
Abstract
Rare loss-of-function variants in ITSN1 were recently reported to confer a high risk for Parkinson’s disease (PD). From our local large exome sequencing dataset of PD cases, we identified five carriers from three families. Clinical features of ITSN1-PD are typical and responsive to standard treatments. Additionally, we discuss whether ITSN1 loss-of-function variants should only be considered as a high-risk factor or a Mendelian PD gene.
Similar content being viewed by others


Main
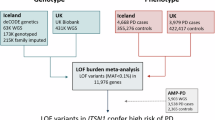
ITSN1 is a multidomain scaffold protein that plays an important role in synaptic vesicle endocytosis, actin cell signaling, cytoskeleton rearrangements, and the regulation of RNA-binding proteins1,2. Heterozygous loss-of-function (LOF) variants in ITSN1 were first reported to cause a childhood-onset disease with global developmental delay, autism spectrum disorder, and intellectual disability3. Recently, two different articles using overlapping large Parkinson’s disease (PD) datasets have reported a strong association between ITSN1 LOF variants and Parkinson’s disease (PD), with odds ratios ranging from 7.3 (95% confidence interval [3.5–15.2])4 to 10.5 (95% confidence interval [5.2–21.34])5. Clinical information was limited, and whether ITSN1-PD is associated with a specific phenotype responsive to treatments remains elusive.
Here, in a cohort of 2076 PD patients who underwent whole-exome sequencing (WES), we report phenotypic and genetic data of five affected carriers from three novel families, indicating that ITSN1 may be considered a Mendelian PD gene with reduced penetrance.
In pedigree A, a proband (III-2) and her mother (II-3) developed PD at 40 years (Fig. 1). The grandmother died at the age of 94 years, and the grandfather at 60 years after an accident without any PD symptoms. Clinical information is limited for II-3. Motor and cognitive features slowly worsened, and she died at the age of 62 years in a bedridden state. For III-2, the disease started with a rapidly progressing tremor of the left hand with a weak response to L-dopa. Cerebral magnetic resonance imaging (MRI) was normal. Deep brain stimulation (DBS) initiated at 49 years of age had a positive effect on the tremor. Clinical examination at 65 years showed dysphonia and walking difficulties. At that time, she had a mild distal tremor, no hypertonia, no urinary impairment, and neither pyramidal nor cerebellar signs. At her last examination at the age of 66 years, she had orthostatic hypotension with frequent falls. DBS remained efficient on motor symptoms. She was treated with 575 mg of L-dopa equivalent daily dose (LEDD), and the Unified Parkinson’s Disease Rating Scale (UPDRS) III motor score was 40/108 (no information whether on or off). WES identified a frameshift duplication of a single nucleotide in ITSN1 (NM_003024.3:c.472dup; p.Leu158ProfsTer3) in individual III-2 located in exon 6/40 (Supplementary Fig. 1). This variant has three occurrences in gnomAD v4.1 (allele frequency: 2.052 × 10−6) and none in a cohort of 4854 healthy French individuals6,7 and is absent from the ClinVar database and literature. WES did not find any pathogenic or likely pathogenic variants in any known PD gene. DNA was not available from any other family member.

The first two pedigrees suggested an autosomal dominant inheritance, while the third suggested an autosomal recessive inheritance. Of note, the two siblings in pedigree C were carriers of the GBA1 p.S403N variant classified as “mild risk” in Gaucher disease. The unaffected individuals II-3 and II-4 carried neither the ITSN1 nor the GBA1 variant. AO age at onset, AP age at passing, AE age at last examination.
In pedigree B, six members were affected with PD. DNA was available for two of them. III-6 is a female who presented at 62 years with a right-hand resting tremor. Cerebral computed tomography scan was normal. The initial response to L-dopa was positive (assessed at 65%), and the disease evolved slowly. At the last examination at the age of 72 years, in addition to tremor, she had an akinetorigid syndrome without any atypical neurological signs. UPDRS III was 13/108 on and 20/108 off, and the Hoehn and Yahr stage was 2/5. She had urinary incontinence and urgenturia. LEDD was 300 mg without limb-induced dyskinesia or motor fluctuations. Her son (IV-6) started at 30 years with an asymmetrical resting tremor of the left upper limb and slowly evolved. At the age of 40 years, he had no cognitive impairment (mini mental status examination score 30/30), no urinary impairment, and no atypical signs. UPDRS III was 24/108 on, and the Hoehn and Yahr stage was 2/5. WES revealed that they both carry a frameshift duplication of one nucleotide in ITSN1 (NM_003024.3:c.2263dup; p.Ser755LysfsTer3) located in exon 19/40 (Supplementary Fig. 1). This variant is absent from gnomAD v4.1 and from a cohort of 4854 healthy French individuals6,7, ClinVar database, and literature. WES did not find any pathogenic or likely pathogenic variants in any known PD gene.
