC complexes, particularly complex I, are impaired in PD with decreases in both protein level and activity292,293,294. α-syn-mediated complex I dysfunction decreases ΔΨm and increases ROS production214,227,228,295,296, resulting in increased oxidative damage297,298,299, including damage to nuclear DNA300 and mtDNA301. α-syn also disrupts calcium homeostasis: the formation of calcium-permeable pores in cell membranes increases cytosolic calcium302,303, which promotes mitochondrial calcium overload and opening of the mPTP214,231,295. Dysregulation of MERCS by α-syn is reported, although conflicting evidence makes it difficult to determine whether α-syn promotes or impairs MERCS formation288.
As in senescence, ETC dysfunction in PD perturbs cellular metabolism. ATP production is reduced in the skeletal muscle of PD patients304, in cells exposed to α-syn296,305, and in a Drosophila model of PD306. Like AD, the role of AMPK in PD is controversial. Both an increase and a decrease in AMPK have been reported in PD. AMPK activation may have beneficial effects by inducing autophagy and promoting α-syn clearance, or may promote α-syn aggregation by mediating α-syn phosphorylation240,307. In addition to ATP, NAD+ levels are reduced in animal and cell models of PD242, and in skeletal muscle of PD patients304. A reduction in NAD+ may contribute to impaired mitophagy as sirtuins are known mitophagy regulators308 and NAD+ activates mitophagy in cell models of PD309. Mutations in mitophagy-regulating genes encoding PINK1 and Parkin are similarly implicated in familial forms of PD310. In addition, exposure to α-syn affects autophagy and mitophagy, typically increasing activity in an effort to clear α-syn and defective mitochondria311,312. Changes in mitophagy are also linked to mitochondrial fragmentation, another feature of PD313. Transfection of cells with α-syn causes changes in expression of DRP1 and OPA1 in favor of fission and mitochondrial fragmentation311, which may partially explain the increase in mitophagy observed in PD. Finally, like AD, and unlike cellular senescence, mitochondrial biogenesis declines in PD models314 and PGC-1α-mediated mitochondrial biogenesis displays neuroprotective effects in dopaminergic neurons of PD cell and mouse models315,316.
Neuroinflammation has long been recognized as a feature of PD257,317, with recent research beginning to implicate mitochondria as a source of inflammation. Mitochondrial complex I inhibition in microglia activates NLRP3 inflammasome activity and results in dopaminergic neuron loss318. In PD patients, circulating levels of mtDNA are higher319, and mtDNA-induced cGAS-STING pathway activity occurs in different PD mouse models320,321,322. Furthermore, activation of TLR9 signaling through administration of mtDNA to the substantia nigra of mice leads to activation of microglia and loss of dopaminergic neurons323. These results suggest that an investigation into the potential involvement of other mitochondrial-derived DAMPs in PD-associated neuroinflammation is warranted.
Although less extensively studied than AD, evidence also exists linking PD to senescence of neural cells324. In PD patient tissue, the substantia nigra displays increased expression of p16 and SASP factors as well as loss of Lamin B1 in astrocytes325. Treatment of cultured human astrocytes with paraquat, an herbicide associated with the onset of PD, induces senescence325. Likewise, administering mice paraquat induces senescence in astrocytes and causes phenotypic changes resembling PD, including loss of dopaminergic neurons in the substantia nigra and deficits in motor function; genetic ablation of p16 expressing cells ameliorates these effects325. Another study found signs of senescence in post-mortem midbrain tissue from PD patients, including reduced levels of HMGB1 and Lamin B1, along with an increase in p21, but not p16, expression326. α-syn preformed fibril treatment in isolated cortical neurons, astrocytes, and microglia also induces a senescent phenotype, although less dramatically in cortical neurons326. Injection of α-syn preformed fibrils into the striatum of mice decreased HMGB1 and Lamin B1 expression and increased p21 expression in astrocytes and microglia in the substantia nigra compacta, striatum, and cortex326. Overexpression of human α-syn in the brain of mice causes DNA damage327, suggesting this may be one of the mechanisms whereby it induces senescence. Mirroring findings made in mouse models, midbrain organoids produced from induced pluripotent stem cells (iPSCs) of patients with familial PD exhibit dopaminergic neuron loss and the presence of astrocyte senescence328. Providing evidence that mitochondrial dysfunction plays an important role in senescence development in PD, metformin was found to delay astrocyte senescence in a mouse model of PD329. Metformin elevated MFN2 expression, which decreased mtDNA release and reduced the SASP through decreased cGAS-STING signaling, ultimately preventing loss of dopaminergic neurons. As senescence data in PD patients remains correlative, clinical trials of senolytics are needed to robustly implicate senescence in PD.
Mitochondrial dysfunction and cellular senescence in Huntington’s disease
HD is an inherited autosomal dominant condition caused by an expanded CAG nucleotide repeat of variable size in the HTT gene encoding the protein huntingtin. This mutant form of huntingtin (mHTT) contains polyglutamine repeats, which are prone to aggregation and cause neuronal dysfunction and death, particularly in the striatum330. As with AD and PD, mitochondrial dysfunction is a prominent feature of HD331,332,333. mHTT is produced within cells and interacts with mitochondria, particularly at the outer mitochondrial membrane, where it interferes with mitochondrial function334,335. In HD patients, the activity of ETC complexes II–IV is reduced in the caudate nucleus, an area of the brain that experiences significant neurodegeneration in HD336. This impairment in ETC complexes may explain the reduction of ΔΨm found in lymphoblasts of HD patients and in mouse models expressing mutant mHTT334. Mitochondrial ROS production is also higher in skin fibroblasts from HD patients and the striatum of the YAC128 mHTT-expressing HD mouse model337, which can drive oxidative damage337,338,339,340, including to nuclear DNA and mtDNA341. Impaired mitochondrial calcium handling is also characteristic of HD: medium spiny neurons in YAC128 mice are prone to calcium overload and subsequent mPTP opening342, potentially due to the action of mHTT, which has been demonstrated to lower the calcium threshold needed for mPTP opening343. The tendency for calcium overload may be caused by mHTT interacting with inositol 1,4,5-trisphosphate receptors (IP3R), calcium channels located in the ER, to increase their responsiveness and promote calcium uptake by mitochondria344.
Consistent with AD, PD, and cellular senescence, OXPHOS dysfunction in HD leads to deficits in energy metabolism. Decreased ATP levels are observed in both the YAC128345 and R6/2346 HD mouse models and in the cerebrum347 and skeletal muscle348 of human PD patients. In general, AMPK overactivation is observed in HD mouse models240,349, although activation of AMPK has also been reported as beneficial in HD models350,351,352, making it difficult to draw broad conclusions about its role in HD. In Drosophila HD models, depletion of NAD+ levels correlates with HD progression353,354, and supplementation with NAD+ precursors has shown therapeutic potential in preclinical models242. Beneficial effects of NAD+ supplementation may be due to SIRT1-PGC-1α-induced mitochondrial biogenesis355. Indeed, mHTT interferes with the PGC-1α promoter to repress its expression, likely impairing mitochondrial biogenesis356, and PGC-1α upregulation has therapeutic effects in mHTT-expressing mouse models356,357. Mitochondrial morphology is also impaired in HD, exhibiting fragmentation. HD patients display increased expression of DRP1 and FIS1 and decreased expression of MFN1, MFN2, and OPA1 in the striatum, consistent with mitochondrial fission358. In addition, mHTT interacts with DRP1 to enhance its enzymatic activity and promote mitochondrial fission359. Increased DRP1-mediated mitochondrial fission in primary striatal neurons of the R6/1 HD mouse model reduces MERCS and impairs mitochondrial calcium handling360. mHTT also inhibits mitophagy by interfering with the formation of complexes necessary for autophagy initiation and impairing mitophagy receptors361.
