
Abstract
Antibiotic susceptibility tests (ASTs) often fail to predict treatment outcomes because they do not account for biofilm-specific tolerance mechanisms. In the present study, we explored alternative approaches to predict tobramycin susceptibility of Pseudomonas aeruginosa biofilms that were experimentally evolved in physiologically relevant conditions. To this end, we used four analytical methods – whole-genome sequencing (WGS), matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), isothermal microcalorimetry (IMC) and multi-excitation Raman spectroscopy (MX-Raman). Machine learning models were trained on data outputs from these methods to predict tobramycin susceptibility of our evolved strains and validated with a collection of clinical isolates. For minimal inhibitory concentration (MIC) predictions of the evolved strains, the highest accuracy was achieved with MALDI-TOF MS (97.83%), while for biofilm prevention concentration (BPC) predictions, Raman spectroscopy performed best with an accuracy of 80.43%. Overall, all analytical methods demonstrated comparable predictive performance, showing their potential for improving biofilm AST.
Similar content being viewed by others

Introduction
Antibiotic susceptibility tests (ASTs) guide the selection of antimicrobial agents for treating infections1,2. These ASTs typically evaluate the response to antibiotics of planktonic bacteria in suspension (e.g., broth microdilution test3) or bacteria grown on agar surfaces (e.g., disk diffusion test4). However, the conditions of these in vitro tests do not represent the in vivo microenvironment, where bacteria often occur as biofilms, i.e., aggregates encased in extracellular matrix derived from either the bacteria or the host5. Biofilm-growing bacteria exhibit distinct physiological characteristics that confer increased tolerance and resistance to antibiotics compared to their planktonic counterparts6,7 and they can withstand antibiotic concentrations up to 100-1,000 times higher than those effective against planktonic cells8,9,10,11. As a result, conventional ASTs frequently fail to predict treatment success, as the biofilm phenotype is not considered8,12,13. To improve in vitro ASTs, culture media that more closely mimic the in vivo microenvironment can be used. An example of such a medium is synthetic cystic fibrosis medium 2 (SCFM2), which simulates the lung environment of cystic fibrosis (CF) patients14. In SCFM2, Pseudomonas aeruginosa forms biofilm microaggregates that closely resemble those found in the sputum of CF patients15 and exhibit similar gene expression profiles16. P. aeruginosa biofilm growth in SCFM2 was found to be highly reproducible15,17,18,19,20 and this medium can be used to determine biofilm susceptibility10.
However, biofilm-based ASTs remain challenging to implement and are time consuming. In the present study we explored alternative methods to predict the antibiotic susceptibility of P. aeruginosa biofilms, using whole-genome sequencing (WGS), matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), isothermal microcalorimetry (IMC) and multi-excitation Raman spectroscopy (MX-Raman). Each of these analytical techniques provides unique insights into different bacterial properties. WGS allows the prediction of antibiotic susceptibility by confirming the presence of known resistance genes or mutations21,22,23; however, it may be less suitable to detect poorly understood and/or novel tolerance and resistance mechanisms. MALDI-TOF MS, widely used in clinical microbiology laboratories for rapid pathogen identification, also holds potential for antibiotic susceptibility testing24,25,26. Machine learning algorithms trained on large MALDI-TOF MS datasets have been successfully used to classify isolates as susceptible or resistant27. IMC measures heat production associated with metabolic processes. Microbial metabolism plays an important role in antimicrobial susceptibility6,28, and measuring metabolic activity with IMC can be used to evaluate the activity of antibiotics29,30,31. Lastly, Raman spectroscopy (RS) measures the inelastic scattering of light due to molecular vibrations, thereby offering information about the molecular composition of a sample32. In MX-Raman, multiple wavelengths are used to excite the sample, providing a more comprehensive fingerprint of bacterial cells33. Several studies have demonstrated the potential of RS to differentiate between resistant and susceptible bacteria34,35,36. Combined, these approaches allow to interrogate complementary aspects of bacterial biology, i.e., genomic differences (WGS), proteomic fingerprints (MALDI-TOF), metabolic activity (IMC), and overall biochemical composition (RS) and as such provide independent and machine learning–ready datasets for susceptibility prediction.
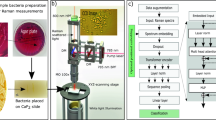
Using WGS, MALDI-TOF MS, IMC and MX-Raman data obtained from experimentally evolved P. aeruginosa strains, we developed an ordinal regression model to predict the minimal inhibitory concentration (MIC, as a measure of susceptibility of planktonic cells) and the biofilm prevention concentration (BPC) of tobramycin. For this study we focused on the aminoglycoside antibiotic tobramycin, as it is frequently used to treat exacerbations of P. aeruginosa infections in people with CF. In addition, several formulations are available for inhalation therapy, which means that high local concentrations of this antibiotic can be achieved at the site of the infection37,38, The BPC was defined as the lowest concentration of antimicrobial agent that prevented at least 90% of biofilm growth compared to the growth control in SCFM213. For MIC predictions, all techniques achieved an accuracy±1 > 89% (allowing a margin of error of one dilution), with MALDI-TOF MS achieving the highest performance by predicting the MIC with an accuracy±1 of 97.83%. For BPC predictions, all techniques showed an accuracy±1 > 73%; the best BPC predictions were obtained with Raman spectroscopy at 532 nm, achieving an accuracy±1 of 80.43%. After training, the model was validated with a set of P. aeruginosa isolates recovered from CF patients. For tobramycin MIC predictions, IMC and MALDI-TOF MS scored above random predictions, with IMC showing the best performance. For BPC predictions, only IMC performed above random predictions. An overview of the workflow is presented in Fig. 1.

The minimal inhibitory concentration (MIC) and the biofilm prevention concentration (BPC) of tobramycin were determined for 46 experimentally evolved P. aeruginosa strains (derived from six P. aeruginosa reference strains). These isolates were characterised using four analytical approaches, i.e. whole-genome sequencing (WGS), matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), isothermal microcalorimetry (IMC) and multi-excitation Raman spectroscopy (MX-Raman). The resulting data were used to train ordinal regression machine learning models to predict MIC or BPC values. Model performance was assessed by evaluating the accuracy ( ± 1) and the concordance index ( ± 1). After developing the prediction model, it was validated with an independent dataset of clinical P. aeruginosa isolates. Created in BioRender.
Results
Antibiotic susceptibility of experimentally evolved P. aeruginosa biofilms in SCFM2
We used an experimental evolution approach to generate a collection of evolved P. aeruginosa isolates derived from six reference strains. For each P. aeruginosa strain, eight independent cultures were set up in SCFM2 (except for CF1 with six cultures), resulting in 46 lineages that were maintained for 15 cycles. Half of the lineages were exposed to tobramycin and the other half served as untreated controls. While the amount of CFU/mL remained stable in control lineages, it significantly increased over time in all tobramycin-exposed lineages (except for AA2-1) (Supplementary Fig. 1). At cycle 15, the experiment was concluded and the MIC and BPC of tobramycin were determined (Fig. 2). The strains exposed to tobramycin during evolution showed significantly higher BPC values compared to those evolved without antibiotic exposure, indicating that exposure during evolution led to a reduced susceptibility to tobramycin in biofilms (Supplementary Fig. 2; increase in median BPC from 4 to 16 µg/mL). There was also a higher median MIC in tobramycin-exposed strains (median MIC of 2 µg/mL) compared to those evolved without antibiotic exposure (median MIC of 1 µg/mL), but this difference was not statistically significant.

a Minimal Inhibitory Concentration (MIC) and (b) Biofilm Prevention Concentration (BPC) of tobramycin (µg/mL) for all evolved strains (blue squares: control, red triangles: exposed to tobramycin during evolution) and their respective wild-types (WT) (green dots). Data points represent the median value of three biological replicates per sample.
