
Abstract
Quantitative pathology-specific biomarkers are needed for patients with Parkinson’s disease (PD). We estimated the α-syn seeding dose giving 50% of positive seed amplification assay (SAA) reactions (SD50) in serially diluted samples from 260 PD participants, of whom 54 had longitudinal samples. We then evaluated the associations between SD50 values and demographic and clinical parameters, including motor and cognitive scales, REM sleep behaviour disorder (RBD), and hyposmia. Higher SD50 values were significantly associated with older age, longer disease duration, worse motor and cognitive scores, and presence of RBD and visual hallucinations. Baseline SD50 values predicted the development of motor wearing-off and severe cognitive impairment. In participants with longitudinal samples, SD50 values remained substantially stable over time. Quantification of α-syn through endpoint dilution SAA may serve as a potential surrogate marker of LB pathological burden, which may support prognostication and patient stratification.
Similar content being viewed by others

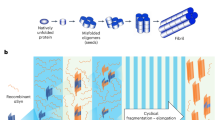

Introduction
The clinicopathological heterogeneity in Lewy body disorders (LBD), including Parkinson’s disease (PD) and Dementia with Lewy bodies (DLB), underscores the need for pathology-specific biomarkers to facilitate accurate diagnosis and predict disease trajectories. Recently, the development of ultrasensitive seed amplification assays (SAA), capable of detecting minute amounts of misfolded alpha-synuclein (α-syn) in cerebrospinal fluid (CSF) and other tissues, has drastically improved the identification of patients with LBD in vivo, even at preclinical or prodromal stages1,2,3,4,5. This has led to the proposal of new biologically based research criteria for this prevalent disorder6,7. While the current assays’ qualitative dichotomous seeding response (positive vs. negative seeding) is valuable for stratifying and enriching cohorts for α-syn pathology in general, more quantitative parameters reflecting the Lewy body (LB) pathology load would be essential. These could be particularly helpful in predicting dynamics of disease progression, trajectories until reaching disease milestones and might serve as exploratory outcome measures in clinical trials targeting α-syn. Recent evidence from the analysis of brain homogenates suggests that SAA kinetic parameters, which describe the dynamics of α-syn aggregation and amplification, could provide quantitative data that indirectly reveals the concentration of α-syn seeds present in biological samples and, possibly, the LB pathology burden8. Initial attempts to translate these findings using biofluids have identified the number of positive outcomes in multiple replicates and the time to threshold (TTT) or lag phase (i.e. the time required for the fluorescent signal to reach the positivity threshold) as the most promising kinetic variables for α-syn seeds quantification in CSF samples, with some studies reporting associations of these parameters, at both baseline and longitudinally, with parameters of clinical progression9,10,11. However, evidence also indicates that pre-analytical and analytical factors, such as matrix composition and batch-to-batch variations in the critical assay reagents, may affect these parameters, reducing their discrimination power12,13.
New perspectives on SAA implementation are currently emerging, primarily driven by the application of serial sample dilution methods. Specifically, in analogy to endpoint dilution animal bioassays, the endpoint dilution (ED) SAA involves testing serial dilutions of samples and statistically estimating the seeding dose, which yields positive responses in 50% of replicate reactions (SD50). Preliminary evidence suggests that this approach facilitates a more accurate estimation of α-syn seeding activity, thereby minimizing dependence on the kinetics of the aggregation reaction14. For this calculation, it was recently shown that data analysis using the midSIN algorithm provides the most consistent and statistically significant discrimination of different seed concentrations14. We hypothesised that higher CSF α-syn SD50 values may indicate a higher seed load, serving as a proxy for more accelerated or widespread LB pathology. This, in turn, could be associated with faster progression to clinical disease milestones such as cognitive impairment. To evaluate this hypothesis, we investigated the correlation between SD50 values and clinical severity and progression measures. Furthermore, we investigated the variation of quantitative parameters over time in a subset of participants with longitudinal CSF samples available.
Results
Overall findings by standard and endpoint dilution α-syn SAA
The initial screened cohort consisted of 333 participants, comprising 285 patients with sporadic PD and 48 with GBA mutations (PDGBA). All available CSF samples were initially tested undiluted by α-syn SAA.