In pedigree C, two males (II-2 and II-7) from a three-generation pedigree were affected. Their parents died at the ages of 86 and 74 years without any PD symptoms and were not related. The disease started in their sons at the ages of 48 years (II-2) and 47 years (II-7). For the proband (II-7), the disease started with a left upper and lower limb akinesia. Cerebral MRI was normal. Rapidly, tremor and rigidity were present. He was on 150 mg LEDD of dopaminergic agonists. Hoehn and Yahr stage was 1/5. Nine years after disease onset at 56 years, he suffered from an akinetorigid syndrome. No atypical sign was noted. UPDRS III was 8/108 off. At that time, he had 400 mg LEDD. For II-2, the disease started with an asymmetrical akinesia and then slowly worsened. DBS was initiated 13 years after disease onset at 61 years. Four years later, at 65 years, he had an additional tremor and rigidity. No atypical sign had developed; however, he had cognitive impairment (no score from any scale available) and urinary incontinence. LEDD was 200 mg with dyskinesia and motor fluctuations. At that time, UPDRS III score was assessed at 10/108 on, and the Hoehn and Yahr stage was still at 1/5. WES in both individuals identified a frameshift heterozygous deletion NM_003024.3:c.2842_2843del p.Met948ValfsTer41 located in exon 23/40 (Supplementary Fig. 1). Allele count in gnomAD v4.1 was eight (allele frequency: 5.472 × 10−6) and none in a cohort of 4854 healthy French individuals6,7, and this variant was absent from the ClinVar database. This variant was already reported as being de novo in an individual with autism spectrum disorder8. Both affected individuals from pedigree C also carried a missense mutation in GBA1 (NM_000157.4:c.1208G>A; p.S403N). Although this mutation was already reported in six publications in the MDSgene database (website visited on 02/10/25), five of them report the affected siblings described in this study; therefore, only one other individual has been reported in the literature9,10. This mutation is considered “mild” in the GBA1 classification11. Subsequent segregation analysis revealed that the unaffected individuals II-3 and II-4, who are now in their 70s, carried neither the ITSN1 nor the GBA1 variants (Supplementary Fig. 1). DNA was not available from any other family member.
Initially, Skuladottir et al. identified 18 PD carriers of ITSN1 LOF variants4. Subsequently, Spargo et al. replicated this finding in overlapping cohorts, providing additional evidence of an association between ITSN1 LOF variants and PD in Drosophila and in vitro assays5. This study reports the first ITSN1 multiplex pedigrees, provides details on the phenotype, and further discusses whether ITSN1 is a risk factor or a Mendelian gene for PD.
On the clinical level, no atypical symptoms for PD were identified. Instead, ITSN1-PD motor phenotype appears to be typical, with a good response to dopaminergic treatments. DBS in two cases was efficient on motor symptoms, suggesting DBS might be a good therapeutic option for ITSN1-PD. Non-motor symptoms are not at the forefront of ITSN1-PD phenotype. Disease course is slow, since after about 10 years, the available UPDRS motor score ranged from 8 to 25 (pedigrees B and C). Interestingly, this pattern is similar to the one reported in three ITSN1-PD cases by Spargo et al.5. The mean ages at onset of PD in carriers reported by Skuladottir et al. and Spargo et al. were 62 and 64 years, respectively, earlier than non-carriers (70 years, p = 0.022 and p = 0.05, respectively)4,5. Here, median age at onset is 47 years, with great variability (range 30–62). Interestingly, pedigrees A and C show a high concordance in age at onset (and disease course as well). In contrast, the gap in pedigree B is 32 years between the two affected carriers. Therefore, ITSN1-PD shows intrafamilial and interfamilial variability. Other factors, genetics or environmental, likely play a role.