As with AD and PD, neuroinflammation characterizes HD362, with recent findings suggesting a potential role for mitochondrial DAMPs in promoting inflammation. cGAS, phosphorylated STING, and cGAS-dependent inflammatory genes are upregulated in striatal cells of HD patients363. In R6/2 mice, increased cytosolic mtDNA triggers cGAS-STING activation, and a reduction in cytosolic mtDNA release by administration of melatonin or the NAD+ precursor nicotinamide riboside reduces inflammation364. Less evidence exists for NLRP3 inflammasome activation in HD, although increased expression of NLRP3 in peripheral blood mononuclear cells of HD patients365 suggests further investigation is warranted.
The study of the role cellular senescence plays in HD is still in its infancy, although senescence has begun to be considered as a potential contributor366. Like AD and PD, models of HD exhibit an increase in senescent markers. In the striatum of HD knock-in mouse models, gene pathways associated with cellular proliferation, DDR, and senescence are differentially regulated367. iPSC-derived neural stem cells of HD patients showcase increased p16 expression and SA-β-gal activity; when these cells are differentiated into medium spiny neurons, markers of senescence remain, suggesting HD pathology may be driving senescence368. Levels of inflammatory cytokines and matrix metalloproteinases in the plasma of HD patients and R6/2 mice are elevated, including the prominent SASP factors IL-6 and MMP-9369. Senescence-focused interventions to target HD are still generally lacking. Fisetin, a flavonoid polyphenol and known senolytic agent370, suppresses HD pathology in a Drosophila and mouse model of HD371, although, since this study was performed before fisetin’s discovery as a senolytic, no analysis of a possible role for senescence was explored. The many shared hallmarks of mitochondrial dysfunction in both senescence and HD, as well as AD and PD, point to a common etiology. This suggests a fruitful line of research investigating whether the mitochondrial dysfunction characteristic of neurodegenerative disease induces senescence and subsequently targeting mitochondria as the nexus between cellular senescence and neurodegenerative disease.
Conclusion
The accumulation of senescent cells with age is now recognized as a major contributor to age-related decline. Current evidence strongly supports the idea that mitochondrial dysfunction is not just a consequence of senescence, but a central driver of its onset and its associated proinflammatory phenotype. Key mitochondrial defects, including ETC dysfunction, increased ROS production, calcium accumulation, changes in energy metabolism, dysregulated mitochondrial dynamics, reduced mitochondrial quality control, and release of DAMPs are all known to play a role in senescence. Notably, these same mitochondrial abnormalities are also prominent features of neurodegenerative disease, suggesting a mechanistic link between mitochondrial dysfunction, the induction of senescence, and neurodegeneration.
This growing overlap highlights mitochondrial pathways as a promising target of both anti-senescence and neurodegeneration therapies. Indeed, evidence suggests that many current anti-senescence interventions exert their effects by modulating mitochondrial function. Deepening our understanding of how mitochondrial function is impaired in senescent cells and how impaired mitochondria in turn influence senescence will likely prove valuable for precise and effective anti-senescence therapies. Ultimately, targeting these mechanisms may yield broad benefits in combating both aging and neurodegenerative diseases.
Data availability
No datasets were generated or analyzed during the current study.
References
Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 (1955).
Harman, D. The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 20, 145–147 (1972).
Fleming, J. Is cell aging caused by respiration-dependent injury to the mitochondrial genome?. Gerontology 28, 44–53 (1982).
Lambert, A. J. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell 6, 607–618 (2007).
Shi, Y. Comparative studies of oxidative stress and mitochondrial function in aging. Integr. Comp. Biol. 50, 869–879 (2010).
Shields, H. J., Traa, A. & Van Raamsdonk, J. M. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol. 9, 628157 (2021).
Munro, D. & Pamenter, M. E. Comparative studies of mitochondrial reactive oxygen species in animal longevity: Technical pitfalls and possibilities. Aging Cell 18, 13009 (2019).
Sadowska-Bartosz, I. & Bartosz, G. Effect of antioxidants supplementation on aging and longevity. BioMed. Res. Int. 2014, 404680 (2014).
Gusarov, I. et al. Dietary thiols accelerate aging of C. elegans. Nat. Commun. 12, 4336 (2021).
Dutta, N. et al. Investigating impacts of the mycothiazole chemotype as a chemical probe for the study of mitochondrial function and aging. GeroScience 46, 6009–6028 (2024).
Dutta, N., Garcia, G. & Higuchi-Sanabria, R. Hijacking cellular stress responses to promote lifespan. Front. Aging 3, 860404 (2022).
Lustgarten, M., Muller, F. L. & Van Remmen, H. An Objective Appraisal of the Free Radical Theory of Aging. in Handbook of the Biology of Aging (Seventh Edition) (eds Masoro, E. J. & Austad, S. N.) 177–202 (Academic Press, 2011).
Ma, Y., Chen, M., Huang, K. & Chang, W. The impact of cysteine on lifespan in three model organisms: a systematic review and meta-analysis. Aging Cell 24, e14392 (2025).
Sohal, R. S. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 40, 480–487 (2006).
Van Remmen, H. et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37 (2003).
Muller, F. L. Trends in oxidative aging theories. Free Radic. Biol. Med. 43, 477–503 (2007).
Pérez, V. I. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75 (2009).
Lennicke, C. & Cochemé, H. M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 81, 3691–3707 (2021).
Gladyshev, V. N. The free radical theory of aging is dead. long live the damage theory! Antioxid. Redox Signal. 20, 727–731 (2014).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Campisi, J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522 (2005).
Campisi, J. & Fagagna, F. D. A. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740 (2007).
Huang, W., Hickson, L. J., Eirin, A., Kirkland, J. L. & Lerman, L. O. Cellular senescence: the good, the bad and the unknown. Nat. Rev. Nephrol. 18, 611–627 (2022).
Fagagna, F. D. A. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522 (2008).
Lee, B. Y. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5, 187–195 (2006).
Kuilman, T. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Hu, L. et al. Why senescent cells are resistant to apoptosis: an insight for senolytic development. Front. Cell Dev. Biol. 10, 822816 (2022).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Hall, B. M. et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884 (2017).
Cohn, R. L., Gasek, N. S., Kuchel, G. A. & Xu, M. The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol. 33, 9–17 (2023).
Wechter, N. et al. Single-cell transcriptomic analysis uncovers diverse and dynamic senescent cell populations. Aging 15, 2824–2851 (2023).
Coppé, J.-P., Desprez, P.-Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 5, 99–118 (2010).
Salminen, A. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 24, 835–845 (2012).