SAA demonstrated α-syn seeding activity in 271 participants (228 with sporadic PD and 43 with PDGBA), and 62 negative outcomes, resulting in an overall sensitivity of 81.4% (80.0% in sporadic PD and 89.6% in PDGBA). For each participant, the baseline was defined as the timepoint of the first available SAA-positive CSF sample. In six participants (2.3%), the baseline did not correspond to the time of the first available CSF sample, as they tested positive only at a follow-up sample (i.e., they converted from negative to positive status).
ED SAA values were obtained in 260 participants (dilution cohort), as limited CSF volume was available in 11 patients (3 PDGBA and 8 sporadic PD).
Among them, 89 showed 8/8 or 7/8 positive replicates at baseline (undiluted sample), 89 had 6/8 or 5/8 positive replicates, and 72 yielded 4/8, 3/8 or 4/12 positive replicates. The remaining 10 patients were identified as low positives (see Methods for criteria). Of the positive samples, 57 (21.9%) were positive only undiluted, 59 (22.7%) remained positive up to a dilution of 1:2, 64 (24.6%) up to a 1:4 dilution, 62 up to 1:8 (23.8%), 15 (5.8%) up to 1:16, 2 (0.8%) up to 1:32, and only 1 (0.4%) up to a 1:64 dilution. This resulted in log10SD50/ml values ranging from 0.47 to 2.07, with a median of 1.14, consistent with previously reported values14,15.
When analysing the trend of the number of positive replicates (Nrep) across increasing dilutions, most participants showed a consistent decrease or, in some cases, temporary stability. In only a minority of cases (16 participants, 6.1%), the assay yielded an increase in Nrep ( ≥ 50% compared to the previous dilution), followed by a stable decreasing trend.
Demographic, clinical, and biomarker data of the 260 participants in the dilution cohort are reported in Table 1. The Supplementary Table 1 lists all the different GBA1 variants identified in the 40 participants of the dilution cohort. Demographic, clinical and biomarker data of the participants converting from a negative to a positive status are reported in Supplementary Table 2.
SD50 values are associated with clinical characteristics cross-sectionally
At baseline, higher SD50 values showed statistically significant correlations with older age at sampling, older age at onset, longer disease duration, higher Hoehn and Yahr scale (H&Y) scores, higher Unified Parkinson’s Disease Rating Scale part III (UPDRS part III) scores, lower Montreal Cognitive Assessment (MoCA) scores (Table 2 and Fig. 1), and the presence of REM sleep behaviour disorder (RBD), visual hallucinations, and motor wearing-off (Table 3). Further, a higher SD50 score was associated with more severe autonomic symptoms, including constipation and orthostatic dysfunction, as defined by Unified Multiple System Atrophy Rating Scale (UMSARS) (Table 2).

Scatter plots indicating the correlation between log10SD50/ml values and selected clinical or demographic variables: age at sampling (a), MoCA score (b), UPDRS part III score (c) and Hoehn and Yahr score (d). Blue dots represent single observations. Red lines are derived from univariable linear regression models. Correlation coefficients (Rho) and p values are reported in each graph. Abbreviations: H&Y Hoehn and Yahr, MoCA Montreal Cognitive Assessment, UPDRS III Unified Parkinson’s Disease Rating Scale part III.
The association with higher UPDRS part III scores was retained when considering only participants with MDS-UPDRS scores available (Rho = 0.22, p = 0.001).
Interestingly, the association between SD50 values and clinical scores was even stronger (higher UPDRS part III score, Rho = 0.23, p = 0.014; lower MoCA scores, Rho = -0.27, p = 0.005) when the analysis was limited to the participants with early-stage PD (disease duration ≤ 5 years), despite the significantly reduced number per group (Supplementary Table 3).
Univariable linear regression models confirmed the significant correlation between SD50 values and H&Y stage (β = 0.38, 95% CI 0.14−0.62, p = 0.002), UPDRS part III scores (β = 7.03, 95% CI 2.22−11.85, p = 0.004), and lower scores at the MoCA scale (β = -3.01, 95%CI -4.82–-1.20, p = 0.001) (Supplementary Table 4). After correction for age and disease duration, the association between SD50 values and UPDRS part III scores retained statistical significance (β = 5.03, 95% CI 0.19−9.87, p = 0.041), even when only considering participants with MDS-UPDRS part III scores available (β = 6.42, 95% CI 1.34−11.50, p = 0.013). In contrast, the association with MoCA scores reached only a trend of significance (β = -1.67, 95% CI -3.38–0.04, p = 0.056) (Supplementary Table 4).