Development of a machine learning model to predict antimicrobial susceptibility
A machine learning model was developed to predict the MIC or BPC of experimentally evolved strains based on WGS, MALDI-TOF MS, IMC or MX-Raman data. In what follows, a single sample denotes one evolved lineage, for which one prediction was made. Given the ordinal nature of the prediction targets (MIC or BPC values), an ordinal regression model was used. Prediction plots that visualise the probability at which each ordinal class was predicted, can be found in Supplementary Fig. 3. Prediction quality was assessed in terms of accuracy and concordance index (C-index). The accuracy measures the percentage of samples for which the correct MIC or BPC was predicted. The C-index measures the probability that two randomly chosen predictions are ranked correctly. For both of these metrics, an additional version is computed that allows misprediction up to one 2-fold dilution step: accuracy±1 and C-index±1, respectively (for more information, see Methods). To assess the prediction quality of the machine learning models, performances are compared to ‘random’ predictions. Here, ‘random’ performance consists of the score obtained when every prediction would be the MIC or BPC category that occurred the most often in the training dataset. More formally, the predictions \(P\left(y={k|x}\right)\) correspond to the probability \(P\left(y=k\right)\), unconditioned on \(x\). Hence, the score of a random prediction model corresponds to the setting where no relevant signal is present in the input data. This score was computed for both the accuracy and the accuracy±1. For the C-index and C-index±1, this random score is always 0.5, making these metrics more useful when comparing scores between MIC and BPC predictions. Models trained on MIC and BPC values have different ‘random’ performances as there are only four different MIC values present in our dataset (i.e., four ordinal categories), whereas the BPC dataset consists of six ordinal categories. A higher number of ordinal categories generally results in a more challenging prediction scenario and a lower random performance. Furthermore, the distribution of values also affects the random performance; evenly distributed categories make it harder to guess the correct value, whereas skewed distributions can increase the random performance score. It is therefore mandatory to compare a model’s performance to the random performance score and a model’s performance should be judged by how much it exceeds the performance of a random model.
Statistical tests typically require repeated measurements of performance obtained from independent test data sets. Given the nature of the evaluation metrics (which delivers a single estimate of performance on the whole data set), it is impossible to test for statistically significant differences between the different methods without violating test assumptions39. Instead, in what follows, model performances for different data sources are compared using the aforementioned notion of random predictions. Informally, a (higher) performance level above random indicates the more obvious presence of signals indicative of antimicrobial resistance in the data.
Whole-genome sequencing
Whole-genome sequences were obtained for 46 experimentally evolved P. aeruginosa strains. By comparing these sequences to that of the unevolved WT, the mutational landscape could be mapped (Supplementary Data 1). Next, a machine learning model was developed to predict the MIC and BPC values based on the detected DNA variants. This unbiased approach integrates all observed DNA variants, enabling the model to estimate which mutations affect MIC and BPC values, without requiring prior knowledge of determinants of susceptibility. While this approach is effective for experimentally evolved strains with a known WT ancestor, it is not directly applicable to clinical isolates as the choice of a ‘WT’ reference genome is inherently ambiguous (or even impossible) in that case. The model predicted the MIC from WGS data with an accuracy±1 of 89.13% (random performance: 82.61%), while the BPC was predicted with an accuracy±1 of 76.09% (random performance: 69.57%). The C-index±1 was 71.77% and 80.16%, for MIC and BPC predictions, respectively (Fig. 3, Supplementary Table 1), indicating that the model is slightly better in predicting the BPC than the MIC from WGS data.
WGS has been widely used to predict antimicrobial susceptibility, typically by screening for well-studied, known resistance genes22,40,41,42,43. For instance, Eyre et al. used this approach to predict the MIC and MIC ± 1 2-fold dilution of several antibiotics in Neisseria gonorrhoeae with accuracies of 53% and 93%, respectively40. However, focusing only on known resistance genes poses a major limitation, as many tolerance and resistance mechanisms remain poorly understood. To address this challenge, we included all detected DNA variants as input for our machine learning model, regardless of prior knowledge on the role of specific gene products. Other studies have tackled these limitations by using entire genome sequences as input for prediction algorithms44,45,46. For example, Nguyen et al. used this method to predict MIC values ± 1 2-fold dilution for several antibiotics in Salmonella, achieving an accuracy±1 of 95%, which is comparable to our results44. In the present study, using entire genome sequences as input for the machine learning model was not possible due to the high-dimensional nature of this approach, which required a large sample size to be effective. Instead, we used DNA variants detected by mapping the entire genome sequences against a reference genome as input for the machine learning model. Nonetheless, integrating entire genome sequences with machine learning models would be an ideal strategy for future studies with larger datasets.

(a) Scatterplot showing the performance for predicting the MIC (squares) or the MIC with an allowed margin of error of one 2-fold dilution (dots) (b) Scatterplot showing the performance for predicting the BPC (squares) or the BPC with an allowed margin of error of one 2-fold dilution (dots). The blue lines indicate scores based on random predictions. Horizontal blue dashes: random accuracy performance. Horizontal blue full line: random accuracy±1 performance. Vertical blue full line: random C-index and C-index±1.
MALDI-TOF MS
MALDI-TOF mass spectra were collected for 46 experimentally evolved strains (Fig. 4a), and a machine learning model was developed to predict MIC or BPC values directly from these spectra, using them as unique spectral fingerprints. The model predicted the correct MIC with an accuracy±1 of 97.83% (random performance: 82.61%), while the BPC was predicted with an accuracy±1 of 73.91% (random performance: 69.57%). The model obtained a C-index±1 of 86.12% and 76.92% for MIC and BPC predictions, respectively (Fig. 3, Supplementary Table 1). These results indicate that MALDI-TOF mass spectra allow a more precise prediction of the MIC than of the BPC. Overall, these results demonstrated that MALDI-TOF mass spectra can provide valuable insights into antibiotic susceptibility. Recent studies have demonstrated the potential of machine learning models in classifying isolates as susceptible or resistant based on mass spectra27,47,48,49. For example, Weis et al. classified Staphylococcus aureus isolates as susceptible or resistant with an area under the receiver operating characteristic curve (AUROC) of 80%27. Similarly, Nguyen et al. classified P. aeruginosa isolates resistant to ceftazidime/avibactam and ceftolozane/tazobactam, achieving AUROCs of 86.9% and 85.6%, respectively47. The AUROC metric is equivalent to the C-index used in our study, but is specific to binary predictions. These studies predict whether the MIC is above or below a certain breakpoint (EUCAST or CLSI), but as such breakpoints are not yet established for biofilms, it is currently not possible to translate a BPC value into a classification as ‘resistant’ or ‘susceptible’. Our study addresses this limitation by training machine learning models to predict exact BPC values from the mass spectra, enabling compatibility with potential future biofilm breakpoints based on epidemiological cut-off values or other measures13. Using this approach, our models obtained a C-index±1 of 86.12% for MIC predictions, comparable to the aforementioned studies, and a C-index±1 of 76.92% for BPC predictions. Optimizing MALDI-TOF MS for AST could offer several advantages, as these devices are already widely used in hospitals for pathogen identification. Additionally, the technique has a high throughput, allowing the analysis of up to 96 samples in just minutes, making it a fast and efficient tool in clinical settings.

Data is shown of evolved P. aeruginosa AA2-1 L26 (orange) and L32 (green), and LES B58 L08 (blue). (a) MALDI-TOF mass spectra (b) Thermograms measuring the heatflow (µW) over time. Several parameters can be derived from thermograms, such as the time to peak (TTP) and the maximum metabolic rate (MMR) (c) Raman spectra obtained at excitation wavelength 532 nm (d) and 785 nm.
Isothermal microcalorimetry
In IMC, the heat produced by microbial metabolic processes is measured in real time. Using this method, we determined the metabolic profiles of 24 h old biofilms in SCFM2 for all experimentally evolved strains (Fig. 4b). A machine learning model was trained to predict MIC and BPC values based on thermograms obtained with IMC. The model predicted the correct MIC with an accuracy±1 of 93.48% (random performance: 82.61%) and the BPC with an accuracy±1 of 76.09% (random performance: 69.57%). The model obtained a C-index±1 of 83.25% and 64.78%, for MIC and BPC predictions, respectively (Fig. 3, Supplementary Table 1). These results suggested that IMC data were more predictive for the MIC than for the BPC. Nonetheless, both MIC and BPC predictions outperformed random predictions, indicating that even without antibiotic exposure, metabolic profiles of biofilms can reveal meaningful insights into their antibiotic susceptibility. Previous studies that explore the use of IMC for AST typically measured the metabolic activity of planktonic cultures or biofilms exposed to antibiotics29,30,31,50. These approaches mirror the principle of broth microdilution MIC determination, but benefit from IMC’s high sensitivity (detection limit: 104 CFU/mL51) leading to faster results. Using this approach, Tellapragada et al. predicted the MIC of amikacin in planktonic P. aeruginosa with an essential agreement (number of results that were within one doubling dilution of the matching MIC determined by reference methods) of 97.4%29. In contrast, our setup measures untreated biofilms, eliminating the need to test multiple antibiotic concentrations and increasing throughput. To our knowledge, this is the first study that combines IMC data from untreated biofilms with machine learning algorithms to predict the MIC or BPC.
