
Abstract
Circulating tumor DNA (ctDNA) has potential as a prognostic factor for predicting relapse in high-risk breast cancer (BC). This study investigates the utility of ctDNA assessment using a tumor-informed assay, in patients with high-risk BC treated with neoadjuvant chemotherapy (NAC). Thirty newly diagnosed patients with various high-risk BC subtypes participated, providing serial blood samples at multiple time points, including baseline, during NAC, and during follow-up. ctDNA was detected at baseline in 29/29 patients for whom an assay panel could be designed, with detection sensitivity reaching 0.0083% (variant allele frequency). Among patients with detectable baseline ctDNA, 94% showed clearance during treatment, correlating with improved outcomes. Additionally, ctDNA detection post-surgery or during follow-up predicted disease recurrence. These findings suggest that serial ctDNA monitoring throughout NAC and follow-up can effectively identify residual disease in BC and correlate with clinical outcomes.
Similar content being viewed by others

Introduction
Neoadjuvant chemotherapy (NAC) is the standard of care in high-risk early-stage and locally advanced breast cancer (BC), particularly in HER2-positive and triple-negative breast cancer (TNBC) cases1. Current protocols combine NAC with HER2-targeted antibodies or immune checkpoint inhibitors, respectively. NAC may also be indicated in hormone receptor-positive (HR+/HER2-) BC to facilitate breast conservation and avoid an axillary dissection. The efficacy of NAC is primarily assessed through clinical and radiological examinations, and the degree of pathological response after surgery.
Pathological complete response (pCR) is currently the main prognostic factor guiding adjuvant therapies in HER2-positive and TNBC patients after NAC, with treatment escalation recommended in cases of non-pCR2,3,4. While pCR is strongly correlated with improved outcomes, recurrences can still occur. Similarly, not achieving a pCR is far from being an absolute predictor of recurrence, as many patients with residual disease remain at low risk of future events2. Moreover, only the local and/or regional tumor response is evaluated, without assessing the presence of micrometastases. Therefore, there is a need to improve patient stratification during and after NAC to better individualize future treatment escalation or de-escalation.
Circulating tumor DNA (ctDNA) detection and monitoring has been shown to be a strong prognostic factor for relapse. Conventional NGS-based assays and non-tumor-informed assays are constrained by their detection sensitivity (~0.1% detection)5, limiting their ability to provide real-time monitoring of NAC efficacy through ctDNA. Despite these limitations, ctDNA monitoring during follow-up has been demonstrated to be a powerful biomarker for metastatic relapse, with a median lead time between ctDNA detection and clinical relapse of 6–12 months, depending on the cancer subtype and the ctDNA assay used6,7,8,9,10,11,12.
Here, we present the results of a study evaluating the clinical validity of a tumor-informed ctDNA assay for detection and monitoring of ctDNA minimal residual disease (MRD) during treatment in a diverse cohort of high-risk BC patients undergoing NAC.
Results
Patient characteristics
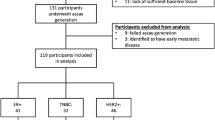
The study cohort included 30 patients with various subtypes of high-risk BC, all treated with NAC. One patient was excluded due to the failure of ctDNA panel design (Fig. 1). Among the remaining 29 patients, 12 (41%) had HER2-positive BC, 11 (38%) had TNBC, and 6 (21%) had HR+/HER2- breast cancer (Table 1). Median patient age was 49 years old. Most patients (69%) were premenopausal, and 55%, 38%, and 7% had T2, T3, or T4 breast cancer, respectively, with 72% presenting with node-positive disease. All patients underwent breast cancer surgery, and a pCR was achieved in 11 (38%) cases (17% HR/HER2-, 36% TNBC, 50% HER2-positive). Additionally, 21 (72%) patients received adjuvant chemotherapy. After a median follow-up of 45.2 months, 5 metastatic relapses were observed (17%) including one patient with also a local recurrence.

CONSORT flow diagram of the study.
ctDNA panel design and detection
For the 29 patients with successfully designed ctDNA panels, each panel targeted a median of 48 variants (ranging 21–51). Following QC and the exclusion of germline and clonal haematopoiesis (CH) variants, the panels retained a median of 42 variants for targeted analysis (ranging 11–48, IQR [28–45]). A total of 184 plasma samples were analyzed from these patients, including baseline/pretreatment plasma samples available for 24 of them (Fig. 1).
ctDNA detection at baseline/pre-treatment
ctDNA was detected in all patients with available samples at baseline (24/24), with a median estimated variant allele fraction (eVAF) of 0.31% (ranging: 0.0083%–8.91%) (Fig. 2). Pretreatment ctDNA levels showed no significant correlation with receptor subtype (p = 0.4), stage (II vs. III, p = 0.19), tumor stage (T2 vs T3-4, p = 0.4), node positivity (p = 0.31), grade (p = 0.1) or pCR (p = 0.86) (Fig. 2). ctDNA levels were significantly predictive of later relapse (p = 0.019, Fig. 2), and baseline eVAF, dichotomized by the median value, was associated with Disease-Free Survival (DFS) (p = 0.018, Fig. 3A).