Baseline SD50 values were significantly higher in participants showing RBD (p = 0.002) and visual hallucinations (p = 0.028) at baseline than in those without these clinical features (Table 3). Logistic regression models confirmed the associations in the univariable analysis with motor wearing-off (OR = 5.44, 95% CI 1.38−24.25, p = 0.019), RBD (OR = 3.43, 95% CI 1.53−8.02, p = 0.003) and visual hallucinations (OR = 2.85, 95% CI 1.18−7.13, p = 0.02). The association with repeated falls showed a significant trend but missed statistical significance (OR = 2.76, 95% CI 0.99−8.12, p = 0.057) (Supplementary Table 5). After correction for age and disease duration, the association between SD50 values on the risk of having RBD at baseline was retained (OR = 2.76, 95% CI 1.20−6.57, p = 0.019), while the association with the risk of having motor wearing-off reached a trend of significance (OR = 5.33, 95% CI 1.02−32.47, p = 0.057) (Supplementary Table 5).
When compared to participants in the lowest baseline SD50 tertile, those in the highest tertile were older (69.5 years vs. 63.9 years; p = 0.003), had a higher prevalence of RBD (55.2% vs. 34.9%; p = 0.0006), motor wearing-off (17.2% vs. 5.8%; p = 0.011) and freezing of gait at sampling (14.9% vs. 7.0%; p = 0.042). Additionally, patients in the highest tertile showed a faster TTT (17.8 h vs. 21.0 h; p < 0.0001). The prevalence of participants carrying a GBA1 variant was equally distributed in the highest and lowest SD50 tertiles (18.4% vs. 15.1%; p = 0.564).
Prognostic value of baseline SD50 values
Higher baseline SD50 values showed statistically significant correlations with a higher risk of motor deterioration, as indicated by the progression to H&Y stage ≥ 3 (HR 5.09, 95% CI 1.73−14.95, p = 0.003) (Fig. 2a). The effect was almost independent of examined covariates (HR 3.07, 95% CI 0.85−11.11, p = 0.088) (Table 4). Higher baseline SD50 also significantly predicted the onset of severe cognitive impairment, as measured by a MoCA score ≤ 18 (HR 14.31, 95% CI 2.87−71.21, p = 0.001) (Fig. 2b). The association retained statistical significance in the multivariable analysis (HR 9.21, 95% CI 1.54−55.22, p = 0.015) (Table 4). Baseline SD50 values also predicted the appearance of motor wearing-off (HR 2.62, 95% CI 1.08–6.33, p = 0.032), even after accounting for covariates (HR 2.94, 95% CI 1.16−7.47, p = 0.023) (Table 4). The positive association between baseline SD50 and the risk of developing repeated falls (HR 2.90, 95% CI 1.04–8.09, p = 0.043) disappeared after adjustment in the multivariable model (Table 4). No significant predictive effect was found for the occurrence of visual hallucinations (p = 0.252). Hazard ratios and p values of all prognostic analyses are shown in Table 4 and Supplementary Table 6. Additional survival curves are shown in Supplementary Fig. 1.

Kaplan-Meier curves showing the probability of developing postural instability (a) or severe cognitive impairment, as defined by MoCA score ≤18 (b), according to baseline log10SD50/ml tertiles. Time is expressed in months. Abbreviations: H&Y Hoehn and Yahr, MoCA Montreal Cognitive Assessment.
Longitudinal trajectories of SD50 values
Among the 260 participants in the dilution cohort, 54 had longitudinal samples available (number of available CSF samples: median 2.0, IQR 2.0–3.0; sampling time duration: median 30.5 months, IQR 22.5–49.0 months).