Raman spectroscopy
Raman spectroscopy provides a rapid and label-free method to obtain specific molecular fingerprints. In the present study, biofilms were grown overnight in SCFM2, and the resulting pellet from that overnight culture was applied to a fused quartz slide for analysis under a Raman microscope at two different excitation wavelengths, i.e., 532 and 785 nm (Fig. 4c, d). The spectral data from these individual wavelengths were also combined to create multi-excitation Raman spectra (MX-Raman)33. Using both single-wavelength (RS 532 nm or RS 785 nm) and MX-Raman spectra, a machine learning model was trained to predict MIC and BPC values. For MIC predictions, all three input types – RS 532 nm, RS 785 nm and MX-Raman – achieved an accuracy+-1 of 91.30% (Fig. 3, Supplementary Table 1). However, RS 785 nm yielded the highest C-index±1 (86.60%), followed by RS 532 nm (81.34%), and MX-Raman with the lowest C-index±1 (69.06%). While Lister et al. previously reported that combining multiple wavelengths improved bacterial strain identification using a support vector machine (SVM) model, our results do not show a clear advantage for MX-Raman in susceptibility predictions33. For BPC predictions, values for accuracy±1 were similar across methods; i.e., 80.43% for RS 532 nm and 76.09% for RS 785 nm and MX-Raman. Likewise, C-index±1 scores were similar across methods (85.63%, 84.01% and 81.16% for RS 532 nm, RS 785 nm and MX-Raman, respectively). Overall, the C-index±1 scores for both MIC and BPC predictions were quite comparable, except for MX-Raman’s notably lower performance on MIC predictions (69.06% vs. 81.16% for BPC). Nonetheless, this score is still substantially above random (50%), suggesting that Raman spectroscopy is a good predictor for both MIC and BPC. When the Raman spectra from samples sharing the same MIC or BPC value were averaged, distinct differences in peak intensities emerged among the resulting group averages, correlating with the respective MIC or BPC values (Supplementary Fig. 4). Previous studies have demonstrated the potential of Raman spectroscopy for differentiating resistant and susceptible bacteria based on unique spectral signatures34,35. Lister et al. classified resistant bacterial strains with 100% accuracy and susceptible strains with 98.89% accuracy using an SVM model34. Similarly, Lu et al. distinguished a susceptible Acinetobacter baumannii strain from five resistant strains with 99.92% accuracy using a random forest model, though their approach could not identify to which antibiotics the strains were resistant35. Our study is the first to combine Raman spectroscopy with machine learning to predict BPC values, demonstrating its potential as a valuable tool in biofilm susceptibility testing.
Combining data from different analytical approaches does not increase the performance of MIC or BPC predictions
To evaluate if there is an added value in combining results from multiple analytical approaches, a stacking model that integrated data from all sources was used. This stacking approach used predictions based on the different types of data as inputs for a final model52,53. Using the stacking model, MIC values were predicted with an accuracy±1 of 91.30% (random performance: 82.61%), while the highest individual performance was achieved with MALDI-TOF (accuracy±1 of 97.83%). A C-index±1 of 82.30% was obtained with the stacking model, while the best-performing individual technique (RS 785 nm) had a C-index±1 of 86.60%. For BPC predictions, the stacking model achieved an accuracy±1 of 71.74% (random performance 69.57%), whereas the highest accuracy±1 was obtained with RS 532 nm (80.43%). The C-index±1 for BPC predictions with the stacking model was 80.57%, while the best-performing method (RS 532 nm) achieved a C-index±1 of 85.63% (Fig. 3, Supplementary Table 1). Overall, these results showed that combining data sources through stacking did not lead to a higher performance than the best-performing individual data source alone (Fig. 3). Nonetheless, the stacking model can provide insights into complementary data sources by analysing learned ordinal model coefficients \(\beta \in {{\mathbb{R}}}^{f\times 1}\) (Supplementary Fig. 5). For both the MIC and BPC predictions, we observed highly positive coefficients for MALDI-TOF MS and WGS. The fact that both techniques showed a high variable importance indicated that they provided complementary, non-overlapping information. This finding suggested that, while we could not establish a performance gain with this combination in this study, future work with different datasets or additional refinement may reveal that combining WGS and MALDI-TOF MS data can further enhance prediction accuracy.
Differences in prediction accuracy depending on the MIC or BPC value
We subsequently addressed the question whether predictions are equally accurate regardless of whether the MIC or BPC is high or low. For this we used the RS 785 data, as our prediction models consistently perform well for this data set. In order to do so, we constructed two ‘confusion matrices’ (one for the MIC and one for the BPC) in which the probability that a true value is correctly predicted for every MIC or BPC value is shown (Supplementary Fig. 6). This representation allows to estimate the accuracy for different MICs and BPCs; in case of no or limited differences in accuracy, the highest values would be on the diagonal. This analysis suggests that MIC predictions might be better for cases in which the actual MIC is 1 or 2 µg/ml. For the prediction of the BPC, differences in accuracy are less pronounced, although there might be a slight trend towards ‘overpredicting’ higher BPC values (≥ 8 µg/ml). There are however some caveats with this type of analysis on this particular dataset. First of all, the size of the datasets for the individual subsets becomes quite small. Secondly, this type of analysis is inherently biased by the number of cases in each group. In the MIC dataset the number of cases with MIC = 2 µg/ml is highest and in the BPC dataset this is the case for the groups with BPCs between 8 and 32 µg/ml. This uneven distribution of isolates over different categories at least partially explains the bias in accuracy observed. Finally, this type of analysis does not take into account the generally accepted variability of the assays (e.g., replicate MIC measurements can differ by a two-fold dilution54). So while there may be differences in accuracy depending on whether isolates have a high or a low MIC (and to a lesser extent, a high or a low BPC), the current dataset does not allow to distinguish between a real bias, and a bias related to the uneven distribution of values and/or small numbers in each subgroup.
Validation of machine learning model with clinical isolates
The prediction model was initially trained on data from experimentally evolved strains. To evaluate its ability to predict the MIC or BPC of clinical P. aeruginosa isolates, we analysed MALDI-TOF MS, IMC and MX-Raman data obtained from 30 CF isolates. For MIC predictions, the best performance was observed for IMC, with an accuracy±1 of 80% (random performance: 60%) and a C-index±1 of 86.90% (Fig. 5, Supplementary Table 2). Predictions based on MALDI-TOF MS achieved an accuracy±1 of 76.67% and a C-index±1 of 58.93%. Using RS 532 nm, the accuracy±1 was 63.33%, but the C-index±1 of 10.71% was far below the random predictions of 50%. RS 785 nm had an accuracy±1 of 60% - equal to random performance – while its C-index±1 reached 53.57%, only slightly exceeding the random performance of 50%. MX-Raman achieved an accuracy±1 of 60% - equal to random performance – and a C-index±1 of 47.02%, which fell below random predictions. For BPC predictions, IMC again performed best, achieving a very high accuracy±1 of 95.24% (random performance: 80.95%) and C-index±1 of 97.73%. Meanwhile, MALDI-TOF MS, RS 532 nm and MX-Raman all performed at or below random performance levels, with accuracy±1 scores of 66.67%, 76.19% and 80.95%, respectively, and C-index±1 scores of 43.18%, 31.82% and 45.45%. RS 785 nm achieved the second-highest C-index±1 of 77.27% (well above the 50% random prediction level), although its accuracy±1 of 80.95% was the same as that of random predictions. In summary, for MIC predictions, IMC and MALDI-TOF MS were the only techniques scoring above random predictions for both accuracy and C-index, with IMC showing the best performance. For BPC predictions, only IMC performed above random predictions for both accuracy and C-index. These results indicate that IMC data have the highest predictive power when it comes to predicting antimicrobial susceptibility, particularly for predicting the BPC. In contrast, the other techniques showed reduced performance when models trained on data from evolved strains were externally validated with clinical isolates. This discrepancy highlights the challenges of translating model performances across different datasets, a limitation also observed in other studies. For instance, Weis et al. and Ren et al. reported reduced performance when isolates from one dataset were tested on models that were trained on another dataset27,48. We hypothesize that the IMC data were easier for the model to interpret because the thermograms displayed clear trends (Supplementary Fig. 7). As the MIC or BPC increased, the time to peak (TTP) also increased, while the maximum metabolic rate (MMR) generally decreased. This particularly resulted in a high C-index, as this is a ranking-based metric and the thermograms exhibited a clear ranking pattern. Despite visual differences between thermograms of clinical isolates and those of the experimentally evolved strains (Supplementary Fig. 7), the model was still able to interpret them effectively. Furthermore, the observed IMC changes related to susceptibility are consistent with recent findings linking changes in bacterial metabolism to reduced susceptibility28,55.

