
Abstract
Genome sequencing (GS) was applied to 3317 individuals from 1452 Korean families with suspected rare genetic disorders to assess diagnostic yield and clinical utility. Patients were categorized into 16 clinical subgroups with curated phenotypes, and variant interpretation was refined by post-analytic phenotype matching. A molecular diagnosis was achieved in 46.2% of families, influencing clinical management in 18.5% of diagnosed cases. Family-based GS had a higher yield than singleton testing (48.5% vs. 41.5%). Neuromuscular and neurodevelopmental disorders showed the highest yields. GS-specific variant types, including deep intronic, noncoding, complex structural variants, and tandem repeat expansions, accounted for 14.6% of diagnoses. Secondary findings were identified in 4.3% of individuals. Novel disease-associated genes such as RYBP, DNAJA3, CAMK2D, and small nuclear RNA genes were also reported. These results highlight the diagnostic power of GS and support its use as a first-tier test, especially in underrepresented populations.
Similar content being viewed by others

Introduction
With the advent of next-generation sequencing (NGS), approximately 5000–8000 rare genetic diseases have been identified, and ongoing research continues to uncover additional disease-causing genes1,2. Because the identification of a causative gene is directly linked to a definitive diagnosis and appropriate clinical management, NGS has served as a crucial tool in clinical genetics for over a decade. Recent technological advances and declining sequencing costs have accelerated the adoption of exome sequencing (ES) and genome sequencing (GS) as first-tier diagnostic tools for individuals with suspected Mendelian genetic disorders3,4.
ES remains a widely used diagnostic method and an efficient research platform, supported by large-scale variant databases and mature analytical pipelines. Reported diagnostic yields for ES in patients with suspected Mendelian disorders range from 20% to 60%, depending on the cohort characteristics, including phenotype specificity and ethnic background5,6,7. Although many cases remain undiagnosed, routine reanalysis of ES data has been shown to provide an additional 10% of diagnoses in a cost-effective manner8,9.
However, an increasing number of studies have highlighted the contribution of structural variations (SVs) or noncoding variants, which are poorly identified by ES but detectable through GS, in the etiology of rare diseases10,11. GS has consistently demonstrated a higher diagnostic yield than ES across multiple recent studies12,13. Moreover, GS enables comprehensive detection of copy number variations (CNVs) even at exon-level resolution, supporting a streamlined, one-step diagnostic approach that can replace traditional multistep testing strategies, such as chromosome microarrays or other targeted genetic testing followed by ES.
Given the accelerating development of analytic pipelines and the ongoing reduction in sequencing costs, GS is emerging as a highly competitive and potentially superior diagnostic modality for rare genetic disorders. Despite the growing consensus that GS may soon become a first-line diagnostic test, there remains a lack of large-scale, real-world studies demonstrating its clinical utility across diverse phenotypic presentations, particularly in non-Western populations.
To address this gap, we conducted a comprehensive analysis of GS data from 3317 individuals in 1452 Korean families with suspected rare genetic disorders affecting a broad range of organ systems. By integrating detailed clinical phenotypes with genome-wide variant data, we aimed to provide robust evidence for the diagnostic utility and future potential of GS in routine clinical practice.
Results
Demographic profile and molecular diagnosis
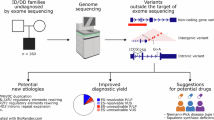
The characteristics of all participants are summarized in Fig. 1 and Table 1. A total of 3317 individuals from 1452 families were enrolled in this study, including 1452 index cases (808 males and 644 females). The age of the index patients ranged from 0 to 71 years (median 10.0 ± 11.7 years), with the majority being pediatric cases (1142/1452, 78.7%). All families were classified into 16 disease categories. Neurologic and neurodevelopmental disorders accounted for the largest proportion (689/1452, 47.5%), followed by neuromuscular disorders (197/1452, 13.6%). The dataset included 785 trios (54.1%, proband and both parents), 161 duos (11.1%, proband and one parent), 37 quads and pentas (2.5%, proband, both parents, and additional affected family members such as a sibling), and 469 proband-only samples (32.3%). Each family had 1–8 clinical phenotypes, described using HPO terms. As part of the inclusion criteria, all enrolled patients had undergone at least one form of genetic testing prior to GS (Supplementary Table 1). Among them, 73.4% had previously received either targeted gene panel sequencing or clinical exome sequencing. Other commonly performed tests included chromosomal microarray analysis and single-gene testing. When stratified by prior testing history, the diagnostic yield of GS was 44.9% in patients who had undergone panel or exome sequencing, compared with 50.7% in those without such prior NGS testing.