pCR pathological complete response.

Disease-free survival according to ctDNA detection, in months, at A baseline (N = 24) B post-surgery or follow-up (N = 29). ctDNA circulating tumor DNA.
ctDNA dynamics during NAC and pre-surgery
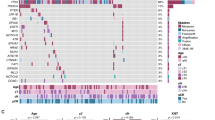
During NAC, ctDNA was detected in 7 out of 21 patients, with eVAF declining in all patients to a median of 0.01% (Figs. 4 and 5, and Supplementary Fig. 1). Among the patients with ctDNA detected prior to the initiation of NAC, 17 out of 18 (94%) showed clearance of ctDNA by the pre-surgery timepoint. In one instance, ctDNA dropped to levels below 1 part per million (=0.0001%). Among the 11 patients who achieved pCR, 9 (82%) had non-detectable ctDNA during NAC. It is noteworthy that the timing of last ctDNA detection differed between these subsets: pCR patients were last positive more than 120 days before surgery, while non-pCR patients showed positivity within 120 days of surgery. However, these patterns should be interpreted cautiously given the small sample size and other factors, such as disease subtype and treatment schedules that may confound the analysis (Supplementary Fig. 2). Clinical outcomes varied by clearance status, the single patient without ctDNA clearance experienced relapse, while 76% (13/17) of patients with clearance remained recurrence-free. Statistical analyzes of associations between post-NAC ctDNA status and clinical outcomes (pCR or disease-free survival) were not performed due to only one patient showing ctDNA positivity post-NAC.

Swimmer plot of patients illustrating ctDNA dynamics according to subtype. TNBC Triple-negative breast cancer, HR Hormone-receptor, pCR pathological complete response.
ctDNA detection post-surgery or during follow-up
ctDNA was detected in one patient after surgery (3%), and in 3 out of 29 patients (10%) after surgery or during the follow-up period. Among the 5 patients who experienced disease recurrence, 3 (60%) showed ctDNA positivity, with one positive throughout all timepoints (pre- and post-treatment). For the other 2 positive patients, ctDNA was first detected in follow-up plasma collected at 4.4 and 9.8 months prior to clinical recurrence (Figs. 4 and 5, and Supplementary Fig. 1). The detection of MRD positivity post-surgery or during follow-up was significantly associated with shorter DFS, Hazard ratios (HR) = 52 95% confidence intervals (CI) (5–522), (Fig. 3B), while the association between MRD positivity only after surgery was not analyzed because only one patient had ctDNA positivity at this timepoint. Two patients with recurrence showed no ctDNA detection at any time point post-surgery. The first (patient 12) had a ctDNA negative follow up timepoint collected 196 days prior to distant relapse, though subsequent analysis revealed subclinical signals just below the detection threshold in both the original and repeat testing of separate plasma aliquots, potentially indicating early relapsing disease, as indicated by the eVAF “trajectory” (Supplementary Fig. 3). The other patient (patient 14) had an oligometastatic exclusive ovarian recurrence. Notably, all patients who remained recurrence-free showed consistently negative ctDNA results during follow-up, yielding 100% specificity for the assay.

Summary of ctDNA dynamics over time in the context of treatment. Patients that recurred are indicated by red boxes. MRD was detected in 60% (3 out of 5). Date of surgery is indicated by a vertical red dashed line.
Discussion
Our study validated the utility of serial ctDNA monitoring as a highly sensitive and specific approach for assessing MRD in patients with high-risk BC undergoing NAC.