a Scatterplot showing the performance of predicting MIC (squares) or the MIC with an allowed margin of error of one 2-fold dilution (dots). b Scatterplot showing the performance of predicting BPC (squares) or the BPC with an allowed margin of error of one 2-fold dilution (dots). The blue lines indicate scores based on random predictions. Horizontal blue dashes: random accuracy performance. Horizontal blue full line: random accuracy±1 performance. Vertical blue full line: random C-index and C-index±1.
Does the MIC allow to predict the BPC?
When data for all isolates (controls, evolved isolates, clinical isolates) are pooled, a significant correlation between MIC (determined in MH) and BPC (determined in SCFM2) (Kendall’s Tau correlation coefficient of 0.695, p < 0.001) is found (Fig. 6), which is in line with results of an earlier smaller-scale study10. While the data seems to suggest that higher MIC values typically translate into higher BPC values, the analysis also illustrates that predicting the BPC based on the MIC is not obvious. For example, isolates with MIC value of 1 µg/ml show BPC values between 1 and 32 µg/ml and isolates with MIC value of 8 µg/ml show BPC values between 16 and 128 µg/ml. So while there is a positive correlation between MIC and BPC, it does not allow for accurate prediction of BPC based on the MIC. This observation is in line with observations in several other studies that show that there may be an overall positive correlation between planktonic and biofilm susceptibility measurements, but that in many cases the reduced susceptibility observed in biofilms is independent of resistance in planktonic cells13.

XY-scatter plot showing the correlation between tobamycin MIC (determined in MH broth) and BPC (determined in SCFM2 medium) for all isolates investigated in the present study.
Learned model coefficients suggest an important role for Anr and NppA1A2BCD mutations in tobramycin susceptibility
All experimentally evolved strains were genome-sequenced and compared to their ancestral wild-type (WT) strain. The observed mutations are listed in Supplementary Data 1. Overall, we found that many global regulators and two-component systems (TCS), such as lasR, pqsR, bfmR/bfmS and gacS/gacA56,57 acquired mutations. Additionally, numerous mutations were observed in proteins involved in 3’,5’-cyclic diguanylic acid (c-di-GMP) metabolism, including wspF, dipA, morA and rbdA, all of which play a role in biofilm formation58. Interestingly, only one gene that has a known association with tobramycin resistance, i.e., the efflux pump regulator mexZ59, was mutated in two tobramycin-evolved lineages. Learned model coefficients can provide valuable insights into which genes might be correlated with increased MICs and BPCs. However, these correlations should be interpreted with caution, as some strongly correlated mutations may not directly correspond to causal biological mechanisms. For instance, two mutations might occur together within a single isolate with a high MIC. In such cases, it is possible that only one mutation is the actual causal (i.e., biologically relevant) factor driving the higher MIC value, while the other mutation merely co-evolved. Despite this, the model might estimate both mutations as being correlated with an increased MIC, even though only one exerted a true biological effect. Therefore, careful consideration is necessary when interpreting these results. Nevertheless, in large datasets, learned model coefficients can serve as valuable tools for identifying candidate genes that merit further investigation. The model assigned a highly positive coefficient to mutations in anr, an anaerobic transcriptional regulator that controls the expression of genes essential for survival in low-oxygen environments. Previous studies have shown that regulation of the anaerobic respiratory pathway can reduce intracellular accumulation of aminoglycosides, contributing to adaptive resistance in P. aeruginosa60. In the present study, mutations in anr were identified in CF1 populations evolved in the presence of tobramycin. Isolates with the amino acid substitution Phe→ Ser 107 show a reduced susceptibility to the aminoglycosides tobramycin, gentamicin and amikacin. Compared to the CF1 WT (with an MIC of 1, 2, and 2 µg/mL for tobramycin, gentamicin and amikacin, respectively), the CF1 anr mutant displayed increased MICs of 4, 8 and 16 µg/mL for these antibiotics. Several genes within the same cluster, encoding the ABC transporter NppA1A2BCD, also exhibited highly positive coefficients (Supplementary Fig. 8). A previous study has shown that in P. aeruginosa NppA1A2BCD is required for the uptake of peptidyl nucleoside antibiotics61. However, while we hypothesized that mutations in the Npp transporter also reduce uptake of the aminoglycoside tobramycin, we observed no differences in MIC or BPC of tobramycin between PA14 WT and the ΔnppBCD knockout mutant (MIC = 2 µg/mL, BPC = 8 µg/mL). When evaluating the minimal biofilm inhibitory concentration (MBIC) of tobramycin, a two-fold increase was observed in the knockout mutant (16 µg/mL in the PA14 WT vs. 32 µg/mL in the ΔnppBCD strain). Other mutated genes with high positive coefficients include rbdA (PA0861), bfmR (PA4101) and pmrA (PA4776), although these were not experimentally investigated in the present study. The rbdA gene encodes a regulator of biofilm dispersal with phosphodiesterase (PDE) activity, and its inactivation in PAO1 has been demonstrated to lead to hyperbiofilm formation62. The bfmR gene encodes a two-component system response regulator (TCS RR) that regulates genes involved in biofilm maturation and mediates the transition from acute to chronic virulence63. Mutations in bfmR may indirectly influence antibiotic susceptibility by altering biofilm development or virulence pathways. The TCS RR pmrA regulates genes responsible for modifying the bacterial outer membrane (OM). Mutations in pmrA confer resistance to polymyxins by promoting OM modifications that reduce binding of cationic antibiotics64. Furthermore, mutations in the PmrAB TCS have been shown to elicit cross-resistance to aminoglycosides65.
Discussion
Using WGS, MALDI-TOF MS, IMC and RS data obtained from a collection of experimentally evolved P. aeruginosa strains, we were able to train machine learning algorithms that allowed to predict the MIC and BPC of tobramycin. All analytical approaches used demonstrated a predictive power that was higher than that of random predictions, confirming that each data type contained relevant information about antimicrobial susceptibility. We showed that an unbiased approach to predict susceptibility was possible with MALDI-TOF MS, IMC and MX-Raman data, as these methods do not require prior knowledge of mechanisms of susceptibility. Machine learning models successfully identified patterns in spectral data and thermograms, even without exposing the bacterial strains to antibiotics. This proof-of-concept study highlights the potential of alternative methods for predicting MIC and BPC values.
While our study provides evidence that several innovative analytical approaches can be used to predict biofilm susceptibility, not all of these approaches are currently available outside the research setting, which precludes their implementation in clinical susceptibility testing at this point. In addition, the present study focused on a single species and a single antibiotic and conclusions from our work can at present not be extrapolated to other species and/or antibiotics, especially when considering antibiotics with other another mode of actions and/or organisms in which inducible resistance mechanisms are important. In addition, this study focused on P. aeruginosa biofilms formed in SCFM2, and while there is ample evidence that this medium allows formation of biofilms that closely resemble those found in vivo14,15,16, translation of findings obtained with SCFM2 to other experimental settings and/or to real-world settings may be difficult. Finally, future research should focus on expanding datasets to include more strains and a wider range of MIC and BPC values to further validate our results.
Methods
Bacterial strains, culture conditions and chemicals
The following P. aeruginosa strains were used: AA2-1 (a lasR+ isolate derived from strain AA2; LMG 27630), LES B58 (LMG 27622), LES 431 (LMG 27624), UCBPP-PA14 (LMG 27639), IST27 (LMG 27643)66 and CF1 (a Danish CF isolate belonging to sequence type 560). Strains derived from these wild type (WT) strains through experimental evolution are listed in Supplementary Data 2. In addition, isolates recovered from chronically infected CF patients at Ghent University Hospital were used (Supplementary Data 2); the collection of these strains was approved by the Ethics Committee of Ghent University Hospital, registration number B670201836204. The PA14 ΔnppBCD knockout mutant was previously described in ref. 61. The CF1 anr mutant (Phe→Ser 107) was isolated in the present study from an experimentally evolved CF1 strain (Supplementary Data 2). Bacteria were stored at −80 °C in 8% DMSO or using Microbank vials (Pro Lab Diagnostics, Canada) and were cultured at 37 °C on Tryptone Soy Agar (TSA) plates and in Tryptone Soy Broth (TSB) (Neogen, UK). Stock solutions of tobramycin (TCI Europe, Belgium), gentamicin (Sigma-Aldrich, USA) and amikacin (Sigma-Aldrich, USA) at 10 mg/mL were prepared in MilliQ water (filter sterilized, PES, 0.22 µm, VWR, Belgium). Synthetic cystic fibrosis medium (SCFM2) was prepared as described before14, with the modification that mucin (a solution of 25 mg/ml) was sterilized by autoclaving (121 °C, 15 min) instead of UV exposure. Previous work has shown that this does not meaningfully alter mucin structure or function67,68.