In the subgroup of participants with longitudinal CSF samples available, there was no significant change in SD50 values over time (β = 0.0003, 95% CI -0.001–0.002, p = 0.682) (Fig. 3). Similarly, there were no significant variations in the SD50 values over time when stratifying for participants with or without severe cognitive impairment (as measured by a MoCA score ≤ 18) across the entire clinical history (with severe cognitive impairment: β = -0.001, 95% CI -0.005–0.003, p = 0.554; without severe cognitive impairment: β = 0.0007, 95% CI -0.001–0.002, p = 0.460).

A single grey dot represents the observation of a single participant over time. Grey lines connect repeated longitudinal samples from the same participant. The blue line represents the overall longitudinal trend of log10SD50/ml values, as predicted by the linear mixed-effects model. The shaded band represents the 95% CI. Time is expressed in months.
When stratifying participants according to the baseline SD50 values, those in the low and middle tertile showed stable values over time (low tertile: β = 0.002, 95% CI -0.0005–0.005, p = 0.119; middle tertile: β = 0.0001, 95% CI -0.001–0.002, p = 0.906), while in those in the high tertile a mild longitudinal decreasing trend in SD50 values was observed (β = -0.003, 95% CI -0.004–-0.001, p = 0.005).
A longitudinal stability of SD50 values was observed in participants with early-stage PD at baseline (β = -0.0005, 95% CI -0.001–0.002, p = 0.641).
Correlations between SD50 and other quantitative SAA kinetic parameters at baseline
As expected, SD50 values showed a strong correlation with Lewy fold pathology (LFP) scores (Rho = 0.99, p < 0.0001), representing the total Nrep across the dilution series. Consequently, SD50 and LFP values showed almost identical significant associations with clinical variables, with only minimal variations in statistical significance. Differences in significance threshold between the two variables were limited to the correlations with constipation, which was only reached by SD50 (p = 0.049), and the association with repeated falls, which was only evident with LFP (p = 0.041) (Tables 2 and 3 for SD50, Supplementary Tables 7 and 8 for LFP). TTT and SD50 values were also significantly correlated (Rho = -0.31, p < 0.0001). In contrast, TTT showed no significant associations with demographics and clinical variables (Supplementary Tables 9 and 10).
Analysis of the interrelation between SD50 and TTT revealed two patient subgroups: (1) the “high and fast” group defined by the top tertile of dilution parameter (SD50) and the bottom tertile of the kinetic parameter (TTT). (2) the “low and slow” group defined by the bottom tertile of dilution parameters (SD50) and the top tertile of the kinetic parameter (TTT) (Supplementary Fig. 2).
“High and fast” seeding participants showed higher UPDRS part III scores (High and fast: median 30.0, IQR 19.0–39.3; Low and slow: median 24.0, IQR 19.0–29.0; p = 0.048), and a higher prevalence of RBD (67.6% vs. 40.5%; p = 0.022) and urinary urge at sampling (71.8% vs. 48.7%; p = 0.037) compared to the “low and slow” seeding group (Supplementary Table 11 and Supplementary Fig. 3).
Discussion
The development of a refined “quantitative” α-syn SAA reflecting the burden of the underlying neuropathological process, to be correlated with clinical parameters of disease severity and progression, and to be tested as a marker of treatment response, is a research priority for PD and other disorders belonging to the LBD spectrum. In a recent study, we demonstrated that the ED SAA, executed on serially diluted ventricular post-mortem CSF samples, reliably estimates the α-syn seed concentration, which positively correlates with the LBD load and disease stage, as evaluated through immunohistochemistry16. In the present work, we extended this approach to the clinical scenario by testing the association between the ED SAA quantitative parameter SD50 and clinical variables using in vitam collected CSF from PD patients. The results show a significant positive association between the SD50 values and clinical scores of motor deterioration and motor wearing-off, as well as with key non-motor symptoms such as hallucinations and cognitive decline. Importantly, participants with higher SD50 values also exhibited a higher frequency of RBD, independently of age and disease duration. Higher SD50 scores also correlated with the age at onset and disease duration. Overall, these results are consistent with the idea that higher SD50 values indicate a higher load of LB pathology, thus validating the ED SAA as a semiquantitative pathology-associated assay. The finding of an even stronger association between SD50 values and clinical scores in participants with early-stage PD supports the idea that the LB pathology burden is a major contributor of clinical disease severity and progression, which notoriously does not depend only on disease duration.