Tumor-informed ctDNA assays have demonstrated high sensitivity and very high specificity for monitoring patients during follow-up in breast cancer and predict tumor relapse5,8,9,11,13,14. Due to the low levels of ctDNA in early-stage disease, the sensitivity of an assay is crucial14. It has been shown that the number of variants monitored is an important factor for sensitivity, as a higher number of variants increases the likelihood of detecting ctDNA14,15. In this context, RaDaR, the tumor-informed assay used in the present study, monitors up to 48 variants identified by whole exome sequencing of the tumor tissue, offering a higher number of variants compared to other assays. The assay demonstrated remarkable sensitivity, detecting VAF as low as 0.0083% at baseline and below 1 part per million in one patient during NAC. This performance notably exceeds that reported in the largest study evaluating ctDNA in patients treated with NAC, the I-SPY2 trial, where baseline detection rates were 69% and 91% for HR+/HER2- and TNBC, respectively, using a 16-variant panel13. Another study using the RaDaR assay found that the baseline ctDNA detection rate was 77.2%, and persistent ctDNA detection was associated with outcome16. Ultrasensitive ctDNA assays using whole-genome sequencing to track more variants also demonstrated their ability to detect almost all patients at baseline17,18.
Among the five patients who developed metastasis in this study, ctDNA was not detected prior to clinical relapse in two cases, while the two patients with concomitant samples were detected. This could be explained by the lead time between ctDNA positivity and clinical relapse, highlighting the need for frequent monitoring even with a sensitive assay8,10,14. One patient, who did not have signal detected prior to relapse, showed potential ctDNA signal just below the conservative calling threshold of the assay 167 days prior to relapse. It may be that a follow-on sample collected days or weeks later would have yielded a positive call as ctDNA levels rise with returning disease. The other patient without ctDNA detected during follow-up had an isolated metastatic relapse in the ovaries, which is considered a sanctuary site for metastasis. ctDNA shedding from tumors housed at this site is not well understood, potentially explaining this negative ctDNA result 223 days before the clinical relapse. These findings emphasize the importance of more frequent monitoring intervals and understanding site-specific metastatic patterns.
Between 5 and 10% of patients who achieved pCR experienced disease recurrence within 3 years after surgery. This creates the significant clinical challenge of identifying the subset of high-risk patients who would benefit from intensified treatment while avoiding overtreatment in those with favorable outcomes. In this context, our study demonstrated the prognostic value of ctDNA monitoring during treatment. We found that nearly all patients achieved ctDNA clearance either before or after surgery, and this clearance correlated with improved outcomes. These findings suggest that ctDNA monitoring could provide more precise patient risk stratification compared to conventional pCR assessment alone13,18,19,20.
Looking ahead, the detection of MRD through ctDNA analysis could inform new clinical trials, particularly those investigating treatment escalation strategies for MRD-positive cases. One example is the ongoing TRAK-ER trial (NCT04985266), where ctDNA molecular relapse could indicate the need to add a CDK4/6 inhibitor in HR+/HER2- BC21. Improving ctDNA detection sensitivity will be crucial for clinical trials based on MRD to guide treatment de-escalation or escalation. In this context, although they are more costly and require large studies to demonstrate excellent specificity, ultrasensitive assays using whole-genome sequencing show promise17,18.
While the results are promising, the study acknowledges certain limitations, particularly the small cohort, sample size, limited follow-up, and the heterogeneity of cancer subtypes, which may affect the generalizability of the findings. Future studies with larger cohorts and longer follow-up periods are essential to validate these preliminary findings. Additionally, integrating ctDNA analysis with other emerging biomarkers and imaging techniques could improve the predictive accuracy and reliability of MRD assessments.
In conclusion, this study provides promising evidence that ctDNA detection using a sensitive tumor-informed ctDNA assay could be a valuable biomarker for monitoring MRD in high-risk BC during NAC and follow-up.
Methods
Study design and population
This study was conducted under the umbrella MSK-LINC (Memorial Sloan Kettering-Liquid biopsy for INterception of Cancer) study and enrolled patients with newly diagnosed stage II-III breast cancer who consented to the MSK genomic biospecimen protocols (NCT01775072). Patients were enrolled if they were node-positive and/or had T2 TNBC or HER2-positive breast cancer, or T3 HR+/HER2- primary tumors. A total of 30 patients across various subtypes i.e., triple-negative, HER2-positive, and HR+/HER2- from this cohort were selected.
Sample collection
Serial blood samples were collected longitudinally at several critical timepoints: baseline (prior to NAC initiation), on NAC, pre-operatively, 2–4 weeks post-operatively, and every 4–6 months post-surgery until disease progression, when feasible. Samples, including plasma and buffy coat, were stored at −80 °C to preserve DNA integrity.
