
Abstract
Colorectal cancer (CRC) is a leading cause of cancer mortality worldwide and is increasingly recognized as the outcome of complex host–microbe interactions. Beyond established genetic and environmental drivers, the gut microbiome has emerged as a causal and mechanistic contributor to CRC initiation, progression, and therapy response. This review synthesizes current molecular, ecological, and translational evidence to explain how gut microbial communities reprogram immune, metabolic, neural, and endocrine networks within the tumor microenvironment. CRC-associated dysbiosis is characterized by enrichment of pathobionts such as Fusobacterium nucleatum, pks⁺ Escherichia coli, and enterotoxigenic Bacteroides fragilis, and by loss of protective, short-chain-fatty-acid-producing commensals. These microbes promote carcinogenesis through genotoxin-induced DNA damage, epithelial barrier disruption, metabolic rewiring, and chronic inflammation that collectively sustain immune suppression and tumor growth. Defined mutational signatures from bacterial metabolites, including colibactin, cytolethal distending toxin, and indolimines, now directly link microbial exposures to human cancer genomes. By integrating these findings, this review conceptualizes CRC as a biofilm-structured, microbiome-driven ecosystem disease, where polymicrobial consortia coordinate barrier breakdown, immune evasion, and metabolic cooperation. Finally, we highlight emerging microbiota-targeted strategies, including dietary modulation, pre- and probiotics, postbiotics, bacteriophage therapy, engineered live biotherapeutics, and fecal microbiota transplantation, that translate these insights into precision prevention and therapy. Through this integrative framework, the review aims to reposition the microbiome from a correlative feature to a tractable determinant of CRC pathogenesis and treatment response.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is not only a major global health burden but also a disease increasingly understood as the product of complex host–microbe interactions. It ranks as the third most common malignancy and the second leading cause of cancer-related mortality, with an increasing incidence in early-onset cases, particularly in industrialized nations and transitioning economies1. Early-life microbial colonization critically shapes immune development and long-term disease risk. Birth mode, breastfeeding, maternal microbiota, and early antibiotics influence microbial diversity during a sensitive period, where disruptions can lead to persistent dysbiosis, impaired tolerance, and higher susceptibility to inflammatory and neoplastic diseases later in life2,3,4.
The human gastrointestinal tract harbors a complex microbial ecosystem exceeding 3.8 × 10¹³ microorganisms, predominantly belonging to the phyla Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, Fusobacteriota, and Verrucomicrobiota, which collectively maintain intestinal homeostasis, nutrient metabolism, and immune regulation5. Disruption of this balance, or dysbiosis, has been associated with shifts toward pathogenic or pro-inflammatory taxa, leading to chronic inflammation, epithelial barrier dysfunction, and exposure of host tissues to microbial toxins and metabolites with oncogenic potential6,7,8,9,10. In line with emerging conceptual frameworks, the microbiome has been proposed as a hallmark of cancer, underscoring its central role in shaping key tumor-enabling processes such as inflammation, immune evasion, metabolic rewiring, and genomic instability11,12. This conceptual shift repositions the microbiome from a peripheral risk factor to a core component of tumor biology, integrated within the evolving hallmarks of the cancer paradigm.
While strong associations between specific microbial taxa and CRC have been extensively documented, establishing a direct causal relationship remains a major challenge in microbiome-cancer research. Classic Koch’s postulates provide a conceptual foundation for defining causative pathogens; however, their direct application to the complex, polymicrobial structure of the gut microbiota is inherently limited13,14. To address this, modern adaptations of these criteria increasingly rely on integrating multi-omics datasets, microbial gene knockouts, and gnotobiotic or humanized murine models to move from correlation toward causality. A paradigmatic example is pks⁺ Escherichia coli, for which well-defined mutational signatures attributable to colibactin exposure have been identified in human CRC genomes and reproduced in preclinical models8. Likewise, enterotoxigenic Bacteroides fragilis and polymicrobial biofilms have demonstrated tumor-promoting effects in longitudinal murine studies, providing functional evidence of microbe-driven oncogenesis15,16,17,18,19,20. Collectively, these findings underscore that the mere presence or absence of a microorganism is insufficient to drive disease and highlight the need for rigorous mechanistic, longitudinal, and functional studies to establish robust causal inference in CRC.
CRC-associated microbiota alterations are consistently characterized by the enrichment of Fusobacterium nucleatum, pks⁺ E. coli, enterotoxigenic Bacteroides fragilis (ETBF), Streptococcus gallolyticus, Enterococcus faecalis, and specific Porphyromonas spp., alongside a decrease in beneficial butyrate-producing genera like Faecalibacterium prausnitzii and Roseburia21,22,23,24,25,26,27,28. These microbial shifts are not merely correlative but have been shown to influence tumorigenesis through multiple, often interlinked mechanisms. Pathogens such as F. nucleatum promote tumor growth via adhesins (FadA, Fap2) that activate β-catenin signaling, modulate E-cadherin-mediated adhesion, and suppress anti-tumor immune responses29,30. ETBF secretes B. fragilis toxin (BFT), which triggers E-cadherin cleavage, activates NF-κB, and drives pro-inflammatory cytokine release29,31. Pks⁺ E. coli produces the genotoxin colibactin, inducing double-strand DNA breaks, chromosomal instability, and mutational signatures found in human CRC32,33,34. Other microbial metabolites, including secondary bile acids, polyamines, and hydrogen sulfide, can promote proliferation, inhibit apoptosis, and contribute to a mutagenic microenvironment35.
A particularly relevant mechanistic axis involves mitochondrial dysfunction as a mediator between microbial genotoxicity and CRC progression. Microbial toxins such as colibactin and cytolethal distending toxin (CDT) may not only damage nuclear DNA but also induce mutations in mitochondrial DNA (mtDNA), compromising oxidative phosphorylation and enhancing reactive oxygen species (ROS) production—conditions that promote genomic instability and malignant transformation36,37,38. This mitochondrial-genotoxicity axis represents a novel mechanistic bridge between microbial exposure and the metabolic reprogramming characteristic of tumor cells.
The tumor microenvironment (TME) is both shaped by and responsive to microbiome alterations. Certain bacteria modulate immune infiltration, promoting tumor tolerance by inducing regulatory T cells, myeloid-derived suppressor cells, or by inhibiting cytotoxic lymphocyte activity31,39. For example, F. nucleatum can drive immune evasion via Fap2–TIGIT interactions, suppressing natural killer (NK) cell function, while microbial metabolites such as formate (produced by F. nucleatum) activate AhR signaling, enhance cancer stemness, and promote invasive phenotypes30,40. Additionally, microbial communities influence the balance between tumor-promoting inflammation and anti-tumor immunity, as observed with polyamine-rich environments that fuel epithelial proliferation versus short-chain fatty acids (SCFAs) like butyrate, which have anti-inflammatory and anti-neoplastic effects35,41.
Importantly, microbial metabolites exhibit context-dependent duality. SCFA-producing taxa such as Lactobacillus, Bifidobacterium, and Faecalibacterium reinforce epithelial integrity and immune tolerance, inducing apoptosis in cancer cells35,42. However, even metabolites traditionally considered beneficial, such as butyrate, can exhibit dual roles depending on context; for instance, overgrowth of specific Porphyromonas spp. in CRC patients has been shown to induce cellular senescence and pro-inflammatory responses via excessive butyrate secretion, accelerating tumorigenesis in murine models21,43. This context-dependent effect underscores the complexity of host-microbe metabolic interactions in cancer biology.
Mechanistically, the interplay between microbiota and CRC converges on canonical oncogenic pathways, including Wnt/β-catenin, NF-κB, STAT3, and MAPK signaling cascades29,31. Dysbiotic microbiota can also influence epigenetic landscapes by altering DNA methylation, histone modifications, and non-coding RNA profiles, further modulating gene expression in ways that favor tumor initiation and progression. Moreover, recent experimental studies have provided compelling mechanistic evidence that polymicrobial biofilms act as functional tumor-promoting units within the colonic mucosa. These structured microbial communities induce epithelial IL-6/STAT3 activation, disrupt E-cadherin, and enhance proliferation17, while gnotobiotic models show that biofilms formed by ETBF, E. coli, and F. nucleatum accelerate tumorigenesis in ApcMin/+ mice16. Imaging and metagenomic analyses further reveal spatial metabolic zonation, oxygen gradients, and cooperative virulence strategies among biofilm-associated taxa18,19,20, collectively establishing biofilms as dynamic, inflammation-sustaining ecosystems that drive CRC progression.
From an ecological perspective, microbial genotoxins not only damage host DNA but also shape the gut microbial community through interbacterial antagonism, altering competition dynamics and potentially selecting for more virulent or inflammation-adapted strains33. This dynamic ecosystem-level interaction suggests that CRC-associated microbiomes may evolve toward a stable yet pathogenic state that perpetuates tumor-promoting conditions. Furthermore, oral pathogens such as Fusobacterium, Porphyromonas, and Prevotella, often linked to periodontal disease, have been identified within colorectal tumors, supporting the concept of an oral-gut axis in CRC pathogenesis22,34,44. Such translocation events may be facilitated by systemic inflammation, immune dysregulation, or microbiota-mediated breakdown of mucosal barriers.
Finally, multi-omics approaches integrating metagenomics, metatranscriptomics, metabolomics, and host genomic analyses are refining our understanding of the microbiome-CRC interface. These approaches are revealing disease-specific microbial signatures with potential diagnostic and prognostic utility30,39. Several bacterial taxa and their metabolic products are being explored as non-invasive biomarkers for early CRC detection, complementing current screening modalities such as colonoscopy and fecal occult blood testing39,42. Collectively, the gut microbiome emerges as an active driver of colorectal carcinogenesis, modulating genetic, epigenetic, metabolic, and immunological pathways throughout the adenoma–carcinoma sequence. This review outlines a conceptual roadmap integrating microbial signatures, mechanistic insights, and translational strategies to define the microbiome’s role in CRC pathogenesis.
Genotoxin-producing bacteria and host mutational signatures
To move from association to causation, this section outlines microbial genotoxins that leave recognizable imprints in human CRC genomes, linking specific taxa to defined DNA lesions and mutational signatures. We first contextualize how colibactin, BFT, CDT, typhoid toxin, and emerging indolimines converge on replication stress, double-strand breaks (DSBs), and repair pathway rewiring, then summarize complementary small-molecule metabolites that broaden this mutagenic spectrum.
Colibactin
Colibactin, a hybrid nonribosomal peptide–polyketide genotoxin encoded by the pks genomic island of certain Escherichia coli strains, was first identified by Nougayrède et al. as a bacterial factor capable of inducing DNA DSBs in mammalian cells45. Since then, it has emerged as a potent microbial driver of colorectal carcinogenesis through its capacity to induce defined mutational signatures in host cells21,46. The pks island encompasses a suite of clb genes (clbA–clbS) orchestrating a modular biosynthetic pathway that yields a structurally unstable, cyclopropane-containing metabolite capable of alkylating DNA and generating interstrand crosslinks47,48. These lesions, particularly when unrepaired, precipitate DSBs and promote single-base substitution (SBS)-pks and small insertion/deletion (ID-pks) mutational patterns, now recognized as molecular hallmarks of colibactin exposure in human colorectal tumors48,49. Structural instability of the molecule has hindered direct purification; however, chemical biology approaches and synthetic analogs have elucidated the cyclopropane warhead’s reactivity toward adenine-rich sequences in duplex DNA, providing a mechanistic link between microbial metabolite chemistry and carcinogenic mutagenesis50,51.
Epidemiologically, pks⁺ E. coli strains are enriched in CRC patients compared with healthy controls, with meta-analyses indicating a statistically significant elevation in CRC risk among colonized individuals52,53,54. Spatial mapping of tumor-associated microbiota reveals a predilection of colibactin-producing E. coli for distal colon and rectal tumors, aligning with dietary and lifestyle factors, such as Western dietary patterns, that foster expansion of these strains53,55,56. Preclinical murine models corroborate these associations: monoassociation or co-colonization of pks⁺ E. coli with ETBF accelerates adenoma–carcinoma progression, particularly in the context of concurrent inflammatory stimuli or APC loss15. In vitro, exposure of human epithelial and fibroblast cells to colibactin-producing bacteria triggers senescence, chromosomal aberrations, and activation of DNA damage response (DDR) pathways, including ATM/ATR signaling, yet fails to elicit robust p53-mediated apoptosis, favoring survival of genomically unstable clones47,48.
At the genome level, Dziubańska-Kusibab et al. first defined a distinct colibactin-associated mutational signature characterized by SBS-pks and ID-pks patterns57, while Pleguezuelos-Manzano et al. demonstrated causality by exposing human intestinal organoids to pks⁺ E. coli, reproducing the same mutational imprint observed in human colorectal cancers8. Consequently, mutational signature analyses of human CRC specimens have identified recurrent SBSs at AT-rich motifs and crosslink-associated rearrangements attributable to colibactin, thereby positioning this genotoxin as one of the few bacterially derived agents with a defined imprint in human cancer genomes49,51. These insights extend beyond etiologic attribution, offering potential diagnostic leverage: colibactin-specific mutational signatures could stratify tumors by microbial exposure history, informing surveillance strategies in high-risk populations21,55. Moreover, recent findings suggest that colibactin tolerance in tumor cells is not static; chronic exposure selects for clones with restored homologous recombination (HR) proficiency, conferring resistance to DNA-damaging agents such as irinotecan and its active metabolite SN3853. This cross-resistance underscores a translational paradox—while colibactin may initiate tumorigenesis through genotoxic stress, persistent exposure can drive therapeutic refractoriness by enriching DNA repair–competent subpopulations.
Mechanistically, this HR-mediated tolerance likely reflects adaptive rewiring of DNA repair networks under chronic microbial genotoxic pressure, akin to chemoresistance evolution in response to platinum drugs46,53. Such adaptive dynamics expand the oncogenic role of colibactin from a mutagenic initiator to a microbe-derived selective force sculpting tumor evolutionary trajectory. The interplay between colibactin and host DNA repair proficiency also raises the possibility of synthetic lethality–based interventions: inhibiting HR in pks⁺ tumors could resensitize cells to both microbial and therapeutic DNA-damaging agents52,53.
From a host–microbiome interaction standpoint, colibactin’s influence is not confined to local mutagenesis. DNA damage–induced senescence and associated secretory phenotypes may remodel the tumor microenvironment, fostering pro-inflammatory, immunosuppressive niches that synergize with microbial community shifts to accelerate malignant progression48,52. Additionally, genotoxin-induced epithelial barrier disruption can enhance systemic dissemination of bacterial metabolites and microvesicles, potentially priming distant tissues for metastasis21,50,58. These systemic sequelae position colibactin as a multifaceted contributor to both the initiation and propagation of CRC.
Therapeutically, the identification of colibactin biosynthetic genes and regulatory elements presents opportunities for targeted microbiome modulation. Strategies under investigation include bacteriophage-mediated depletion of pks⁺ strains, competitive exclusion using engineered commensals, and small-molecule inhibitors of key biosynthetic enzymes such as ClbP peptidase47,55. Such interventions could attenuate genotoxic exposure in at-risk individuals or mitigate tumor evolution in established disease. Furthermore, dietary and probiotic interventions aimed at shifting gut microbial composition away from pks⁺ populations offer preventive potential, particularly in genetically predisposed cohorts48,52.
Collectively, the evidence positions colibactin as a paradigm of microbe–host co-carcinogenesis, distinguished by its chemical specificity, defined genomic imprint, and dual role in both tumor initiation and therapeutic resistance. Its study exemplifies the broader principle that bacterial secondary metabolites can act as direct modulators of cancer genome evolution, bridging molecular microbiology with oncology and opening translational avenues for microbiota-targeted prevention and therapy.