Experimental evolution
Six P. aeruginosa reference strains were experimentally evolved under biofilm conditions for 15 cycles, with or without exposure to tobramycin (Fig. 7). For each strain, four control lineages and four tobramycin-treated lineages were included (except for CF1, three lineages each), resulting in 46 independently evolved lineages. To this end, overnight cultures of P. aeruginosa were diluted in SCFM2 to approximately 5 × 107 CFU/mL. 100 µL of the resulting suspension was added to a flatbottom 96-well plate (VWR, USA) and incubated for 24 h at 37 °C under aerobic conditions (without shaking). After 24 h, biofilms were treated with 100 µL of tobramycin solution or 100 µL of SCFM2 medium (for untreated controls). Tobramycin concentrations were selected based on preliminary experiments that resulted in a 2–3 log reduction in the number of CFU in a 24 h old biofilm; i.e., 16 µg/mL for CF1, 32 µg/mL for UCBPP-PA14 and IST27, 64 µg/mL for AA2-1 and LES 431 and 128 µg/mL for LES B58. After an additional 24 h incubation at 37 °C, biofilm aggregates were disrupted by vortexing (5 min, 900 rpm) (Titramax 1000, Heidolph Scientific Products GmbH, Germany) and sonication (40 kHz, 5 min) (Branson 3510, Branson Ultrasonics, USA) and the number of CFU/mL was quantified by plating on TSA. A frozen stock of each lineage was stored at −80 °C in cryovials with 8% DMSO in TSB. An overnight culture was prepared by inoculating 5 mL TSB and incubating for 24 h at 37 °C while shaking. The next day, a new cycle was initiated by inoculating with 5 × 107 CFU/mL from that overnight culture. This process was repeated for 15 cycles.

100 µL of a P. aeruginosa culture is added to the well of a 96-well plate. After 24 h, suspended biofilm aggregates are formed and these are either treated with 100 µL of tobramycin, or 100 µL fresh SCFM2 medium is added. After 24 h, the biofilm is disrupted and a part of the bacterial suspension is used to inoculate an overnight culture to allow the start of a new cycle. The number of surviving cells after each cycle is quantified by plating on TSA. The samples are stored at −80 °C. Created with BioRender.
Antibiotic susceptibility testing
MICs of tobramycin, gentamicin and amikacin were determined following the EUCAST guidelines using the broth microdilution method3. MICs were defined as the lowest concentration of an antimicrobial agent that inhibited at least 90% of microbial growth after 24 h of incubation. The BPC of tobramycin was determined using serial dilutions of the antibiotic in SCFM2 medium and inoculating bacteria at a final concentration of 5 × 107 CFU/mL in SCFM210. After 24 h incubation at 37 °C, the contents of the wells were plated and after 24 h of incubation colonies were counted. The BPC was defined as the lowest concentration of antimicrobial agent that prevented at least 90% of biofilm growth compared to the growth control after 24 h of incubation. For selected isolates, the minimal biofilm inhibitory concentration (MBIC) of tobramycin was determined. The MBIC is the lowest concentration of tobramycin that resulted in at least 90% reduction of biofilm growth compared to the growth control). To this end, 50 µL of a 5 × 107 CFU/mL bacterial suspension in SCFM2 was added to round bottom 96-well plates. After 24 h incubation at 37 °C, double concentrated tobramycin solutions prepared in SCFM2 were added to each well. After 24 h of treatment at 37 °C, biofilms were disrupted by vortexing and sonicating (40 kHz, Branson 3510, Branson Ultrasonics, USA) the plate for 5 min each. The contents of the wells were plated on TSA, and after 24 h incubation at 37 °C, colonies were counted. All experiments were performed in biological triplicate.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics (version 29). The normality of the data was verified with a Shapiro-Wilk test. If the data were normally distributed, a two-sided independent samples t-test (to compare differences between two timepoints or two treatment groups) or ANOVA with Bonferroni correction for multiple testing (to compare differences between multiple groups) were performed. If the data were not normally distributed, a two-sided nonparametric Mann-Whitney U test was performed. Kendall’s Tau correlation was calculated to assess the relationship between MIC and BPC values. Graphs were constructed using GraphPad Prism (version 10.6.0).
DNA extraction
Overnight liquid cultures were centrifuged to obtain a pellet. The pellet was resuspended in 200 µL 10 mM TE-buffer (10 mM Tris-HCl pH 8, 1 mM EDTA pH 8), after which 100 µL was transferred to a 2 mL microcentrifuge tube containing approximately 500 µL acid-washed glass beads ( ≤ 106 μm; Sigma Aldrich, USA) and 500 µL lysis buffer (50 mM Tris-HCl pH 8, 70 mM EDTA pH 8, 1% SDS) with 0.5 mg/mL Pronase (Roche, Germany). Samples were vortexed vigorously for 5–10 s, incubated at 37 °C for 30–60 min, and centrifuged at 13,000 rpm for a short spin. Following the addition of 200 µL of saturated ammonium acetate, samples were vortexed vigorously for 5–10 s and centrifuged again at 13,000 rpm for 2 min. To separate phases, 600 µL chloroform was added, and samples were vortexed horizontally for 5–10 s, and centrifuged for 5 min at 13,000 rpm. Then, 400 µL of clear aqueous top phase was transferred to an Eppendorf LoBind microcentrifuge tube (Eppendorf AG, Germany) containing 1 mL 100% ethanol. The tubes were mixed by inversion to precipitate the DNA, followed by centrifugation at 13,000 rpm for 5 min. Afterwards, the supernatant was discarded and the pellet was washed with 500 µL 70% ethanol. After a short spin, the ethanol was removed by pipetting and the pellet was air-dried. The dry pellet was dissolved in 300 µL low EDTA TE-buffer (10 mM Tris-HCl pH 8, 0.1 mM EDTA, 0.5 µg/mL RNase) and incubated at 37 °C for 60 min. DNA concentrations were determined using the BioDrop µLITE (BioDrop, UK).
Whole-genome sequencing and data analysis
A PCR-free library preparation was performed using the NEBNext Ultra II Library Prep Kit for Illumina, following a size selection protocol using AMPure XP beads after adapter ligation. Samples were sequenced on the Illumina NextSeq 500 System, generating 75 bp single-end reads. The reads were analysed with CLC Genomics Workbench and mapped to reference genomes of P. aeruginosa AA2 (NZ_CP051547.1), LES B58 (NC_011770.1), LES 431 (NC_023066.1), UCBPP-PA14 (NZ_CP034244.1) or to the 28 contigs of IST27 (whole genome shotgun sequencing project MCMX01000001 to MCMX01000028), or the 36 contigs of CF1. Reads were mapped using a local alignment and filtered based on a 50% length fraction and 80% similarity fraction. The basic variant detection tool was used to detect single nucleotide polymorphisms (SNPs) with a minimum frequency of 10%, minimum count of 5, minimum quality of 20 and minimum forward/reverse balance of 0.3. All SNPs were manually screened to remove false positives. Insertions and deletions were detected using the InDels and Structural Variants tool, with a minimum sequence complexity of 0.2 and a minimum count of 5. Entries meeting minimum requirements were further manually filtered for false positives derived from sequencing and mapping errors. The raw reads generated in this study are available in the ArrayExpress database under the accession number E-MTAB-11894.
MALDI-TOF mass spectrometry
Pure cultures were plated in biological duplicate on TSA. Confluent growth was collected with a sterile loop and suspended in 300 µL MQ. After vortexing, 900 µL of 100% ethanol was added and the tubes were homogenized by inversion. After centrifuging for 3 min at 4 °C, the ethanol was discarded and evaporated. Next, the cell pellet was suspended in 40 µL formic acid and the samples were vortexed. Then, 40 µL acetonitrile was added and the samples were vortexed. After centrifugation, 1 µL supernatant (containing the protein extract) was spotted in duplicate on the target plate. Subsequently, 1 µL matrix solution (10 mg/mL alpha-cyano-4-hydroxycinnamic acid) was spotted on the plate. A bacterial test standard (BTS) was included for calibration. Mass spectra were obtained using the Biotyper Microflex LT/SH MALDI-TOF MS system (Bruker Daltonik GmbH, Germany). The experiments were performed in biological and technical duplicate.