Whole exome sequencing
Whole-exome recapture of MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) libraries was performed using leftover tumor (diagnostic biopsy) and normal sequencing libraries derived from primary breast tumor formalin-fixed paraffin-embedded (FFPE) tissue and matched normal blood, as described previously22. For 26 tumor-normal pairs, libraries were recaptured using the xGen Exome Research Panel v2.0 (Integrated DNA Technologies) and sequenced at a target depth of 250× for tumor and 150× for normal. For the remaining cases, the leftover MSK-IMPACT libraries were captured by using either the xGen Exome Research Panel v1.0 or the Agilent Exome Kit (v4) and sequenced at a target depth of 200–250× (average received coverage: 186x-272x) for tumor and 70–150× (average received coverage: 76x-217x) for normal.
ctDNA analysis
The personalized RaDaR tumor-informed assay (NeoGenomics) was utilized to retrospectively detect ctDNA in patient plasma (Supplementary Fig. 4). The RaDaR assay has been described elsewhere in detail23,24. Briefly, patient-specific panels, each targeting up to 48 tumor-specific somatic variants, were designed based on mutations identified by WES of tissue. Each panel underwent a panel QC step, in which the panel was applied to tissue (for all but one) and buffy coat DNA (for all) to confirm panel performance and that the targeted mutations were not germline in nature, or the result of CH. Panels passed this step if at least 8 variants were confirmed as being tumor-specific. Subsequently, panels were applied to plasma with analysis confined to the remaining informative variants. To ascertain ctDNA detection, a statistical framework evaluates the cumulative statistical score of the apparent somatic signal and determines whether it exceeds a predetermined threshold. The limit of detection (LoD95) of the assay has previously been described as 0.001%. For each sample called as “ctDNA detected”, ctDNA levels are quantified as an eVAF. Further in-depth details of the statistical model to determine the ctDNA status of each sample, the pre-set statistical thresholds and the process of calculating the eVAF are all a core part of proprietary code and fully IP protected and, as such, cannot be disclosed. NeoGenomics was blinded to clinical outcomes during the analysis phase of the study.
Statistical analysis and monitoring
To compare the distribution of continuous variables between two independent groups, the Wilcoxon rank-sum test was applied, while the Kruskal–Wallis test was used for comparisons involving more than two independent groups.
DFS was defined as the time from the date of surgery until the first occurrence of cancer relapse or death from any cause, whichever occurred first. Patients who had not experienced any of these events were censored at the time of the last follow-up. DFS was estimated using the Kaplan–Meier method, and survival curves were compared using the log-rank test. HR and 95% CI were calculated using Cox proportional hazards regression models. All statistical analyzes were performed using R (version 4.3.2) with a significance threshold at p < 0.05.
Data availability
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
Loibl, S. et al. Early breast cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 35, 159–182 (2024).
Yau, C. et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: a multicentre pooled analysis of 5161 patients. Lancet Oncol. 23, 149–160 (2022).
Schmid, P. et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N. Engl. J. Med. 386, 556–567 (2022).
von Minckwitz, G. et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 380, 617–628 (2019).
Bronkhorst, A. J. & Holdenrieder, S. The changing face of circulating tumor DNA (ctDNA) profiling: factors that shape the landscape of methodologies, technologies, and commercialization. Medizinische Genetik 35, 201–235 (2023).
Coombes, R. C. et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin. Cancer Res. 25, 4255–4263 (2019).
Lipsyc-Sharf, M. et al. Circulating tumor DNA and late recurrence in high-risk hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer. J. Clin. Oncol. 40, 2408–2419 (2022).
Shaw, J. A. et al. Serial postoperative circulating tumor DNA assessment has strong prognostic value during long-term follow-up in patients with breast cancer. JCO Precis. Oncol. 8, e2300456 (2024).
Radovich, M. et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: preplanned secondary analysis of the BRE12-158 Randomized Clinical Trial. JAMA Oncol. 6, 1410–1415 (2020).
Garcia-Murillas, I. et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncol. 5, 1473–1478 (2019).
Loi, S. et al. Prognostic utility of ctDNA detection in the monarchE trial of adjuvant abemaciclib plus endocrine therapy (ET) in HR+, HER2-, node-positive, high-risk early breast cancer (EBC). JCO 42, LBA507–LBA507 (2024).
Turner, N. C. et al. Detection of circulating tumor DNA following neoadjuvant chemotherapy and surgery to anticipate early relapse in ER positive and HER2 negative breast cancer: analysis from the PENELOPE-B trial. JCO 41, 502–502 (2023).