Mansfield, L. et al. Emerging insights in senescence: pathways from preclinical models to therapeutic innovations. npj Aging 10, 53 (2024).
Jeyapalan, J. C., Ferreira, M., Sedivy, J. M. & Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 128, 36–44 (2007).
Idda, M. L. Survey of senescent cell markers with age in human tissues. Aging 12, 4052 (2020).
Tuttle, C. S. Cellular senescence and chronological age in various human tissues: a systematic review and meta-analysis. Aging Cell 19, 13083 (2020).
Ovadya, Y. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 1–15 (2018).
Yousefzadeh, M. J. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Wang, B., Han, J., Elisseeff, J. H. & Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 25, 958–978 (2024).
Acosta, J. C. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Kuilman, T. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Nelson, G. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012).
Acosta, J. C. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Baker, D. J. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Baker, D. J. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Xu, M. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Chaib, S., Tchkonia, T. & Kirkland, J. L. Cellular senescence and senolytics: the path to the clinic. Nat. Med. 28, 1556–1568 (2022).
Chen, P., Wang, Y. & Zhou, B. Insights into targeting cellular senescence with senolytic therapy: The journey from preclinical trials to clinical practice. Mech. Ageing Dev. 218, 111918 (2024).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Cheng, N., Kim, K.-H. & Lau, L. F. Senescent hepatic stellate cells promote liver regeneration through IL-6 and ligands of CXCR2. JCI Insight 7, e158207 (2022).
Sugrue, M. M. Reduced mitochondrial membrane potential and altered responsiveness of a mitochondrial membrane megachannel in p53-induced senescence. Biochem. Biophys. Res. Commun. 261, 123–130 (1999).
Passos, J. F. et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLOS Biol. 5, e110 (2007).
Moiseeva, O. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 29, 4495–4507 (2009).
Passos, J. F. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347 (2010).
Amaral, S. et al. Testicular aging involves mitochondrial dysfunction as well as an increase in UCP2 levels and proton leak. FEBS Lett. 582, 4191–4196 (2008).
Rangarajan, S. et al. Mitochondrial uncoupling protein-2 reprograms metabolism to induce oxidative stress and myofibroblast senescence in age-associated lung fibrosis. Aging Cell 21, e13674 (2022).
Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta BBA-Bioenerg. 1859, 940–950 (2018).
Miwa, S. et al. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 5, 3837 (2014).
Allen, R. Differences in electron transport potential, antioxidant defenses, and oxidant generation in young and senescent fetal lung fibroblasts (WI-38. J. Cell. Physiol. 180, 114–122 (1999).
Lee, A. C. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274, 7936–7940 (1999).
Takahashi, A. Mitogenic signalling and the p16 INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 8, 1291–1297 (2006).
Ahmed, E. K. Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell 9, 252–272 (2010).
Ademowo, O. S. Lipid (per) oxidation in mitochondria: an emerging target in the ageing process?. Biogerontology 18, 859–879 (2017).
Uchida, K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 42, 318–343 (2003).
Yoon, Y.-S. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J. Biol. Chem. 278, 51577–51586 (2003).
Stöckl, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 41, 674–682 (2006).
Zhang, J. Down-regulation of mitochondrial cytochrome c oxidase in senescent porcine pulmonary artery endothelial cells. Mech. Ageing Dev. 123, 1363–1374 (2002).
Xin, M.-G. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech. Ageing Dev. 124, 911–919 (2003).
Pircher, H. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. J. Biol. Chem. 290, 6755–6762 (2015).
Etemad, S. Oxaloacetate decarboxylase FAHD1–a new regulator of mitochondrial function and senescence. Mech. Ageing Dev. 177, 22–29 (2019).
Petit, M. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Exp. Gerontol. 92, 7–12 (2017).
Takenaka, Y., Inoue, I., Hirasaki, M., Ikeda, M. & Kakinuma, Y. Temporal inhibition of the electron transport chain attenuates stress-induced cellular senescence by prolonged disturbance of proteostasis in human fibroblasts. FEBS J. 290, 3843–3857 (2023).
Vizioli, M. G. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445 (2020).
Dou, Z. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017).
Glück, S. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 (2017).
Yang, H. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 114, 4612–4620 (2017).
Zheng, Z. et al. Cytosolic DNA initiates a vicious circle of aging-related endothelial inflammation and mitochondrial dysfunction via STING: the inhibitory effect of Cilostazol. Acta Pharmacol. Sin. 45, 1879–1897 (2024).
Blander, G., Oliveira, R. M. de, Conboy, C. M., Haigis, M. & Guarente, L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J. Biol. Chem. 278, 38966–38969 (2003).
Velarde, M. C., Flynn, J. M., Day, N. U., Melov, S. & Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 4, 3–12 (2012).
Dutta, R. K. et al. Catalase-deficient mice induce aging faster through lysosomal dysfunction. Cell Commun. Signal. 20, 192 (2022).
Sun, Y., Zheng, Y., Wang, C. & Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 9, 1–15 (2018).
Song, Y. et al. Glutathione peroxidase 3 is essential for countering senescence in adipose remodelling by maintaining mitochondrial homeostasis. Redox Biol. 77, 103365 (2024).
Li, Z. TGF-β induces corneal endothelial senescence via increase of mitochondrial reactive oxygen species in chronic corneal allograft failure. Aging 10, 3474 (2018).
Abdeahad, H. et al. MitoQ treatment mitigates senescence burden in DOXO-induced senescent endothelial cells through reductions in mtROS and DNA damage. Physiology 39, 1989 (2024).
Serra, V. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J. Biol. Chem. 278, 6824–6830 (2003).
Zglinicki, T. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 28, 64–74 (2000).
Xiong, Z.-M. et al. Anti-aging potentials of methylene blue for human skin longevity. Sci. Rep. 7, 2475 (2017).
McFarland, G. A. & Holliday, R. Retardation of the senescence of cultured human diploid fibroblasts by carnosine. Exp. Cell Res. 212, 167–175 (1994).
Fujisawa, H. Aminoguanidine supplementation delays the onset of senescence in vitro in dermal fibroblast-like cells from senescence-accelerated mice. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 54, 276–282 (1999).
Wang, P., Zhang, J., Zhang, Z. & Tong, T. Aminoguanidine delays the replicative senescence of human diploid fibroblasts. Chin. Med. J. 120, 2028 (2007).
Kang, H. T., Lee, H. I. & Hwang, E. S. Nicotinamide extends replicative lifespan of human cells. Aging Cell 5, 423–436 (2006).
Sadowska-Bartosz, I. & Bartosz, G. Effect of antioxidants on the fibroblast replicative lifespan in vitro. Oxid. Med. Cell. Longev. 2020, 6423783 (2020).
Lee, H.-C., Yin, P.-H., Chi, C.-W. & Wei, Y.-H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 9, 517–526 (2002).
Fielder, E. P. et al. Mild uncoupling of mitochondria synergistically enhances senolytic specificity and sensitivity of BH3 mimetics. Aging Biol. 1, 20240022 (2024).
Correia-Melo, C. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 (2016).
Pezze, P. D. et al. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLOS Comput. Biol. 10, e1003728 (2014).
Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011).
Kelly, G. et al. Suppressed basal mitophagy drives cellular aging phenotypes that can be reversed by a p62-targeting small molecule. Dev. Cell 59, 1924–1939 (2024).
