
Abstract
Genetic studies of Parkinson’s disease (PD) have focused on single nucleotide variants (SNVs), with limited attention to copy number variants (CNVs). This study investigates CNVs in PD using candidate PD-related genes and genome-wide approaches. We identified CNVs from the ProtectMove project genotyping data of 2364 PD patients and 2909 controls using PennCNV. We validated 119 of 137 detected CNVs in PD-related genes (87%) using MLPA/qPCR, including 104 in PRKN, six in PARK7, four in SNCA, and others in LRRK2, RAB32, and VPS35. CNVs were present in 2.4% of patients and 1.5% of controls. Notably, 0.9% of patients carried potentially disease-causing CNVs compared to 0.1% in controls. CNVs were enriched in patients (OR = 1.67, p = 0.03) due to PRKN CNVs, particularly in early-onset cases. These results highlight the importance of CNVs in PD, particularly in PRKN, and suggest that rare CNVs in LRRK2 and RAB32 may contribute to disease risk and diagnostic potential.
Similar content being viewed by others
Introduction
The etiology of sporadic Parkinson’s disease (PD) is complex and multifactorial, shaped by a dynamic interplay between genetic susceptibility and environmental exposures1,2,3. While the majority of PD cases are sporadic, genetic factors have emerged as important contributors to both disease risk and disease progression. These genetic influences range from rare, highly penetrant variants to the cumulative effect of common risk alleles acting additively across the genome. Approximately 5% of PD cases are attributed to monogenic forms, which arise from rare pathogenic variants in genes following either autosomal dominant (SNCA, LRRK2, VPS35, CHCHD2, and RAB32) or recessive (PRKN, PINK1, and PARK7) inheritance patterns, while GBA1 risk variants are identified in about 10% of patients3,4,5,6.
In addition to single-nucleotide variants (SNVs), structural variants, particularly copy number variants (CNVs), are increasingly recognized as a significant source of genetic risk in PD. CNVs, which encompass large genomic rearrangements such as deletions and duplications, can disrupt gene dosage and genomic architecture, thereby contributing to disease mechanisms7,8,9.
Pathogenic CNVs affecting PD-related genes have been described in both familial and sporadic cases. Duplications and triplications of the SNCA locus are known causes of autosomal dominant PD10,11. Deletions in PINK112 and PARK713 have been described in familial forms of PD, although they are less common than those in PRKN14,15,16,17. These deletions are frequently observed in patients with early-onset PD. Of particular note, PRKN harbors a genomic region highly prone to rearrangements18, making CNVs in this gene a common event among PD patients.
Despite the growing recognition of CNVs in PD genetics, most studies to date have been limited by small sample sizes or targeted approaches focusing solely on a predefined set of genes. More recently, a comprehensive genome-wide CNV burden analysis in a Latin American cohort comprising 747 PD patients and 632 neurologically healthy controls demonstrated a significant enrichment of CNVs affecting PD-related genes in patients compared to controls19. These findings underscore the relevance of CNVs as an underappreciated yet important component of genetic risk in PD and highlight the need for further studies in larger and more diverse populations.
Long-read sequencing has revealed complex structural variants (SVs) in PD, including a 7 Mb PRKN inversion missed by standard methods9 and three genome-wide significant SVs associated with PD risk20. Tools like CNV-Finder enable scalable CNV detection in PD genes using array data and deep learning21, while large-scale brain sequencing studies link SVs to gene regulation, advancing our understanding of PD genetics22.
We investigated the impact of CNVs on PD risk using both a candidate PD-related genes approach and a genome-wide burden analysis in European samples from the ProtectMove project (https://protect-move.de/). We identified 22 PD patients with potentially disease-causing CNVs and observed a significant enrichment of CNVs overlapping with PRKN in PD patients compared to controls.
Results
Study cohort and overview of CNV findings
After genotyping QC, we included data from 5273 individuals of European ancestry (2364 PD patients and 2909 controls) (Supplementary Tables S1, S2). Among those, 443 PD patients exhibited EOPD (mean age at onset (AAO) 42.7 ± 7.4 years, mean age at assessment 53.6 ± 9.8 years).