Magbanua, M. J. M. et al. Clinical significance and biology of circulating tumor DNA in high-risk early-stage HER2-negative breast cancer receiving neoadjuvant chemotherapy. Cancer Cell S1535-6108(23)00134–4 https://doi.org/10.1016/j.ccell.2023.04.008 (2023)
Coakley, M. et al. Comparison of circulating tumor DNA assays for molecular residual disease detection in early-stage triple-negative breast cancer. Clin. Cancer Res. 30, 895–903 (2024).
Avanzini, S. et al. A mathematical model of ctDNA shedding predicts tumor detection size. Sci. Adv. 6, eabc4308 (2020).
Elliott, M. J. et al. Longitudinal evaluation of circulating tumor DNA in patients undergoing neoadjuvant therapy for early breast cancer using a tumor-informed assay. Nat. Commun. 16, 1837 (2025).
Garcia-Murillas, I. et al. Whole genome sequencing powered ctDNA sequencing for breast cancer detection. Ann. Oncol. 36, 673–681 (2025).
Parsons, H. A. et al. Circulating tumor DNA association with residual cancer burden after neoadjuvant chemotherapy in triple-negative breast cancer in TBCRC 030. Ann. Oncol. 34, 899–906 (2023).
Stecklein, S. R. et al. ctDNA and residual cancer burden are prognostic in triple-negative breast cancer patients with residual disease. NPJ Breast Cancer 9, 10 (2023).
Cailleux, F. et al. Circulating tumor DNA after neoadjuvant chemotherapy in breast cancer is associated with disease relapse. JCO Precis. Oncol. 6, e2200148 (2022).
Aj, M. et al. Molecular residual disease in breast cancer: detection and therapeutic interception. Clin. Cancer Res. 29, 4540–4548 (2023).
Jonsson, P. et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 571, 576–579 (2019).
Flach, S. et al. Liquid BIOpsy for MiNimal RESidual DiSease detection in head and neck squamous cell carcinoma (LIONESS)-a personalised circulating tumour DNA analysis in head and neck squamous cell carcinoma. Br. J. Cancer 126, 1186–1195 (2022).
van Dorp, J. et al. High- or low-dose preoperative ipilimumab plus nivolumab in stage III urothelial cancer: the phase 1B NABUCCO trial. Nat. Med. 29, 588–592 (2023).
Acknowledgements
We thank the members of the Translational Oncology Partnership Program, Breast Disease Management Team, Center for Molecular Oncology, Integrated Genomics Operation, and Bioinformatics Core at MSKCC for their support in sample preparation, library construction, sequencing, and computational infrastructure.This research was funded in part through NeoGenomics, Susan G. Komen Foundation, and the NIH/NCI Cancer Center Support Grant P30 CA008748.
Author information
Authors and Affiliations
Contributions
L.C., J.A.A., H.K.K., C.G.S., and P.R. drafted the manuscript. L.C., C.G.S. conducted the data analysis. P.R. conceived and designed the work. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
CGS and AC are employees of NeoGenomicsMER COI declared uncompensated advisory for Foundation Medicine SC has received institutional funding from Daiichi-Sankyo and AstraZeneca, Share options in Totus Medicines, and consultation/Ad board/Honoraria from Daiichi-Sankyo, AstraZeneca, Lilly, Casdin Capital, and Pathos AI. S.C. is supported by grants from the BCRF and the NCI.PR reports Consulting/Ad Board/Advisory for Novartis, AstraZeneca, BioNTechLilly, Tempus, Prelude Therapeutics, NeoGenomics, Regor Pharmaceuticals, Natera, SAGA Diagnostics, Guardant Health, Myriad Genetics, Foresight Diagnostics and Institutional grant/funding support from Grail, Novartis, AstraZeneca, Biothernostics, Tempus, Sophia Genetics, Biovica, Guardant Health, Personalis, Myriad, Foresight Diagnostics, SAGA Diagnostics, Biodesix, Haystack, Roche. P.R. is supported by grants from Susan G. Komen, NCI, MBCure. JZD has received institutional grants/funding from Merck, AstraZeneca, Bolt Therapeutics, Jazz Pharmaceutics and Orum therapeutics, and has performed consulting/advisory work for AstraZeneca, Genagon, AmMax Bio, NuProbe, Revelio Therapeutics, MEDACorp, and BAIRD conferencesLC, JAA, HKK, JK, TE, EM, EF, AS, KN, KS, LN, AX, AJK, BTL, GP declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cabel, L., An, J.AR., Kim, HK. et al. Monitoring of circulating tumor DNA in patients with stage II-III breast cancer treated with neoadjuvant chemotherapy. npj Breast Cancer 12, 22 (2026). https://doi.org/10.1038/s41523-025-00878-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00878-w