Parone, P. A. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLOS One 3, 3257 (2008).
Twig, G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008).
Mai, S. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 123, 917–926 (2010).
Rizza, S. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 115, 3388–3397 (2018).
Ahmad, T. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 29, 2912–2929 (2015).
García-Prat, L. Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016).
Guo, Y. et al. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 41, 101915 (2021).
Li, S. et al. BRG1 accelerates mesothelial cell senescence and peritoneal fibrosis by inhibiting mitophagy through repression of OXR1. Free Radic. Biol. Med. 214, 54–68 (2024).
Zhang, S. et al. TIN2-mediated reduction of mitophagy induces RPE senescence under high glucose. Cell. Signal. 119, 111188 (2024).
Hong, Y.-X. et al. Cardiac senescence is alleviated by the natural flavone acacetin via enhancing mitophagy. Aging 13, 16381–16403 (2021).
Yang, B., et al. NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 20, e13329 (2021).
Cho, S. I., Jo, E.-R. & Song, H. Urolithin A attenuates auditory cell senescence by activating mitophagy. Sci. Rep. 12, 7704 (2022).
Cinat, D. et al. Mitophagy induction improves salivary gland stem/progenitor cell function by reducing senescence after irradiation. Radiother. Oncol. 190, 110028 (2024).
Hou, X., Li, Z., Higashi, Y., Delafontaine, P. & Sukhanov, S. Insulin-like growth factor I prevents cellular aging via activation of mitophagy. J. Aging Res. 2020, 4939310 (2020).
Ha, M. H. et al. PTEN-induced kinase 1 is associated with renal aging, via the cGAS-STING pathway. Aging Cell 22, e13865 (2023).
Park, S. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 5, 1–14 (2015).
Zhou, R. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Gupta, S., Cassel, S. L., Sutterwala, F. S. & Dagvadorj, J. Regulation of the NLRP3 inflammasome by autophagy and mitophagy. Immunol. Rev. 329, e13410 (2025).
Joy, J. Proteostasis failure and mitochondrial dysfunction leads to aneuploidy-induced senescence. Dev. Cell 56, 2043–2058 (2021).
Xu, D. & Finkel, T. A role for mitochondria as potential regulators of cellular life span. Biochem. Biophys. Res. Commun. 294, 245–248 (2002).
Mao, G.-X. Salidroside delays cellular senescence by stimulating mitochondrial biogenesis partly through a miR-22/SIRT-1 pathway. Oxid. Med. Cell. Longev. 12, 5276096 (2019).
Lee, S. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 282, 22977–22983 (2007).
Lin, J.-R. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler. Thromb. Vasc. Biol. 35, 1413–1422 (2015).
Zhang, J.-J. miR-21-5p/203a-3p promote ox-LDL-induced endothelial cell senescence through down-regulation of mitochondrial fission protein Drp1. Mech. Ageing Dev. 164, 8–19 (2017).
Park, Y.-Y. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J. Cell Sci. 123, 619–626 (2010).
Szymański, J. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 18, 1576 (2017).
Ziegler, D. V., Martin, N. & Bernard, D. Cellular senescence links mitochondria-ER contacts and aging. Commun. Biol. 4, 1–14 (2021).
Puebla-Huerta, A. et al. Calcium (Ca2+) fluxes at mitochondria-ER contact sites (MERCS) are a new target of senolysis in therapy-induced senescence (TIS). Npj Aging 11, 1–7 (2025).
Abrisch, R. G., Gumbin, S. C., Wisniewski, B. T., Lackner, L. L. & Voeltz, G. K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 219, e201911122 (2020).
Nguyen, T. T. & Voeltz, G. K. An ER phospholipid hydrolase drives ER-associated mitochondrial constriction for fission and fusion. eLife 11, e84279 (2022).
Jiménez, R. et al. Quercetin preserves mitochondria-endoplasmic reticulum contact sites improving mitochondrial dynamics in aged myocardial cells. Biogerontology 26, 1–17 (2025).
Wiel, C. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 5, 1–10 (2014).
Borodkina, A. V. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging 8, 3400 (2016).
Ziegler, D. V. et al. Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat. Commun. 12, 720 (2021).
Bernardi, P. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099 (2006).
Biasutto, L. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta BBA-Mol. Cell Res. 1863, 2515–2530 (2016).
Rottenberg, H. & Hoek, J. B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16, 943–955 (2017).
Zorov, D. B. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950 (2014).
E, Y. et al. Exogenous H2S alleviates senescence of glomerular mesangial cells through up-regulating mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1870, 119568 (2023).
Protasoni, M. et al. Cyclophilin D plays a critical role in the survival of senescent cells. EMBO J. 43, 5972–6000 (2024).
Chen, Y. et al. Senolysis by GLS1 inhibition ameliorates kidney aging by inducing excessive mPTP opening through MFN1. J. Gerontol. Ser. A 80, glae294 (2025).
Wiley, C. D. & Campisi, J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat. Metab. 3, 1290–1301 (2021).
Lee, S.-M. et al. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology 13, 525–536 (2012).
Veer, E. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 282, 10841–10845 (2007).
Ma, C. et al. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting sirt1. PLOS One 12, e0170930 (2017).
Lisa, F. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 276, 2571–2575 (2001).
Girbal, L. & Soucaille, P. Regulation of Clostridium acetobutylicum metabolism as revealed by mixed-substrate steady-state continuous cultures: role of NADH/NAD ratio and ATP pool. J. Bacteriol. 176, 6433–6438 (1994).
Wiley, C. D. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 (2016).
Nacarelli, T. NAD+ metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 21, 397–407 (2019).
Hardie, D. G. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Jäger, S. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. 104, 12017–12022 (2007).
Cantó, C. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 (2010).
Marin, T. L. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 10, eaaf7478 (2017).
Hurtado-Bagès, S. The taming of PARP1 and its impact on NAD+ metabolism. Mol. Metab. 38, 100950 (2020).
Efimova, E. V. Poly (ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res 70, 6277–6282 (2010).
Lagunas-Rangel, F. A. Current role of mammalian sirtuins in DNA repair. DNA Repair 80, 85–92 (2019).
Hayakawa, T. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLOS One 10, 0116480 (2015).
Grabowska, W. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 18, 447–476 (2017).
Hafner, A. V. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914 (2010).
Zwerschke, W. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 376, 403–411 (2003).
Jones, R. G. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 (2005).
Wang, W. I. ncreasedA. M. P. ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J. Biol. Chem. 278, 27016–27023 (2003).
Wang, W. Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol. Cell. Biol. 21, 5889–5898 (2001).
Peyton, K. J. Activation of AMP-activated protein kinase inhibits the proliferation of human endothelial cells. J. Pharmacol. Exp. Ther. 342, 827–834 (2012).
Lee, J. H. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53- dependent pathway. FEBS J. 282, 361–371 (2015).
Chen, M. et al. Metformin alleviates oxidative stress-induced senescence of human lens epithelial cells via AMPK activation and autophagic flux restoration. J. Cell. Mol. Med. 25, 8376–8389 (2021).
Han, X. et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD+ elevation. Aging Cell 15, 416–427 (2016).