Bacteroides fragilis toxin (BFT)
BFT, produced by ETBF, is a zinc-dependent metalloprotease that plays a central role in colorectal carcinogenesis through multifaceted effects on the epithelium, immune system, and tumor microenvironment. Mechanistically, BFT cleaves the extracellular domain of E-cadherin, disrupting adherens junction integrity and releasing β-catenin into the cytoplasm, thereby enabling its nuclear translocation and transcriptional activation of genes linked to proliferation, inflammation, and survival59,60,61. This initial epithelial barrier perturbation not only primes the mucosa for inflammatory infiltration but also fosters conditions conducive to oncogenic transformation. In human and murine models, BFT-mediated E-cadherin degradation has been shown to act as a trigger for pro-inflammatory chemokine production, notably IL-8, through both β-catenin–dependent and Stat3-driven transcriptional programs, with Stat3 activation emerging as a critical mediator in CRC cells6,9,61,62.
A seminal study by Wu et al. provided the first in vivo evidence that ETBF, via BFT, drives colonic tumorigenesis in ApcMin/+ mice through an IL-17–dependent mechanism. In this model, ETBF colonization selectively induces distal colon tumors by activating an IL-17–Stat3–NF-κB inflammatory axis that bridges epithelial and immune compartments25. A defining feature of ETBF-induced tumorigenesis is the IL-17–Stat3–NF-κB signaling relay between colonic epithelial cells (CECs) and mucosal immune populations. Following BFT-induced epithelial injury, IL-17–producing T helper cells amplify NF-κB activity in CECs, leading to the transcriptional upregulation of C-X-C chemokines such as CXCL1, which drive the recruitment of CXCR2+ polymorphonuclear immature myeloid cells to tumor-prone regions of the distal colon9,63. These infiltrating myeloid cells acquire an immunosuppressive, pro-tumorigenic phenotype, largely monocytic myeloid-derived suppressor cells (MO-MDSCs), under the combined influence of BFT-induced epithelial stress signals and IL-17–driven cytokines63. This myeloid signature not only supports tumor initiation but also sustains a chronic inflammatory microenvironment that accelerates progression.
Beyond immune modulation, BFT orchestrates oncogenic processes through microRNA regulation. ETBF infection downregulates miR-149-3p via METTL14-mediated N6-methyladenosine modification, impairing its maturation and facilitating the derepression of targets such as PHF5A, which in turn modulates alternative splicing of KAT2A to activate SOD2 transcription. This axis promotes oxidative stress adaptation and enhances the survival of transformed cells. Importantly, miR-149-3p is also packaged into exosomes, enabling paracrine modulation of T helper 17 (Th17) cell differentiation, thereby reinforcing the inflammatory circuitry initiated by BFT64. The oncogenic relevance of miR-149-3p suppression is further substantiated by its regulation through the lncRNA LRP11-AS1, which acts as a molecular sponge to sequester miR-149-3p, leading to upregulation of CDK4 and cell cycle progression65. This reveals a broader paradigm in which BFT-induced noncoding RNA networks contribute to sustained proliferative signaling and immune dysregulation in colorectal carcinogenesis.
Epidemiological and clinical studies underscore the strong association between ETBF carriage and CRC risk. Prevalence rates of ETBF, and specifically the bft gene, are significantly higher in CRC patients compared with healthy individuals, with bft-2 frequently enriched in advanced-stage tumors59. Tissue-based analyses have revealed mucosal colonization rates approaching 85% in CRC patients, highlighting ETBF as not merely a bystander but a potential driver of oncogenesis60. Moreover, ETBF presence correlates with distinct transcriptional and immunological profiles in tumor tissues, including heightened pro-inflammatory gene expression and altered immune cell composition63,64.
Recent work has expanded the functional spectrum of B. fragilis beyond tumor initiation to include modulation of therapeutic response. B. fragilis, including BFT-producing strains, has been implicated in chemoresistance to agents such as 5-fluorouracil and oxaliplatin. This effect is mediated by bacterial surface proteins, such as SusD/RagB, which bind to Notch1 on tumor cells, activating EMT and stemness programs that blunt chemotherapy-induced apoptosis. Strikingly, targeted elimination of B. fragilis using a bacteriophage (VA7) restores chemosensitivity in preclinical CRC models, positioning BFT-producing B. fragilis as both a therapeutic target and a biomarker for treatment stratification66.
The pathogenic potential of BFT is further amplified in polymicrobial contexts. Synergistic interactions between ETBF and other toxin-producing bacteria, notably pks⁺ Escherichia coli, have been documented within colonic biofilms, particularly in familial adenomatous polyposis and sporadic CRC. These co-colonizations enable parallel genotoxic and pro-inflammatory insults, with colibactin from pks⁺ E. coli inducing direct DNA DSBs, while BFT from ETBF disrupts epithelial integrity and drives immune dysregulation15. Such microbial networking exemplifies how BFT acts within a consortium of carcinogenic microbes to potentiate disease.
Importantly, the pathogenicity of B. fragilis is strain-specific, as non-toxigenic B. fragilis (NTBF) lacks BFT but may still influence early neoplasia through other pro-inflammatory mechanisms, including lipopolysaccharide-mediated TLR4 activation67. However, the distinct oncogenic footprint of ETBF is characterized by its ability to integrate epithelial barrier disruption, chronic Th17 inflammation, myeloid cell recruitment, and transcriptional reprogramming into a coherent pro-carcinogenic axis.
Collectively, the evidence positions BFT as a multifactorial oncotoxin whose effects span from epithelial perturbation to systemic immune modulation and therapy resistance. Its carcinogenic influence is mediated through: (1) cleavage of E-cadherin and β-catenin activation; (2) IL-17–Stat3–NF-κB inflammatory loops first defined in ETBF-colonized ApcMin/+ mice25; (3) recruitment and polarization of MDSCs; (4) suppression of tumor-suppressive miRNAs and activation of oncogenic lncRNA circuits; (5) promotion of oxidative stress adaptation; and (6) potentiation of microbial synergy in biofilms. These findings not only reinforce BFT as a compelling microbial target in CRC prevention but also suggest its utility as a biomarker for early detection, risk stratification, and therapeutic decision-making. Targeted interventions, ranging from microbiome modulation and vaccination to phage therapy, may hold promise in mitigating BFT-driven oncogenesis and overcoming associated treatment resistance9,65.
Cytolethal distending toxin
CDT, a tripartite AB-type genotoxin produced by diverse Gram-negative pathogens, such as Escherichia coli, Campylobacter jejuni, Aggregatibacter actinomycetemcomitans, Haemophilus ducreyi, and Helicobacter hepaticus, exerts profound genotoxic effects that directly link bacterial infection to carcinogenesis10,68 Structurally, CDT comprises two binding subunits, CdtA and CdtC, and a catalytic subunit, CdtB, the latter sharing strong structural homology with DNase I and functioning as the active nuclease responsible for DNA cleavage69,70 Upon receptor-mediated endocytosis, the toxin undergoes retrograde trafficking to the endoplasmic reticulum, enabling CdtB translocation into the host cell nucleus where it induces DNA lesions71. Historically, CDT was classified as a potent inducer of DNA DSBs based on γ-H2AX activation, ATM/ATR checkpoint signaling, and recruitment of repair factors such as the MRN complex72. However, recent mechanistic studies reveal that CDT predominantly generates single-strand breaks (SSBs) at sublethal doses, which are converted into replication-dependent DSBs during S-phase, thereby coupling genotoxicity to proliferative status10,73,74. This mode of damage inflicts chronic replication stress and triggers prolonged G2/M arrest, leading either to apoptosis or to survival with persistent genomic alterations that can fuel oncogenic transformation72,75.
The mutational imprints of CDT intoxication align with patterns of chromosomal instability and specific SBS signatures observed in gastrointestinal and hepatobiliary cancers, supporting its role in shaping host mutational landscapes10,70,76. In Helicobacter hepaticus-driven murine models, CDT is indispensable for the development of dysplastic nodules, as CdtB-deficient strains fail to initiate neoplastic lesions, confirming its necessity for bacterial oncogenicity71. In C. jejuni infection, CDT activity correlates with increased DNA damage markers in intestinal crypt cells and inflammatory-driven tumor promotion, implicating both direct genotoxic and indirect pro-inflammatory pathways68,69. Additionally, oral pathogens like A. actinomycetemcomitans exploit CDT to impair lymphocyte proliferation, thereby weakening immune surveillance and facilitating chronic infection–tumorigenesis cycles in head-and-neck malignancies72.
Cellular responses to CDT-induced damage involve complex engagement of DNA repair pathways. Both HR and non-homologous end joining (NHEJ) contribute to DSB resolution, while SSB repair (SSBR) plays a pivotal role in mitigating primary CDT lesions. Notably, the Fanconi anemia (FA) pathway is activated to stabilize stalled replication forks and resolve interstrand crosslinks arising from CDT-induced replication stress, indicating that CDT provokes a multifaceted DDR beyond classical nuclease-mediated DSBs72. Failure or saturation of these repair systems results in persistent DNA damage foci, micronuclei formation, and chromothripsis-like rearrangements, which can act as drivers of malignant progression70,75.
At the mutational signature level, CDT exposure generates a bias toward deletions at short repeats and clustered substitutions, potentially linked to aberrant repair of replication-associated breaks10,69. These genomic scars parallel patterns found in colorectal tumors enriched with CDT-producing E. coli and in hepatocellular carcinomas associated with H. hepaticus exposure10,70,71. Beyond direct genotoxicity, CDT exerts epigenetic influence; prolonged DDR activation leads to transcriptional reprogramming via p53-dependent and -independent pathways, altering cell cycle regulators, DNA repair gene expression, and pro-survival signaling72,73. Furthermore, CDT promotes a senescence-associated secretory phenotype (SASP) in surviving cells, characterized by chronic secretion of pro-inflammatory cytokines such as IL-6 and IL-8, which can reinforce a tumor-permissive microenvironment68,75.
The oncogenic potential of CDT is amplified in the context of dysbiosis and co-infection. For example, in murine models, co-colonization with CDT-producing E. coli and pro-inflammatory pathobionts accelerates adenoma formation via synergistic effects on DNA damage and inflammatory signaling10,69. In humans, CDT genes have been detected at higher prevalence in stool metagenomes from patients with CRC than in matched controls, suggesting their utility as microbial biomarkers for early cancer risk stratification70 Moreover, CDT-producing strains may interact with other microbial genotoxins, such as colibactin, compounding mutational burden and complicating the attribution of specific mutational signatures to single toxins10.
From a therapeutic perspective, CDT’s reliance on active proliferation for maximal genotoxicity presents an exploitable vulnerability: pharmacologic induction of cell cycle arrest can attenuate toxin-induced mutagenesis, while targeted inhibition of CDT receptor interactions or retrograde trafficking may block nuclear translocation of CdtB71,73. Additionally, emerging strategies propose leveraging CDT-induced DDR as a synthetic lethality trigger in tumors deficient in specific repair pathways, such as BRCA1/2-mutant cancers, although the mutagenic risk necessitates careful translational consideration72.
In summary, CDT exemplifies how bacterial genotoxins integrate direct DNA damage, replication stress, immune modulation, and inflammatory signaling to reshape the mutational and microenvironmental landscape of the host. Its multifaceted pathogenicity, spanning from acute cell cycle blockade to chronic genome destabilization, places it among the most consequential microbial effectors in cancer biology. Future research integrating multi-omics mutational profiling with longitudinal microbiome surveillance will be critical to definitively map CDT-associated mutational signatures and to design precision interventions that mitigate its carcinogenic potential10,72.
Typhoid toxin
Typhoid toxin also referred to as Salmonella cytolethal distending toxin (S-CDT), is a structurally unique A₂B₅ bacterial genotoxin produced by Salmonella enterica serovars Typhi and Paratyphi A, as well as multiple nontyphoidal serovars, including S. javiana, S. schwarzengrund, and S. bredeney. Its composition is functionally bipartite: the “A” moiety contains CdtB, a DNase I-like nuclease responsible for DNA cleavage, and PltA, an ADP-ribosyltransferase that modifies host G-protein α subunits, altering intracellular signaling. The “B” moiety forms a pentamer of PltB or its homolog ArtB, which dictates receptor specificity by recognizing glycans terminating in N-acetylneuraminic acid. This assembly allows the toxin to couple precise host cell targeting with the capacity to damage DNA directly and modulate host signaling29,77,78,79.
Following receptor binding, typhoid toxin is internalized via clathrin-dependent endocytosis and traffics retrogradely through the Golgi apparatus to the endoplasmic reticulum. From there, CdtB translocates into the nucleus, where it induces DNA SSBs and DSBs through a magnesium-dependent nuclease activity. These lesions activate the DDR, characterized by ATM/ATR kinase signaling, γH2AX phosphorylation, and 53BP1 recruitment. Notably, the damage is replication-dependent, meaning that actively cycling cells, particularly those in S-phase, are most susceptible to genotoxic insult80,81,82,83.
A hallmark of S-CDT intoxication is its sublethal nature in many cell types. Rather than inducing immediate apoptosis, affected cells frequently undergo prolonged G₂/M arrest and develop a distended morphology, giving the toxin its name. Surviving cells often carry unresolved DNA lesions, chromosome breaks, and micronuclei, hallmarks of genomic instability. Over time, this persistence can lead to the fixation of mutations through error-prone repair pathways such as NHEJ and microhomology-mediated end joining (MMEJ), which are particularly active when HR is compromised78,83,84.
Mutational signature analyses suggest that chronic S-CDT exposure may contribute to SBS patterns and small insertion/deletion profiles like COSMIC signatures SBS3 and ID6, which are associated with defective HR repair and alternative end-joining mechanisms. These associations are supported by experimental systems in which prolonged intoxication results in deletions at short repeats, clustered base substitutions, and structural rearrangements, all of which could serve as molecular fingerprints of toxin exposure in cancer genomes36,80,81.
The pathogenicity of typhoid toxin extends beyond genotoxicity. PltA’s ADP-ribosylation activity has been shown to disrupt heterotrimeric G-protein signaling, potentially dampening host immune responses and altering epithelial barrier function. In animal models, S-CDT-positive strains demonstrate enhanced systemic dissemination, with CdtB activity linked to increased bacterial loads in the liver and spleen. In S. javiana infections, this systemic tropism occurs without significantly altering intestinal colonization, suggesting a role for the toxin in niche adaptation and organ-specific persistence77,78.
Epidemiological and experimental data together point toward a role for typhoid toxin in cancer risk, particularly in chronic carriage states. In endemic regions, S. Typhi DNA and typhoid toxin genes have been detected in gallbladder tissues from patients with carcinoma, consistent with a model in which repeated genotoxic insult within the biliary epithelium promotes accumulation of driver mutations. Similar patterns are observed in hepatobiliary malignancies associated with chronic S. paratyphi and S. javiana infections. Although these associations have been primarily described in non-CRC contexts, the underlying mechanisms, chronic sublethal genotoxic stress, persistent DDR activation, and SASP induction, are mechanistically analogous to those implicated in colorectal carcinogenesis. This highlights the broader relevance of bacterial genotoxins, including typhoid toxin, in shaping tumor-promoting microenvironments across gastrointestinal tissues. The tissue specificity of these associations is likely due to both the distribution of PltB/ArtB-binding glycans and the proliferative status of epithelial targets78,83,84.