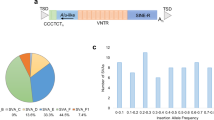
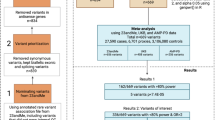
Initial CNV calls identified 99,759 deletions and 44,451 duplications (Supplementary Table S2), which were subsequently filtered (Fig. 1A, Supplementary Table S2) to 87,450 deletions and 36,986 duplications. Of these, 535 CNVs overlapped with PD-related genes, with 137 CNVs (Table 1, Supplementary Table S3) exceeding 500 bp in length. Validation using MLPA/qPCR confirmed 119 CNVs across 56 PD patients and 43 controls, yielding an 87% validation rate. These CNVs largely involved the PRKN gene (104 CNVs), six in PARK7, four in SNCA, two in LRRK2, two in RAB32, and one in VPS35 (Table 1). No CNVs were identified in GBA1 and CHCHD2.

A Overview of the study: schematic representation of the analysis workflow. B. Visualisation of CNVs in the PRKN gene. C-D Forest plot showing the CNV burden of validated CNVs in PD-related genes and PRKN (C) and the genome-wide CNV burden (D) in PD patients compared to controls. The sample size for each comparison was included in the figures showing the number of corresponding carriers and the total number of samples. Logistic regression was used to estimate odds ratios (ORs) and p-values for each CNV category and were adjusted for age at assessment, sex, and the first five components of PCA. (*) significant p-values were adjusted using FDR methods.
Characterization of PRKN CNVs
In PRKN, the validation rate was particularly high (95.4%). A total of 104 CNVs (87 unique CNVs) were validated in 48 PD patients (2.0% of patients) and 36 controls (1.2%). 18 individuals (13 PD and 5 controls) carried at least two distinct PRKN CNVs. The most frequent PRKN CNVs were Exon 2 duplication (n = 33 (32%), observed in 19 PD patients and 14 controls) and Exon 4 deletions (n = 18 (18%), observed in 15 PD patients and three controls, Supplementary Table S3 and Fig. 1B). PD patients with validated PRKN CNVs had a significantly earlier AAO (51.9 ± 17.9 years) compared to non-PRKN CNV carriers (60.9 ± 11.6 years, padj = 7e-07).
The majority of PRKN CNV carriers were heterozygous (58 CNVs in 43 patients (1.8%) and 41 CNVs in 36 controls (1.2%)), while five patients had homozygous CNVs (mean AAO = 40.2 ± 9.6 years, Supplementary Table S3). Twelve PD patients carried both a validated CNV (eleven heterozygous, one homozygous) and a rare SNV in PRKN, of which nine SNVs were classified as pathogenic or likely pathogenic (Supplementary Table S3). These twelve patients had a mean AAO of 34.3 ± 21.3 years, including four with juvenile PD (onset before age 21 years). This was significantly earlier than the AAO in PD patients having only a heterozygous PRKN CNV but no additional rare PRKN SNV (mean AAO of 59.5 ± 11.2 years, padj = 0.01, Supplementary Table S3). In comparison, control individuals with a heterozygous PRKN CNV had a mean age at assessment of 65.0 ± 6.4 years.
In total, of the 48 PD patients with PRKN CNVs, 16 (~0.7% of all PD patients) were carriers of homozygous CNVs and possible disease-causing compound heterozygous CNV-SNV combinations. Therefore, the frequency of PD patients with only heterozygous PRKN CNVs is reevaluated at 1.4% (46 CNVs in 32 patients), which is similar to the frequency observed in the controls (1.2%). No significant AAO difference was observed between PRKN deletion and duplication carriers (padj = 0.1).
CNVs in other PD-related genes and overall burden
Additionally, eleven individuals (10 PD patients, one control) were identified to carry four different heterozygous CNVs in PINK1 (Supplementary Table S3), but MLPA and qPCR did not confirm these findings. In PARK7, six heterozygous CNVs were identified (three in PD patients, three in controls), all of which were validated. Regarding the PD genes linked to autosomal dominantly inherited PD, two heterozygous LRRK2 duplications were identified, one in an EOPD patient and one in a 77-year-old control. We estimated the frequency of LRRK2 duplications in gnomAD23, where four whole-gene duplications were identified, each observed in a unique individual with European ancestry, with frequencies ranging from 4.31e−6 and 2.15e−6. We also identified two individuals with the same 19 kb duplication in RAB32, a 70-year-old healthy control and a PD patient with AAO of 73 years. Of note, a RAB32 duplication has been reported in 12 European individuals in gnomAD (frequency: 3.6e−05). Additional biomaterials of these patients were not available to test for expression and functional changes. We validated four CNVs in SNCA, two duplications and two deletions, among the five identified CNVs (Supplementary Table S3). These were found in three PD patients and one 79-year-old control. Additionally, we identified and confirmed a whole-gene duplication of VPS35 in a 70-year-old control (Supplementary Table S3).
