
Abstract
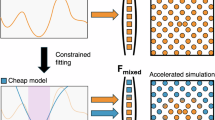
Machine learning interatomic potentials (MLIP) are powerful tools for using large-scale molecular dynamics (MD) to evaluate material properties, including the performance of solid-state electrolytes (SSEs). While there are many efforts for constructing universal big MLIP models, their accuracies and speeds of inference still need to be improved for many practical applications. Another approach is to develop a system-specific MLIP model relying on active learning strategy. Although much cheaper than training a big model, using the conventional procedure, it still requires large numbers of active learning loops and the corresponding DFT calculations to ensure convergency. Here, we propose a single-shot workflow that significantly accelerates small MLIP model development by leveraging the capabilities of the big model (using MACE as one example) and requiring only a few hundred additional DFT calculations. Our workflow comprises two stages, first the MACE model itself is fine-tuned to make it more accurate for the given system, second a smaller MLIP model (using NEP as one example) is distilled from the fine-tuned MACE model. We employed a MACE-driven sampling strategy, carried out additional DFT calculations without relying on active learning iterations. We show that fine-tuned MACE model can inherit the stability of the pretrained model, and fine-tuning the pretrained MACE model is much more DFT data efficient comparing to training a start-from-scratch NEP model. In the second stage, the fine-tuned MACE model provides the dataset to train the NEP model, allows the final NEP model to carry out large scale MD simulations with competitive accuracy. This integrated workflow establishes a systematic pathway for rapid MLIP development via small additional DFT dataset, with potential applications to many material systems.
Similar content being viewed by others

Data availability
The computational data to support the findings of this study is available from the corresponding author on reasonable request.
Code availability
The code used for the MACE training is accessible from https://github.com/hyjwpk/ELoRA. The code used for NEP training is available from https://github.com/LonxunQuantum/MatPL.
References
Janek, J. & Zeier, W. G. A solid future for battery development. Nat. Energy 1, 16141 (2016).
Chen, D. et al. Superionic ionic conductor discovery via multiscale topological learning. J. Am. Chem. Soc. 147, 20888–20898 (2025).
Zhang, W. et al. Rapid mining of fast ion conductors via subgraph isomorphism matching. J. Am. Chem. Soc. 146, 18535–18543 (2024).
He, X. et al. Crystal structural framework of lithium super-ionic conductors. Adv. Energy Mater. 9, 1902078 (2019).
Jun, K. et al. Lithium superionic conductors with corner-sharing frameworks. Nat. Mater. 21, 924–931 (2022).
Jun, K., Chen, Y., Wei, G., Yang, X. & Ceder, G. Diffusion mechanisms of fast lithium-ion conductors. Nat. Rev. Mater. 9, 887–905 (2024).
Zhang, W. et al. Revealing morphology evolution of lithium dendrites by large-scale simulation based on machine learning force field. Adv. Energy Mater. 13, 2202892 (2023).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Zhang, Y. et al. DP-GEN: a concurrent learning platform for the generation of reliable deep learning based potential energy models. Comput. Phys. Commun. 253, 107206 (2020).
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csanyi, G. MACE: higher order equivariant message passing neural networks for fast and accurate force fields. Adv. Neural Inf. Process. Syst. 35, 11423–11436 (2022).
Batatia, I. et al. The design space of E(3)-equivariant atom-centred interatomic potentials. Nat. Mach. Intell. 7, 56–67 (2025).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Wang, R., Gao, Y., Wu, H. & Zhong, Z. Pre-training, fine-tuning, and distillation (PFD): automatically generating machine learning force fields from universal models. Phys. Rev. Mater. 9, 113802 (2025).
Radova, M., Stark, W. G., Allen, C. S., Maurer, R. J. & Bartók, A. P. Fine-tuning foundation models of materials interatomic potentials with frozen transfer learning. npj Comput. Mater. 11, 237 (2025).
Jain, A. et al. Commentary: The materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Fu, X. et al. Learning smooth and expressive interatomic potentials for physical property prediction. in Proceedings of the 42nd International Conference on Machine Learning 17875–17893 (PMLR, 2025).
Rhodes, B. et al. Orb-v3: atomistic simulation at scale. Preprint at https://doi.org/10.48550/arXiv.2504.06231 (2025).