Maharajan, N. et al. Licochalcone D ameliorates oxidative stress-induced senescence via AMPK activation. Int. J. Mol. Sci. 22, 7324 (2021).
Kim, S. G., Sung, J. Y., Kim, J.-R. & Choi, H. C. Nifedipine-induced AMPK activation alleviates senescence by increasing autophagy and suppressing of Ca2+ levels in vascular smooth muscle cells. Mech. Ageing Dev. 190, 111314 (2020).
Meyer, A. Mitochondria: an organelle of bacterial origin controlling inflammation. Front. Immunol. 9, 536 (2018).
Zhang, Q. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Fang, C. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 7, 11–16 (2016).
Iyer, S. S. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 (2013).
McGuire, P. J. Mitochondrial dysfunction and the aging immune system. Biology 8, 26 (2019).
Rodríguez-Nuevo, A. & Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 3, 195 (2019).
Picca, A. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 21, 350–359 (2018).
Latz, E. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 5, 190–198 (2004).
Zhang, Q. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34, 55–59 (2010).
Zhang, J.-Z. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int. J. Mol. Med. 33, 817–824 (2014).
West, A. P. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Pinti, M. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging. Eur. J. Immunol. 44, 1552–1562 (2014).
Victorelli, S. et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 622, 627–636 (2023).
Shimada, K. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Xin, W. A new mechanism of inhibition of IL-1β secretion by celastrol through the NLRP3 inflammasome pathway. Eur. J. Pharmacol. 814, 240–247 (2017).
García, J. A. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor- α and blocks the septic response in mice. FASEB J. 29, 3863–3875 (2015).
Zhong, Z. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
Arivazhagan, P. Cardiolipin induces premature senescence in normal human fibroblasts. Biochem. Biophys. Res. Commun. 323, 739–742 (2004).
Chaung, W. W. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int. J. Mol. Med. 30, 199–203 (2012).
Little, J. P. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 60, 88–96 (2014).
Julian, M. W. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE-and TLR9-responsive plasmacytoid dendritic cells. PLOS One 8, 72354 (2013).
Schirmer, R. H. Lest we forget you—methylene blue. Neurobiol. Aging 32, 2325 2327–2325 2325 (2011).
Tucker, D. From mitochondrial function to neuroprotection—an emerging role for methylene blue. Mol. Neurobiol. 55, 5137–5153 (2018).
Atamna, H. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 22, 703–712 (2008).
Atamna, H. Combined activation of the energy and cellular-defense pathways may explain the potent anti-senescence activity of methylene blue. Redox Biol. 6, 426–435 (2015).
Heidari, R. Methylene blue improves mitochondrial function in the liver of cholestatic rats. Trends Pharm. Sci. 6, 73–86 (2020).
Ommati, M. M. Methylene blue treatment enhances mitochondrial function and locomotor activity in a C57BL/6 mouse model of multiple sclerosis. Trends Pharm. Sci. 6, 29–42 (2020).
Kuro-o, M. Klotho and the aging process. Korean J. Intern. Med. 26, 113 (2011).
Zhang, X. et al. Klotho-derived peptide 1 inhibits cellular senescence in the fibrotic kidney by restoring Klotho expression via posttranscriptional regulation. Theranostics 14, 420–435 (2024).
Li, M. et al. Klotho regulates club cell senescence and differentiation in chronic obstructive pulmonary disease. Cell Prolif. 58, e70000 (2025).
Miao, J. Klotho retards renal fibrosis through targeting mitochondrial dysfunction and cellular senescence in renal tubular cells. Physiol. Rep. 9, 14696 (2021).
Shaker, M. R., Aguado, J., Chaggar, H. K. & Wolvetang, E. J. Klotho inhibits neuronal senescence in human brain organoids. npj Aging Mech. Dis. 7, 18 (2021).
Fontana, L. Extending healthy life span—from yeast to humans. Science 328, 321–326 (2010).
Fontana, L. Caloric restriction and cellular senescence. Mech. Ageing Dev. 176, 19–23 (2018).
Aversa, Z. et al. Calorie restriction reduces biomarkers of cellular senescence in humans. Aging Cell 23, e14038 (2024).
Lanza, I. R. et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 16, 777–788 (2012).
Chinnapaka, S., Malekzadeh, H., Tirmizi, Z. & Ejaz, A. Caloric restriction mitigates age-associated senescence characteristics in subcutaneous adipose tissue-derived stem cells. Aging 16, 7535–7552 (2024).
Noh, S.-E., Lee, S. J., Lee, T. G., Park, K.-S. & Kim, J. H. Inhibition of cellular senescence hallmarks by mitochondrial transplantation in senescence-induced ARPE-19 cells. Neurobiol. Aging 121, 157–165 (2023).
Hou, Y. et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Federico, A. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 322, 254–262 (2012).
Martínez-Cué, C. & Rueda, N. Cellular senescence in neurodegenerative diseases. Front. Cell. Neurosci. 14, 16 (2020).
Wang, Y. et al. The role of cellular senescence in neurodegenerative diseases. Arch. Toxicol. 98, 2393–2408 (2024).
Przedborski, S. Series introduction: neurodegeneration: what is it and where are we? J. Clin. Invest. 111, 3–10 (2003).
Abramov, A. Y. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 45, 1025–1033 (2017).
Watts, M. E. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front. Mol. Neurosci. 11, 216 (2018).
Perez Ortiz, J. M. & Swerdlow, R. H. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176, 3489–3507 (2019).
Ashleigh, T., Swerdlow, R. H. & Beal, M. F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement 19, 333–342 (2023).
D’Alessandro, M. C. B., Kanaan, S., Geller, M., Praticò, D. & Daher, J. P. L. Mitochondrial dysfunction in Alzheimer’s disease. Ageing Res. Rev. 107, 102713 (2025).
Moawad, M. H. E. D. et al. Exploring the mechanisms and therapeutic approaches of mitochondrial dysfunction in Alzheimer’s disease: an educational literature review. Mol. Neurobiol. 62, 6785–6810 (2025).
Swerdlow, R. H. & Khan, S. M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20 (2004).
Knopman, D. S. et al. Alzheimer disease. Nat. Rev. Dis. Prim. 7, 33 (2021).
Hansson Petersen, C. A. et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 105, 13145–13150 (2008).
Torres, A. K., Jara, C., Olesen, M. A. & Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 11, 4448 (2021).
Casley, C. S. et al. Beta-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 10, 258–267 (2002).
Bobba, A. et al. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 13, 298–311 (2013).
Rhein, V. et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 106, 20057–20062 (2009).
Angelova, P. R. & Abramov, A. Y. Alpha-synuclein and beta-amyloid–different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 483, 1110–1115 (2017).
Bustamante-Barrientos, F. A. et al. Mitochondrial dysfunction in neurodegenerative disorders: potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J. Transl. Med. 21, 613 (2023).
Wang, J., Xiong, S., Xie, C., Markesbery, W. R. & Lovell, M. A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962 (2005).
Cascella, R. & Cecchi, C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 22, 4914 (2021).
Matuz-Mares, D., González-Andrade, M., Araiza-Villanueva, M. G., Vilchis-Landeros, M. M. & Vázquez-Meza, H. Mitochondrial calcium: effects of its imbalance in disease. Antioxidants 11, 801 (2022).