From a prognostic standpoint, higher baseline SD50 values were independently associated with faster progression to key clinical milestones, including motor deterioration (H&Y stage ≥ 3) and the appearance of motor wearing-off and repeated falls, as well as the onset of significant cognitive decline (MoCA ≤ 18). Some of these associations remained significant after adjusting for confounding variables, reinforcing the potential of SD50 as a predictive biomarker of disease trajectory.
Regarding the longitudinal variations of α-syn seeding activity, we found that, as a general trend, SD50 values remain stable over time, regardless of cognitive status, with even a tendency to decrease over time in those in the highest tertile of baseline values. This could reflect a plateau effect, where CSF seeding activity stops increasing once a critical pathological threshold is reached, as is the case for some Alzheimer’s disease markers17,18. Alternatively, it may indicate a limited analytical sensitivity of α-syn SAA in detecting small changes in LB pathology burden at the clinical stage. Moreover, the absence of a significant longitudinal increase may also partially be influenced by the relatively short sampling intervals in our cohort.
However, most patients with multiple samples showed consistent results by SAA, maintaining either positivity or negativity depending on the result obtained at baseline. A small number of “converters” (n = 6) shifted from negative to a positive status during follow-up, possibly indicating the emergence of more widespread α-syn pathology. These rare transitions may reflect the dynamic nature of α-syn aggregation and its gradual spread across the central nervous system throughout disease progression.
The strong correlations between SD50 values and other kinetic parameters, particularly the LFP score and TTT, further support the internal consistency of the assay. The identification of distinct “high and fast” vs. “low and slow” seeding profiles suggests that different PD subgroups, possibly reflecting different underlying pathophysiological mechanisms or disease trajectories, may be characterized by specific α-syn SAA signatures. Patients in “high and fast” group exhibited more severe motor impairment (as expressed by UPDRS part III), higher RBD prevalence, and greater urinary urge, features commonly associated with a more diffuse LB pathology.
Despite extensive efforts by several groups19,20,21, early attempts to establish correlations between α-syn SAA-derived measures and clinical indicators of disease severity in cross-sectional analysis have been inconsistent. However, more recent studies have reported significant correlations between specific α-syn SAA kinetic parameters (e.g., TTT, Nrep) and disease progression, particularly in relation to the development of cognitive decline over time9,10,11. However, in our cohort, the TTT failed to show meaningful associations with the clinical variables considered, limiting its value as a marker of disease burden or trajectory outside highly controlled experimental conditions. In contrast, SD50 emerged as a substantially more informative and reliable parameter, offering a quantitative estimate of seeding activity that better captured the clinical heterogeneity of PD. The SD50 calculation is more time-consuming and technically demanding than the qualitative assay, as one quantitative SAA corresponds to running at least five standard assays. However, its superior clinical relevance strongly supports its potential adoption in diagnostic settings.
Recently, Bernhardt et al. 15 reported significant clinical associations, using the LFP score, calculated by ED SAA as a surrogate marker of α-syn burden. Their cross-sectional results were encouraging, but the longitudinal component of their work was conducted on a considerably smaller cohort, which limits the interpretability and statistical strength of these preliminary findings. Notably, a closer examination of their raw data reveals that, in some cases, the expected monotonic decrease in positive replicates with increasing dilution was not observed, with some samples showing an unexpected increase in positive replicates at higher dilutions. This raises the possibility of spontaneous aggregation or other technical factors influencing the results, which were not explicitly addressed in that study. In contrast, in our study, ED determination was performed under optimized and validated assay conditions in an established SAA laboratory, where a consistent monotonic trend was observed. Furthermore, our study benefits from a substantially larger cohort, which enhances the reliability of cross-sectional correlations between SD50 values and multiple indicators of disease severity. The overall scale and design of our study provide a stronger foundation for establishing SD50 as a sensitive and direct biomarker of α-syn burden, potentially offering improved detection of clinically relevant differences compared to other kinetic parameters.
A major strength of this study is the inclusion of a large, clinically well-characterized cohort of PD patients with a long mean follow-up time, including 54 patients with serial longitudinal CSF samples. Another key strength is the robustness of our α-syn SAA, as shown in previous works.