From a molecular perspective, the sublethal DNA damage profile induced by typhoid toxin is well-suited to carcinogenic progression. The persistent DDR signaling it triggers can induce a SASP, characterized by elevated IL-6, IL-8, and other pro-inflammatory mediators. This chronic inflammatory milieu may enhance tumor initiation by providing survival and growth signals to genetically altered cells, while simultaneously impairing immune surveillance. In addition, the toxin’s effects on DNA repair gene expression and checkpoint regulation suggest that it may function synergistically with other bacterial effectors or host-derived mutagens, compounding the mutational load80,81,82.
The evolutionary conservation of typhoid toxin across diverse Salmonella lineages hints at its adaptive value. In host-pathogen dynamics, the toxin may serve primarily as a persistence factor, enabling long-term colonization without causing acute lethality. However, in the context of prolonged exposure, such as in chronic carriers, its genotoxic activity becomes a double-edged sword, simultaneously benefiting the pathogen and driving host genome alterations that can culminate in cancer. This duality mirrors the paradigm seen with other bacterial genotoxins, where fitness advantages for the microbe translate into oncogenic risk for the host29,77,78.
Therapeutically, the distinct features of typhoid toxin open possibilities for targeted intervention. Blocking the glycan–PltB interaction with competitive analogs or monoclonal antibodies could prevent host cell entry. Alternatively, disruption of retrograde trafficking might impair nuclear delivery of CdtB. Given that its genotoxicity depends on cell proliferation, temporary pharmacological arrest of susceptible epithelial populations could reduce mutational risk during acute infection. However, such strategies would need careful evaluation to balance antimicrobial efficacy with preservation of host tissue integrity80,81,82.
In summary, typhoid toxin exemplifies how bacterial genotoxins can intertwine precise molecular targeting, sublethal DNA damage, and immune modulation to shape host mutational landscapes. By coupling a potent nuclease with a signaling-modifying ADP-ribosyltransferase, and delivering both through a highly selective receptor-binding system, S. Typhi and related serovars have evolved a virulence factor capable of both promoting bacterial persistence and driving host genome instability. As multi-omics approaches refine our understanding of toxin-associated mutational signatures in human cancers, typhoid toxin stands out as a compelling biomarker for microbial carcinogenesis and a potential target for cancer-preventive interventions in high-risk populations29,78,85.
Indolimines
Indolimines represent a newly identified class of bacterial genotoxins derived from tryptophan metabolism, discovered in Morganella morganii, a gut pathobiont frequently associated with inflammatory bowel disease (IBD) and CRC. In a seminal study, Cao et al. systematically screened gut bacterial isolates from patients with IBD and identified M. morganii strains harboring a previously uncharacterized biosynthetic gene cluster (BGC) responsible for the production of small indole–imine metabolites with potent genotoxic activity86. Disruption of the indolimine BGC abrogated DNA damage and abolished tumor-promoting effects in mouse models, thereby establishing a direct causal link between microbial metabolism and intestinal carcinogenesis86. At the cellular level, indolimines induce DNA damage and activate canonical DNA-damage response pathways, including phosphorylation of H2AX (γH2AX) and activation of the ATM–Chk2–p53 signaling cascade86. Unlike colibactin, which generates interstrand crosslinks, indolimines appear to form monoadducts and oxidative DNA lesions, leading to replication stress and genomic instability86. These findings position indolimines within a broader class of microbiota-derived metabolites capable of directly modifying host DNA. However, their precise chemical structure, mutational signatures, and distribution among gut taxa remain incompletely defined, highlighting an emerging area for biochemical and genomic investigation.
From a microbiome–ecological standpoint, M. morganii carrying the indolimine BGC is enriched in dysbiotic communities under inflammatory conditions, suggesting that intestinal inflammation facilitates both bacterial persistence and host exposure to genotoxins86. This observation aligns with the broader concept that mucosal inflammation and altered nutrient availability, particularly tryptophan metabolism, create ecological niches favoring genotoxin-producing bacteria33,87. Indeed, Patel et al. reported that indolimines can also activate aryl hydrocarbon receptor (AHR) signaling, linking their production to host immune and metabolic regulation87. Such dual activity, genotoxic and signaling, underscores the multifaceted role of tryptophan-derived metabolites in host–microbe interactions.
Therapeutically, the discovery of indolimines has opened new microbiome-based avenues for cancer prevention. Targeting the indolimine pathway through small-molecule inhibition of biosynthetic enzymes, bacteriophage-mediated gene disruption, or competitive exclusion by non-producing commensals has shown preliminary efficacy in experimental models86. More broadly, these findings emphasize the need to incorporate microbial metabolic profiling and genotoxin screening into CRC risk assessment frameworks, as previously overlooked commensals can function as direct mutagens and tumor promoters33. Collectively, indolimines highlight the expanding landscape of microbial metabolites that integrate into the genomic and immunometabolic circuitry of the host, demonstrating how a single commensal species can reconfigure mucosal homeostasis, induce DNA damage, and promote carcinogenesis. As mechanistic dissection continues, indolimines stand as a compelling example of microbe-derived small molecules bridging metabolism and mutagenesis within the human gut ecosystem33,86,87.
Other genotoxic metabolites
Beyond well-characterized bacterial genotoxins such as colibactin or cytolethal distending toxin, several gut-associated microbes produce additional small-molecule metabolites capable of inducing DNA damage, altering host cell signaling, and shaping the mutational landscape of colorectal and other gastrointestinal cancers. Among these, secondary bile acids, phenolic derivatives, hydrogen sulfide (H₂S), and certain tryptophan catabolites have emerged as important contributors to host genomic instability. Microbiota-mediated deconjugation, dehydroxylation, and epimerization of primary bile acids generate cytotoxic species such as deoxycholic acid (DCA) and lithocholic acid (LCA), which can trigger oxidative stress, lipid peroxidation, and DNA DSBs via ROS accumulation88. These metabolites also function as signaling ligands for farnesoid X receptor (FXR) and G-protein-coupled bile acid receptor 1 (TGR5), altering epithelial proliferation and apoptosis pathways in ways that may promote tumorigenesis89,90. In murine models, high-fat diet–induced enrichment of Ruminococcus gnavus and Ileibacterium valens was shown to drive non-classical amino acid conjugation of bile acids, generating unique adducts that impair intestinal stem cell regulation, suggesting a diet–microbiome–metabolome axis in carcinogenesis90.
Hydrogen sulfide, primarily generated by sulfate-reducing bacteria such as Desulfovibrio spp. and certain Fusobacterium strains, represents another genotoxic microbial metabolite. At physiological concentrations, H₂S has signaling functions, but at higher levels it inhibits cytochrome c oxidase, disrupts mitochondrial respiration, and induces DNA damage through oxidative and alkylative stress mechanisms. Chronic mucosal exposure to elevated H₂S can inhibit cytochrome c oxidase, disrupt mitochondrial respiration, and trigger DNA damage through oxidative and alkylative stress, impairing colonocyte butyrate oxidation and creating a pro-inflammatory microenvironment that enhances mutagenic risk91,92. In parallel, aromatic amines and phenolic metabolites, produced via proteolytic fermentation of dietary proteins, can undergo host-mediated N-oxidation to yield electrophilic intermediates that covalently bind DNA, forming promutagenic adducts. Elevated concentrations of aromatic metabolites such as p-cresol, phenol, and 4-ethylphenol have been detected in the stool and serum of CRC patients, correlating with an enrichment of proteolytic taxa such as Bacteroides fragilis and Clostridium spp., and with downregulation of host DNA repair pathways93. These phenolic and aromatic amine derivatives exert mutagenic activity through oxidative and alkylative DNA damage, promoting genomic instability and enhancing tumor-promoting inflammation39,42,94.
Beyond canonical bacterial toxins, microbial metabolites, including secondary bile acids, hydrogen sulfide (H₂S), phenolic derivatives, aromatic amines, and tryptophan catabolites, constitute critical, though often underappreciated, drivers of mutagenesis in the colonic epithelium. Secondary bile acids such as DCA and LCA induce oxidative stress, DNA DSBs, and mutational signatures resembling SBS17a/b, establishing a mechanistic link between high-fat diets, microbial metabolism, and genomic instability in colorectal carcinogenesis. H₂S produced by sulfidogenic bacteria like Desulfovibrio spp. or Fusobacterium disrupts mitochondrial respiration and causes oxidative DNA damage, thereby amplifying mutational burden. Likewise, phenolic compounds and aromatic amines derived from proteolytic fermentation, such as p-cresol and 4-ethylphenol, form promutagenic adducts and impair DNA repair responses. Tryptophan-derived indole catabolites further modulate immune tone and epithelial barrier integrity, potentially increasing susceptibility to mutagenic insults under dysbiotic conditions. These metabolites act synergistically, linking dietary exposures, microbial shifts, and host mutational processes, offering new targets for microbiota-based prevention strategies95,96.
Microbiota-driven inflammation and immune suppression in CRC
Having established direct genotoxic mechanisms, we next examine how dysbiosis sustains a chronic inflammatory milieu and an immunosuppressive TME that enables clonal selection and tumor escape. This section integrates pattern-recognition signaling, myeloid and lymphoid remodeling, and metabolite-mediated immune editing to explain reduced cytotoxic surveillance and variable response to therapy.
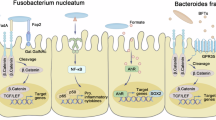
CRC arises within a complex ecological niche where gut microbial communities and host tissues engage in reciprocal interactions that can profoundly shape tumor initiation, progression, and therapeutic outcomes. The human gut harbors a vast and diverse microbiota, comprising trillions of microorganisms, whose collective genome encodes an array of metabolic, immunomodulatory, and signaling capacities that can either sustain epithelial homeostasis or foster malignant transformation97,98. Dysbiosis, characterized by an imbalance between beneficial commensals and pathogenic taxa, has emerged as a hallmark of CRC, driving chronic inflammation and sculpting an immunosuppressive TME that supports tumor survival and immune evasion99,100 (Table 1 and Fig. 1).

This figure illustrates the interactions between CRC-associated microbial taxa, their bioactive metabolites, and genotoxins, integrated into four major pathogenic axes: endocrine, neural, metabolic, and immune. It highlights how dysbiosis drives DNA damage, chronic inflammation, metabolic rewiring, epithelial barrier disruption, and tumor microenvironment remodeling, ultimately promoting colorectal tumor initiation and progression. Key representative taxa and molecular effectors are indicated in each pathway.
Pro-tumorigenic bacteria such as Fusobacterium nucleatum, ETBF, and pks⁺ E. coli exemplify microbiota members that promote inflammation-driven oncogenesis through converging molecular pathways. Seminal studies have demonstrated that F. nucleatum activates Toll-like receptor 4 (TLR4) signaling, leading to NF-κB–dependent transcription of pro-inflammatory cytokines, including IL-6 and IL-8, which sustain a tumor-promoting inflammatory environment101,102. Moreover, F. nucleatum expresses the adhesin FadA, which binds E-cadherin on colonocytes, disrupts adherens junction integrity, and triggers β-catenin nuclear translocation and transcriptional activation of oncogenic targets7. Consistently, high intratumoral levels of F. nucleatum have been associated with increased myeloid infiltration, reduced CD8⁺ T cell density, and poorer therapeutic outcomes in CRC patients103,104. ETBF secretes the metalloprotease toxin BFT, which cleaves E-cadherin, compromises epithelial barrier function, and initiates STAT3-driven IL-17 responses, amplifying Th17-mediated inflammation105. Meanwhile, pks⁺ E. coli produces colibactin, a genotoxin that induces DNA DSBs, initiating mutagenic events that complement inflammatory signaling in driving tumor progression45,106.
These microbes orchestrate a chronic inflammatory milieu characterized by elevated levels of IL-6, IL-1β, TNF-α, and IL-17, which not only sustain epithelial proliferation but also recruit MDSCs, tumor-associated macrophages, and regulatory T cells (Tregs) into the TME107,108. Such immune cell infiltration contributes to a feed-forward loop where inflammatory mediators promote angiogenesis, extracellular matrix remodeling, and epithelial-to-mesenchymal transition, thereby enhancing tumor invasiveness85. Notably, microbiota-derived metabolites, including secondary bile acids, hydrogen sulfide, and polyamines, exert context-dependent effects on inflammation and immunity. While some SCFAs, such as butyrate, possess anti-inflammatory properties via histone deacetylase inhibition and Foxp3+ Treg induction, dysbiotic shifts in microbial metabolism during CRC often reduce SCFA levels and increase pro-inflammatory metabolites, tipping the balance toward immune suppression100,109,110.
Immune evasion in CRC is facilitated by multiple microbiota-dependent mechanisms that subvert both innate and adaptive immune surveillance. Beyond FadA–E-cadherin interactions, F. nucleatum directly impairs NK cell cytotoxicity by engaging the inhibitory receptor TIGIT, thereby limiting antitumor immune clearance40,103. Other bacterial taxa modulate antigen-presenting cell (APC) function to dampen CD8⁺ T cell activation109. Disruption of the gut epithelial barrier by pathogenic taxa increases microbial translocation and systemic endotoxemia, which skews myeloid cell polarization toward an immunosuppressive phenotype99,111. Furthermore, chronic activation of pattern recognition receptors (PRRs) by bacterial ligands can lead to immune tolerance rather than activation, reducing the effectiveness of tumor-targeting immune responses97,107,112.
The bidirectional relationship between microbiota-driven inflammation and immune suppression has direct implications for CRC therapy. Inflammation-associated immune suppression reduces the efficacy of immune checkpoint blockade (ICB) by limiting effector T cell infiltration into the TME85,109. Conversely, modulating the gut microbiota, via antibiotics, probiotics, dietary interventions, or FMT, has been shown in preclinical and clinical studies to reshape immune landscapes and enhance therapeutic responsiveness85,100,113,114. For example, selective depletion of F. nucleatum restores CD8⁺ T cell activity and improves chemotherapeutic sensitivity, whereas enrichment of beneficial commensals such as Bifidobacterium and Lactobacillus augments antitumor immunity and synergizes with ICB103,115.
At the molecular level, microbiota-induced inflammation in CRC is mediated by sustained activation of intracellular signaling cascades such as NF-κB, STAT3, and MAPK pathways, which regulate cytokine transcription, cell survival, and proliferation105. These signaling networks converge with DDRs induced by bacterial genotoxins, amplifying mutational burden and clonal selection within the tumor epithelium106. Moreover, persistent exposure to microbial ligands and metabolites remodels the epigenetic landscape of immune cells and epithelial cells alike, influencing chromatin accessibility at loci encoding immune checkpoint molecules, inflammatory mediators, and antigen-presentation machinery107,109,116.
The immunosuppressive TME in CRC is further reinforced by the recruitment of regulatory immune subsets under the influence of microbial cues. IL-10–producing Tregs suppress effector T cell proliferation, while arginase-1-expressing MDSCs deplete local arginine pools, impairing T cell receptor (TCR) signaling100,107,117. TAMs polarized toward an M2-like phenotype by microbial metabolites and cytokines facilitate tissue remodeling and tumor growth through secretion of vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs)85. The resulting immune landscape is one of functional paralysis, in which tumor cells proliferate unchecked despite the presence of immune infiltrates.
An emerging dimension of microbiota-driven immune modulation in CRC involves the crosstalk between the gut microbiome and the intratumoral microbiome. While the gut serves as the primary reservoir of inflammatory and immunomodulatory bacteria, microbial translocation into the tumor site introduces localized microbial-immune interactions that may differ from those occurring in the intestinal lumen85. Low-biomass intratumoral bacteria have been detected within cancer cells and infiltrating immune cells, where they may influence local cytokine production, antigen presentation, and therapeutic sensitivity85,109,118.