Isothermal microcalorimetry
Overnight cultures were diluted in SCFM2 medium to a final concentration of 5 × 107 CFU/mL, and 100 µL of the bacterial suspension was transferred to plastic inserts (calVials, Symcel, Sweden) for biofilm growth over 24 h at 37 °C. The next day, 100 µL of fresh SCFM2 medium was added to the biofilms. The plastic inserts were then transferred to titanium cups and microcalorimetric measurements were conducted using the calScreener device (Symcel, Sweden). The resulting thermograms (in which heat production is plotted over time) were analysed with calView 2.0 software (Symcel). All experiments were performed in technical duplicates.
Raman spectroscopy
Bacterial cultures were grown overnight at 37 °C in SCFM2 medium and were centrifuged for 10 min at 4000 rpm. Pellets were washed with ddH2O, and after another round of centrifugation the resulting pellet was applied to a fused quartz microscopic slide (UQG Optics, UK) and dried on a heater. Samples were excited with a 532 nm or a 785 nm lasers using the Renishaw InVia Raman Microscope (Renishaw, UK). Spectra were acquired over three accumulations with a 5 s exposure time. Three biological repeats of each evolved lineage were prepared, with 50 spectra collected for each biological repeat such that 150 Raman spectra were obtained for each strain and experimental condition. Cosmic rays were removed from all spectra using the Renishaw WiRe 5.5 software. Multi-excitation spectra were obtained by manually merging the 532 nm spectrum to the end of the 785 nm spectrum.
Data preprocessing for machine learning
Four different data types were collected, i.e., DNA variants, MALDI-TOF mass spectra, thermograms, and Raman spectra. To prepare these data for machine learning modeling, data were preprocessed to fixed-length feature vectors. In what follows, a ‘sample’ denotes a single evolved lineage. For all data sources – aside from DNA variants – multiple technical replicates per sample were generated. In data preprocessing, technical replicates were considered independently, every technical replicate becoming a separate row in the final machine learning input matrix. All preprocessed data are available at the following GitHub repository: https://github.com/gdewael/biofilm-amr. Considering all possible variants in the genome constitutes an infeasibly big feature space, and for that reason DNA variant data were collated on a per-gene basis. The DNA variant input for the machine learning model, hence, was defined as ‘whether a variant was found in gene X’, for any gene X. The feature space was further reduced by eliminating loci for which no variant was found in any sample. Further, as machine learning cannot learn generalizing patterns for features present in a single sample, genes with variants in a single sample were similarly eliminated. The resulting feature vector for every sample was of binary nature, with every feature consisting of a single gene, indicating whether a variant in it was found in said sample. For MALDI-TOF data, spectra were preprocessed according to standard practices27: (1) square-root transformation of intensities, (2) smoothing with a Savitzky-Golay filter using a half-window size of 10, (3) baseline correction with the 20 iterations of the SNIP algorithm, and (4) trimming to the 2000–20,000 Da range. To reduce the feature space, peaks were detected using the persistence transformation algorithm, keeping only the top 128 for every spectrum69. A fixed-length feature vector was then obtained by placing the detected peaks in 3Da-interval bin features (ranging from 2000 to 20,000 Da, 6000 bins overall). To retain congruence with DNA variant data, bins were binarized, hence, each feature in the input vector indicates whether a peak was found in a bin. Features were similarly eliminated if they were found in either no spectra, or only one spectrum in the entire dataset. Raman spectra were preprocessed similarly to the MALDI-TOF mass spectra, using iRootlab70. Briefly, the following steps were performed: (1) square-root transformation of intensities, (2) smoothing with a Savitzky-Golay filter using a half-window size of 10, (3) baseline correction with the 20 iterations of the SNIP algorithm, and (4) trimming to the 600–1700 cm−1 range. After, peaks were similarly detected using the persistence transformation algorithm, keeping only the top 128 peaks for every spectrum. Peaks were then binned in 1 cm−1 intervals and binarized. Features were eliminated if they were found in either no, or only one spectrum in the entire dataset. For MX-Raman, the final feature representation constitute the features for the 532 nm and 785 nm spectra, taken together by concatenation. Microcalorimetry data are presented as thermograms, i.e., heatflow (µW) over time. Heatflow measurements were taken at regular intervals in time (every 10 min), so these data are already in fixed-length feature format (every feature being the heatflow in µW at a specific time point). Data were normalized to a range between zero and one by dividing all heatflow numbers by 250 (the highest heatflow number encountered rounded up to ten).
Machine learning modeling
The prediction targets (MIC or BPC values) are of ordinal nature: 1, 2, 4, … (µg/mL). For this reason, an ordinal regression model is used to predict MIC or BPC values based on one of the data types described above. More formally, let us denote an input data sample as \(x\in {{\mathbb{R}}}^{f}\), with f number of input predictors. Its corresponding MIC or BPC value is given by \(y\in \left\{{2}^{n}{|n}{\mathbb{\in }}{\mathbb{Z}}\right\}\). The cumulative logistic link function71, then, models \(y\) as a function of \(x\) as follows (Eqs. 1–3):
Where \(k\) denotes the ordinal classes numbered 1 to K. These ordinal classes map to the distinct MIC or BPC values in the data. For MIC, these correspond to: {1, 2, 4, 8}. For BPC values, these correspond to: {1, 2, 4, 8, 16, 32}. In essence, this model linearly transforms the input data to one dimension via learnable coefficients \(\beta \in {{\mathbb{R}}}^{f\times 1}\). This one-dimensional space is then ‘cut up’ into ordinal classes via learned (strictly increasing) cutpoints \({c}_{\left\{1,\ldots ,K-1\right\}}\). The ordinal model can be used to either predict the probability that a sample will have a certain MIC or BPC: \(P\left(y={k|x}\right)\), or predict a single MIC or BPC value for a sample: arg m axk \(P\left(y={k|x}\right)\). Both the coefficients \(\beta\) and cutpoints \({c}_{\left\{1,\ldots ,K-1\right\}}\) were jointly optimized to minimize the negative log-likelihood (Eq. 4):
Models were trained using gradient descent. Every training iteration considered the full training data set as batch. All code to train models and fully reproduce all experiments is available at https://github.com/gdewael/biofilm-amr.
Hyperparameter tuning
Models were tuned separately for every data source and target type (MIC or BPC). Hyperparameter tuning was performed through nested leave-one-out cross validation (CV). In case of technical replicates (present for all data sources, except DNA variant data), this data splitting scheme was adjusted to leave-one-group-out CV, with a group constituting all replicates corresponding to one sample. The outer CV loop served to obtain predictions. Hyperparameter tuning is performed for every iteration in the outer loop through an additional CV inner loop. Optimal hyperparameters are found through grid search, using the following possible values for hyperparameters: learning rate: {0.1, 0.5, 1}, number of iterations: {250, 750}, L2 regularization of coefficients: {0.0001, 0.001}. Quality of every hyperparameter configuration was determined using the concordance±1 index on every held-out sample of the inner CV. After every inner CV loop, a final model was re-trained using the optimal hyperparameters on the full validation and training set. This final model was used to make predictions for the sample left out of the outer loop. For the Raman spectroscopy dataset, an exception was made to this procedure because of the size of the dataset (150 replicates per sample). Whereas other data sources used leave-one-(group)-out CV both in the inner and outer CV loops, for Raman spectroscopy, the inner (tuning) CV loop used 5-fold CV. In total, 347 760 machine learning models were trained to produce the main results of the study shown in Fig. 3. This number is derived as follows: for both MIC and BPC, separate models were trained for each data type. To produce performance estimates for each of these, nested CV was performed, consisting of iteratively setting one of 46 samples aside for evaluation, and using the other 45 for training. For each of these 46 iterations, an inner CV loop was performed using the same procedure to search for optimal hyperparameters (out of 12 possible different combinations). In total, these numbers add up to: 2 (MIC or BPC) * 7 (data obtained with different analytical approaches) * 46 (outer CV loop) * 45 (inner CV loop) * 12 (different hyperparameter configurations) (equals 347 760).