Bochkarev, A., Lysogorskiy, Y. & Drautz, R. Graph atomic cluster expansion for semilocal interactions beyond equivariant message passing. Phys. Rev. X 14, 021036 (2024).
Batatia, I. et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 163, 184110 (2025).
Riebesell, J. et al. A framework to evaluate machine learning crystal stability predictions. Nat. Mach. Intell. 7, 836–847 (2025).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Aono, H., Sugimoto, E., Sadaoka, Y., Imanaka, N. & Adachi, G. Ionic conductivity of the lithium titanium phosphate (Li1 + XMXTi2 − X(PO4)3, M = Al, Sc, Y, and La) systems. J. Electrochem. Soc. 136, 590 (1989).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Qi, J., Ko, T. W., Wood, B. C., Pham, T. A. & Ong, S. P. Robust training of machine learning interatomic potentials with dimensionality reduction and stratified sampling. npj Comput. Mater. 10, 43 (2024).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Deng, B. et al. Systematic softening in universal machine learning interatomic potentials. npj Comput. Mater. 11, 1–9 (2025).
Frey, N. C. et al. Neural scaling of deep chemical models. Nat. Mach. Intell. 5, 1297–1305 (2023).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the matbench test set and automatminer reference algorithm. npj Comput. Mater. 6, 138 (2020).
Fan, Z. et al. GPUMD: a package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations. J. Chem. Phys. 157, 114801(2022).
Wang, C., Hu, S., Tan, G. & Jia, W. ELoRA: low-rank adaptation for equivariant GNNs. in Forty-Second International Conference on Machine Learning (ICML, 2025).
Fu, X. et al. Forces are not enough: benchmark and critical evaluation for machine learning force fields with molecular simulations. in Transactions on Machine Learning Research (TMLR, 2022).
He, X., Zhu, Y. & Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 8, 15893 (2017).
Qi, J. et al. Bridging the gap between simulated and experimental ionic conductivities in lithium superionic conductors. Mater. Today Phys. 21, 100463 (2021).
Zhang, Y., Luo, J.-D., Yao, H.-B. & Jiang, B. Size dependent lithium-ion conductivity of solid electrolytes in machine learning molecular dynamics simulations. Artif. Intell. Chem. 2, 100051 (2024).
Wang, S., Liu, Y. & Mo, Y. Frustration in super-ionic conductors unraveled by the density of atomistic states. Angew. Chem. Int. Ed. 62, e202215544 (2023).
Yang, G. et al. Li-rich channels as the material gene for facile lithium diffusion in halide solid electrolytes. eScience 2, 79–86 (2022).
Hjorth Larsen, A. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Fan, Z., Siro, T. & Harju, A. Accelerated molecular dynamics force evaluation on graphics processing units for thermal conductivity calculations. Comput. Phys. Commun. 184, 1414–1425 (2013).
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 12, 398 (2021).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996).
Zhang, B., Lin, Z., Dong, H., Wang, L.-W. & Pan, F. Revealing cooperative Li-ion migration in Li1+xAlxTi2−x(PO4)3 solid state electrolytes with high Al doping. J. Mater. Chem. A 8, 342–348 (2020).
Mo, Y., Ong, S. P. & Ceder, G. First principles study of the Li10GeP2S12 lithium super ionic conductor material. Chem. Mater. 24, 15–17 (2012).
Fu, Z.-H. et al. The chemical origin of temperature-dependent lithium-ion concerted diffusion in sulfide solid electrolyte Li10GeP2S12. J. Energy Chem. 70, 59–66 (2022).
Huang, J. et al. Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors. J. Chem. Phys. 154, 094703 (2021).
Klimeš, J., Bowler, D. R. & Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter 22, 22201 (2009).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Acknowledgements
This research was funded by the National Key Research and Development Program of China (Grant No. 2024YFA1408200).
Author information
Authors and Affiliations
Contributions
W.Z.: Conceptualization, model development, data analysis, writing-original draft. X.W. and C.W.: Model development. S.H. and Y. L.: Data analysis. L.W.: Writing and editing, data analysis, and funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater (2026). https://doi.org/10.1038/s41524-026-02023-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-026-02023-y