In summary, microbiota-driven inflammation and immune suppression in CRC represent a tightly interwoven network of microbial, immune, and epithelial interactions. Pathogenic taxa initiate chronic inflammation through barrier disruption, PRR activation, and genotoxin production, while simultaneously fostering an immunosuppressive TME that facilitates tumor escape from immune surveillance. The dual capacity of the microbiota to both incite tumor-promoting inflammation and attenuate antitumor immunity underscores its pivotal role in CRC pathogenesis and therapeutic resistance. Harnessing microbiota modulation strategies to dismantle this inflammatory-immunosuppressive axis holds considerable promise for improving CRC prevention and treatment in the era of precision oncology98,109.
Metabolic rewiring of the tumor microenvironment by gut microbes
Building on inflammatory-immune circuits, we consider how microbial communities reprogram tumor and stromal metabolism, shifting SCFA profiles, amino-acid and bile-acid flux, and redox balance, to support proliferation and dampen antitumor immunity. We map microbe–host co-metabolism and nutrient competition that collectively sculpt an energetically favorable niche for CRC.
Emerging evidence highlights the profound impact of gut microbes on the metabolic architecture of the TME in CRC, where microbially derived metabolites and signaling cues reshape nutrient fluxes, redox balance, and immune cell energetics to favor tumor progression. In the healthy colon, the gut microbiota sustains mucosal metabolic homeostasis through the fermentation of complex polysaccharides into SCFAs such as butyrate, propionate, and acetate, which fuel colonocytes, regulate histone acetylation, and maintain epithelial barrier function110,119. In CRC, however, compositional and functional shifts in the microbiome reprogram these pathways toward tumor-supportive phenotypes, often by depleting butyrate-producing taxa (e.g., Faecalibacterium prausnitzii) and enriching species capable of generating oncometabolites, such as polyamines, secondary bile acids, and lactate120,121,122. This metabolic reshaping is not a passive consequence of dysbiosis; rather, it reflects a dynamic crosstalk between microbial consortia, transformed epithelial cells, and infiltrating immune cells that compete for and modify shared nutrient pools97,123 (Table 1 and Fig. 1).
A hallmark of this microbe-driven metabolic rewiring is the altered SCFA profile in the TME. While butyrate exhibits anti-proliferative and pro-apoptotic effects on CRC cells via GPR109A activation and HDAC inhibition, tumor-associated microbiota often shifts fermentation toward acetate and propionate, which may feed into acetyl-CoA and lipogenesis pathways that support rapid cell proliferation119,124,125. Concurrently, specific pathobionts such as Fusobacterium nucleatum and enterotoxigenic Bacteroides fragilis can modulate host glycolytic flux through inflammatory signaling and NF-κB activation, driving a Warburg-like phenotype in epithelial and immune cells126,127,128. This metabolic bias is reinforced by lactate accumulation from both tumor cells and certain lactic acid–producing bacteria, acidifying the TME and impairing cytotoxic T lymphocyte function129,130. Excess lactate not only serves as an energy source for tumor-associated fibroblasts but also promotes regulatory T cell (Treg) expansion and myeloid-derived suppressor cell polarization, deepening local immune suppression123,131.
Microbiota-driven modulation of amino acid metabolism further illustrates the metabolic plasticity of CRC TMEs. For example, Peptostreptococcus anaerobius and E. coli strains harboring the pks island can increase tryptophan metabolism toward kynurenine via IDO1 induction, an immunoregulatory pathway that promotes T cell exhaustion121,122,132. Similarly, microbial catabolism of arginine can deprive effector T cells of this essential substrate, while favoring arginase-expressing suppressive myeloid populations97. Methionine and serine metabolic pathways are also influenced by microbial metabolites, fueling one-carbon metabolism and nucleotide biosynthesis necessary for rapid tumor cell proliferation119,120. Notably, dysregulated bile acid metabolism, driven by 7α-dehydroxylating bacteria, generates secondary bile acids such as deoxycholic acid, which act as both genotoxins and signaling molecules that activate FXR and TGR5, modulating epithelial proliferation and inflammatory tone122,127.
These metabolic perturbations are not restricted to tumor epithelium; they extend to the stromal and immune compartments of the TME. Microbial metabolites can directly reprogram macrophage polarization, with butyrate favoring M1 anti-tumor phenotypes and lactate or succinate driving M2 pro-tumor phenotypes123,129,131. Moreover, microbiota-derived inosine and formate have been shown to enhance oxidative phosphorylation in activated T cells under certain conditions, yet in CRC their availability is often curtailed by tumor-microbe metabolic competition119,124,133. The depletion of beneficial metabolites and accumulation of immunosuppressive by-products thus creates a metabolic bottleneck that selectively disadvantages anti-tumor immune cells while preserving tumor fitness.
From a systems perspective, the microbial contribution to CRC metabolic rewiring can be conceptualized as a multi-layered network of direct enzymatic transformations, host-microbe co-metabolism, and immune-metabolic feedback loops119,120,126. Direct effects include bacterial enzymes that convert dietary substrates into tumor-active metabolites, while co-metabolic processes involve sequential transformations between microbial and host enzymes—exemplified by polyamine synthesis, where microbial ornithine decarboxylase activity synergizes with host pathways to generate spermidine, a molecule linked to tumor growth and immune evasion97,121,134,135. Feedback loops arise when microbial metabolites modulate immune cell metabolism, which in turn shapes the microbial community structure through altered cytokine and nutrient profiles123,129.
This metabolic interdependence underscores the therapeutic potential of targeting microbiota-mediated pathways in CRC. Strategies under investigation include dietary modulation to enrich SCFA-producing bacteria, prebiotics that shift fermentation profiles toward anti-tumor metabolites, engineered probiotics that deliver metabolic inhibitors directly into the TME, and FMT to restore eubiosis122,124,136. Pharmacological interventions aimed at disrupting microbial production of oncometabolites, such as inhibitors of bacterial bile salt hydrolases or colibactin biosynthesis, represent another promising avenue119,127. Given that metabolic rewiring also dictates the efficacy of ICB and other immunotherapies, integrating microbiome modulation into oncologic treatment regimens could synergistically restore anti-tumor immunity120,129.
Overall, gut microbes are not passive bystanders in CRC pathogenesis but active architects of the tumor metabolic niche. Through precise modulation of carbohydrate, amino acid, lipid, and bile acid metabolism, they influence the availability of nutrients, the pH and redox state of the TME, and the functional programming of infiltrating immune cells. This metabolic rewiring reinforces tumor survival and immune evasion, making the microbiota both a driver of disease progression and a tractable target for therapeutic innovation. As our understanding of these intricate host-microbe-metabolism interactions deepens, interventions that selectively disrupt tumor-promoting microbial pathways while restoring metabolic balance may redefine the landscape of CRC therapy97,120,124.
Neuro-microbial crosstalk in colorectal carcinogenesis
We then expand to neuromodulatory axes, detailing how enteric and extrinsic neural circuits interact with microbial signals to drive neuritogenesis, perineural invasion, and immune reprogramming. This section highlights bidirectional pathways, neurotransmitters, neurotrophins, and microbially derived metabolites, that integrate stress, motility, and inflammation into tumor progression.
The intricate interplay between the nervous system and the gut microbiota has emerged as a pivotal determinant in CRC pathogenesis, reshaping our understanding of tumor biology beyond traditional genetic and environmental paradigms. Within the colorectal TME, neural elements and microbial consortia engage in bidirectional communication that modulates oncogenic signaling, immune responses, and stromal remodeling. The enteric nervous system (ENS), in concert with extrinsic sympathetic and parasympathetic inputs, orchestrates local neurochemical landscapes via neurotransmitters, neuropeptides, and neurotrophins, which not only regulate epithelial proliferation and barrier integrity but also shape microbial community composition and function137,138,139. Microbial metabolites, such as SCFAs, secondary bile acids, and tryptophan derivatives, can influence neuronal excitability and synaptic transmission within the ENS, thereby altering gut motility, mucosal secretions, and immune cell trafficking89,140,141,142. These metabolites, in turn, impact tumor-promoting inflammation, angiogenesis, and metastatic competence (Table 1 and Fig. 1).
CRC progression is frequently accompanied by neuroplastic changes, including increased intratumoral nerve density and aberrant neuritogenesis, which have been correlated with enhanced invasion, perineural spread, and poor prognosis143,144,145. Mechanistically, tumor-derived neurotrophic factors such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) recruit and sustain neuronal fibers within tumor niches, creating a feed-forward loop wherein nerves supply tumor-promoting neurotransmitters like norepinephrine and acetylcholine146,147. These transmitters activate β-adrenergic and muscarinic receptors on epithelial and stromal cells, leading to downstream activation of oncogenic pathways, including MAPK, PI3K-AKT, and STAT3 signaling, thereby promoting proliferation, migration, and resistance to apoptosis111,148. Concurrently, specific gut bacteria, including Fusobacterium nucleatum and Bacteroides fragilis, potentiate neuronal remodeling by stimulating pro-neurogenic cytokines such as IL-6 and CXCL12, further integrating microbial and neural axes in tumor evolution7,138,149.
Within CRC, a compelling biglycan–IL-10–microbiota axis has emerged in which host extracellular matrix composition shapes the abundance and function of Bacteroides thetaiotaomicron, thereby reprogramming mucosal immunity toward either tumor suppression or promotion. In murine models, biglycan (BGN) is abundantly expressed in enteric neurons within CRC tissue; BGN deficiency leads to impaired gastrointestinal motility, altered tumor distribution, and a marked upregulation of IL-10 secretion from enteric neurons, which in turn suppresses tumor growth and inhibits cancer cell migration150. Mechanistically, BGN may compete with B. thetaiotaomicron for chondroitin sulfate binding, partially regulating its abundance and, by extension, its effects on the tumor microenvironment150. B. thetaiotaomicron is not a passive passenger in this system: its methylmalonyl-CoA mutase pathway produces propionate, which promotes goblet cell differentiation via GPR41 signaling, strengthening the mucus barrier and mitigating DSS-induced colitis151. These barrier-protective and anti-inflammatory effects are relevant in the context of CRC, where chronic inflammation drives tumor progression. Clinically, Bacteroides species, including B. thetaiotaomicron, have been enriched in young-onset CRC152, and proposed as candidate biomarkers in early CRC screening153. Metatranscriptomic analyses show Bacteroides dominance in reactive oxygen species–detoxification pathways, indicating niche-specific adaptation that modulates epithelial injury and immune tone154. Therapeutically, FMT has been shown to increase B. thetaiotaomicron abundance and enhance anti-PD-1 immunotherapy efficacy, potentially via dendritic cell IL-10 induction and propionate-mediated tumor suppression155. At the population scale, CRC-associated bacteria, including B. thetaiotaomicron, are widely distributed in global microbiomes and act as drivers of precancerous change156. Collectively, these findings underscore the translational potential of targeting the BGN–IL-10–B. thetaiotaomicron axis through microbiota modulation, via pro-, pre-, or postbiotics, antibiotics, or FMT, to tilt the tumor microenvironment away from tolerance and toward anti-tumor immunity157.
Neuro-immune crosstalk represents a critical node in this interaction. Neural innervation of immune organs, including the gut-associated lymphoid tissue, enables the nervous system to modulate immune cell function within the TME158,159. Sympathetic activation can skew macrophage polarization toward an M2-like, tumor-supportive phenotype, suppress cytotoxic T lymphocyte activity, and expand regulatory T cell populations, all of which contribute to immune evasion140,158. Conversely, microbial dysbiosis in CRC can modulate vagal tone and sympathetic output via the gut-brain axis, thereby indirectly reprogramming immune surveillance89,144,160. Certain bacterial metabolites, including butyrate and indole derivatives, exhibit neuroactive properties that dampen pro-inflammatory responses, while microbial-associated molecular patterns (MAMPs) such as lipopolysaccharide activate TLR-expressing neurons, driving neurogenic inflammation that fuels tumor progression111,137,161.
The anatomical proximity of colonic nerves and the mucosal microbiota facilitates direct interaction. Electron microscopy and single-cell transcriptomics have revealed specialized neuroepithelial circuits in the colon where enteroendocrine cells synapse with sensory neurons to rapidly transmit luminal microbial cues to the central nervous system139,143,146. These circuits enable real-time modulation of local immunity, epithelial turnover, and vascular tone in response to microbial fluctuations. In CRC, however, malignant transformation disrupts these homeostatic reflexes, replacing them with maladaptive signaling loops that sustain oncogenesis149. Notably, stress-induced activation of hypothalamic-pituitary-adrenal (HPA) axis pathways elevates systemic glucocorticoids, which suppress anti-tumor immunity and alter microbial ecology, thus creating a permissive neuro-microbial milieu for tumor growth158.
Emerging evidence also implicates enteric glial cells (EGCs) as mediators of neuro-microbial-tumor cross-communication. EGCs, traditionally regarded as structural support for neurons, are now recognized as immunomodulatory hubs that respond to microbial metabolites and inflammatory cytokines by releasing neurotrophic factors and pro-tumorigenic mediators144,162. In CRC, EGC activation has been linked to increased myofibroblast proliferation, extracellular matrix remodeling, and enhanced metastatic niche preparation. Certain gut microbes can directly influence EGC function via G-protein coupled receptor (GPCR) signaling, thus providing a molecular bridge between dysbiosis and neural reprogramming of the TME89,111,159.
At the systemic level, gut microbes modulate afferent sensory signaling to brainstem nuclei, influencing central autonomic outputs that regulate colonic motility, vascular perfusion, and immune readiness140,146. This gut-brain feedback loop is particularly relevant in CRC patients, where dysbiotic shifts in microbial communities alter nociceptive thresholds and visceral sensitivity, potentially contributing to cancer-associated pain and cachexia137,143,163. Moreover, the vagus nerve emerges as a key anti-inflammatory conduit; its cholinergic anti-inflammatory reflex can be modulated by microbial cues, with implications for controlling tumor-associated inflammation and improving therapeutic outcomes158.
Therapeutically, targeting neuro-microbial crosstalk offers novel opportunities in CRC management. β-adrenergic blockade, vagal nerve stimulation, and microbiota modulation through probiotics, prebiotics, or FMT are under investigation for their ability to disrupt pro-tumor neuro-microbial signaling loops111,138,140. Precision approaches that integrate neural circuit mapping, microbiome profiling, and metabolomic signatures may enable personalized interventions that attenuate tumor-promoting neural activity while restoring microbial homeostasis144,149,164. However, challenges remain in delineating causal pathways from associative findings, as well as in translating preclinical insights into clinically effective strategies.
Taken together, neuro-microbial crosstalk constitutes a dynamic and multifaceted driver of colorectal carcinogenesis, integrating neuronal remodeling, microbial dysbiosis, and immune suppression into a unified pathogenic framework. The reciprocal modulation between gut microbes and neural networks not only shapes the tumor microenvironment but also dictates systemic cancer trajectories89,144,146. Deciphering this dialogue at cellular, molecular, and systems levels will be crucial for developing innovative therapies that exploit the vulnerabilities of the neuro-microbial axis in CRC.
Endocrine pathways modulated by the intestinal microbiome
Parallel to neural inputs, endocrine–microbiome interactions shape CRC risk and behavior via SCFAs, bile-acid receptors, the estrobolome, insulin/IGF-1 signaling, HPA-axis tone, and thyroid-metabolic coupling. Here we synthesize how microbial metabolism perturbs hormone production, receptor activation, and downstream transcriptional programs in epithelial and immune compartments.