A Overall diagnostic outcome, showing the proportion of patients categorized as diagnosed, possibly diagnosed, or undiagnosed following genome sequencing. B Diagnostic rates across different family structures: singleton, duo, trio, and quad + penta. C Diagnostic rates stratified by disease categories.
Among the 1452 families, 46.2% had pathogenic or likely pathogenic variants, with 632 (43.5%) classified as the “diagnosed” and 40 (2.8%) as the “possibly diagnosed” group with highly plausible candidate variants (Fig. 1A, Supplementary Tables 2 and 3). Notably, as shown in Fig. 1B, sequencing involving family members, such as duo, trio, or extended family analyses, resulted in a significantly higher diagnostic yield compared with singleton testing (P = 0.015). The yield varied by disease category, with the highest rates in neuromuscular (123/197, 62.4%, P = 1.47 × 10–6) and neurologic and neurodevelopmental disorders (339/689, 49.2%, P = 0.039). Although statistical significance was not reached due to small sample sizes, high diagnostic rates were also noted in tumor syndromes (15/23, 65.2%) and ophthalmologic disorders (25/40, 62.5%). By contrast, lower diagnostic yields were observed in hearing and ear disorders (10/50, 20.0%, P = 2.64 × 10–4), immunologic disorders (9/41, 22.0%, P = 2.61 × 10–3), and dysmorphic and congenital anomaly syndromes (56/148, 37.8%, P = 0.037). Although not statistically significant, low diagnostic yields were also observed in cardiovascular disorders (4/18, 22.2%) and endocrine disorders (4/16, 25.0%) (Fig. 1C).
Multiple pathogenic variants were identified in 4.3% (29/672) of the diagnosed patients. Secondary findings were observed in 4.3% (143/3317) of study participants, most commonly in BRCA2 (n = 18), followed by BRCA1 (n = 15), PCSK9 (n = 14), LDLR (n = 13), MYBPC3 (n = 13), MYH7 (n = 10), PALB2 (n = 8), and TTN (n = 8) (Supplementary Table 4). These variants were primarily associated with cardiomyopathy/arrhythmia (42.8%), cancer predisposition (36.6%), and familial hypercholesterolemia (18.6%).
Genetic distribution and properties of diagnosed variants
Among the total diagnosed patients involving 462 different disorders, 60.5% (279/462) exhibited autosomal dominant inheritance, 24.5% (113/462) autosomal recessive, and 12.6% (58/462) X-linked inheritance (Fig. 2A). Variants associated with neurologic and neurodevelopmental disorders were the most frequently identified, reflecting the high proportion of patients presenting with neurological symptoms. Consistently, variants in genes such as DMD, MECP2, SMN1, and RNU4-2 were commonly reported (Supplementary Table 5). In particular, 43.1% (302/701) of all identified causal variants were de novo (Fig. 2B).

A Mode of inheritance among patients with diagnosed and possibly diagnosed findings. B Variant segregation patterns observed in diagnosed and possibly diagnosed patients. C Distribution of causative variant types identified in diagnosed and possibly diagnosed patients. D Proportion of patients diagnosed exclusively through genome sequencing (GS). The blue bars represent the proportion of patients whose diagnoses were made exclusively through GS. The inset box (navy bars) further breaks down the variant types identified exclusively through GS, with deep intronic variants, exonic deletions/duplications, and noncoding variants being the most common. E Distribution of causative genes based on the year of their initial gene–disease association discovery.
Although family-based sequencing can aid variant interpretation, diagnoses were largely achieved through singleton testing, with family testing being essential in only 7.5% (51/672) of diagnostic cases, contributing to a 3.5% (51/1452) increase in overall yield. Among 127 variants from 84 probands and 11 duos further evaluated by family testing, 54 were presumed de novo, 57 were confirmed to be in trans, and 3 were inherited from a similarly affected parent; six were inherited from an unaffected parent, likely due to incomplete penetrance. In addition, 6 variants on the X chromosome and 1 variant in the mitochondrial genome were inherited from unaffected mothers. Of these, 56 variants in 51 patients were critical for diagnosis, primarily due to a reclassification based on the confirmation of de novo or in trans compound heterozygous inheritance patterns.