Du, H. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. 107, 18670–18675 (2010).
Du, H. & Yan, S. S. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 1802, 198–204 (2010).
Ge, M. et al. Role of calcium homeostasis in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 18, 487–498 (2022).
Hedskog, L. et al. Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 110, 7916–7921 (2013).
Pérez-Leaños, C. A., Romero-Campos, H. E., Dupont, G. & González-Vélez, V. Reduction of ER-Mitochondria Distance: a Key Feature in Alzheimer’s and Parkinson’s Disease, and During Cancer Treatment. in 2021 43rd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC) 4412–4415 (IEEE, 2021). https://doi.org/10.1109/EMBC46164.2021.9631090.
Marinho, D. et al. Reduction of class I histone deacetylases ameliorates ER-mitochondria cross-talk in Alzheimer’s disease. Aging Cell 22, e13895 (2023).
Eckert, A. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 5, 157–159 (2008).
Orr, A. L. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J. Alzheimers Dis. 68, 991–1011 (2019).
Domise, M. & Vingtdeux, V. AMPK in neurodegenerative diseases. AMP-Activated Protein Kinase 153, 177 (2016).
Assefa, B. T., Tafere, G. G., Wondafrash, D. Z. & Gidey, M. T. The bewildering effect of AMPK activators in Alzheimer’s disease: review of the current evidence. BioMed. Res. Int. 2020, 9895121 (2020).
Lautrup, S. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
Blacher, E. et al. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 78, 88–103 (2015).
Hu, Y. et al. Aβ promotes CD38 expression in senescent microglia in Alzheimer’s disease. Biol. Res. 55, 10 (2022).
Kerr, J. S. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 40, 151–166 (2017).
Fang, E. F. Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy 15, 1112–1114 (2019).
Reddy, P. H. & Oliver, D. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8, 488 (2019).
Mary, A., Eysert, F., Checler, F. & Chami, M. Mitophagy in Alzheimer’s disease: molecular defects and therapeutic approaches. Mol. Psychiatry 28, 202–216 (2023).
Martín-Maestro, P. Mitophagy failure in APP and tau overexpression model of Alzheimer’s disease. J. Alzheimers Dis. 70, 525–540 (2019).
Manczak, M., Calkins, M. J. & Reddy, P. H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509 (2011).
Manczak, M. & Reddy, P. H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547 (2012).
Wang, X. et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103 (2009).
DuBoff, B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632 (2012).
Wang, X., Su, B., Fujioka, H. & Zhu, X. Dynamin-Like Protein 1 Reduction Underlies Mitochondrial Morphology and Distribution Abnormalities in Fibroblasts from Sporadic Alzheimer’s Disease Patients. Am. J. Pathol. 173, 470–482 (2008).
Sheng, B. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 120, 419–429 (2012).
Singulani, M. P. et al. Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp. Gerontol. 133, 110882 (2020).
Glass, C. K. Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934 (2010).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015).
Govindarajulu, M. et al. Role of cGAS–sting signaling in Alzheimer’s disease. Int. J. Mol. Sci. 24, 8151 (2023).
Xie, X. et al. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nat. Aging 3, 202–212 (2023).
Hou, Y. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS–STING. Proc. Natl. Acad. Sci. USA 118, e2011226118 (2021).
Gulen, M. F. et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 620, 374–380 (2023).
Tauber, S. C. Stimulation of Toll-like receptor 9 by chronic intraventricular unmethylated cytosine-guanine DNA infusion causes neuroinflammation and impaired spatial memory. J. Neuropathol. Exp. Neurol. 68, 1116–1124 (2009).
Lin, M. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 43, 1–9 (2022).
Yu, H. et al. Mitochondrial DAMPs: Key mediators in neuroinflammation and neurodegenerative disease pathogenesis. Neuropharmacology 264, 110217 (2025).
Mishra, Y., Kumar, A. & Kaundal, R. K. Mitochondrial Dysfunction is a Crucial Immune Checkpoint for Neuroinflammation and Neurodegeneration: mtDAMPs in Focus. Mol. Neurobiol. 62, 6715–6747 (2025).
Quan, S. et al. The neuroimmune nexus: unraveling the role of the mtDNA-cGAS-STING signal pathway in Alzheimer’s disease. Mol. Neurodegener. 20, 25 (2025).
Saez-Atienzar, S. & Masliah, E. Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat. Rev. Neurosci. 21, 433–444 (2020).
Liu, R.-M. Aging, cellular senescence, and Alzheimer’s disease. Int. J. Mol. Sci. 23, 1989 (2022).
Zhu, J., Wu, C. & Yang, L. Cellular senescence in Alzheimer’s disease: from physiology to pathology. Transl. Neurodegener. 13, 55 (2024).
Zhang, P. et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728 (2019).
Musi, N. et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840 (2018).
Dehkordi, S. K. Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology. Nat. Aging 1, 1107–1116 (2021).
Herdy, J. R. et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell 29, 1637–1652 (2022).
Fancy, N. N. et al. Characterisation of premature cell senescence in Alzheimer’s disease using single nuclear transcriptomics. Acta Neuropathol. 147, 78 (2024).
Karabag, D. et al. Characterizing microglial senescence: Tau as a key player. J. Neurochem. 166, 517–533 (2023).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Porkka, K. et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome–positive leukemia. Blood 112, 1005–1012 (2008).
Ferri, P. et al. Enhancement of flavonoid ability to cross the blood–brain barrier of rats by co-administration with α-tocopherol. Food Funct. 6, 394–400 (2015).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Yao, M. et al. Senolytic therapy preserves blood-brain barrier integrity and promotes microglia homeostasis in a tauopathy model. Neurobiol. Dis. 202, 106711 (2024).
Gonzales, M. M. et al. Senolytic therapy in mild Alzheimer’s disease: a phase 1 feasibility trial. Nat. Med. 29, 2481–2488 (2023).
Millar, C. L. et al. A pilot study of senolytics to improve cognition and mobility in older adults at risk for Alzheimer’s disease. eBioMedicine 113, 105612 (2025).
Garbarino, V. R. et al. Evaluation of exploratory fluid biomarkers from a phase 1 senolytic trial in mild Alzheimer’s disease. Neurotherapeutics 0, e00591 (2025).
Bose, A. & Beal, M. F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 139, 216–231 (2016).
Gao, X.-Y., Yang, T., Gu, Y. & Sun, X.-H. Mitochondrial dysfunction in Parkinson’s disease: from mechanistic insights to therapy. Front. Aging Neurosci. 14, 885500 (2022).
Moradi Vastegani, S. et al. Mitochondrial dysfunction and Parkinson’s disease: pathogenesis and therapeutic strategies. Neurochem. Res. 48, 2285–2308 (2023).
Sohrabi, T. et al. Common mechanisms underlying α-synuclein-induced mitochondrial dysfunction in Parkinson’s disease. J. Mol. Biol. 435, 167992 (2023).
Poewe, W., et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013 (2017).
Nakamura, K. et al. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 28, 12305–12317 (2008).