The intestinal microbiome exerts profound regulatory effects on endocrine signaling pathways that influence CRC pathogenesis, acting through bidirectional interactions with host hormonal systems. Microbial communities within the gut metabolize dietary and host-derived compounds into bioactive metabolites, such as SCFAs, bile acid derivatives, indoles, and polyamines, that can modulate endocrine function both locally in the intestinal mucosa and systemically through the circulation165,166. SCFAs, particularly butyrate, not only regulate epithelial proliferation and differentiation but also influence hormone secretion by enteroendocrine cells, modulating glucagon-like peptide-1 (GLP-1), peptide YY (PYY), and serotonin release, which in turn affect gut motility, nutrient absorption, and energy homeostasis167,168. This endocrine modulation is critical in CRC because altered microbial fermentation profiles can shift enteroendocrine signaling toward pro-inflammatory and pro-tumorigenic states, including enhanced insulin resistance and aberrant activation of the insulin/IGF-1 axis, a well-established driver of colorectal tumorigenesis169 (Table 1 and Fig. 1).
Microbial metabolism of primary bile acids into secondary bile acids, such as deoxycholic acid and lithocholic acid, represents another endocrine-linked mechanism relevant to CRC. These metabolites can activate nuclear receptors, including farnesoid X receptor (FXR) and pregnane X receptor (PXR), and membrane-bound receptors such as Takeda G-protein receptor 5 (TGR5), which modulate not only metabolic homeostasis but also inflammatory responses in the colonic epithelium170,171. Dysbiosis-associated accumulation of certain secondary bile acids has been shown to promote DNA damage, oxidative stress, and Wnt/β-catenin pathway activation, while concurrently impairing FXR-mediated tumor-suppressive signaling119,172. The gut microbiota also participates in estrogen metabolism through the “estrobolome,” the aggregate of bacterial genes capable of deconjugating estrogens via β-glucuronidase activity, thereby regulating circulating estrogen levels and receptor-mediated signaling in colonic tissues173. Elevated estrogen reactivation can engage estrogen receptor beta (ERβ) pathways, which have been associated with anti-proliferative effects in normal colonocytes but may exhibit context-dependent roles in malignant transformation174,175.
Sex-specific differences in microbiome-endocrine interactions have emerged as a key dimension in CRC risk modulation. The “microgenderome” concept describes the reciprocal regulation between sex hormones and gut microbial communities, wherein estrogens tend to promote a microbial composition enriched in anti-inflammatory taxa, while androgens may facilitate expansion of pro-tumorigenic species176,177. These compositional differences have functional consequences: for instance, estrogen-associated microbiota produce metabolites that attenuate NF-κB–driven inflammation, whereas androgen-associated profiles may increase microbial products that activate TLRs and enhance IL-6/STAT3 signaling, fostering an immunosuppressive tumor microenvironment178. The gut microbiota, in turn, can modulate sex hormone bioavailability via enzymatic transformation or by influencing enterohepatic circulation of steroid hormones, ultimately affecting endocrine receptor activation within colonic epithelial and stromal cells176,179.
Another critical layer of endocrine regulation involves microbial modulation of the HPA axis through production of neurotransmitter precursors and neuromodulatory metabolites, such as tryptophan-derived indoles and γ-aminobutyric acid (GABA). These metabolites can influence cortisol secretion, stress responses, and systemic inflammatory tone, indirectly shaping CRC progression via glucocorticoid receptor-mediated transcriptional programs in immune and epithelial cells167,169. Chronic stress-associated dysbiosis has been linked to elevated corticosterone levels, reduced intestinal barrier integrity, and augmented translocation of microbial products into the circulation, which perpetuate systemic low-grade inflammation and enhance tumor-promoting endocrine-immune crosstalk165,170.
Thyroid hormone metabolism also intersects with gut microbial activity in CRC. Certain gut bacteria can deiodinate thyroxine (T4) and triiodothyronine (T3) precursors, altering thyroid hormone homeostasis, which is known to influence colonic epithelial turnover and mitochondrial metabolism119. Hypothyroid states have been correlated with altered gut microbial diversity and increased CRC risk, potentially due to impaired regulation of oxidative metabolism and epithelial renewal173. Similarly, microbial metabolites can engage peroxisome proliferator-activated receptors and aryl hydrocarbon receptor, which integrate metabolic and endocrine signals to control epithelial proliferation, immune surveillance, and xenobiotic detoxification174,178.
The endocrine–microbiome axis in CRC is further shaped by diet, lifestyle, and pharmacological interventions. High-fat diets increase bile acid flux and favor microbial communities capable of producing carcinogenic bile acid derivatives, thereby altering FXR/TGR5-mediated hormonal signaling170. Conversely, fiber-rich diets enhance SCFA production, promoting GLP-1 and PYY release and improving insulin sensitivity, which can indirectly mitigate CRC-promoting endocrine pathways167. Antibiotic exposure, hormonal replacement therapy, and probiotic interventions can each shift the microbial endocrine landscape, with variable effects on CRC risk depending on the context173,176,179. Collectively, the intestinal microbiome functions as a dynamic endocrine organ that communicates bidirectionally with host hormonal systems, influencing CRC initiation, progression, and therapeutic responsiveness. By modulating hormone metabolism, receptor activation, and downstream signaling pathways, gut microbes integrate metabolic, inflammatory, and endocrine cues into the tumor microenvironment119,170. Disentangling these complex interactions is essential for identifying microbiome-targeted strategies that harness endocrine modulation to prevent or treat CRC.
Integrative roles of microbial consortium in tumor progression
Moving from single taxa to ecological organization, this section frames CRC as an outcome of coordinated microbial consortia whose functional redundancy and cross-feeding amplify barrier disruption, DNA damage, and immune evasion. We emphasize consortium dynamics across tumor stages and their implications for diagnostics and control points for intervention.
The concept of microbial consortia as coordinated biological entities influencing tumor progression has emerged as a pivotal paradigm in cancer microbiome research. Unlike single-species effects, these complex communities act through intricate, multi-layered interactions within the TME, integrating metabolic, inflammatory, and immunological pathways to shape cancer initiation, growth, and dissemination. Evidence from metagenomic and functional studies reveals that CRC-associated microbial consortia often comprise synergistic assemblages of pathogenic, commensal, and opportunistic taxa whose cooperative activities can potentiate tumorigenesis beyond the additive effects of individual members99,180,181,182. A critical ecological feature of these polymicrobial communities is their frequent organization within structured biofilms adherent to the colonic mucosa. These matrix-embedded microbial aggregates enable spatial and metabolic cooperation, facilitating horizontal gene transfer, quorum-sensing communication, and metabolic exchange between species such as Fusobacterium nucleatum, Bacteroides fragilis, and Escherichia coli. Biofilm-associated consortia establish hypoxic, inflammatory niches that promote chronic epithelial injury, persistent IL-6/STAT3 and NF-κB pathway activation, and immune evasion, ultimately sustaining tumor growth. The extracellular polymeric matrix protects constituent taxa from immune clearance and antimicrobial stress, favoring the persistence of genotoxin-producing pathobionts and the amplification of tumor-promoting signals15,16,18,183.
Beyond structural and metabolic cooperation, temporal regulation is increasingly recognized as a critical layer shaping consortium behavior. Circadian oscillations at both the host and microbial levels synchronize metabolic activity, immune tone, and epithelial renewal, directly influencing the spatial organization and functional output of CRC-associated consortia. Bidirectional interactions between circadian clocks and the gut microbiome regulate the timing of metabolite production, quorum sensing, and virulence expression, thereby modulating inflammatory and oncogenic signaling cascades within the TME. Disruption of circadian–microbial synchrony, whether through lifestyle factors or tumor-driven metabolic reprogramming, has been shown to exacerbate dysbiosis, fuel chronic inflammation, and facilitate tumor progression184,185,186,187,188.
This cooperative dynamic is exemplified by the co-occurrence of Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis, and colibactin-producing Escherichia coli, which collectively amplify epithelial barrier disruption, DNA damage, and pro-tumor immune modulation through parallel yet converging mechanisms15,189,190. Within such consortia, niche partitioning ensures sustained colonization: while F. nucleatum mediates adhesion to epithelial cells and immune evasion, B. fragilis modulates local cytokine profiles, and E. coli induces genotoxic stress—together establishing a pro-carcinogenic micro-niche that supports tumor cell proliferation and invasion191,192.
Integrative analyses of large-scale multi-omics datasets demonstrate that these consortia are not static; their composition and functional gene repertoire evolve alongside tumor progression, adapting to shifts in nutrient availability, oxygen gradients, and host immune pressure156,193,194. In early lesions, metabolic cross-feeding within consortia fosters a pro-inflammatory milieu rich in short-chain fatty acid derivatives, polyamines, and secondary bile acids, which can stimulate epithelial proliferation and modulate oncogenic signaling pathways such as Wnt/β-catenin and NF-κB99,181. As the tumor advances, oxygen depletion and necrosis-associated nutrient release select for facultative and obligate anaerobes capable of thriving in hypoxic cores, thereby reinforcing tumor-favorable metabolic states156,180. These adaptations not only sustain tumor-promoting inflammation but also reshape the immune landscape: certain consortium members secrete metabolites that impair dendritic cell maturation, skew macrophages toward an M2-like phenotype, and induce regulatory T cell expansion, collectively dampening anti-tumor immunity189.
Experimental models highlight that disrupting consortium integrity, either by targeted antimicrobial strategies or through competitive colonization by beneficial taxa, can attenuate tumor growth and restore immune surveillance191,192. For example, synthetic microbial consortia engineered to preserve butyrate-producing bacteria while reducing inflammation-associated Bacteroides species have been shown to limit polyp formation and improve epithelial barrier function in carcinogen-induced CRC models195,196. These findings suggest that functional redundancy within tumor-associated consortia confers resilience to perturbation, underscoring the need for precision approaches that selectively dismantle pro-tumor networks while maintaining beneficial community functions193,195. Emerging therapeutic strategies are increasingly leveraging these consortium-level vulnerabilities through microbiome engineering, phage therapy, and rational design of synthetic consortia to modulate specific ecological interactions rather than inducing broad microbial depletion. For example, remodeling the consortium using engineered commensals has successfully restored epithelial integrity, dampened inflammatory signaling, and enhanced immune surveillance in preclinical CRC models195,197,198.
From a systems biology perspective, microbial consortia operate as distributed information-processing networks within the TME, sensing and responding to host-derived cues such as pH, hypoxia, and immune mediators189,192,199. This adaptive capability enables synchronized modulation of host pathways at multiple levels, from epigenetic reprogramming of epithelial cells to metabolic rewiring of stromal and immune compartments181,190,194,200. For instance, certain consortia upregulate tumor-associated fibroblast activation via microbiome-induced SASPs, thereby enhancing extracellular matrix remodeling and tumor invasion156. Similarly, quorum sensing within bacterial networks can regulate the timing and magnitude of virulence factor release, optimizing their impact on tumor-supportive inflammation and angiogenesis99,180.
Recent evidence underscores that the microbiome not only shapes primary tumorigenesis but also orchestrates metastatic dissemination. Intratumoral and gut-resident microbial communities regulate epithelial–mesenchymal transition, angiogenesis, immune escape, and the establishment of pre-metastatic niches, thereby actively participating in the metastatic cascade. Microbial signals are increasingly recognized as modulators of organotropism, where microbial-derived metabolites and microvesicles remodel distant tissues through matrix remodeling, immune priming, and metabolic preconditioning to favor organ-specific colonization. Spatial multi-omic studies have revealed structured microbial ecosystems within metastatic sites, challenging the notion of sterile tumors and positioning microbes as active co-drivers of metastasis58,201,202.
From a clinical perspective, acknowledging that tumor progression is governed by the dynamic interplay of entire microbial communities rather than single taxa fundamentally reshapes microbiome-targeted intervention strategies. Biomarker panels capturing consortium-level interactions have demonstrated superior predictive accuracy for CRC risk stratification compared with single-species indicators27,156,193. Beyond diagnostics, the rational design of programmable synthetic consortia is emerging as a transformative therapeutic platform, capable of tumor-specific recognition and localized delivery of anticancer agents while minimizing off-target effects. Incorporating multiple sensing and regulatory modules into bacterial chassis such as E. coli Nissle 1917 has enabled the controlled activation of therapeutic payloads within the TME, with enhanced efficacy when coupled to ecological modulation of the resident microbiota195,198. Crucially, the influence of microbial consortia extends beyond the primary tumor site. Circulating microbial metabolites and extracellular vesicles originating from tumor-associated communities can precondition distant organs, modulate systemic immune responses, and facilitate metastatic colonization181,191. This systemic dimension positions microbial consortia as potential architects of the “pre-metastatic niche,” creating functional links between gut microbial ecology, metastatic organotropism, and patient prognosis180,189.
Overall, the integrative actions of microbial consortia in tumor progression represent a shift from reductionist models toward a holistic ecological framework. In this view, the TME is a dynamic ecosystem co-constructed by tumor cells, host stroma, immune components, and complex bacterial communities whose interactions collectively determine disease trajectory. Dissecting the structure-function relationships within these consortia, leveraging high-resolution spatial metagenomics, metabolomics, and in situ functional assays, will be essential for translating mechanistic insights into precision diagnostics and microbiome-based therapeutics99,156,190. By harnessing or re-engineering these microbial networks, future strategies may transform the microbiome from an accomplice of cancer progression into an ally of cancer prevention and therapy.
Translational insights and microbiota-targeted therapeutic strategies
Finally, we translate these mechanistic layers into clinical opportunity—covering diet, pre/pro/postbiotics, FMT, and engineered consortia, as well as intratumoral microbiome considerations that modulate drug metabolism and immunotherapy efficacy. Figure 2 presents a hypothetical working model integrating microbiome-targeted therapeutic strategies designed to modulate these axes for CRC prevention and treatment. We foreground biomarker-guided patient selection and systems multi-omics to operationalize precision microbiome modulation in CRC.

This figure presents an integrative mechanistic and therapeutic model illustrating how modulation of the gut microbiome can reprogram neural, endocrine, immune, and metabolic pathways to counteract CRC progression. The neural pathway highlights microbiota–neuro–immune crosstalk where microbial disruption of epithelial junctions activates β-catenin, NF-κB, and STAT3 signaling, driving neuroinflammation and tumor-promoting innervation; therapeutic strategies include β-adrenergic blockade, SCFA-producing probiotics and postbiotics, vagal activation, and FMT restoring IL-10-producing commensals to reinforce epithelial integrity. The endocrine pathway emphasizes dysregulated GLP-1/PYY/5-HT signaling and bile acid metabolism, where interventions such as high-fiber and polyphenol-rich diets, FXR/TGR5 agonists, β-glucuronidase-inhibiting probiotics, metformin, and chrononutrition restore hormonal balance and barrier function. The immune pathway describes chronic inflammation, immune evasion, and MDSC recruitment driven by taxa like Fusobacterium nucleatum, ETBF, and pks⁺ E. coli; targeted depletion, immunostimulatory probiotics, FMT combined with checkpoint blockade, postbiotics, and anti-inflammatory nutrients (omega-3s, curcumin, resveratrol) reactivate cytotoxic immunity and suppress tumor-promoting inflammation. Finally, the metabolic pathway depicts mitochondrial inhibition, Warburg-like reprogramming, and oncometabolite accumulation through AhR activation and SASP induction; therapeutic strategies include prebiotics and engineered probiotics to enhance butyrate/propionate, colibactin synthesis inhibitors, modulation of microbial AhR ligands, antioxidants, and metabolic interventions to restore redox and immune–metabolic balance. Collectively, these interventions illustrate a systems-level therapeutic framework where microbiome-directed strategies reestablish epithelial, metabolic, immune, and neuroendocrine homeostasis, attenuating colorectal tumorigenesis.