Model evaluation
Model prediction quality was evaluated using the outer loop of a nested leave-one-out CV. To evaluate predictions for data sources with differing number of technical replicates on equal footing, predictions for technical replicates were collated to a single sample-level prediction. To obtain predicted probabilities at the sample level, a prediction was made for every technical replicate, and their probabilities for each ordinal category were averaged across replicates. All model evaluations occurred on these sample-level predictions. Model prediction quality was assessed through the accuracy score, the accuracy±1 score, the concordance index, and the concordance±1 index. The accuracy score indicates how often the MIC/BPC category with the highest predicted probability was the correct one. The accuracy±1 score indicates how often a predicted MIC/BPC belonged to the correct ordinal category or one category higher or lower (i.e. the correct MIC/BPC or within one 2-fold dilution step, which is the accepted variability of phenotypic AST)2,44,72. The concordance index evaluates the overall ranking quality of predictions. It evaluates every pair of samples with different MIC/BPC values and counts the proportion of pairs for which the higher MIC/BPC value also had a higher predicted MIC/BPC value. Because of its pairwise evaluation, it is numerically more stable in small sample sizes than the accuracy score. The concordance±1 extends this metric by only evaluating pairs of samples with MIC/BPC values differing by at least two 2-fold dilution steps. Here, the variant of the concordance index was applied that does not count ties as ‘half-correct’73. To test how much information the machine learning models learned from data, performances are compared to ‘random’ predictions. Here, ‘random’ performance consists of the score obtained when every prediction would be the MIC or BPC value that occurred most often in the training data set. This score was computed for both the accuracy and accuracy±1. For the concordance and concordance±1 indices, this score always corresponds to 0.5.
External validation of models with clinical isolates
To externally validate the trained models, data obtained with 30 CF-derived P. aeruginosa isolates were used. The same training and tuning procedures as described above were used, the only difference being that in this case an external test set was used. Because of this, it was not necessary to perform nested cross validation to obtain unbiased estimates of model performance. Instead, only the previously described inner cross-validation loop was used to obtain optimal hyperparameters. To establish random predictions for the clinical isolate test set, the same procedure as previously described was used, i.e., ‘random’ predictions are taken as the most frequently occurring class in the training data set (consisting of evolved strains).
Combining data sources through stacking
To combine information from various data sources, stacking models were used. In stacking models, predictions from previous models are used as input for a second model. Per sample, to construct a single feature for every data source, the weighted sums of predicted probabilities of their previously trained models were computed:
where \(j\) denotes one of the four used data sources. After data set construction, all stacking models were trained and tuned identically as previously described (i.e., using nested leave-one-(group)-out CV). As all previously trained models used leave-one-(group)-out CV as their splitting strategy, no specialized strategies were necessary to prevent leakage of information from training to evaluation sets.
Data availability
All data necessary for supporting the findings of this study are enclosed in this manuscript. The raw sequencing reads generated in this study are available in the ArrayExpress database under the accession number E-MTAB-11894. The reference genomes used during sequencing analysis can be found in the GenBank database with the accession numbers NZ_CP051547.1 (AA2), NC_011770.1 (LES B58), NC_023066.1 (LES 431), NZ_CP034244.1 (UCBPP-PA14) and MCMX01000001 (IST27). All preprocessed data are available at the following GitHub repository: https://github.com/gdewael/biofilm-amr.
Code availability
Code used in this study is available at https://github.com/gdewael/biofilm-amr.
References
Leclercq, R. et al. EUCAST expert rules in antimicrobial susceptibility testing. Clin. Microbiol Infect. 19, 141–160 (2013).
Jorgensen, J. H. & Ferraro, M. J. Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin. Infect. Dis. 49, 1749–1755 (2009).
Hasselmann, C. & Microbiology, E. S. C Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin. Microbiol. Infect. 9, 9–15 (2003).
Matuschek, E., Brown, D. F. & Kahlmeter, G. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol Infect. 20, O255–O266 (2014).
Bjarnsholt, T. et al. The in vivo biofilm. Trends Microbiol 21, 466–474 (2013).
Ciofu, O. & Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents-How P. aeruginosa Can Escape Antibiotics. Front Microbiol 10, 913 (2019).
Ciofu, O., Moser, C., Jensen, P. O. & Hoiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol 20, 621–635 (2022).
Macia, M. D., Rojo-Molinero, E. & Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol Infect. 20, 981–990 (2014).
Thoming, J. G. & Haussler, S. Pseudomonas aeruginosa Is More Tolerant Under Biofilm Than Under Planktonic Growth Conditions: A Multi-Isolate Survey. Front Cell Infect. Microbiol 12, 851784 (2022).
De Bleeckere, A. et al. High throughput determination of the biofilm prevention concentration for Pseudomonas aeruginosa biofilms using a synthetic cystic fibrosis sputum medium. Biofilm 5, 100106 (2023).
Ceri, H. et al. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol 37, 1771–1776 (1999).
LiPuma, J. J. The Sense and Nonsense of Antimicrobial Susceptibility Testing in Cystic Fibrosis. J. Pediatr. Infect. Dis. Soc. 11, S46–S52 (2022).
Coenye, T. Biofilm antimicrobial susceptibility testing: where are we and where could we be going?. Clin. Microbiol Rev. 36, e0002423 (2023).
Turner, K. H., Wessel, A. K., Palmer, G. C., Murray, J. L. & Whiteley, M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl Acad. Sci. USA 112, 4110–4115 (2015).
Bove, M., Bao, X., Sass, A., Crabbe, A. & Coenye, T. The Quorum-Sensing Inhibitor Furanone C-30 Rapidly Loses Its Tobramycin-Potentiating Activity against Pseudomonas aeruginosa Biofilms during Experimental Evolution. Antimicrob. Agents Chemother. 65, e0041321 (2021).
Palmer, K. L., Aye, L. M. & Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189, 8079–8087 (2007).
Bao, X., Bove, M. & Coenye, T. Organic Acids and Their Salts Potentiate the Activity of Selected Antibiotics against Pseudomonas aeruginosa Biofilms Grown in a Synthetic Cystic Fibrosis Sputum Medium. Antimicrob. Agents Chemother. 66, e0187521 (2022).
Cai, Y. M. et al. Echinacoside reduces intracellular c-di-GMP levels and potentiates tobramycin activity against Pseudomonas aeruginosa biofilm aggregates. NPJ Biofilms Microbiomes 11, 40 (2025).
Darch, S. E. et al. Phage Inhibit Pathogen Dissemination by Targeting Bacterial Migrants in a Chronic Infection Model. mBio. 8. https://doi.org/10.1128/mBio.00240-17. (2017)
Duncan, R. P. et al. RNA-seq reproducibility of Pseudomonas aeruginosa in laboratory models of cystic fibrosis. Microbiol Spectr. 13, e0151324 (2025).
Khaledi, A. et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol. Med. 12, e10264 (2020).
Cortes-Lara, S., Barrio-Tofino, E. D., Lopez-Causape, C., Oliver, A. & Group, G.-S. R. P. S Predicting Pseudomonas aeruginosa susceptibility phenotypes from whole genome sequence resistome analysis. Clin. Microbiol Infect. 27, 1631–1637 (2021).
Ellington, M. J. et al. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee. Clin. Microbiol Infect. 23, 2–22 (2017).
Idelevich, E. A. & Becker, K. Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry for Antimicrobial Susceptibility Testing. J. Clin. Microbiol 59, e0181419 (2021).
Florio, W. et al. Detection of Antibiotic-Resistance by MALDI-TOF Mass Spectrometry: An Expanding Area. Front Cell Infect. Microbiol 10, 572909 (2020).
Yoon, E. J. & Jeong, S. H. MALDI-TOF Mass Spectrometry Technology as a Tool for the Rapid Diagnosis of Antimicrobial Resistance in Bacteria. Antibiotics (Basel) 10, 982 (2021).
Weis, C. et al. Direct antimicrobial resistance prediction from clinical MALDI-TOF mass spectra using machine learning. Nat. Med 28, 164–174 (2022).
Crabbe, A., Jensen, P. O., Bjarnsholt, T. & Coenye, T. Antimicrobial Tolerance and Metabolic Adaptations in Microbial Biofilms. Trends Microbiol 27, 850–863 (2019).
Tellapragada, C. et al. Isothermal microcalorimetry minimal inhibitory concentration testing in extensively drug resistant Gram-negative bacilli: a multicentre study. Clin. Microbiol Infect. 26, 1413 e1411–1413.e1417 (2020).
Butini, M. E. et al. Real-Time Antimicrobial Susceptibility Assay of Planktonic and Biofilm Bacteria by Isothermal Microcalorimetry. Adv. Exp. Med Biol. 1214, 61–77 (2019).
Sultan, A. R. et al. Real time monitoring of Staphylococcus aureus biofilm sensitivity towards antibiotics with isothermal microcalorimetry. PLoS One 17, e0260272 (2022).