Among diagnosed and possibly diagnosed variants, 39.3% were novel (Supplementary Table 6), with the highest rates seen in skeletal disorders (53.3%), followed by neurologic and neurodevelopmental disorders (42.8%), and dysmorphic and congenital anomaly syndromes (42.2%). Most causative variants were SNVs or indels (78.7%), followed by CNVs (15.4%), short tandem repeat (STR) expansions (3.4%), and SVs (2.6%) (Fig. 2C). Notably, 14.6% (98/672) of diagnosed families required GS for diagnosis to detect complex structural variants, inversions, and noncoding variants (Fig. 2D and Supplementary Table 7). In addition, nine cases (1.3%) required long-read sequencing to confirm STR expansions, including those in ATXN8OS (n = 3), NOTCH2NLC (n = 2), and FGF14, NOP56, and NUTM2B-AS1 (n = 1 each). Figure 3A presents an example of an STR expansion identified through nanopore Cas9-targeted sequencing (nCATS). This patient presented with progressive muscle weakness and a repeat expansion in NUTM2B-AS1, a gene associated with oculopharyngeal myopathy with leukoencephalopathy 1 (OMIM #618637). It was suggested by ExpansionHunter and subsequently confirmed through nCATS.

A Integrative Genomics Viewer screenshots showing nanopore Cas9-targeted sequencing data for a short tandem repeat expansion in the NUTM2B-AS1 gene. B Pedigree analysis of patients carrying RYBP, DNAJA3, and CAMK2D variants. The figure shows family segregation for the de novo RYBP variant (c.152_54del and c.116C>A) in patients SNU04963 and SNU04602, respectively, alongside DNAJA3 and CAMK2D variants identified in compound heterozygous and maternally inherited patterns in SNU05211 and SNU04584, respectively. C Schematic illustration of the RYBP gene, showing the distribution of both previously reported and newly identified variants in relation to the functional domains. D Prevalence of major recurrently identified genes in diagnosed patients with neurology and neurodevelopmental disorder (n = 339). The numbers in parentheses (n/339) indicate the count of patients in whom the respective gene variant was found, and the blue bar chart represents their proportion. RNU-related genes are specifically highlighted with navy bars to show their internal distribution; RNU4-2 (9/15, 60.0%), RNU2-2P (4/15, 26.0%), and RNU5B-1 (2/15, 13.3%).
Time-dependent diagnostic yield: highlighting the need for reanalysis
We annotated each gene identified in this study with the year it was first listed in Online Mendelian Inheritance in Man (OMIM) as disease-associated and simulated cumulative diagnostic yields over time. As shown in Fig. 2E, only about 60% of the identified genes would have been detectable using knowledge available up to 2010, whereas this proportion increased to approximately 90% by 2020. Although the rate of novel gene discovery has slowed in recent years, new diagnostic genes continue to emerge, with 7.5% of the genes in our study first reported after 2021. These findings underscore the importance of periodic reanalysis of genomic data as knowledge in the field evolves.
Clinical utility of genome sequencing in guiding therapeutic decisions
Genetic diagnosis can enable curative or disease-modifying therapeutic interventions. In our cohort, long-term treatment plans were implemented in 18.5% (124/672) of diagnosed patients, including stem cell transplantation for those with IL10RA variants and selumetinib treatment for patients with neurofibromatosis type 1. To isolate the impact of genetic diagnosis, we reviewed follow-up data and found that in 4.0% (25/632) of cases, treatment decisions were clearly prompted by genetic diagnoses (Supplementary Table 8). These included initiation of treatment for spinal muscular atrophy (SMA) in 14 patients, supplementation for mitochondrial disease in 3 patients, and further management changes in one patient each with CoQ10 deficiency, congenital myasthenia, pustular psoriasis, and Gitelman syndrome.
Emerging genetic disorders: representative clinical cases
RYBP-related neurodevelopmental disorder
In our cohort, we identified several patients with recently characterized genetic disorders, including two cases of RYBP-related neurodevelopmental disorder, a condition marked by global developmental delay, syndromic features, and multisystem involvement14. The first case (SNU04963) was a 17-year-old female harboring a de novo heterozygous in-frame indel in the RYBP gene (c.52_54del; p.Ser18del). She exhibited global developmental delay, severe intellectual disability, spasticity, microcephaly, short stature, and distinct craniofacial dysmorphism. She was born at term with a low birth weight of 2.26 kg, and was diagnosed with tetralogy of Fallot, which was surgically corrected in infancy. Multiple congenital anomalies were noted, including bilateral camptodactyly, proximal radioulnar synostosis, and left equinovarus deformity. At 6 months of age, she experienced a right middle cerebral artery infarction. Over time, she was also diagnosed with hypothyroidism and experienced recurrent otitis media, for which she underwent multiple tympanostomy tube insertions.