In total, 22 CNVs found in PD patients (~0.9%) were considered potentially disease-causing. These included five in autosomal-dominant genes (three in SNCA, one in LRRK2, and one in RAB32) and 17 in autosomal-recessive genes (16 in PRKN and one in PARK7), either as homozygous CNVs or in combination with a rare SNV. In contrast, only four CNVs were detected in controls (~0.1%), affecting SNCA, LRRK2, RAB32, and VPS35.
Genome-wide CNV burden analysis
Overall, validated CNVs in PD-related genes were significantly enriched in PD patients compared to controls (OR = 1.67[1.09–2.55], padj = 0.03). This association was driven by PRKN CNVs (ORPRKN = 1.65[1.05–2.61], padj = 0.04, Fig. 1C). We also found a substantial increase of CNVs in EOPD patients compared to controls (OR = 4.04[2.13–7.50], padj = 7.4e−05, Fig. 1C). Among 443 EOPD patients, 20 (4.5%) had PRKN CNVs (ORPRKN = 4.03[2.02–7.78], padj = 1.3e−04) versus 1.2% of controls. No significant burden was observed in LOPD patients compared to controls in PD genes (OR = 0.97[0.58−1.62], padj = 0.9) and PRKN (ORPRKN = 1.03[0.59–1.79], padj = 0.8, Fig. 1C).
Genome-wide CNV analysis and identification of novel loci
Next, we performed a genome-wide burden analysis with filtering steps to exclude false-positive CNV calls based on SNV coverage, CNV length, and density (Fig. 1A, Supplementary Table S2). No significant differences were found between PD patients and controls for genome-wide CNV burden (OR = 1.10 [0.97–1.24], padj = 0.2), duplications (OR = 0.99[0.89–1.09], padj = 0.8), or deletions (OR = 1.09[0.98–1.22], padj = 0.2, Fig. 1D), nor for non-PD-related genes (OR = 1.09[0.98–1.21], padj = 0.8) or large CNVs (OR = 1.06[0.79–1.41], padj = 0.8) (Fig. 1D).
Notably, three loci containing genes not yet linked to PD were significantly enriched for duplications in PD patients. These were located on chromosome 16p13.3 (577,717-634,136 (GRCh37), CAPN15, C16orf11, NHLRC4 and PIGQ) and two adjacent regions on chromosome 19p13.3 (851,014-921,015, ELANE, CFD, MED16, R3HDM4, and KISS1R) and (925,781-1,228,428, ARID3A, WDR18, GRIN3B, TMEM259, CNN2, ABCA7, and STK11). However, these associations were no longer significant after multiple test corrections (Supplementary Table S4). Carriers of these CNVs are listed in Supplementary Table S5, along with their age and carrier status. Among controls, three individuals carried these CNVs and were all over 60 years old at the time of assessment. No genes were significantly enriched for deletions or in the control group.
Discussion
In this genome-wide CNV analysis of over 5,000 PD patients and controls based on genotyping data, we identified and experimentally confirmed 119 CNVs in PD-related genes, achieving an overall validation rate of 87%. While PRKN accounted for the majority of these CNVs, we also identified rare CNVs in PARK7, SNCA, and VPS35, as well as in LRRK2 and RAB32, where no previously reported (heterozygous) CNVs had been experimentally confirmed. Among these, 22 CNVs identified in PD patients were considered potentially disease-causing, representing a substantially higher burden compared to controls. This classification was based on gene content, known pathogenic mechanisms, and frequency in patients versus controls. However, we acknowledge that the functional impact of some duplications, particularly those involving LRRK2 and RAB32, remains uncertain, as SNP-array data cannot determine their genomic context, regulatory influence, or expression consequences. These findings expand the spectrum of CNVs in PD-associated genes and highlight both established and potential novel contributors to disease risk, while also underscoring the need for functional validation, especially of duplications.