Advances in microbiome science have rapidly transitioned from descriptive associations to actionable translational strategies aimed at improving cancer prevention, prognosis, and therapy. Preclinical and clinical evidence has established that the gut microbiota, as well as tumor-resident microbial communities, can modulate virtually every step of oncogenesis and treatment response, making microbial modulation a promising axis in precision oncology85,203,204,205. At the mechanistic level, specific commensals such as Akkermansia muciniphila, Bifidobacterium longum, and Faecalibacterium prausnitzii have been shown to augment antitumor immunity by enhancing antigen presentation, increasing cytotoxic T-cell infiltration, and optimizing interferon-γ signaling, thereby potentiating the efficacy of immune checkpoint inhibitors113,206,207,208. Conversely, pathobionts like Fusobacterium nucleatum and colibactin-producing Escherichia coli can drive tumor progression and therapy resistance via chronic inflammation, DNA alkylation, and modulation of autophagy pathways, underscoring the therapeutic imperative of targeted microbial control85,209,210,211.
From a translational perspective, dietary modulation, prebiotics, probiotics, postbiotics, and engineered bacterial therapeutics are emerging as clinically viable interventions. Diet-based approaches leveraging high-fiber, polyphenol-rich, and omega-3-enriched regimens have been linked to shifts in microbial metabolism toward increased production of SCFAs, which exert anti-inflammatory and epigenetic effects that may attenuate tumor progression200,212,213,214. Probiotic supplementation with strains such as Clostridium butyricum MIYAIRI 588 has demonstrated capacity to maintain epithelial integrity, suppress pro-oncogenic cytokine cascades, and extend progression-free survival in metastatic cancer patients undergoing immunotherapy, exemplifying the bench-to-bedside translation of microbial therapeutics85,215. In parallel, postbiotic administration, defined as the use of non-viable microbial products or metabolites, has shown potential to reduce systemic inflammation and modulate immune checkpoints without the colonization risks associated with live microbes210,213.
FMT represents one of the most potent microbiota-targeted strategies currently under clinical investigation. Early-phase trials have revealed that FMT from immunotherapy responders can reprogram the gut microbiome of non-responders, restoring sensitivity to PD-1 blockade and enhancing tumor regression203,207,208,216. Such findings have catalyzed efforts to refine FMT protocols, including donor selection based on microbial consortia signatures predictive of therapeutic benefit and the integration of pathogen-free, standardized consortia to improve safety and reproducibility206,208. Synthetic ecology approaches take this a step further, enabling the programmable assembly of microbial consortia tailored to deliver targeted payloads within the TME. Engineered E. coli Nissle 1917 and other chassis organisms have been outfitted with biosensors responsive to hypoxia, pH, or quorum-sensing signals, triggering localized release of cytotoxins, immune modulators, or oncolytic peptides with minimal systemic exposure85,204,216.
Therapeutic exploitation of microbial consortia requires an integrative approach that considers ecological interactions, temporal regulation, and host-microbiota crosstalk. Clinical translation is increasingly focusing on engineered bacterial chassis capable of sensing and responding to TME cues, synchronized with host circadian signals to optimize delivery and minimize off-target effects. Beyond classical probiotics, synthetic consortia and targeted phage cocktails are being developed to dismantle tumor-promoting networks while preserving beneficial microbial niches. These strategies align with advances in next-generation immunotherapy, including checkpoint blockade and adoptive cell transfer, underscoring the synergistic potential of microbiome–immune co-modulation217,218.
Beyond gut-targeted interventions, growing recognition of the intratumoral microbiome has opened a new translational frontier. Tumor-resident bacteria can directly metabolize chemotherapeutic agents, as observed with Gammaproteobacteria in pancreatic tumors inactivating gemcitabine, or modulate immune infiltration patterns that dictate treatment response118,194,209,210. This insight is informing adjuvant strategies such as localized antimicrobial delivery or the use of microbial enzyme inhibitors to preserve drug bioavailability. Likewise, modulation of microbial extracellular vesicles and metabolite signaling pathways offers a means to reprogram systemic immunity and metastatic niche formation, bridging local interventions to systemic outcomes85,204,207.
Clinical translation is also shaped by advances in multi-omics, which have enabled the integration of metagenomics, metatranscriptomics, metabolomics, and immunoprofiling to identify mechanistic biomarkers of therapeutic response200,219. Consortium-level biomarkers, rather than single taxa, are emerging as more robust predictors of treatment efficacy and adverse event risk, facilitating patient stratification for microbiota-guided therapy205,206,208. Integrating host pharmacogenomic and molecular biomarker networks with microbiome-derived signatures further refines this precision-oncology framework, linking microbial ecology to tumor genetics and drug response220. This systems-level perspective is driving a paradigm shift toward personalized microbial modulation, where therapeutic regimens are adapted not only to tumor genomics but also to individual microbial ecotypes203,207,212.
Despite these advances, challenges remain in standardizing interventions, mitigating off-target effects, and understanding long-term ecological consequences. Regulatory hurdles for live biotherapeutics, variability in manufacturing, and donor-to-donor microbiome heterogeneity in FMT underscore the need for stringent quality control and mechanistic validation204,212,213. Moreover, patient-specific factors, including baseline microbiome composition, antibiotic history, diet, and host genetics, must be integrated into therapeutic design to maximize efficacy and minimize risks203,209. Addressing these complexities will require adaptive trial designs, real-time microbiome monitoring, and iterative feedback loops between preclinical modeling and clinical implementation.
Looking ahead, the convergence of microbiome engineering, immuno-oncology, and precision medicine holds transformative potential. Hybrid strategies that combine microbial modulation with existing treatments, such as checkpoint blockade, targeted therapy, or radiotherapy, may achieve synergistic tumor control while reducing toxicity85,208,210. In parallel, next-generation probiotics and synthetic microbial consortia are being developed to deliver combination payloads that couple immune activation with metabolic rewiring of the TME, creating hostile conditions for tumor survival204,207,212. Ultimately, microbiota-targeted interventions are poised to become integral components of cancer care, shifting from adjunctive measures to central pillars of therapeutic regimens. The path forward will depend on rigorous mechanistic studies, biomarker-driven patient selection, and the seamless translation of multi-omics insights into clinically actionable strategies—ensuring that microbiome modulation fulfills its promise as a precision tool in oncology.
Conclusions and future perspectives
Over the past decade, the integration of microbiome science into oncology has progressed from descriptive associations to a mechanistic understanding that positions host–microbe interactions as a central axis in cancer biology. Cumulative evidence now establishes that the interplay between the microbiome and host mutational processes is not a peripheral phenomenon but a fundamental driver of oncogenesis49,55. Specific microbial taxa can directly shape the tumor microenvironment through the production of genotoxins, pro-inflammatory metabolites, and immune-modulatory molecules, thereby generating mutational landscapes that foster tumor initiation, progression, and therapeutic resistance176,195,203. These molecular alterations include defined mutational signatures, such as DNA DSBs, alkylation damage, and oxidative lesions, that map predictably onto cancer genomes, underscoring the need to incorporate microbiome analysis into standard genomic profiling of tumors to refine etiological classification and inform precision prevention strategies. At the same time, the broader microbial community exerts profound effects on tumor immunity and therapy responsiveness, acting as both a modulator of immune tone and a determinant of treatment outcomes55,77,221.
Advances in high-resolution metagenomics, metabolomics, spatial transcriptomics, and single-cell sequencing now enable the delineation of microbial signatures within the TME and at distant mucosal sites, revealing previously unrecognized niches of microbial colonization and metabolic activity9,10. These technologies have been instrumental in identifying how specific bacterial metabolites and toxins contribute to carcinogenesis, while also highlighting that cancer-associated dysbiosis is not merely a bystander but an active participant in tumor biology—deserving recognition alongside the canonical hallmarks of cancer12,222,223,224. Nevertheless, translating these insights into clinical oncology faces significant barriers, foremost among them the heterogeneity of microbial composition across individuals, driven by geography, diet, host genetics, antibiotic exposure, and comorbidities225. This variability complicates the search for universal microbial biomarkers and mandates precision-microbiome strategies capable of stratifying patients into biologically meaningful subgroups.
A persistent challenge remains the disentanglement of causality from correlation. While bacterial presence in tumors is well documented, definitive proof of direct carcinogenic activity in humans is still limited to a few pathogens, like Helicobacter pylori and certain Escherichia coli pathobionts, despite growing preclinical evidence for additional drivers. Longitudinal, geographically diverse cohort studies, combined with integrative multi-omics approaches, will be essential to establish temporal and mechanistic causality, particularly in cancers where bacterial contributions are suspected but not yet validated. These studies must also address the temporal dynamics of microbial colonization during tumor evolution, ensuring that interventions target relevant windows of susceptibility75,121.
From a translational perspective, microbial dysbiosis is increasingly recognized as a modifiable driver of carcinogenesis rather than a static correlate. Preclinical models demonstrate that interventions such as diet modulation, targeted probiotics, bacteriophage therapy, and small-molecule inhibitors of genotoxin biosynthesis can attenuate mutagenic pressure within the gut milieu144,158. In parallel, microbiome modulation is emerging as a dual therapeutic strategy—attenuating tumor-promoting bacteria while enriching taxa that potentiate antitumor immunity49,226. Early-phase clinical trials suggest that fecal microbiota transplantation, engineered probiotics, and dietary interventions can enhance responses to ICB, although inter-patient variability and regulatory uncertainties currently temper broad clinical adoption10. Moreover, bacterially derived toxins, such as Clostridium perfringens enterotoxin, are being repurposed as targeted cytotoxic agents that exploit tumor-specific receptor overexpression, offering a novel layer to precision oncology226.
The rapid convergence of synthetic biology and microbiome research offers unprecedented opportunities to design rationally engineered microbial consortia capable of delivering therapeutic payloads, producing immune-modulatory metabolites in situ, or competitively excluding oncogenic taxa within the TME. CRISPR-based editing platforms further expand these possibilities, allowing targeted in situ gene disruption of bacterial virulence factors or metabolic pathways implicated in carcinogenesis. Such living biotherapeutics, tailored to an individual’s tumor–microbiome ecology, could function as dynamic, self-sustaining adjuncts to conventional cancer therapies. While technically compelling, these approaches demand rigorous biosafety evaluation, particularly to mitigate risks of horizontal gene transfer, off-target ecological effects, and unanticipated immunogenicity55,98,121,225.
In diagnostics, microbial signatures hold considerable promise as minimally invasive biomarkers detectable in stool, saliva, or circulating tumor DNA–microbe hybrids, enabling earlier cancer detection and real-time monitoring of treatment responses9,77. The major challenge will be to distinguish clinically actionable microbial shifts from transient fluctuations unrelated to disease progression. Here, AI-driven analytics integrating host genomics, immune profiling, and microbial data could yield robust predictive models for cancer risk and therapeutic outcomes225. Importantly, incorporating spatial microbiome profiling within tumor tissues, rather than relying exclusively on fecal samples, will refine our understanding of microbe-tumor cell interactions, especially at invasive fronts and within metastatic niches.
Future research must expand its scope beyond bacteria to encompass the broader tumor-associated microbiome, including fungi, viruses, and archaea, whose roles in mutagenesis, immune modulation, and therapy resistance remain underexplored75,221. In oncology, circadian rhythms regulated by the suprachiasmatic nucleus and peripheral clocks influence immune surveillance, metabolism, and tissue homeostasis, while their disruption can promote tumor progression. The spatial organization of tumor-associated microbes, within biofilms, aggregates, or intracellular niches, further shapes cancer biology and therapy response. Integrating chronobiology with high-resolution spatial omics offers a novel avenue for microbiome-targeted and circadian-based interventions to restore rhythmicity, modulate tumor–microbe interactions, and enhance cancer treatment outcomes10,184,227,228.
Standardizing sample collection, sequencing platforms, and bioinformatic pipelines remains a prerequisite for clinical translation, ensuring cross-study comparability and reproducibility. Equally essential is the development of robust regulatory and ethical frameworks to govern live biotherapeutic products, standardize microbiome sampling protocols, and guarantee equitable access to microbiome-driven diagnostics and therapeutics49,225,229. Achieving these goals will require sustained interdisciplinary collaboration among microbiologists, oncologists, immunologists, bioinformaticians, and regulatory bodies to accelerate the path from proof-of-concept to routine clinical application.
In sum, the microbiome has transitioned from a hidden variable in cancer biology to a central, targetable component of the oncogenic process. Its influence spans the continuum of tumorigenesis—from initiation via genotoxic and metabolic insults, through immune modulation in tumor promotion, to shaping metastatic potential and therapy responsiveness55,77,226. As advances in technology enhance our capacity to profile, modulate, and leverage microbial communities, microbiome-based strategies are set to transition from experimental approaches to core components of personalized oncology. Over the next decade, the field is poised for clinical maturation, contingent upon overcoming translational and regulatory barriers with the same innovation and scientific rigor that have fueled its rapid emergence.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Arrieta, M.-C. et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 7, 307ra152 (2015).
Tamburini, S., Shen, N., Wu, H. C. & Clemente, J. C. The microbiome in early life: implications for health outcomes. Nat. Med. 22, 713–722 (2016).
Bautista, J. & López-Cortés, A. Microbial beginnings: determinants and disruptions of the neonatal microbiome. Front. Microbiol. 16, (2025).
Senchukova, M. A. Microbiota of the gastrointestinal tract: friend or foe?. World J. Gastroenterol. 29, 19–42 (2023).
Wu, S., Lim, K. C., Huang, J., Saidi, R. F. & Sears, C. L. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. USA 95, 14979–14984 (1998).
Rubinstein, M. R. et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206 (2013).
Pleguezuelos-Manzano, C. et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580, 269–273 (2020).
Chung, L. et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 23, 203–214.e5 (2018).
He, Z. et al. Campylobacter jejuni-derived cytolethal distending toxin promotes colorectal cancer metastasis. Cell Host Microbe 32, 2080–2091.e6 (2024).
Fulbright, L. E., Ellermann, M. & Arthur, J. C. The microbiome and the hallmarks of cancer. PLoS Pathog. 13, e1006480 (2017).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Byrd, A. L. & Segre, J. A. Infectious disease. Adapting Koch’s postulates. Science 351, 224–226 (2016).
Schmidt, T. S. B., Raes, J. & Bork, P. The human gut microbiome: from association to modulation. Cell 172, 1198–1215 (2018).
Dejea, C. M. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597 (2018).
Tomkovich, S. et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J. Clin. Invest. 129, 1699–1712 (2019).
Dejea, C. M. et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl. Acad Sci USA 111, 18321–18326 (2014).
Drewes, J. L. et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. npj Biofilms Microbiomes 3, 34 (2017).
Kvich, L. et al. Biofilms and core pathogens shape the tumor microenvironment and immune phenotype in colorectal cancer. Gut Microbes 16, 2350156 (2024).
Johnson, C. H. et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 21, 891–897 (2015).
Okumura, S. et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 12, 5674 (2021).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298 (2012).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 (2012).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022 (2009).
Abdulamir, A. S., Hafidh, R. R. & Abu Bakar, F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J. Exp. Clin. Cancer Res. 30, 11 (2011).
Wirbel, J. et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679–689 (2019).
Wang, T. et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 6, 320–329 (2012).