The second case (SNU04602) was a 13-year-old female with a de novo heterozygous missense variant in RYBP (c.116 C > A; p.Thr39Asn). She presented microcephaly, severe global developmental delay, profound intellectual disability, and dysmorphic facial features. Her clinical picture included autistic behaviors, prominent upper motor neuron signs, and marked lower extremity spasticity. Despite normal findings on brain MRI, her developmental milestones were severely delayed. She achieved independent ambulation only at age 8 years, and her verbal output remained limited to fewer than 10 words. Additional features included camptodactyly and equinus foot deformity.
DNAJA3-associated polyneuropathy
We identified a 20-year-old female (SNU05211) with compound heterozygous variants in DNAJA3 (c.1229 T > C; p.Ile410Thr and c.283 T > C; p.Tyr95His), a gene recently implicated in polyneuropathy15. Since late childhood, she presented with recurrent episodes of burning pain in the soles of her feet, lasting days to weeks and worsening at night. Electrophysiologic studies at the age of 15 years demonstrated sensorimotor polyneuropathy of the distal lower limbs, predominantly of axonal type. She also exhibited intermittent, left-dominant intention tremor and mild distal weakness during episodes, with no disease progression over several years.
CAMK2D-related dilated cardiomyopathy
A recent study linked gain-of-function variants in CAMK2D to dilated cardiomyopathy (DCMP)16. We identified the same missense variant (c.824G>A; p.Arg275His) in a 4-year-old male (SNU04584), inherited from his asymptomatic mother. He presented with congenital severe mitral regurgitation, left atrial enlargement, and DCMP. The patient underwent multiple cardiac surgeries, including mitral and tricuspid valve repairs as well as left atrial reduction plasty. Follow-up echocardiography showed residual mild valvular dysfunction.
Enrichment of pathogenic snRNA gene variants in neurodevelopmental disorders
Another notable feature of this study cohort is the high frequency of pathogenic variants in small nuclear RNA (snRNA) genes. Variants in RNU4-2 (n = 9), RNU2-2P (n = 4), and RNU5B-1 (n = 2) were found in 4.4% (15 out of 339) of diagnosed patients with neurologic and neurodevelopmental disorders, ranking second after MECP2 (4.7%, 16 out of 339) (Fig. 3D). Notably, all three snRNA genes harbored recurrent variants. In RNU4-2, 8 out of 9 patients carried the same variant, n.64_65insT, and all four patients with RNU2-2P variants shared the identical n.4G>A substitution. Similarly, both patients with RNU5B-1 variants carried the same insertion, n.42_43insA. In all 13 patients with available parental data, the variants were confirmed to be de novo.
Discussion
This study demonstrates the clinical utility of GS in diagnosing rare genetic disorders. A definitive or possible diagnosis with phenotype correlation was achieved in 46.2% of our cohort (672/1452), representing a favorable diagnostic yield, particularly considering that many of these patients had previously undergone ES or targeted gene panel testing without a conclusive result.
GS provided a significant diagnostic advantage by detecting a broad spectrum of variant types. Notably, previous studies have similarly demonstrated that GS improves diagnostic yield by identifying complex structural variants, noncoding variants, and other pathogenic alterations that are typically missed by ES or targeted panels17,18. In our study, approximately 14.6% of diagnosed families required GS for diagnosis because their causative variants included complex structural variants, inversions, and STR expansions. Specifically, GS enabled the detection of pathogenic variants in deep intronic or noncoding regions and snRNA genes, including RNU4-2 (n = 9), RNU2-2P (n = 4), and RNU5B-1 (n = 2), aligning with recent studies that have highlighted the role of noncoding RNA genes in rare genetic disorders19,20,21. These findings underscore the distinctive power of GS to interrogate the entire genome, including regulatory and functional noncoding elements, thereby uncovering pathogenic mechanisms inaccessible through traditional sequencing strategies.
In this study, family testing was required to determine variant pathogenicity in only 7.5% of diagnostic cases, contributing to a 3.5% increase in diagnostic yield. A recent study comparing singleton and trio GS indicated an 8% incremental benefit from parental testing22. However, another study found that follow-up family testing after singleton analysis resulted in a 3.5% increase in diagnostic yield, in line with our findings17. This limited added value likely reflects advances in variant databases, in silico prediction tools, and gene–disease associations that enhance the diagnostic power of singleton analysis. These findings support singleton GS as a cost-effective and practical first-tier diagnostic approach, particularly when parental samples are unavailable.