Despite extensive screening, PRKN was the only gene with CNVs significantly associated with PD, reaffirming its role as the predominant driver of CNV burden in PD. The high validation rate of PRKN CNVs (95.4%) underscores the robustness of our detection approach, whereas the absence of validated CNVs in PINK1 suggests that structural variants in this gene may be exceedingly rare and challenging to detect. A previous Latin American study (LARGE-PD)19 confirmed seven carriers in PRKN or SNCA with 100% accuracy using MLPA.
From a methodological perspective, various algorithms detect CNVs from genotyping arrays, with PennCNV being the most used despite its high false-positive rate for small CNVs24,25. Our empirical filtering retained reliable calls, with MLPA and qPCR used as reference methods for validation. Studies validating PennCNV calls with these methods found high concordance rates26,27,28, though they generally involved fewer patients or less stringent filtering.
Notably, on the group level, our study revealed an increased burden of validated CNVs overlapping PD-related genes in PD patients, driven by PRKN CNVs, without a genome-wide CNV burden increase. These findings align with the LARGE-PD study19 and previous findings in familial PD cases of European ancestry26.
We conducted a genome-wide CNV burden analysis to test whether individuals with PD carry a higher load of CNVs than controls. This approach builds on findings from other neurological disorders29,30, where increased CNV burden has been associated with disease risk. By evaluating global CNV load, we aimed to identify both known and novel loci that may contribute to PD susceptibility. We found that, duplications of three genomic regions on chromosomes 16 and 19, not previously linked to PD and not overlapping with significant variants from the most recent SNP-based genome-wide association study31, showed potential enrichment in PD patients. These regions contain over 16 protein-coding genes, including ABCA7, which is linked to late-onset Alzheimer’s disease32. However, the small number of PD patients with these duplications limits conclusions, requiring further studies with larger sample sizes. These CNVs showed nominal enrichment in PD patients but did not survive multiple testing correction. Their small number and presence in controls over 60, beyond typical early PD-onset, limit conclusions, warranting further investigation in larger cohorts. We further investigated the three previously reported PD-associated genome-wide significant deletions20, but did not detect comparable variants in our dataset, possibly due to differences in detection methods, population structure, or sample size.
PRKN homozygous or compound heterozygous deletions and duplications are common in EOPD and familial PD33,34,35. In our study, 2.0% of PD patients (4.5% of EOPD) carried PRKN CNVs, significantly linked to earlier AAO. Our findings align with the prior ROPAD study6, where 66 unique CNVs were detected in PD-related genes, with PRKN carrying the highest burden (42 unique CNVs), followed by SNCA, PARK7, and PINK1. Additionally, a study of 647 PRKN-PD patients14 found deletions and duplications accounted for 43.6% of all variants, with Exon 3 deletions most common and associated with earlier PD onset. In our study, Exon 2 duplications were most frequent in PD patients, whereas exon 3 deletions represented only 9% of all PRKN CNVs. Five PD patients had homozygous PRKN deletions, while most PRKN CNV carriers were heterozygous, with no significant difference between PD patients and controls. Although some studies have suggested that heterozygous loss of PRKN function may increase PD risk and is associated with earlier AAO36,37,38, more recent and larger studies have not supported this hypothesis16,39,40. Screening of heterozygous PRKN CNV carriers for additional PRKN coding SNVs revealed that some PD patients, unlike the controls, carried an additional pathogenic SNV and had early-onset PD, supporting the pathogenic role of compound heterozygous CNV-SNV combinations in PRKN. Furthermore, recent studies have identified PRKN structural variants (SVs) detectable only by long-read sequencing9, suggesting that such SVs may also be present in our cohort, with some individuals possibly harboring a CNV or another hard-to-detect variant on the other allele.
Previous studies have identified CNVs in SNCA10, PARK741 in familial or sporadic PD. One study reported CNVs in LRRK2 in Vietnamese PD patients42 and the ROPAD study reported a duplication in VPS356 and none in RAB32. In our study, we validated CNVs in all five of these genes in eight PD patients and seven controls. Therefore, the significance of these CNVs warrants further elucidation. In LRRK2, we detected a whole-gene duplication in an EOPD patient and a partial duplication in a control. The exact disease mechanism of LRRK2 remains unclear, but most PD-causing variants act through a gain-of-function effect, increasing kinase activity43. In line with this, there is no association between loss-of-function LRRK2 variants and PD44,45. We also identified a duplication in RAB32, the most recently recognized PD gene, where only a pathogenic missense variant (p.Ser71 Arg) has been reported5,46 to date.