The cumulative ITSN1 LOF variant frequency in the various PD cohorts ranges between 0.11%4 and 0.24%5, about 6–13 times greater than the ~0.018% in control populations4. In our cohort, we find a similar frequency of proband carriers of ITSN1 LOF variants (3/2076, 0.14%). When we only consider pedigrees compatible with autosomal dominant inheritance (n = 454), the frequency reaches 0.7%. Several elements show that ITSN1 LOF variants may be considered as a Mendelian PD gene rather than a high-risk factor. First, the odds ratio for ITSN1 LOF variants may be underestimated due to the potential future PD conversion of some unaffected carriers in both studies4,5. Second, in the latest reports of genetic mutations in large PD cohorts12,13,14, GBA1 risk and mild variants as well as LRRK2 G2019S were considered to contribute to monogenic PD, even though the odds ratios for these variants are estimated to be lower than or equivalent to ITSN1 LOF variants, ranging from 1.4 to 7.8 for the former, and about 10 for the latter11,15. Third, although segregation analysis was not performed, six ITSN1-PD cases from the study of Spargo et al. reported a first-degree family history of PD and/or Alzheimer’s disease, among which four specifically reported PD5. The discovery of three novel families provides new evidence for the monogenic hypothesis because all genotyped affected individuals are carriers of ITSN1 LOF variants. However, this hypothesis should be taken with caution for several reasons. First, the existence of several unaffected parents that are likely to be carriers in pedigrees A and C, with ages ranging from 60 to 80 years, points toward a censor effect or an age-dependent and reduced penetrance. Second, the lack of DNA from the affected mother (II-3) in pedigree A did not allow for the determination of her genotype. Third, the age at onset difference between III-6 and IV-6 in pedigree B (30 years) could be the consequence of a different cause, such as biallelic pathogenic variants in a recessive gene for IV-6. However, WES did not identify any additional variants in known AR-EOPD genes. Fourth, a GBA1 variant was identified in the two affected individuals of pedigree C. Although this variant was found only once in an individual affected with PD10, it is known to contribute to Gaucher disease type I (non-neurological) when associated with another pathogenic variant and was demonstrated to result in a marked reduction in enzymatic activity16,17. Therefore, it is possible that this variant plays a role in PD in general and that it may influence the phenotype and penetrance of the ITSN1 LOF variant in this pedigree. Finally, apart from GBA1, we were not able to experimentally demonstrate the postulated haploinsufficiency of ITSN1 LOF variants, either in terms of mRNA expression levels or nonsense-mediated decay involvement, due to a lack of available material.
Nevertheless, this study demonstrates that ITSN1 LOF variants may be responsible for autosomal dominant familial PD with reduced penetrance, which will help genetic counseling of ITSN1-PD cases. The description of additional families with positive segregation is required to further investigate the Mendelian inheritance hypothesis for ITSN1 LOF variants.
Methods
Study participants
Participants were selected based on their early age at onset and their family medical history from a large cohort of 2076 PD probands who underwent WES and were enrolled through the French Parkinson Disease genetics Study Group and international collaborations between 1990 and 202418,19. PD was diagnosed among the cohort by clinical assessment based on the diagnosis criteria of the UK Parkinson's Disease Society Brain Bank.
Written informed consent was obtained from participants, and the local ethics committee CCPPRB (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) of the Groupe Hospitalier Pitié-Salpêtrière, Paris, France approved genetic analyses.
WES and Sanger sequencing
WES was performed on individuals III-2 (pedigree A), III-6 and IV-6 (pedigree B), II-2 and II-7 (pedigree C) as previously described (Fig. 1)20,21. In brief, DNA was extracted from blood. Exons were captured using the Roche Seqcap Ez MedExome (Roche Diagnostics Corporation, Indianapolis, IN) kits, followed by 150-bp paired-end sequencing performed on an Illumina NovaSeq 6000 instrument (Illumina Inc., San Diego, CA). FastQC was used to check the quality of the reads, and low-quality reads were removed using Trimmomatic. The DRAGENTM DNA pipeline v3.8.4 (Illumina) was used to align the reads to the human hg19 reference genome, mark the PCR duplicates, and perform the calling of the copy number variants (CNVs) using the panel of normals approach. Within the data set, each sample’s depth of coverage is first corrected for the GC bias and then normalized against the depth of all the unrelated samples in the same sequencing batch. Only the events passing the default filters were considered for analysis and annotated with AnnotSV v3.1.1. Single-nucleotide variants were analyzed according to ACMG guidelines and CNVs to ACMG/AMP guidelines22,23. ITSN1 LOF variants identified, as well as the GBA1 variant in pedigree C, were confirmed by Sanger sequencing according to the manufacturer's guidelines (Supplementary Fig. 1).
Data availability
Data from this manuscript are available upon request.
References
Jakob, B. et al. Intersectin 1 is a component of the Reelin pathway to regulate neuronal migration and synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 114, 5533–5538 (2017).
Herrero-Garcia, E. & O’Bryan, J. P. Intersectin scaffold proteins and their role in cell signaling and endocytosis. Biochim. Biophys. Acta Mol. Cell Res. 1864, 23–30 (2017).
Bruel, A.-L. et al. ITSN1: a novel candidate gene involved in autosomal dominant neurodevelopmental disorder spectrum. Eur. J. Hum. Genet.30, 111–116 (2022).
Skuladottir, A. T. et al. Loss-of-function variants in ITSN1 confer high risk of Parkinson’s disease. npj Parkinsons Dis. 10, 140 (2024).
Spargo, T. P. et al. Haploinsufficiency of ITSN1 is associated with a substantial increased risk of Parkinson’s disease. Cell Rep. 44, 115355 (2025).