Pezzotti, G. Raman spectroscopy in cell biology and microbiology. J. Raman Spectrosc. 52, 2348–2443 (2021).
Lister, A. P. et al. Multi-Excitation Raman Spectroscopy for Label-Free, Strain-Level Characterization of Bacterial Pathogens in Artificial Sputum Media. Anal. Chem. 94, 669–677 (2022).
Lister, A. et al. Multi-excitation Raman Spectroscopy Complements Whole Genome Sequencing for Rapid Detection of Bacterial Infection and Resistance in WHO Priority Pathogens (Cold Spring Harbor Laboratory, 2022).
Lu, W. et al. Identification of pathogens and detection of antibiotic susceptibility at single-cell resolution by Raman spectroscopy combined with machine learning. Front Microbiol 13, 1076965 (2022).
Highmore, C. et al. Identification and antimicrobial resistance profiling of Pseudomonas aeruginosa using multi-excitation Raman spectroscopy and computational analytics. npj Antimicrob. Resist. 3. https://doi.org/10.1038/s44259-025-00141-z (2025).
Smith, S. & Rowbotham, N. J. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 11, CD001021 (2022).
Taccetti, G. et al. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics (Basel) 10. https://doi.org/10.3390/antibiotics10030338 (2021).
Rainio, O., Teuho, J. & Klen, R. Evaluation metrics and statistical tests for machine learning. Sci. Rep. 14, 6086 (2024).
Eyre, D. W. et al. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. J. Antimicrob. Chemother. 72, 1937–1947 (2017).
Coll, F. et al. Antibiotic resistance determination using Enterococcus faecium whole-genome sequences: a diagnostic accuracy study using genotypic and phenotypic data. Lancet Microbe 5, e151–e163 (2024).
Pesesky, M. W. et al. Evaluation of Machine Learning and Rules-Based Approaches for Predicting Antimicrobial Resistance Profiles in Gram-negative Bacilli from Whole Genome Sequence Data. Front Microbiol 7, 1887 (2016).
Bradley, P. et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 6, 10063 (2015).
Nguyen, M. et al. Using Machine Learning To Predict Antimicrobial MICs and Associated Genomic Features for Nontyphoidal Salmonella. J. Clin. Microbiol. 57, https://doi.org/10.1128/JCM.01260-18 (2019).
Drouin, A. et al. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genomics 17, 754 (2016).
Ayoola, M. B. et al. Predicting Salmonella MIC and Deciphering Genomic Determinants of Antibiotic Resistance and Susceptibility. Microorganisms 12, 134 (2024).
Nguyen, H. A. et al. Predicting Pseudomonas aeruginosa drug resistance using artificial intelligence and clinical MALDI-TOF mass spectra. mSystems 9, e0078924 (2024).
Ren, M., Chen, Q. & Zhang, J. Repurposing MALDI-TOF MS for effective antibiotic resistance screening in Staphylococcus epidermidis using machine learning. Sci. Rep. 14, 24139 (2024).
Lin, T. H. et al. Innovative strategies against superbugs: Developing an AI-CDSS for precise Stenotrophomonas maltophilia treatment. J. Glob. Antimicrob. Resist 38, 173–180 (2024).
Christensen, M. H. et al. Antimicrobial susceptibility testing of bone and joint pathogens using isothermal microcalorimetry. APMIS 132, 814–823 (2024).
Braissant, O., Bachmann, A. & Bonkat, G. Microcalorimetric assays for measuring cell growth and metabolic activity: methodology and applications. Methods 76, 27–34 (2015).
Wolpert, D. H. Stacked Generalization. Neural Netw. 5, 241–259 (1992).
Breiman, L. Stacked regressions. Mach. Learn. 24, 49–64 (1996).
Brennan-Krohn, T., Smith, K. P. & Kirby, J. E. The Poisoned Well: Enhancing the Predictive Value of Antimicrobial Susceptibility Testing in the Era of Multidrug Resistance. J. Clin. Microbiol 55, 2304–2308 (2017).
Stokes, J. M., Lopatkin, A. J., Lobritz, M. A. & Collins, J. J. Bacterial Metabolism and Antibiotic Efficacy. Cell Metab. 30, 251–259 (2019).
Sultan, M., Arya, R. & Kim, K. K. Roles of Two-Component Systems in Pseudomonas aeruginosa Virulence. Int J. Mol. Sci. 22, 12152 (2021).
Lee, J. & Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 6, 26–41 (2015).
Park, S. & Sauer, K. 69-94 (Springer International Publishing, 2022).
Prickett, M. H. et al. Aminoglycoside resistance of Pseudomonas aeruginosa in cystic fibrosis results from convergent evolution in the mexZ gene. Thorax 72, 40–47 (2017).
Karlowsky, J. A., Hoban, D. J., Zelenitsky, S. A. & Zhanel, G. G. Altered denA and anr gene expression in aminoglycoside adaptive resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 40, 371–376 (1997).
Pletzer, D. et al. The Pseudomonas aeruginosa PA14 ABC Transporter NppA1A2BCD Is Required for Uptake of Peptidyl Nucleoside Antibiotics. J. Bacteriol. 197, 2217–2228 (2015).
An, S., Wu, J. & Zhang, L. H. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-Di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl Environ. Microbiol 76, 8160–8173 (2010).
Fan, K., Cao, Q. & Lan, L. Genome-Wide Mapping Reveals Complex Regulatory Activities of BfmR in Pseudomonas aeruginosa. Microorganisms 9, 485 (2021).
Moskowitz, S. M., Ernst, R. K. & Miller, S. I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 186, 575–579 (2004).
Bolard, A. et al. Production of Norspermidine Contributes to Aminoglycoside Resistance in pmrAB Mutants of Pseudomonas aeruginosa. Antimicrob Agents Chemother 63, https://doi.org/10.1128/AAC.01044-19 (2019).
De Soyza, A. et al. Developing an international Pseudomonas aeruginosa reference panel. Microbiologyopen 2, 1010–1023 (2013).
Aristoteli, L. P. & Willcox, M. D. Mucin degradation mechanisms by distinct Pseudomonas aeruginosa isolates in vitro. Infect. Immun. 71, 5565–5575 (2003).
Rickert, C. A., Lutz, T. M., Marczynski, M. & Lieleg, O. Several Sterilization Strategies Maintain the Functionality of Mucin Glycoproteins. Macromol. Biosci. 20, e2000090 (2020).
Weis, C. et al. Topological and kernel-based microbial phenotype prediction from MALDI-TOF mass spectra. Bioinformatics 36, i30–i38 (2020).
Trevisan, J., Angelov, P. P., Scott, A. D., Carmichael, P. L. & Martin, F. L. IRootLab: a free and open-source MATLAB toolbox for vibrational biospectroscopy data analysis. Bioinformatics 29, 1095–1097 (2013).
Pedregosa, F., Bach, F. & Gramfort, A. On the Consistency of Ordinal Regression Methods. J. Mach. Learn. Res. 18, 1–35 (2017).
Jorgensen, J. H. Selection criteria for an antimicrobial susceptibility testing system. J. Clin. Microbiol 31, 2841–2844 (1993).
Yan, G. & Greene, T. Investigating the effects of ties on measures of concordance. Stat. Med 27, 4190–4206 (2008).
Acknowledgements
Part of this work was funded by the Ghent University Special Research Fund (grant BOF20/GOA/002). W.W. received funding from the Flemish Government under the “Flanders AI research program”. We thank Dr. Daniel Pletzer (University of Otago, New Zealand) for providing the PA14 ΔnppBCD knockout mutant.
Author information
Authors and Affiliations
Contributions
T.C. developed the conceptual framework, supervised the project, and contributed to the manuscript. F.V. conducted the experiments, analysed the data, and wrote the manuscript with input from all authors. W.W. and G.D.W. managed the machine learning aspects of the study, with G.D.W. also contributing to the manuscript. F.V.N. made the whole-genome sequencing analysis possible. A.S. analysed the sequencing data and helped with the experimental evolution study. T.B. and M.L. provided expertise and assistance with the isothermal microcalorimetry analysis. J.S.W., S.M., C.H., N.H. and Y.C. contributed to the Raman spectroscopy analysis, while P.V. and M.C. assisted in the MALDI-TOF measurements.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vergauwe, F., De Waele, G., Sass, A. et al. Harnessing machine learning to predict antibiotic susceptibility in Pseudomonas aeruginosa biofilms. npj Biofilms Microbiomes 11, 205 (2025). https://doi.org/10.1038/s41522-025-00833-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00833-4