Despite its large sample size and novel findings, our study has several limitations. Due to methodological constraint24, short CNVs were excluded, potentially underestimating the contribution of smaller CNVs to PD risk. Our approach does not capture complex SVs such as inversions, repeat expansions, or balanced rearrangements, which may contribute to disease risk but require sequencing-based methods for detection. Although CNV–SNV co-occurrence may suggest compound heterozygosity, the pathogenicity of these combinations remains uncertain without functional validation. Phasing could not be determined, underscoring the need for parental genotyping or long-read sequencing in future studies. Moreover, we acknowledge that additional pathogenic variants, such as deep intronic changes, regulatory mutations, or complex structural rearrangements, may remain undetected with this approach. Future studies incorporating long-read sequencing or adaptive sampling strategies could help resolve zygosity in heterozygous CNV carriers and improve detection of complex or cryptic variants in PRKN, which are often missed by MLPA and array-based methods. Additionally, although our validation process helped identify many false positives, it was limited to PD-related genes, leaving the false positive rate in other genomic regions unknown. Furthermore, the proportion of false negatives could not be assessed. Future studies using long-read sequencing could better resolve complex structural variants, particularly in PRKN9, but such approaches currently come at significantly higher costs compared to genotyping arrays.
In conclusion, our genome-wide CNV analysis of over 5000 PD patients and controls of European ancestry showed that CNVs in PRKN are most frequent in PD patients, highlighting their importance in genetic testing for improved disease management and personalized treatment. Further, we demonstrated that, albeit rarer, the study of CNVs in other PD genes is warranted.
Methods
Study population and genotyping data
ProtectMove includes 13,330 individuals with PD, dystonia, X-linked dystonia-parkinsonism and healthy controls. In this study, we selected 8382 individuals of predominantly European descent, including 5811 controls and 2571 PD patients. Patients with early-onset PD (EOPD) were defined as those diagnosed before the age of 50 years47, while all other patients were considered to have late-onset PD (LOPD).
Genotyping and quality control
Samples were genotyped on Illumina’s Infinium Global screening array (GSA) v1.0 (Illumina, San Diego, California) in separate batches. A total of 413,738 markers were available for quality control (QC). We performed genotyping data QC using PLINK v1.948. Samples with a call rate <98% or discordant sex status were excluded. We filtered out variants for genotyping rate <98%, minor allele frequency <0.01 and deviation from Hardy–Weinberg equilibrium (P value < 1e−06). We also removed samples exhibiting an excess of heterozygosity. Next, we tested for relatedness using KING49 and excluded samples with first-degree relatedness. To determine genetic ancestry, we performed linkage disequilibrium (LD) pruning and merged our dataset with samples from the 1000 Genomes Project. We then calculated the principal components (PCs) using PLINK v1.9 on the combined dataset. Individuals were assigned to ancestry groups based on their proximity to 1000 Genomes reference populations in PCA space. Only samples clustering within the European ancestry group (within 3 standard deviations on PC1 and PC2) were retained. In order to ensure that the control group was older than the typical at-risk age for PD, healthy controls under the age of 50 years were excluded from the study, thereby reducing the likelihood of including preclinical individuals.
CNV calling, quality control and filtering
CNVs were detected using PennCNV (v1.0.524,) from custom B-allele frequency (BAF) and GC wave-adjusted log R ratio (LRR) intensity files using GenomeStudio (v2.0.5 Illumina). Initial QC using PennCNV (steps 1–3, Fig. 1A) included merging adjacent CNV calls if their marker overlap was less than 20%, followed by intensity-based QC to exclude low-quality data. Post-QC, all samples had an LRR standard deviation <0.24, waviness factor <0.03, and BAF drift <0.001. Spurious CNV calls in problematic regions (centromeric, telomeric, HLA)24 were removed. Filtered CNVs were annotated for gene content using ANNOVAR (v2020-06-08, hg19 assembly). We classified a CNV as ‘genic’ if it overlapped, either partially or fully, with any part of a protein-coding gene, including promoter regions, exons, or introns. CNVs were further filtered (Fig. 1A) using standard parameters for CNV calling19. CNVs spanning less than 20 SNVs, smaller than 20 kb, or with a SNV density (number of SNVs/length of CNV) below 0.0001 were excluded. Quality scores (QS) were calculated using the method of Macé25, and CNVs with a QS between −0.5 and 0.5 were excluded.