Ghio, S., Kamp, F., Cauchi, R., Giese, A. & Vassallo, N. Interaction of α-synuclein with biomembranes in Parkinson’s disease —role of cardiolipin. Prog. Lipid Res. 61, 73–82 (2016).
Holper, L. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 44, 837–849 (2019).
Schapira, A. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827 (1990).
Keeney, P. M. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 26, 5256–5264 (2006).
Luth, E. S., Stavrovskaya, I. G., Bartels, T., Kristal, B. S. & Selkoe, D. J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507 (2014).
Reeve, A. K. et al. Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 6, e1820 (2015).
Halliwell, B. Oxidative stress and neurodegeneration: where are we now? J. Neurochem. 97, 1634–1658 (2006).
Beal, M. F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 32, 797–803 (2002).
Angelova, P. R. & Abramov, A. Y. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 592, 692–702 (2018).
Zhang, J. et al. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 154, 1423–1429 (1999).
Sanders, L. H. et al. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 70, 214–223 (2014).
Zakharov, S. D. et al. Helical α-synuclein forms highly conductive ion channels. Biochemistry 46, 14369–14379 (2007).
Angelova, P. R. et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801 (2016).
Mischley, L. K. et al. ATP and NAD+ deficiency in Parkinson’s disease. Nutrients 15, 943 (2023).
Shavali, S. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 439, 125–128 (2008).
Clark, I. E. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 (2006).
Curry, D. W., Stutz, B., Andrews, Z. B. & Elsworth, J. D. Targeting AMPK signaling as a neuroprotective strategy in Parkinson’s disease. J. Park. Dis. 8, 161–181 (2018).
Zhou, Z. D. & Tan, E. K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Ageing Res. Rev. 62, 101107 (2020).
Zhou, S. et al. NAD+-boosters improve mitochondria quality control in parkinson’s disease models via mitochondrial UPR. Adv. Sci. 12, e08503 (2025).
Singh, F. & Ganley, I. G. Parkinson’s disease and mitophagy: an emerging role for LRRK2. Biochem. Soc. Trans. 49, 551–562 (2021).
Martinez, J. H. et al. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell. Neurosci. 88, 107–117 (2018).
Liu, J., et al. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 8, e3056 (2017).
Yang, D. Mitochondrial dynamics: a key role in neurodegeneration and a potential target for neurodegenerative disease. Front. Neurosci. 15, 359 (2021).
Kumar, M. et al. Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Rep. 15, 629–645 (2020).
Chen, Y., Jiang, Y., Yang, Y., Huang, X. & Sun, C. SIRT1 protects dopaminergic neurons in Parkinson’s disease models via PGC-1α-mediated mitochondrial biogenesis. Neurotox. Res. 39, 1393–1404 (2021).
Liu, J. et al. Urolithin A protects dopaminergic neurons in experimental models of Parkinson’s disease by promoting mitochondrial biogenesis through the SIRT1/PGC-1α signaling pathway. Food Funct. 13, 375–385 (2022).
Wang, Q., Liu, Y. & Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4, 19 (2015).
Sarkar, S. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. Park. Dis. 3, 1–15 (2017).
Borsche, M. et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 143, 3041–3051 (2020).
Jiang, S.-Y. et al. The cGAS-STING-YY1 axis accelerates progression of neurodegeneration in a mouse model of Parkinson’s disease via LCN2-dependent astrocyte senescence. Cell Death Differ. 30, 2280–2292 (2023).
Liu, Y., Duan, R., Li, P., Zhang, B. & Liu, Y. 3-N-butylphthalide attenuates neuroinflammation in rotenone-induced Parkinson’s disease models via the cGAS-STING pathway. Int. J. Immunopathol. Pharmacol. 38, 03946320241229041 (2024).
Su, Z. et al. Rhapontigenin attenuates neurodegeneration in a Parkinson’s disease model by downregulating mtDNA-cGAS-STING-NF-κB-mediated neuroinflammation via PINK1/DRP1-dependent microglial mitophagy. Cell. Mol. Life Sci. 82, 337 (2025).
Maatouk, L. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat. Commun. 9, 1–15 (2018).
Ma, Y., Erb, M. L. & Moore, D. J. Aging, cellular senescence and Parkinson’s disease. J. Park. Dis. 15, 239–254 (2025).
Chinta, S. J. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940 (2018).
Verma, D. K. Alpha-synuclein preformed fibrils induce cellular senescence in Parkinson’s disease models. Cells 10, 1694 (2021).
Yoon, Y.-S. et al. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 54, 115–128 (2022).
Muwanigwa, M. N. et al. Alpha-synuclein pathology is associated with astrocyte senescence in a midbrain organoid model of familial Parkinson’s disease. Mol. Cell. Neurosci. 128, 103919 (2024).
Wang, M. et al. Metformin normalizes mitochondrial function to delay astrocyte senescence in a mouse model of Parkinson’s disease through Mfn2-cGAS signaling. J. Neuroinflammation 21, 81 (2024).
Bates, G. P. et al. Huntington disease. Nat. Rev. Dis. Prim. 1, 15005 (2015).
Carmo, C., Naia, L., Lopes, C. & Rego, A. C. Mitochondrial Dysfunction in Huntington’s Disease. in Polyglutamine Disorders (eds Nóbrega, C. & Pereira de Almeida, L.) 59–83 (Springer International Publishing, 2018). https://doi.org/10.1007/978-3-319-71779-1_3.
Jurcau, A. & Jurcau, C. M. Mitochondria in Huntington’s disease: implications in pathogenesis and mitochondrial-targeted therapeutic strategies. Neural Regen. Res. 18, 1472–1477 (2022).
Joshi, D. C. et al. The role of mitochondrial dysfunction in Huntington’s disease: implications for therapeutic targeting. Biomed. Pharmacother. 183, 117827 (2025).
Panov, A. V. et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736 (2002).
Hamilton, J., Brustovetsky, T., Khanna, R. & Brustovetsky, N. Mutant huntingtin does not cross the mitochondrial outer membrane. Hum. Mol. Genet. 29, 2962–2975 (2020).
Gu, M. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 39, 385–389 (1996).
Lopes, C. et al. Mitochondrial and redox modifications in early stages of Huntington’s disease. Redox Biol. 56, 102424 (2022).
Johri, A. & Beal, M. F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 1822, 664–674 (2012).
Manoharan, S. et al. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a mini review. Oxid. Med. Cell. Longev. 1, 8590578 (2016).
Kumar, A. & Ratan, R. R. Oxidative stress and Huntington’s disease: the good, the bad, and the ugly. J. Huntingt. Dis. (2016) https://doi.org/10.3233/JHD-160205.
Ayala-Peña, S. Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic. Biol. Med. 62, 102–110 (2013).
Tang, T.-S. et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 102, 2602–2607 (2005).
Choo, Y. S., Johnson, G. V. W., MacDonald, M., Detloff, P. J. & Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420 (2004).
Tang, T.-S. et al. Huntingtin and Huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239 (2003).
Lopes, C. et al. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 49, 1126–1142 (2014).
Mochel, F. et al. Early alterations of brain cellular energy homeostasis in Huntington disease models*. J. Biol. Chem. 287, 1361–1370 (2012).