Alves, I. et al. Human genetic structure in Northwest France provides new insights into West European historical demography. Nat. Commun. 15, 6710 (2024).
POPGEN. PFMG. https://pfmg2025.fr/le-plan/projets-pilotes/popgen/ (2025).
Yuen RK, C. et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611 (2017).
Anheim, M. et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78, 417–420 (2012).
Cristina, T.-P. et al. A genetic analysis of a Spanish population with early onset Parkinson’s disease. PLoS ONE 15, e0238098 (2020).
Parlar, S. C., Grenn, F. P., Kim, J. J., Baluwendraat, C. & Gan-Or, Z. Classification of GBA1 variants in Parkinson’s disease: the GBA1-PD Browser. Mov. Disord. 38, 489–495 (2023).
Westenberger, A. et al. Relevance of genetic testing in the gene-targeted trial era: the Rostock Parkinson’s disease study. Brain 147, 2652–2667 (2024).
Cook, L. et al. Parkinson’s disease variant detection and disclosure: PD GENEration, a North American study. Brain 147, 2668–2679 (2024).
Towns, C. et al. Defining the causes of sporadic Parkinson’s disease in the global Parkinson’s genetics program (GP2). npj Parkinsons Dis. 9, 131 (2023).
Kmiecik, M. J. et al. Genetic analysis and natural history of Parkinson’s disease due to the LRRK2 G2019S variant. Brain 147, 1996–2008 (2024).
Torralba, M. A. et al. Identification and characterization of a novel mutation c.1090G>T (G325W) and nine common mutant alleles leading to Gaucher disease in Spanish patients. Blood Cells Mol. Dis. 27, 489–495 (2001).
Torralba, M. A. et al. Residual enzymatic activity as a prognostic factor in patients with Gaucher disease type 1: correlation with Zimran and GAUSS-I index and the severity of bone disease. QJM 109, 449–452 (2016).
Lesage, S. et al. Characterization of recessive Parkinson disease in a large multicenter study. Ann. Neurol. 88, 843–850 (2020).
Vollstedt, E. J. et al. Using global team science to identify genetic Parkinson’s disease worldwide. Ann. Neurol. 86, 153–157 (2019).
Casse, F. et al. Detection of ATXN2 expansions in an exome dataset: an underdiagnosed cause of parkinsonism. Mov. Disord. Clin. Pract. 10, 664–669 (2023).
Bouhouche, A. et al. Mutation analysis of consanguineous Moroccan patients with Parkinson’s disease combining microarray and gene panel. Front. Neurol. 8, 567 (2017).
Brandt, T. et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 22, 336–344 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Acknowledgements
We would like to thank all participants who donated their time and biological samples to be a part of this study. Part of this work was carried out at the DNA and Cell Bank of the Institut du Cerveau (ICM). We gratefully acknowledge Sylvie Forlani and Ludmila Jornea for sample preparation. Part of this work was carried out through the iGenSeq core facility. We gratefully acknowledge Yannick Marie and Agnes Rastetter for the whole-exome sequencing. We gratefully thank Dr. Emannuelle Génin for giving us information on ITSN1 variant frequencies in the French population of POPGEN. We also thank the project “DBS - From genetic mutations to motor circuit dysfunctions & recovery,” which was supported by the RMF. This work was supported by the Fondation pour la Recherche Médicale (FRM, MND202004011718), the Fondation de France, la Fédération pour la Recherche sur le Cerveau (FRC), France-Parkinson Association, and the French program “Investissements d’avenir” (ANR-10-IAIHU-06), and the fond Richard Mille from the Deep-Brain Stimulation project.
Author information
Authors and Affiliations
Contributions
G.C., C.T., F.C., E.L., S.L., A.B.: designed, reviewed, and critiqued the study. G.C., C.T., L.W., F.C.: performed the analysis of the genomic sequencing data. A.D., M.P., B.D., G.M., F.C.-D.: clinically followed the participants of this study. A.L.: reviewed and critiqued the analysis.
Corresponding author
Ethics declarations
Competing interests
G.C. is supported by the Global Parkinson’s Genetics Program (GP2). GP2 is funded by the Aligning Science Across Parkinson’s (ASAP) initiative and implemented by The Michael J. Fox Foundation for Parkinson’s Research (https://gp2.org). For a complete list of GP2 members, see https://gp2.org. S.L. has received grants from the Fondation de la Recherche Médicale (FRM, MND202004011718).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cogan, G., Tesson, C., Welment, L. et al. Should ITSN1 be considered as a Mendelian Parkinson’s disease gene? Description of three novel families. npj Parkinsons Dis. 11, 295 (2025). https://doi.org/10.1038/s41531-025-01141-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01141-6

{kind=link}