Identification and molecular validation of CNVs and SNVs in PD-related genes
We selected CNVs overlapping the PD-related genes LRRK2, SNCA, VPS35, GBA1, PRKN, PARK7, PINK1 (www.mdsgene.org) as well as CHCHD2 and RAB32. CNVs shorter than 500 bp were excluded due to unreliability of calling24. Validation of CNVs was done by MLPA if respective probe mixes were available, i.e., probe mixes P051 and/or P052 (MRC Holland). For variants in other genes or only intronic variants, we designed primers targeting the central region of the proposed CNV and applied quantitative PCR using a LightCycler (RocheDiagnostics). Samples were analyzed in duplicate, and relative quantification of the region of interest was carried out in comparison with the genomic DNA level of the HBB gene. MLPA was performed according to the manufacturer’s instructions, products were separated on an ABI 3500XL sequencing machine (Applied Biosystems) and quantified using the CoffalyserNet software package (MRC Holland). Breakpoints were not determined. If the same CNV (with the exact same predicted breakpoints) was called in multiple samples, validation was assumed for all samples when qPCR confirmed the CNV in at least one sample and DNA quantity/quality was too low to experimentally evaluate the other samples. Individuals carrying heterozygous CNVs in the recessive PD-related genes PRKN, PINK1, and PARK7 were screened for rare SNVs in these genes, and the identified variants were validated by Sanger sequencing. The pathogenicity of these SNVs were defined according to the MDSGene annotation (http://www.mdsgene.org).
Burden analysis
We calculated the CNV burden associated with PD across five categories: (1) overall genome-wide CNV burden, including CNVs in non-genic regions; (2) large CNVs (≥1 Mb); (3) CNVs overlapping non-PD-related genes; (4) validated CNVs overlapping PD-related genes; and (5) validated CNVs overlapping the PRKN gene.
The CNV burden between PD patients and controls was compared using logistic regression (glm function in R v4.3.1) to estimate odds ratios (OR), 95% confidence intervals and p-values. Disease status was included as the dependent variable, with CNV status as the independent variable, adjusting for sex, age at examination, and the first five population stratification PCs (see Genotyping and Quality Control section). We included five PCs as covariates because the corresponding eigenvalues showed these components captured approximately 83% of the total variance. A scree plot (Supplementary Fig S1) confirmed an elbow at this point, indicating that these components represent the main axes of population structure. We also evaluated potential technical confounders, such as genotyping batch and array type, by inspecting PC plots for systematic clustering. No significant effects were observed beyond the variation captured by the selected PCs. Given our stringent QC pipeline, batch-specific artifacts were minimized, and explicit covariates for batch or array type were unnecessary. The p-values from the logistic regression were further adjusted for multiple testing (11 tests) using the FDR method. The enrichment of filtered CNVs in individual genes, separately for duplications and deletions, was also compared between PD patients and controls using logistic regression, with p-values adjusted for 2684 tests using the FDR method.
Statistical analysis of age at onset (AAO) across PRKN CNV groups
The comparison of AAO of PD was conducted across three groups: (1) carriers vs. non-carriers of PRKN CNVs, (2) carriers of a CNV in PRKN with an additional rare single nucleotide variant (SNV) in PRKN vs. carriers of CNVs in PRKN only, and (3) carriers of PRKN duplications vs. deletions. Linear regression models were used to perform the analysis, adjusting for sex, age, and the first five PCs. To account for multiple comparisons across the three tests, p-values derived from linear models were further corrected using the FDR method.
Ethics approval
The local Ethics Committee of the Universities in Lübeck (04-155 and 16-039) and Kiel approved the collection of patients and controls for genetic investigations. All patients and controls provided written informed consent prior to inclusion in the study. The ethics committee of the Physician’s Board Hesse, Germany (approval no. FF89/2008) approved the Denopa study. The Ethics Committee of the Bolzano Health District approved the GESSPARK protocol on 05 June 2008 (30/2008), with an update approved by the Ethics Committee of the Healthcare System of the Autonomous Province of Bolzano-South Tyrol on 11 October 2017. The Ethics Committee of the Bolzano Health District approved the DISP protocol on 25 July 2012 (62/2012), with an update approved by the Ethics Committee of the Healthcare System of the Autonomous Province of Bolzano-South Tyrol on 11 October 2017.