Golden, L. E., Xu, J., Magnotta, V. A., Nopoulos, P. C. & Schultz, J. L. Decreased cerebral ATP in pre-motor manifest Huntington’s disease: a pilot study. Parkinsonism Relat. Disord. 140, 108040 (2025).
Lodi, R. et al. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 48, 72–76 (2001).
Chou, S.-Y. et al. CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J. Neurochem. 93, 310–320 (2005).
Ma, T. C. et al. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 411, 98–103 (2007).
Fu, J. et al. trans-(−)-ε-viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J. Biol. Chem. 287, 24460–24472 (2012).
Vázquez-Manrique, R. P. et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 25, 1043–1058 (2016).
Singh, V. et al. NMR spectroscopy-based metabolomics of drosophila model of Huntington’s disease suggests altered cell energetics. J. Proteome Res. 16, 3863–3872 (2017).
Bertrand, M., Decoville, M., Meudal, H., Birman, S. & Landon, C. Metabolomic nuclear magnetic resonance studies at presymptomatic and symptomatic stages of Huntington’s disease on a drosophila model. J. Proteome Res. 19, 4034–4045 (2020).
Lloret, A. & Beal, M. F. PGC-1α, Sirtuins and PARPs in Huntington’s disease and other neurodegenerative conditions: NAD+ to rule them all. Neurochem. Res. 44, 2423–2434 (2019).
Cui, L. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69 (2006).
Chandra, A. Enhanced mitochondrial biogenesis ameliorates disease phenotype in a full-length mouse model of Huntington’s disease. Hum. Mol. Genet. 25, 2269–2282 (2016).
Shirendeb, U. et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455 (2011).
Song, W. et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382 (2011).
Cherubini, M., Lopez-Molina, L. & Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 136, 104741 (2020).
Franco-Iborra, S. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 17, 672–689 (2021).
Jia, Q., Li, S., Li, X.-J. & Yin, P. Neuroinflammation in Huntington’s disease: from animal models to clinical therapeutics. Front. Immunol. 13, 1088124 (2022).
Sharma, M., Rajendrarao, S., Shahani, N., Ramírez-Jarquín, U. N. & Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl. Acad. Sci. 117, 15989–15999 (2020).
Jauhari, A. Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Invest. 130, 3124–3136 (2020).
Glinsky, G. V. SNP-guided microRNA maps (MirMaps) of 16 common human disorders identify a clinically accessible therapy reversing transcriptional aberrations of nuclear import and inflammasome pathways. Cell Cycle 7, 3564–3576 (2008).
Almalki, W. H. Cellular Senescence in Huntington’s Disease (HD). in Cellular Senescence and Brain Aging: Mechanisms, Therapeutic Strategies, and Implications for Neurodegenerative Diseases (ed. Gupta, G.) 137–156 (Springer Nature, 2025). https://doi.org/10.1007/978-981-96-8873-9_8.
Bigan, E. et al. Genetic cooperativity in multi-layer networks implicates cell survival and senescence in the striatum of Huntington’s disease mice synchronous to symptoms. Bioinformatics 36, 186–196 (2020).
Voisin, J. et al. FOXO3 targets are reprogrammed as Huntington’s disease neural cells and striatal neurons face senescence with p16INK4a increase. Aging Cell 19, e13226 (2020).
Chang, K.-H., Wu, Y.-R., Chen, Y.-C. & Chen, C.-M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain. Behav. Immun. 44, 121–127 (2015).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 36, 18–28 (2018).
Maher, P. et al. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 20, 261–270 (2011).
Zhou, H. et al. Downregulation of Sirt6 by CD38 promotes cell senescence and aging. Aging 14, 9730–9757 (2022).
Phadwal, K. et al. p53 regulates mitochondrial dynamics in vascular smooth muscle cell calcification. Int. J. Mol. Sci. 24, 1643 (2023).
Suresh, S. et al. SenMayo transcriptomic senescence panel highlights glial cells in the ageing mouse and human retina. npj Aging 10, 60 (2024).
Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344 (2002).
Meyne, J. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl. Acad. Sci. 86, 7049–7053 (1989).
Wang, Z. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLOS Genet 6, 1000951 (2010).
Loon, B. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 9, 604–616 (2010).
Qian, W. et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. 116, 18435–18444 (2019).
Barnes, R. P. et al. Telomeric 8-oxo-guanine drives rapid premature senescence in the absence of telomere shortening. Nat. Struct. Mol. Biol. 29, 639–652 (2022).
Opresko, P. L. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res 33, 1230–1239 (2005).
Steensel, B. TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413 (1998).
Sullivan, D. I. et al. Intact mitochondrial function in the setting of telomere-induced senescence. Aging Cell 22, e13941 (2023).
Min, S. et al. Mitoribosomal deregulation drives senescence via TPP1-mediated telomere deprotection. Cells 11, 2079 (2022).
Fouquerel, E. DNA damage processing at telomeres: the ends justify the means. DNA Repair 44, 159–168 (2016).
Fumagalli, M. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 (2012).
Hewitt, G. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 1–9 (2012).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Kolesar, J. E. et al. Defects in mitochondrial DNA replication and oxidative damage in muscle of mtDNA mutator mice. Free Radic. Biol. Med. 75, 241–251 (2014).
Yu, T. et al. Premature aging is associated with higher levels of 8-oxoguanine and increased DNA damage in the Polg mutator mouse. Aging Cell 21, e13669 (2022).
Laberge, R.-M. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Herranz, N. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Macip, S. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 21, 2180–2188 (2002).
Macip, S. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 23, 8576–8585 (2003).
Kim, Y. Y. p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence. FASEB J. 34, 2451–2464 (2020).
Kim, Y. Y. p53-mediated regulation of mitochondrial dynamics plays a pivotal role in the senescence of various normal cells as well as cancer cells. FASEB J. 35, 21319 (2021).
Hoshino, A. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4, 1–12 (2013).
Manzella, N. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 17, 12811 (2018).
Houten, S. M. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44 (2016).
Yamauchi, S. et al. Mitochondrial fatty acid oxidation drives senescence. Sci. Adv. 10, eado5887 (2024).
Quijano, C. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11, 1383–1392 (2012).
Giuliani, A. et al. Senescent endothelial cells sustain their senescence-associated secretory phenotype (SASP) through enhanced fatty acid oxidation. Antioxidants 12, 1956 (2023).
Ogrodnik, M. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 1–12 (2017).
Lin, T. et al. Disturbance of fatty acid metabolism promoted vascular endothelial cell senescence via Acetyl-CoA-induced protein acetylation modification. Oxid. Med. Cell. Longev. 2022, 1198607 (2022).
Acknowledgements
This work was supported by T32AG052374 from the National Institute of Aging (NIA) and the Graduate Research Fellowships Program (DGE-1842487) from the National Science Foundation to A.J.H. and R01AG079806 from the NIA and 2022-A-010-SUP from the Larry L. Hillblom Foundation to R.H.S.
Author information
Authors and Affiliations
Contributions
A.J.H. designed and wrote the manuscript. R.H.S. supervised and edited the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hruby, A.J., Higuchi-Sanabria, R. Mitochondrial dysfunction in cellular senescence: a bridge to neurodegenerative disease. npj Aging 11, 99 (2025). https://doi.org/10.1038/s41514-025-00291-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41514-025-00291-4