This study also highlights the clinical utility of GS beyond the diagnosis, particularly in facilitating timely therapeutic interventions. Fourteen SMA patients benefited from genetic diagnosis, which enabled the initiation of disease-modifying treatment. Although SMA, particularly type I, is mostly diagnosed through targeted tests based on typical features, the patients in this study had atypical and milder presentations (type II or III), delaying early recognition and treatment. The increasing availability of gene therapy further highlights the importance of early and accurate genetic diagnosis to ensure that patients with specific pathogenic variants can benefit from these targeted treatments23,24,25.
The cohort included a wide range of age groups and disease categories involving multiple organ systems. Pediatric patients accounted for 78.7% of the probands (1142/1452), with a predominance of neurodevelopmental disorders and dysmorphic syndromes. This pediatric-skewed distribution is consistent with previous studies of undiagnosed rare disease cohorts, which have similarly reported high proportions of neurodevelopmental presentations and younger age groups5,12,26,27. It also underscores the persistent diagnostic challenges associated with pediatric-onset neurodevelopmental disorders, many of which remain genetically unsolved.
Our cohort included a broad spectrum of disease groups, enabling the assessment of diagnostic yield by disease categories, which ranged from 20% to 62.5%. Yields for neurodevelopmental, neuromuscular, and ophthalmologic disorders were comparable or higher than previous reports12,28,29,30,31. Despite differences in inclusion criteria and variant interpretation across studies, our results highlight the value of phenotype-driven GS analysis followed by clinical confirmation with a significant impact on diagnostic outcomes. By contrast, some disease groups, such as hearing and ear disorders and immunologic disorders, exhibited relatively low yields. This may reflect characteristics of those categories of higher proportion of nongenetic etiologies with phenotype overlap, which have led to lower diagnostic yields in previous studies32,33,34,35,36,37. Notably, most patients with hearing disorders in our cohort had prior negative targeted NGS results, possibly lowering the yield. These findings support the utility of GS as a first-tier diagnostic test, especially for high-yield disorders and for selected cases with inconclusive prior testing.
Temporal analysis of gene discovery demonstrated a substantial improvement in the diagnostic potential of genomic testing over the past decade (Fig. 2E). Although the pace of novel gene discovery has decelerated, new disease genes continue to emerge. This ongoing expansion of the gene-disease spectrum likely contributed to the diagnostic yield observed in our study, even though 73.4% of patients had previously undergone NGS-based testing. Patients without prior NGS showed a slightly higher diagnostic yield, which is expected since they had not undergone previous genome-wide testing. However, those with prior NGS still achieved a considerable yield, underscoring the incremental value of GS even in extensively tested cohorts. In our study, 7.5% of diagnosed cases involved genes first linked to disease after 2021, particularly including RYBP, DNAJA3, and CAMK2D (Fig. 3B). RYBP-related neurodevelopmental disorder was recently defined based on seven patients14, and our study contributes two additional patients with novel variants located in domains similar to those reported previously (Fig. 3C). In addition, DNAJA3- and CAMK2D-related disorders remain incompletely defined, with only a limited number of published reports15,16. The identification of patients with phenotypes highly consistent with prior reports provides important supporting evidence for the pathogenic relevance of these genes. These findings underscore the critical value of iterative, comprehensive genomic analyses that incorporate newly discovered gene–disease associations. Therefore, periodic reanalysis of genomic data should be considered crucial for improving diagnostic yield in unresolved cases.
This study underscores the clinical utility of GS as a powerful diagnostic tool for rare genetic disorders. By detecting diverse variant types, GS broadens the diagnostic landscape beyond that of conventional methods. Importantly, genetic diagnosis not only facilitates molecular confirmation but also informs therapeutic decisions, including gene-targeted treatments. As precision medicine advances, GS will be central to ensuring timely and accurate diagnoses, particularly when early treatment can alter disease progression and outcomes.
Methods
Study design and participants
The Institutional Review Board (IRB) at Seoul National University Hospital approved the study protocol (IRB No. 2302-060-1403), confirming adherence to relevant guidelines and regulations. Informed consent was obtained from all participants or their legal guardians prior to the study.
This retrospective study included 3317 individuals from 1452 families who met the following inclusion criteria: (i) suspected rare genetic disorders who remained undiagnosed despite one or more conventional genetic tests, (ii) availability of complete GS data, (iii) detailed clinical information annotated using Human Phenotype Ontology (HPO) terms, and (iv) availability of post-analysis discussions with attending clinicians.
Clinical evaluation and structured phenotyping using HPO on all patients were conducted at the Seoul National University Hospital between June 2020 and June 2024. All affected individuals were classified into 16 distinct disease categories (Table 1) based on HPO terms and the initial disease categories from their respective cohorts.