Data availability
Individual-level CNV and genotyping data are available upon request, subject to the consent agreements and data sharing policies of each participating centre.
Code availability
Relevant scripts used in the present work are available on GitHub (https://gitlab.lcsb.uni.lu/genomeanalysis/protectmove/copy_number_variants_analysis).
References
Tanner, C. M. & Ostrem, J. L. Parkinson’s disease. N. Engl. J. Med. 391, 442–452 (2024).
Dorsey, E. R. & Bloem, B. R. Parkinson’s disease is predominantly an environmental disease. JPD 14, 451–465 (2024).
Lim, S.Y. & Klein, C. Parkinson’s disease is predominantly a genetic disease. JPD 14, 467–482 (2024).
Funayama, M. et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol. 14, 274–282 (2015).
Hop, P. J. et al. Systematic rare variant analyses identify RAB32 as a susceptibility gene for familial Parkinson’s disease. Nat. Genet. 56, 1371–1376 (2024).
Westenberger, A. et al. Relevance of genetic testing in the gene-targeted trial era: the Rostock Parkinson’s disease study. Brain 147, 2652–2667 (2024).
La Cognata, V., Morello, G., D’Agata, V. & Cavallaro, S. Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum. Genet. 136, 13–37 (2017).
Miano-Burkhardt, A., Alvarez Jerez, P., Daida, K., Bandres Ciga, S. & Billingsley, K. J. The role of structural variants in the genetic architecture of Parkinson’s disease. IJMS 25, 4801 (2024).
Daida, K. et al. Long-read sequencing resolves a complex structural variant in PRKN Parkinson’s disease. Mov. Disord. 38, 2249–2257 (2023).
Singleton, A. B. et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841–841 (2003).
Ahn, T.B. et al. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70, 43–49 (2008).
Marongiu, R. et al. Whole gene deletion and splicing mutations expand thePINK1 genotypic spectrum. Hum. Mutat. 28, 98–98 (2007).
Bonifati, V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset Parkinsonism. Science 299, 256–259 (2003).
Menon, P. J. et al. Genotype–phenotype correlation in PRKN-associated Parkinson’s disease. npj Parkinsons Dis. 10, 72 (2024).
Pankratz, N. et al. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 73, 279–286 (2009).
Kay, D. M. et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology 75, 1189–1194 (2010).
Grünewald, A., Kasten, M., Ziegler, A. & Klein, C. Next-generation phenotyping using the Parkin example: time to catch up with genetics. JAMA Neurol. 70, 1186 (2013).
Ambroziak, W. et al. Genomic instability in the PARK2 locus is associated with Parkinson’s disease. J. Appl Genet. 56, 451–461 (2015).
Sarihan, E. I. et al. Genome-wide analysis of copy number variation in Latin American Parkinson’s disease patients. Mov. Disord. 36, 434–441 (2021).
Billingsley, K. J. et al. Genome-wide analysis of structural variants in Parkinson disease. Ann. Neurol. 93, 1012–1022 (2023).
Kuznetsov, N. et al. CNV-finder: streamlining copy number variation discovery. Preprint at https://doi.org/10.1101/2024.11.22.624040 (2024).
Billingsley, K. J. et al. Long-read sequencing of hundreds of diverse brains provides insight into the impact of structural variation on gene expression and DNA methylation. Preprint at https://doi.org/10.1101/2024.12.16.628723 (2024).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Wang, K. et al. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674 (2007).
Macé, A. et al. New quality measure for SNP array based CNV detection. Bioinformatics 32, 3298–3305 (2016).
Pankratz, N. et al. Copy number variation in familial Parkinson disease. PLoS ONE 6, e20988 (2011).
Curtis, C. et al. The pitfalls of platform comparison: DNA copy number array technologies assessed. BMC Genomics 10, 588 (2009).
Zhang, D. et al. Accuracy of CNV detection from GWAS data. PLoS ONE 6, e14511 (2011).
Niestroj, L.M. et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 143, 2106–2118 (2020).