Li, S. et al. Tumorigenic bacteria in colorectal cancer: mechanisms and treatments. Cancer Biol. Med. 19, 147–162 (2021).
Ternes, D. et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab. 4, 458–475 (2022).
Lopez, L. R., Bleich, R. M. & Arthur, J. C. Microbiota effects on carcinogenesis: initiation, promotion, and progression. Annu. Rev. Med. 72, 243–261 (2021).
Yang, X. et al. Microbial genotoxin-elicited host DNA mutations related to mitochondrial dysfunction, a momentous contributor for colorectal carcinogenesis. mSystems 9, e0088724 (2024).
Zechner, E. L. & Kienesberger, S. Microbiota-derived small molecule genotoxins: host interactions and ecological impact in the gut ecosystem. Gut Microbes 16, 2430423 (2024).
Tortora, S. C., Bodiwala, V. M., Quinn, A., Martello, L. A. & Vignesh, S. Microbiome and colorectal carcinogenesis: linked mechanisms and racial differences. World J. Gastrointest. Oncol. 14, 375–395 (2022).
Liu, Y., Lau, H. C.-H. & Yu, J. Microbial metabolites in colorectal tumorigenesis and cancer therapy. Gut Microbes 15, 2203968 (2023).
Chen, H.-Y. et al. Mitochondrial injury induced by a Salmonella genotoxin triggers the proinflammatory senescence-associated secretory phenotype. Nat. Commun. 15, 2778 (2024).
Secher, T., Samba-Louaka, A., Oswald, E. & Nougayrède, J.-P. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS ONE 8, e77157 (2013).
Azzi-Martin, L. et al. Cytolethal distending toxin induces the formation of transient messenger-rich ribonucleoprotein nuclear invaginations in surviving cells. PLoS Pathog. 15, e1007921 (2019).
Permain, J., Hock, B., Eglinton, T. & Purcell, R. Functional links between the microbiome and the molecular pathways of colorectal carcinogenesis. Cancer Metastasis Rev. 43, 1463–1474 (2024).
Gur, C. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355 (2015).
Coutzac, C. et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 11, 2168 (2020).
Ahmad, A. et al. Gut microbiota and their derivatives in the progression of colorectal cancer: mechanisms of action, genome and epigenome contributions. Heliyon 10, e29495 (2024).
Belcheva, A. et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288–299 (2014).
Nakatsu, G. et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 6, 8727 (2015).
Nougayrède, J.-P. et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851 (2006).
Rosendahl Huber, A. et al. Improved detection of colibactin-induced mutations by genotoxic E. coli in organoids and colorectal cancer. Cancer Cell 42, 487–496.e6 (2024).
Strakova, N., Korena, K. & Karpiskova, R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development—a systematic review. Toxicon 197, 126–135 (2021).
Zhang, G. & Sun, D. The synthesis of the novel Escherichia coli toxin-colibactin and its mechanisms of tumorigenesis of colorectal cancer. Front. Microbiol. 15, 1501973 (2024).
Berger, H. & Meyer, T. F. Mechanistic dissection unmasks colibactin as a prevalent mutagenic driver of cancer. Cancer Cell 39, 1439–1441 (2021).
Lythgoe, M. P., Mullish, B. H., Frampton, A. E. & Krell, J. Polymorphic microbes: a new emerging hallmark of cancer. Trends Microbiol. 30, 1131–1134 (2022).
Dougherty, M. W. et al. The microbial genotoxin colibactin exacerbates mismatch repair mutations in colorectal tumors. Neoplasia 43, 100918 (2023).
Díaz-Gay, M. et al. Geographic and age variations in mutational processes in colorectal cancer. Nature 643, 230–240 (2025).
Sogari, A. et al. Tolerance to colibactin correlates with homologous recombination proficiency and resistance to irinotecan in colorectal cancer cells. Cell Rep. Med. 5, 101376 (2024).
Gaab, M. E. et al. A meta-analysis on the association of colibactin-producing pks+ Escherichia coli with the development of colorectal cancer. Lab. Med. 54, 75–82 (2023).
Chagneau, C. V. et al. The pks island: a bacterial Swiss army knife? Colibactin: beyond DNA damage and cancer. Trends Microbiol. 30, 1146–1159 (2022).
Arima, K. et al. Western-style diet, pks island-carrying Escherichia coli, and colorectal cancer: analyses from two large prospective cohort studies. Gastroenterology 163, 862–874 (2022).
Dziubańska-Kusibab, P. J. et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 26, 1063–1069 (2020).
Bautista, J., Fuentes-Yépez, M. P., Adatty-Molina, J. & López-Cortés, A. Microbial signatures in metastatic cancer. Front. Med. 12, (2025).
Haghi, F., Goli, E., Mirzaei, B. & Zeighami, H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer 19, 879 (2019).
Scott, N., Whittle, E., Jeraldo, P. & Chia, N. A systemic review of the role of enterotoxic Bacteroides fragilis in colorectal cancer. Neoplasia 29, 100797 (2022).
Purcell, R. V., Permain, J. & Keenan, J. I. Enterotoxigenic Bacteroides fragilis activates IL-8 expression through Stat3 in colorectal cancer cells. Gut Pathog. 14, 16 (2022).
Hwang, S., Gwon, S.-Y., Kim, M. S., Lee, S. & Rhee, K.-J. Bacteroides fragilis toxin induces IL-8 secretion in HT29/C1 cells through disruption of E-cadherin junctions. Immune Netw. 13, 213–217 (2013).
Thiele Orberg, E. et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 10, 421–433 (2017).
Cao, Y. et al. Enterotoxigenic Bacteroidesfragilis promotes intestinal inflammation and malignancy by inhibiting exosome-packaged miR-149-3p. Gastroenterology 161, 1552–1566.e12 (2021).
Wu, Z., Yu, M., Zeng, Y., Huang, Y. & Zheng, W. LRP11-AS1 mediates enterotoxigenic Bacteroides fragilis-related carcinogenesis in colorectal Cancer via the miR-149-3p/CDK4 pathway. Cancer Gene Ther. 32, 184–197 (2025).
Ding, X. et al. Bacteroides fragilis promotes chemoresistance in colorectal cancer, and its elimination by phage VA7 restores chemosensitivity. Cell Host Microbe 33, 941–956.e10 (2025).
Kordahi, M. C. et al. Genomic and functional characterization of a mucosal symbiont involved in early-stage colorectal cancer. Cell Host Microbe 29, 1589–1598.e6 (2021).
Tremblay, W. et al. Cytolethal distending toxin promotes replicative stress leading to genetic instability transmitted to daughter cells. Front. Cell Dev. Biol. 9, 656795 (2021).
Graillot, V. et al. Genotoxicity of cytolethal distending toxin (CDT) on isogenic human colorectal cell lines: potential promoting effects for colorectal carcinogenesis. Front. Cell. Infect. Microbiol. 6, 34 (2016).
Chen, H., Ang, C. J., Crowder, M. K., Brieher, W. M. & Blanke, S. R. Revisiting bacterial cytolethal distending toxin structure and function. Front. Cell. Infect. Microbiol. 13, 1289359 (2023).
Lai, Y.-R. et al. From DNA damage to cancer progression: potential effects of cytolethal distending toxin. Front. Immunol. 12, 760451 (2021).
Bezine, E. et al. Cell resistance to the Cytolethal Distending Toxin involves an association of DNA repair mechanisms. Sci. Rep. 6, 36022 (2016).
Pons, B. J. et al. Chronic exposure to Cytolethal Distending Toxin (CDT) promotes a cGAS-dependent type I interferon response. Cell. Mol. Life Sci. 78, 6319–6335 (2021).
Fedor, Y. et al. From single-strand breaks to double-strand breaks during S-phase: a new mode of action of the Escherichia coli Cytolethal Distending Toxin. Cell. Microbiol. 15, 1–15 (2013).
El Tekle, G. & Garrett, W. S. Bacteria in cancer initiation, promotion and progression. Nat. Rev. Cancer 23, 600–618 (2023).
Jinadasa, R. N., Bloom, S. E., Weiss, R. S. & Duhamel, G. E. Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157, 1851–1875 (2011).
Martin, O. C. B. et al. Influence of the microenvironment on modulation of the host response by typhoid toxin. Cell Rep. 35, 108931 (2021).
Miller, R. A. et al. The typhoid toxin produced by the nontyphoidal Salmonella enterica serotype javiana is required for induction of a DNA damage response in vitro and systemic spread in vivo. MBio 9, e00467-18 (2018).
Song, J., Gao, X. & Galán, J. E. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature 499, 350–354 (2013).
Ibler, A. E. M. et al. Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 10, 4040 (2019).
ElGhazaly, M., Collins, M. O., Ibler, A. E. M. & Humphreys, D. Typhoid toxin hijacks Wnt5a to establish host senescence and Salmonella infection. Cell Rep. 42, 113181 (2023).
Mughini-Gras, L. et al. Increased colon cancer risk after severe Salmonella infection. PLoS ONE 13, e0189721 (2018).
Chang, S.-J., Jin, S. C., Jiao, X. & Galán, J. E. Unique features in the intracellular transport of typhoid toxin revealed by a genome-wide screen. PLoS Pathog. 15, e1007704 (2019).
Stévenin, V. et al. Multi-omics analyses of cancer-linked clinical salmonellae reveal bacterial-induced host metabolic shift and mTOR-dependent cell transformation. Cell Rep. 43, 114931 (2024).
Zhao, L.-Y. et al. Role of the gut microbiota in anticancer therapy: from molecular mechanisms to clinical applications. Signal Transduct. Target. Ther. 8, 201 (2023).
Cao, Y. et al. Commensal microbiota from patients with inflammatory bowel disease produce genotoxic metabolites. Science 378, eabm3233 (2022).
Patel, D. et al. Induction of AHR signaling in response to the indolimine class of microbial stress metabolites. Metabolites 13, 985 (2023).
Glinghammar, B., Inoue, H. & Rafter, J. J. Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors NF-kB and AP-1. Carcinogenesis 23, 839–845 (2002).
Wu, H. et al. Gut microbiome-metabolites axis: a friend or foe to colorectal cancer progression. Biomed. Pharmacother. 173, 116410 (2024).
Fu, T. et al. Paired microbiome and metabolome analyses associate bile acid changes with colorectal cancer progression. Cell Rep. 42, 112997 (2023).
Lin, H. et al. Implications of hydrogen sulfide in colorectal cancer: mechanistic insights and diagnostic and therapeutic strategies. Redox Biol. 59, 102601 (2023).
Attene-Ramos, M. S., Wagner, E. D., Plewa, M. J. & Gaskins, H. R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 4, 9–14 (2006).
Andriamihaja, M. et al. The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free Radic. Biol. Med. 85, 219–227 (2015).
Fike, L. T., Munro, H., Yu, D., Dai, Q. & Shrubsole, M. J. Dietary polyphenols and the risk of colorectal cancer in the prospective Southern Community Cohort Study. Am. J. Clin. Nutr. 115, 1155–1165 (2022).
Liu, Y. et al. Secondary bile acids and tumorigenesis in colorectal cancer. Front. Oncol. 12, 813745 (2022).
Moon, J.-Y., Kye, B.-H., Ko, S.-H. & Yoo, R. N. Sulfur metabolism of the gut microbiome and colorectal cancer: the threat to the younger generation. Nutrients 15, 1966 (2023).
Grellier, N. et al. Gut microbiota in inflammation and colorectal cancer: a potential Toolbox for Clinicians. Best Pract. Res. Clin. Gastroenterol. 72, 101942 (2024).
Hanus, M. et al. Immune system, microbiota, and microbial metabolites: the unresolved triad in colorectal cancer microenvironment. Front. Immunol. 12, 612826 (2021).
Karam, F., El Deghel, Y., Iratni, R., Dakroub, A. H. & Eid, A. H. The gut microbiome and colorectal cancer: an integrative review of the underlying mechanisms. Cell Biochem. Biophys. https://doi.org/10.1007/s12013-025-01683-9 (2025).
Sulit, A. K. et al. Bacterial lipopolysaccharide modulates immune response in the colorectal tumor microenvironment. npj Biofilms Microbiomes 9, 59 (2023).
Liu, H., Redline, R. W. & Han, Y. W. Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4-mediated placental inflammatory response. J. Immunol. 179, 2501–2508 (2007).
Casasanta, M. A. et al. Fusobacterium nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci. Signal. 13, eaba9157 (2020).
Chen, Y. et al. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 8, 31802–31814 (2017).
Mima, K. et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 65, 1973–1980 (2016).
Xing, C. et al. Interaction between microbiota and immunity and its implication in colorectal cancer. Front. Immunol. 13, 963819 (2022).
Ivleva, E. A. & Grivennikov, S. I. Microbiota-driven mechanisms at different stages of cancer development. Neoplasia 32, 100829 (2022).
Brennan, C. A. & Garrett, W. S. Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol. 70, 395–411 (2016).
Sieminska, I. & Baran, J. Myeloid-derived suppressor cells in colorectal cancer. Front. Immunol. 11, 1526 (2020).
Xie, M., Li, X., Lau, H. C.-H. & Yu, J. The gut microbiota in cancer immunity and immunotherapy. Cell. Mol. Immunol. 22, 1012–1031 (2025).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Li, Q. et al. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct. Target. Ther. 9, 266 (2024).
Biswas, S. K. & Lopez-Collazo, E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 (2009).
Sivan, A. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017).
Netea, M. G. et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388 (2020).
Parker, K. H., Beury, D. W. & Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv. Cancer Res. 128, 95–139 (2015).
Geller, L. T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 (2017).
Jyoti & Dey, P. Mechanisms and implications of the gut microbial modulation of intestinal metabolic processes. npj Metab. Health Dis. 3, 24 (2025).
Cao, C., Yue, S., Lu, A. & Liang, C. Host-gut microbiota metabolic interactions and their role in precision diagnosis and treatment of gastrointestinal cancers. Pharmacol. Res. 207, 107321 (2024).
Zhang, W. et al. Gut microbiota-derived metabolites in colorectal cancer: the bad and the challenges. Front. Oncol. 11, 739648 (2021).
Wang, M. et al. Amelioration of AOM/DSS-induced murine colitis-associated cancer by evodiamine intervention is primarily associated with gut microbiota-metabolism-inflammatory signaling axis. Front. Pharmacol. 12, 797605 (2021).
Qu, R. et al. Role of the gut microbiota and its metabolites in tumorigenesis or development of colorectal cancer. Adv. Sci. 10, e2205563 (2023).
Bhalla, P., Rengaswamy, R., Karunagaran, D., Suraishkumar, G. K. & Sahoo, S. Metabolic modeling of host-microbe interactions for therapeutics in colorectal cancer. NPJ Syst. Biol. Appl. 8, 1 (2022).
Elangovan, S. et al. The niacin/butyrate receptor GPR109A suppresses mammary tumorigenesis by inhibiting cell survival. Cancer Res. 74, 1166–1178 (2014).
Cui, W., Hao, M., Yang, X., Yin, C. & Chu, B. Gut microbial metabolism in ferroptosis and colorectal cancer. Trends Cell Biol. 35, 341–351 (2025).
Spanogiannopoulos, P. et al. Host and gut bacteria share metabolic pathways for anti-cancer drug metabolism. Nat. Microbiol. 7, 1605–1620 (2022).
Zheng, X. et al. ANGPTL4-mediated promotion of glycolysis facilitates the colonization of Fusobacterium nucleatum in colorectal cancer. Cancer Res. 81, 6157–6170 (2021).