Sample preparation and data generation
Peripheral blood samples were obtained from each proband and their family members. Genomic DNA was isolated using the QIAamp DNA Blood Mini Kit (Qiagen). GS was performed at 3billion, Inc. and Macrogen, Inc. using the TruSeq DNA PCR–free sample preparation kit (Illumina, San Diego, CA, USA). Libraries were sequenced on the NovaSeq 6000 platform (Illumina, San Diego, CA, USA) to generate 150 base pair (bp) paired-end reads. Raw base call (BCL) files were converted to FASTQ format and demultiplexed using bcl2fastq v2.20.0.422 (Illumina, San Diego, CA, USA). Reads were aligned to the Genome Reference Consortium Human Build 38 (GRCh38) and Revised Cambridge Reference Sequence (rCRS) of the mitochondrial genome. Variant calling for single-nucleotide variants (SNVs) and small insertion/deletion variants (indels) was performed using GATK v4.2.1438,39. Mitochondrial SNVs and indels were called using Mutect2 v4.1.0.040. Structural variants (SVs), including copy number variants (CNVs), inversions, and translocations, were identified using MANTA v1.6.0 and 3bCNV, an in-house tool developed by 3billion, Inc. based on the depth-of-coverage (DOC) metrics41. Aneuploidy detection was performed using DOC information. SMN1 gene deletions were detected using SMA Finder42. Short tandem repeat (STR) expansions in 45 disease-associated genes (AFF2, AR, ARX, ATN1, ATXN10, ATXN1, ATXN2, ATXN3, ATXN7, ATXN8OS, BEAN1, C9ORF72, CACNA1A, CNBP, COMP, DAB1, DIP2B, DMPK, FGF14, FMR1, FOXL2, FXN, GIPC1, GLS, HOXD13, HTT, JPH3, LRP12, MARCHF6, NOP56, NOTCH2NLC, NUTM2B-AS1, PABPN1, PHOX2B, PPP2R2B, PRDM12, RAPGEF2, RFC1, RILPL1, SAMD12, STARD7, TBP, TCF4, XYLT1, and ZIC2) were called using ExpansionHunter v5.0.0 with RepeatCatalogs-v1.0.02243. Mobile element insertions were detected using the Mobile Element Locator Tool (MELT) v2.2.244. AutoMap v1.2 was used for detecting regions of homozygosity (ROH)45.
Variant interpretation and result categorization
Variants were annotated, filtered, and prioritized using the EVIDENCE automated interpretation pipeline developed by 3billion46. Variants previously reported in the literature or listed as pathogenic (P) or likely pathogenic (LP) in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) or the Human Gene Mutation Database (HGMD) professional were excluded from the filtering process47. SNVs/indels with an allele frequency (AF) > 5% in gnomAD v3.0 (https://gnomad.broadinstitute.org/), CNVs with an AF > 1% in gnomAD SVs v2.1 (https://gnomad.broadinstitute.org/) and Database of Genomic Variants (DGV) gold standard (https://dgv.tcag.ca/), and SVs with an AF > 1% in 3billion, Inc. database were filtered initially. The remaining coding and non-coding SNVs/indels and CNVs, including novel variants, were classified as P, LP, variant of uncertain significance (VUS), likely benign (LB), or benign (B) according to a customized framework based on the ACMG/AMP guidelines48,49,50. Structural variants involving breakpoints within known disease-associated genes or large inversions/translocations were considered potentially disease-causing. For STR expansion, repeat counts were interpreted as pathogenic if they met or exceeded disease-specific thresholds reported in the literature, OMIM (https://www.omim.org/), or the STRipy database51,52. Mobile element insertions (MEIs) were considered potentially disease-causing if the insertion occurred within a coding exon or a noncoding region previously associated with disease. To assess the phenotypic relevance of each variant, symptom similarity scoring using the depth of HPO terms was performed by modifying a previously described method38,39,40,41,42,43,44,45,46,48,49,50,52,53,54,55,56,57. Clinical geneticists and genomics scientists conducted a manual review of rare variants to identify those suitable for clinical reporting. All selected variants were confirmed by orthogonal tests, including Sanger sequencing or microarray. For a subset of these variants, segregation analysis was performed on available family members by Sanger sequencing.
The results were classified into three categories following established guidelines and phenotype-genotype correlations: diagnosis, possible diagnosis, and undiagnosed. A diagnosis was made when one or more P or LP variants were identified that were consistent with the patient’s phenotype and matched the known inheritance pattern of a gene-disease association. Variants in genes not yet listed as disease-associated in OMIM, but were repeatedly observed in the literature, were also categorized as diagnostic. A possible diagnosis included cases where a heterozygous or hemizygous VUS was found in a gene associated with autosomal dominant or X-linked inheritance and was considered a good fit for the patient’s phenotype. It also included compound heterozygous variants in autosomal recessive disease genes, where one variant was classified as P or LP and the other as a VUS. In cases where variants were found in genes not yet associated with disease in OMIM but previously reported in the literature, the result was also classified as a possible diagnosis. A case was classified as undiagnosed when no clinically significant variants were identified.