Collins, R. L. et al. A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25 (2022).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
De Roeck, A., Van Broeckhoven, C. & Sleegers, K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 138, 201–220 (2019).
Domingo, A. & Klein, C. Genetics of Parkinson disease. In Handbook of Clinical Neurology vol. 147 211–227 (Elsevier, 2018).
Ahmad, A., Nkosi, D. & Iqbal, M. A. PARK2 microdeletion or duplications have been implicated in different neurological disorders including early onset Parkinson disease. Genes. 14, 600 (2023).
Elfferich, P. et al. Breakpoint mapping of 13 large parkin deletions/duplications reveals an exon 4 deletion and an exon 7 duplication as founder mutations. Neurogenetics 12, 263–271 (2011).
Sun, M. et al. Influence of heterozygosity for Parkin mutation on onset age in familial Parkinson disease: the gene PD study. Arch. Neurol. 63, 826 (2006).
Huttenlocher, J. et al. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet. 24, 5637–5643 (2015).
Klein, C., Lohmann-Hedrich, K., Rogaeva, E., Schlossmacher, M. G. & Lang, A. E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 6, 652–662 (2007).
Yu, E. et al. Analysis of heterozygous PRKN variants and copy-number variations in Parkinson’s disease. Mov. Disord. 36, 178–187 (2021).
Zhu, W. et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease. Brain 145, 2077–2091 (2022).
Kasten, M. et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1. MDSGene systematic review. Mov. Disord. 33, 730–741 (2018).
Do, M. D. et al. Clinical and genetic analysis of Vietnamese patients diagnosed with early-onset Parkinson’s disease. Brain Behav. 13, e2950 (2023).
Kalogeropulou, A. F. et al. Impact of 100 LRRK2 variants linked to Parkinson’s disease on kinase activity and microtubule binding. Biochem. J. 479, 1759–1783 (2022).
Blauwendraat, C. et al. Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol. 75, 1416 (2018).
Beetz, C. et al. LRRK2 Loss-of-function variants in patients with rare diseases: no evidence for a phenotypic impact. Mov. Disord. 36, 1029–1031 (2021).
Gustavsson, E. K. et al. RAB32 Ser71Arg in autosomal dominant Parkinson’s disease: linkage, association, and functional analyses. Lancet Neurol. 23, 603–614 (2024).
Mehanna, R. et al. Age cutoff for early-onset Parkinson’s disease: recommendations from the international parkinson and movement disorder society task force on early onset Parkinson’s disease. Mov. Disord. Clin. Pr. 9, 869–878 (2022).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaSci. 4, 7 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Acknowledgements
This study was supported by the German Research Foundation (DFG, project FOR 2488).
Author information
Authors and Affiliations
Contributions
All the authors contributed to revising the manuscript and approved the submitted version.Specific contributions:1.Research project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique; Z.L. 1A, B, C, 2A, B, 3A. K.L. 1A, B, C, 2C, 3A. E.-J.V. 1B, 1C, 2C, 3B. E.W.-B. 1C, 2C, 3B. L.-M.N. 2A, 2C, 3B. B.-H.L. 1C, 2B, 3B. S.S. 1C, 2B, 3B. A.B. 1C, 2C, 3B. M.B. 1C, 2C, 3B. D.L. 2A, 2C, 3B. A.G. 2A, 2C, 3B. N.B. 1C, 2C, 3B. A.F. 1B, 2C, 3B. A.H. 1B, 2C, 3B. M.K. 1B, 1C, 2C, 3B. K.E.Z 1B, 1C, 2C, 3B. L.M.L. 1C, 2C, 3B. W.L. 1B, 2C, 3B. B.M. 1B, 1C, 2C, 3B. H.P. 1C 2C, 3B. P.P.P. 1B, 2C, 3B. A.C. 1B, 2C, 3B. I.R.K. 1B, 2C, 3B. P.M. 1A, 1B, 2C, 3B. C.K. 1A, 1B, 2C, 3B.
Corresponding author
Ethics declarations
Competing interests
Relevant conflict of interests and financial disclosures are documented in the supplementary materials (authors_disclosure.xlsx).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Landoulsi, Z., Lohmann, K., Vollstedt, EJ. et al. Large-scale copy number variant analysis in genes linked to Parkinson´s disease. npj Parkinsons Dis. 11, 225 (2025). https://doi.org/10.1038/s41531-025-01076-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01076-y