Zhao, L. et al. Impacts and mechanisms of metabolic reprogramming of tumor microenvironment for immunotherapy in gastric cancer. Cell Death Dis. 13, 378 (2022).
Romero-Garcia, S., Moreno-Altamirano, M. M. B., Prado-Garcia, H. & Sánchez-García, F. J. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 7, 52 (2016).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
Gao, J. et al. Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol. 8, 13 (2018).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 8, 80–93 (2015).
Nakamura, A. et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat. Commun. 12, 2105 (2021).
Holbert, C. E., Cullen, M. T., Casero, R. A. & Stewart, T. M. Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat. Rev. Cancer 22, 467–480 (2022).
Chen, C., Su, Q., Zi, M., Hua, X. & Zhang, Z. Harnessing gut microbiota for colorectal cancer therapy: from clinical insights to therapeutic innovations. npj Biofilms Microbiomes 11, 190 (2025).
Alpert, O., Begun, L., Issac, T. & Solhkhah, R. The brain-gut axis in gastrointestinal cancers. J. Gastrointest. Oncol. 12, S301–S310 (2021).
Hou, K. et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 7, 135 (2022).
Kaelberer, M. M. et al. A gut-brain neural circuit for nutrient sensory transduction. Science 361, eaat5236 (2018).
Geng, Z.-H., Zhu, Y., Li, Q.-L., Zhao, C. & Zhou, P.-H. Enteric nervous system: the bridge between the gut microbiota and neurological disorders. Front. Aging Neurosci. 14, 810483 (2022).
Coquenlorge, S. et al. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflammation 11, 202 (2014).
Yano, J. M. et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015).
Zhu, P. et al. 5-hydroxytryptamine produced by enteric serotonergic neurons initiates colorectal cancer stem cell self-renewal and tumorigenesis. Neuron 110, 2268–2282.e4 (2022).
Zeng, Z. et al. Neural influences in colorectal cancer progression and therapeutic strategies. Int. J. Colorectal Dis. 40, 120 (2025).
Marshall, C. L. et al. Perineural invasion correlates with neurogenesis in colorectal cancer. J. Surg. Res. 165, 299–300 (2011).
Wu, H. et al. The gut-brain axis in the context of colorectal cancer. Pharmacol. Res. 217, 107816 (2025).
Cheng, K., Shang, A. C., Drachenberg, C. B., Zhan, M. & Raufman, J.-P. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 8, 21106–21114 (2017).
Coelho, M. et al. β-Adrenergic modulation of cancer cell proliferation: available evidence and clinical perspectives. J. Cancer Res. Clin. Oncol. 143, 275–291 (2017).
Lyu, Y. et al. The nerve cells in gastrointestinal cancers: from molecular mechanisms to clinical intervention. Oncogene 43, 77–91 (2024).
Xu, Y. et al. Biglycan regulated colorectal cancer progress by modulating enteric neuron-derived IL-10 and abundance of Bacteroides thetaiotaomicron. iScience 26, 107515 (2023).
Wang, X. et al. Bacteroides methylmalonyl-CoA mutase produces propionate that promotes intestinal goblet cell differentiation and homeostasis. Cell Host Microbe 32, 63–78.e7 (2024).
Barot, S. V. et al. Distinct intratumoral microbiome of young-onset and average-onset colorectal cancer. EBioMedicine 100, 104980 (2024).
Zhou, P., Yang, D., Sun, D. & Zhou, Y. Gut microbiome: new biomarkers in early screening of colorectal cancer. J. Clin. Lab. Anal. 36, e24359 (2022).
Lamaudière, M. T. F., Arasaradnam, R., Weedall, G. D. & Morozov, I. Y. The colorectal cancer microbiota alter their transcriptome to adapt to the acidity, reactive oxygen species, and metabolite availability of gut microenvironments. mSphere 8, e0062722 (2023).
Huang, J. et al. Metagenomic and metabolomic analyses reveal synergistic effects of fecal microbiota transplantation and anti-PD-1 therapy on treating colorectal cancer. Front. Immunol. 13, 874922 (2022).
Minot, S. S. et al. Colorectal cancer-associated bacteria are broadly distributed in global microbiomes and drivers of precancerous change. Sci. Rep. 14, 23646 (2024).
Fong, W., Li, Q. & Yu, J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene 39, 4925–4943 (2020).
Pu, T., Sun, J., Ren, G. & Li, H. Neuro-immune crosstalk in cancer: mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 10, 176 (2025).
Bautista, J. et al. Neurodegeneration rewires the tumor microenvironment via the neuro-immune-cancer axis. iScience 28, 113550 (2025).
Wang, H. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384–388 (2003).
Voss, U. & Ekblad, E. Lipopolysaccharide-induced loss of cultured rat myenteric neurons—role of AMP-activated protein kinase. PLoS ONE 9, e114044 (2014).
Yuan, R. et al. Enteric glia play a critical role in promoting the development of colorectal cancer. Front. Oncol. 10, 595892 (2020).
Zhang, W. et al. Gut-innervating nociceptors regulate the intestinal microbiota to promote tissue protection. Cell 185, 4170–4189.e20 (2022).
Bautista, J., Cardona Maya, W. D., Gancino, K., Criollo, M. & López-Cortés, A. Reprogramming prostate cancer through the microbiome. Front. Med. 12, https://doi.org/10.3389/fmed.2025.1690498 (2025).
Song, C.-H. et al. Anti-PD-L1 antibody and/or 17β-estradiol treatment induces changes in the gut microbiome in MC38 colon tumor model. Cancer Res. Treat. 55, 894–909 (2023).
Greiner, T. U. & Bäckhed, F. Microbial regulation of GLP-1 and L-cell biology. Mol. Metab. 5, 753–758 (2016).
Yoon, K. & Kim, N. Roles of sex hormones and gender in the gut microbiota. J. Neurogastroenterol. Motil. 27, 314–325 (2021).
Larraufie, P. et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci. Rep. 8, 74 (2018).
Nikolaou, S. et al. The prognostic and therapeutic role of hormones in colorectal cancer: a review. Mol. Biol. Rep. 46, 1477–1486 (2019).
Hawkins, M. L. et al. Endocrine and metabolic diseases among colorectal cancer survivors in a population-based cohort. J. Natl. Cancer Inst. 112, 78–86 (2020).
Zhou, M. et al. Farnesoid-X receptor as a therapeutic target for inflammatory bowel disease and colorectal cancer. Front. Pharmacol. 13, 1016836 (2022).
Payne, C. M. et al. Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis 28, 215–222 (2007).
Wu, Z. et al. Direct and indirect effects of estrogens, androgens and intestinal microbiota on colorectal cancer. Front. Cell. Infect. Microbiol. 14, 1458033 (2024).
Botteri, E. et al. Menopausal hormone therapy and colorectal cancer: a linkage between nationwide registries in Norway. BMJ Open 7, e017639 (2017).
Nguyen-Vu, T. et al. Estrogen receptor beta reduces colon cancer metastasis through a novel miR-205 - PROX1 mechanism. Oncotarget 7, 42159–42171 (2016).
Wu, Z. et al. Sex differences in colorectal cancer: with a focus on sex hormone-gut microbiome axis. Cell Commun. Signal. 22, 167 (2024).
Vemuri, R. et al. The microgenderome revealed: sex differences in bidirectional interactions between the microbiota, hormones, immunity and disease susceptibility. Semin. Immunopathol. 41, 265–275 (2019).
Novoa Díaz, M. B., Carriere, P. & Gentili, C. How the interplay among the tumor microenvironment and the gut microbiota influences the stemness of colorectal cancer cells. World J. Stem Cells 15, 281–301 (2023).
Denos, M. et al. Sex hormones and risk of lung and colorectal cancers in women: a Mendelian randomization study. Sci. Rep. 14, 23891 (2024).
Wong, S. H. & Yu, J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 16, 690–704 (2019).
Kumar, R., Kaur, A. & Rajput, D. An insight on gut flora, colorectal cancer mechanism, and treatment strategies. Adv. Gut Microbiome Res. 2023, 1–16 (2023).
Thomas, A. M. et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678 (2019).
Li, S., Konstantinov, S. R., Smits, R. & Peppelenbosch, M. P. Bacterial biofilms in colorectal cancer initiation and progression. Trends Mol. Med. 23, 18–30 (2017).
Bautista, J., Ojeda-Mosquera, S., Ordóñez-Lozada, D. & López-Cortés, A. Peripheral clocks and systemic zeitgeber interactions: from molecular mechanisms to circadian precision medicine. Front. Endocrinol. 16, 1606242 (2025).
Bautista, J. et al. Bidirectional interactions between circadian rhythms and the gut microbiome. Appl. Microbiol. Biotechnol. 109, 218 (2025).
Fagiani, F. et al. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct. Target. Ther. 7, 41 (2022).
Thaiss, C. A. et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159, 514–529 (2014).
Fellows, R. C. et al. Disruption of the intestinal clock drives dysbiosis and impaired barrier function in colorectal cancer. Sci. Adv 10, eado1458 (2024).
Jing, Z. et al. Interaction between gut microbiota and T cell immunity in colorectal cancer. Autoimmun. Rev. 24, 103807 (2025).
Long, J. et al. Intratumoral microbiota in colorectal cancer: focus on specific distribution and potential mechanisms. Cell Commun. Signal. 22, 455 (2024).
Wu, Y. et al. Dynamic interplay of intratumoral microbiota within the colorectal cancer microenvironment. VIEW 5 (2024).
Li, J.-X., Liu, C.-G. & Chen, J. Gut microbes and their metabolites in relation to the classic tumor suppressor gene P53: a novel narrative. npj Biofilms Microbiomes 11, 135 (2025).
Song, M. & Chan, A. T. Environmental factors, gut microbiota, and colorectal cancer prevention. Clin. Gastroenterol. Hepatol. 17, 275–289 (2019).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Zhou, T. et al. Enhancing tumor-specific recognition of programmable synthetic bacterial consortium for precision therapy of colorectal cancer. npj Biofilms Microbiomes 10, 6 (2024).
Clark, R. L. et al. Design of synthetic human gut microbiome assembly and butyrate production. Nat. Commun. 12, 3254 (2021).
Bautista, J. et al. The human microbiome in clinical translation: from bench to bedside. Front. Microbiol. 16, 1632435 (2025).
Din, M. O. et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536, 81–85 (2016).
López-Cortés, A. et al. The close interaction between hypoxia-related proteins and metastasis in pancarcinomas. Sci. Rep. 12, 11100 (2022).
Ocaña-Paredes, B. et al. The pharmacoepigenetic paradigm in cancer treatment. Front. Pharmacol. 15, 1381168 (2024).
Bautista, J. & López-Cortés, A. Deciphering organotropism reveals therapeutic targets in metastasis. Front. Cell Dev. Biol. 13, 1677481 (2025).
Wang, Y. et al. Pre-metastatic niche: formation, characteristics and therapeutic implication. Signal Transduct. Target. Ther. 9, 236 (2024).
Wang, Y. & Li, H. Gut microbiota modulation: a tool for the management of colorectal cancer. J. Transl. Med. 20, 178 (2022).
Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 21, 1325 (2021).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Kim, J. & Lee, H. K. Potential role of the gut microbiome in colorectal cancer progression. Front. Immunol. 12, 807648 (2021).
Chen, M. et al. Therapeutic approaches to colorectal cancer via strategies based on modulation of gut microbiota. Front. Microbiol. 13, 945533 (2022).
Sun, J. et al. Gut microbiota as a new target for anticancer therapy: from mechanism to means of regulation. npj Biofilms Microbiomes 11, 43 (2025).
Bai, X., Liu, B., Fan, D., Lu, Y. & Zhao, X. Modulating the gut microbiota: a novel perspective in colorectal cancer treatment. Cancer Lett. 612, 217459 (2025).
Roy, B. et al. Advances in gut microbiota-related treatment strategies for managing colorectal cancer in humans. Cancer Biol. Med. 22, 93–112 (2025).
Yu, T. et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170, 548–563.e16 (2017).
Lang, T. et al. Combining gut microbiota modulation and chemotherapy by capecitabine-loaded prebiotic nanoparticle improves colorectal cancer therapy. Nat. Commun. 14, 4746 (2023).
Lu, L. et al. New insights into natural products that target the gut microbiota: effects on the prevention and treatment of colorectal cancer. Front. Pharmacol. 13, 964793 (2022).
O’Keefe, S. J. D. et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 6, 6342 (2015).
Dizman, N. et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat. Med. 28, 704–712 (2022).
Davar, D. et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602 (2021).
Kyriakidis, N. C. et al. Reprogramming cancer immunity with next-generation combination therapies. Front. Cell Dev. Biol. 13, 1652047 (2025).
Bautista, J., Villegas-Chávez, J. A., Bunces-Larco, D., Martín-Aguilera, R. & López-Cortés, A. The microbiome as a therapeutic co-driver in melanoma immuno-oncology. Front. Med. 12, 1673880 (2025).
García-Cárdenas, J. M. et al. Data mining identifies novel RNA-binding proteins involved in colon and rectal carcinomas. Front. Cell Dev. Biol. 11, 1088057 (2023).
López-Cortés, A. et al. Pharmacogenomics, biomarker network, and allele frequencies in colorectal cancer. Pharmacogenomics J. 20, 136–158 (2020).
Bautista, J. & Lopez-Cortes, A. Oncogenic viruses rewire the epigenome in human cancer. Front. Cell. Infect. Microbiol. 15, 1617198 (2025).
Zhu, B. et al. A multi-omics spatial framework for host-microbiome dissection within the intestinal tissue microenvironment. Nat. Commun. 16, 1230 (2025).
Zhang, N., Kandalai, S., Zhou, X., Hossain, F. & Zheng, Q. Applying multi-omics toward tumor microbiome research. iMeta 2, e73 (2023).
Hsieh, W.-C. et al. Spatial multi-omics analyses of the tumor immune microenvironment. J. Biomed. Sci. 29, 96 (2022).
El Tekle, G., Andreeva, N. & Garrett, W. S. The role of the microbiome in the etiopathogenesis of colon cancer. Annu. Rev. Physiol. 86, 453–478 (2024).
Pahle, J. et al. Rapid eradication of colon carcinoma by Clostridium perfringens Enterotoxin suicidal gene therapy. BMC Cancer 17, 129 (2017).
Pérez-Villa, A. et al. Integrated multi-omics analysis reveals the molecular interplay between circadian clocks and cancer pathogenesis. Sci. Rep. 13, 14198 (2023).
Bautista, J. & López-Cortés, A. Chronobiome medicine: circadian regulation of host–microbiota crosstalk in systemic physiology. Front. Endocrinol. 16, 1691172 (2025).
Bautista, J., Maldonado-Noboa, I., Maldonado-Guerrero, D., Reinoso-Quinga, L. & López-Cortés, A. Microbiome influence in gastric cancer progression and therapeutic strategies. Front. Med. 12, 1681824 (2025).
Acknowledgements
This work was supported by Universidad de Las Américas (UDLA) from Quito, Ecuador.
Author information
Authors and Affiliations
Contributions
J.B. and A.L.C. conceived the subject and the conceptualization of the study. All authors did data curation, gave conceptual advice, and valuable scientific input. A.L.C. supervised and did funding acquisition. Finally, all authors wrote, reviewed, edited, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bautista, J., Lamas-Maceiras, M., Hidalgo-Tinoco, C. et al. Gut microbiome–driven colorectal cancer via immune, metabolic, neural, and endocrine axes reprogramming. npj Biofilms Microbiomes 12, 21 (2026). https://doi.org/10.1038/s41522-025-00883-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00883-8