Validation of STR expansion through long-read sequencing
To validate STR expansions, we applied nCATS. Genomic DNA extracted from blood samples was processed using the ONT Cas9 sequencing kit and sequenced on either the GridION or MinION platform. Reads were aligned to the GRCh38 reference genome using minimap258, and repeat counts were estimated using Straglr59. Detailed methodologies have been described in our previous study60.
Statistical analysis
Statistical analyses were performed using the chi-square test to evaluate differences in diagnostic rates across various patient subgroups. A P-value of <0.05 was considered statistically significant. All statistical analyses were conducted using SPSS version 25.0 (IBM Corp., Armonk, NY, USA) or equivalent software.
Data availability
Genomic data from this study are not publicly available due to institutional restrictions. De-identified data may be provided upon reasonable request to the corresponding author.
References
Boycott, K. M., Vanstone, M. R., Bulman, D. E. & MacKenzie, A. E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 14, 681–691 (2013).
Nguengang Wakap, S. et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173 (2020).
Petillo, R. et al. Genomic testing in adults with undiagnosed rare conditions: improvement of diagnosis using clinical exome sequencing as a first-tier approach. Clin. Genet. https://doi.org/10.1111/cge.14715 (2025).
Nisar, H. et al. Whole-genome sequencing as a first-tier diagnostic framework for rare genetic diseases. Exp. Biol. Med. (Maywood) 246, 2610–2617 (2021).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N. Engl. J. Med. 369, 1502–1511 (2013).
Han, H. et al. Exome sequencing of 18,994 ethnically diverse patients with suspected rare Mendelian disorders. NPJ Genom. Med. 10, 6 (2025).
Yavarna, T. et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 134, 967–980 (2015).
Ewans, L. J. et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet. Med. 20, 1564–1574 (2018).
Liu, P. et al. Reanalysis of clinical exome sequencing data. N. Engl. J. Med. 380, 2478–2480 (2019).
Hawkes, G. et al. Whole-genome sequencing analysis identifies rare, large-effect noncoding variants and regulatory regions associated with circulating protein levels. Nat. Genet. 57, 626–634 (2025).
Pagnamenta, A. T. et al. Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Med. 15, 94 (2023).
Wojcik, M. H. et al. Genome sequencing for diagnosing rare diseases. N. Engl. J. Med. 390, 1985–1997 (2024).
Marchant, R. G. et al. Genome and RNA sequencing boost neuromuscular diagnoses to 62% from 34% with exome sequencing alone. Ann. Clin. Transl. Neurol. 11, 1250–1266 (2024).
Weisz-Hubshman, M. et al. De novo variants in RYBP are associated with a severe neurodevelopmental disorder and congenital anomalies. Genet. Med. 27, 101369 (2025).
Patra, M. et al. A novel variant of the human mitochondrial DnaJ protein, Tid1, associates with a human disease exhibiting developmental delay and polyneuropathy. Eur. J. Hum. Genet. 27, 1072–1080 (2019).
Rigter, P. M. F. et al. Role of CAMK2D in neurodevelopment and associated conditions. Am. J. Hum. Genet. 111, 364–382 (2024).
Khang, R. et al. Genome sequencing of rare disease patients through the Korean Regional Rare Disease Diagnostic Support program. Hum. Mutat. 2025, 6096758 (2025).
Investigators, G. P. P. et al. 100,000 Genomes pilot on rare-disease diagnosis in health care—preliminary report. N. Engl. J. Med. 385, 1868–1880 (2021).
Greene, D. et al. Mutations in the small nuclear RNA gene RNU2-2 cause a severe neurodevelopmental disorder with prominent epilepsy. Nat. Genet. https://doi.org/10.1038/s41588-025-02159-5 (2025).
Chen, Y. et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 632, 832–840 (2024).
Jackson, A. et al. Analysis of R-loop forming regions identifies RNU2-2 and RNU5B-1 as neurodevelopmental disorder genes. Nat. Genet. https://doi.org/10.1038/s41588-025-02209-y (2025).
Kaschta, D. et al. Evaluating genome sequencing strategies: trio, singleton, and standard testing in rare disease diagnosis. Genome Med. 17, 100 (2025).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Miller, T. M. et al. Trial of antisense oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 387, 1099–1110 (2022).
Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Al-Dewik, N. et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: the Qatari experience. Am. J. Med. Genet. A 179, 927–935 (2019).
Sanchis-Juan, A. et al. Genome sequencing and comprehensive rare-variant analysis of 465 families with neurodevelopmental disorders. Am. J. Hum. Genet. 110, 1343–1355 (2023).
van der Sanden, B. et al. The performance of genome sequencing as a first-tier test for neurodevelopmental disorders. Eur. J. Hum. Genet. 31, 81–88 (2023).
Topf, A. et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 22, 1478–1488 (2020).
Weisschuh, N. et al. Diagnostic genome sequencing improves diagnostic yield: a prospective single-centre study in 1000 patients with inherited eye diseases. J. Med. Genet. 61, 186–195 (2024).
Similuk, M. N. et al. Clinical exome sequencing of 1000 families with complex immune phenotypes: toward comprehensive genomic evaluations. J. Allergy Clin. Immunol. 150, 947–954 (2022).
Elsink, K. et al. Implementation of early next-generation sequencing for inborn errors of immunity: a prospective observational cohort study of diagnostic yield and clinical implications in Dutch Genome Diagnostic Centers. Front. Immunol. 12, 780134 (2021).
Thaventhiran, J. E. D. et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 583, 90–95 (2020).
Sheppard, S. et al. Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genet. Med. 20, 1663–1676 (2018).
Perry, J. et al. Exome sequencing expands the genetic diagnostic spectrum for pediatric hearing loss. Laryngoscope 133, 2417–2424 (2023).
Kim, J. A. et al. Systematic genetic assessment of hearing loss using whole-genome sequencing identifies pathogenic variants. Exp. Mol. Med. https://doi.org/10.1038/s12276-025-01428-x (2025).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Weisburd, B. et al. Diagnosing missed cases of spinal muscular atrophy in genome, exome, and panel sequencing data sets. Genet. Med. 27, 101336 (2024).
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Gardner, E. J. et al. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929 (2017).
Quinodoz, M. et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 12, 518 (2021).
Seo, G. H. et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 98, 562–570 (2020).
Stenson, P. D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003).
McCormick, E. M. et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum. Mutat. 41, 2028–2057 (2020).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
McKusick, V. A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 80, 588–604 (2007).
Halman, A., Dolzhenko, E. & Oshlack, A. STRipy: A graphical application for enhanced genotyping of pathogenic short tandem repeats in sequencing data. Hum. Mutat. 43, 859–868 (2022).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Wright, C. F., FitzPatrick, D. R., Ware, J. S., Rehm, H. L. & Firth, H. V. Importance of adopting standardized MANE transcripts in clinical reporting. Genet. Med. 25, 100331 (2023).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 e524 (2019).
Kohler, S. et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 85, 457–464 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Chiu, R., Rajan-Babu, I. S., Friedman, J. M. & Birol, I. Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences. Genome Biol. 22, 224 (2021).
Lee, S. et al. Prevalence and characterization of NOTCH2NLC GGC repeat expansions in Koreans: from a hospital cohort analysis to a population-wide study. Neurol. Genet. 10, e200147 (2024).
Acknowledgements
This study was supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (Grant No. RS-2022-II220333) and Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Health and Welfare, Ministry of Science and ICT, Ministry of Trade Industry and Energy, Korea Disease Control and Prevention Agency (Grant No. 2020M3E5D7086780, The National Project of Bio Big Data). This study was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No. RS-2023-00265923).
Author information
Authors and Affiliations
Contributions
Conceptualization— S.L., S.Y.K., J.C. Data Curation—S.L., S.Y.K., G.H.S., S.S.J., S.J., S.C., H.C., S.J.L., D.E.O., S.W.R., and J.K. Formal analysis—G.H.S., S.S.J., D.M., S.J., H.H., and H.L. Funding acquisition—B.C.K. and J.C. Project administration—S.L., G.H.S., S.Y.K., B.C.L., J.M., and H.L. Investigation—S.L., G.H.S., S.Y.K., S.J., S.C., H.J., S.J.L., D.E.O. S.W.R., J.K., D.M., and S.J. Writing—original draft—S.L., G.H.S., and S.Y.K. Writing—review and editing—all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S., Seo, G.H., Kim, S.Y. et al. Clinical utility of genome sequencing in rare diseases: lessons from a single-center study of 1,452 Korean families. npj Genom. Med. 11, 2 (2026). https://doi.org/10.1038/s41525-025-00538-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41525-025-00538-9