As a potential limitation, it is worth mentioning that the extended timeframe we required to analyse the large number of samples may have introduced variability and slightly reduced the overall reproducibility of the quantitative parameters of the assay. Specifically, the analysis of this large cohort required multiple batches of recombinant α-syn over time, which might have influenced assay performance. In addition, the use of pooled α-syn SAA negative CSF as diluent, although ensuring a physiologically relevant matrix compared to PBS, may have introduced potential matrix effects or reduced intra-individual variability. Future investigations conducted under more standardized and strictly controlled experimental conditions may help optimize the precision of quantitative measurements. Moreover, these findings are specific to the CSF α-syn SAA protocol applied here and, therefore, additional studies using alternative assay versions are warranted to assess the generalizability of the results.
On another issue, the ED SAA analysis of in vitam collected CSF samples showed significantly lower log10SD50 scores (median 1.14, IQR 0.89–1.39, range 0.47–2.07) compared with those we previously obtained in post-mortem CSF (median 3.93, IQR 2.77–4.51, range 0.50–5.58)16. Although this may be partly explained by the higher mean age and more advanced LBD stage in the latter group, the finding of markedly lower and less variable seeding activity in the clinical cohort likely reflects a combination of lower levels of pathological α-syn in the CSF taken in vivo by lumbar puncture and the relative homogeneity of the present cohort, including only clinically diagnosed PD patients, compared to our previous study which included the entire spectrum of LBD pathology stages. Other possible explanations should also be considered, such as a lower analytical sensitivity of the assay in the present study, potentially due to the use of different batches of recombinant α-syn, the possibility that post-mortem CSF may be contaminated with brain tissue as the brain undergoes rapid decay after death, or the presence of active in vivo mechanisms that limit the levels of α-syn seeds in CSF, which may be lost post-mortem.
Overall, our findings demonstrate that quantitative α-syn SAA parameters, particularly the SD50, offer insight into both the current clinical status and future disease trajectory of patients with PD. This highlights the utility of SD50 not only in diagnosing PD but also in stratifying patients by clinical phenotype and risk of progression. SD50 quantification outperformed other kinetic parameters (i.e., TTT and Imax) in stratifying patients by clinical phenotype, providing a potential translational tool for personalized disease monitoring and therapeutic development. Further longitudinal and neuropathological studies are needed to elucidate the biological mechanisms underlying distinct seeding profiles and to confirm their prognostic relevance in different clinical settings.
Methods
Participants and clinical investigation
The initial cohort comprised 333 consecutive participants with sporadic PD or PDGBA (66% male, mean age 65 years) who had at least one CSF sample available and were evaluated at the outpatient clinic or ward for Parkinson’s disease at the University Hospital of Tübingen between 2002 and 2024. We excluded patients with rare mutations in LRRK2, Parkin, PINK1, and SNCA to reduce genetic confounding effects, given the limited sample size. Repeated longitudinal CSF samples were available from 63 participants. All participants were examined by a movement disorders specialist, and most of them (n = 278, 83.5%) were followed up longitudinally for an average of 5.1 years, with a median time of 51.5 months (IQR 14.5–102.8). Diagnosis of PD was defined according to UK Brain Bank Society Criteria22.
Disease stage was categorized by the modified H&Y scale23. Postural instability was defined as H&Y ≥ 3. The following scores at clinical scales at baseline were also collected, when available: UPDRS part III (for patients evaluated between 2002 and 2008: UPDRS part III, old version; other patients: MDS-UPDRS part III24), UMSARS items 9–12 (orthostatic, urinary, sexual, and bowel function), and Beck Depression Inventory-II (BDI II). Olfactory performance was assessed using the Sniffin’ sticks test and expressed as the percentage of sticks correctly identified out of the total presented. Hyposmia was defined as the correct identification of less than 75% of the presented sticks. RBD was assessed using Question 25 of the PD NMS Questionnaire25. Cognitive function was assessed using MoCA score26 and/or the Mini-Mental State Examination (MMSE)27. Since the MoCA was available from 2009, previously obtained MMSE scores were converted into MoCA equivalents28. Cognitive impairment was defined according to criteria reported by Hoops et al. (MoCA ≤25; point of maximum combined sensitivity and specificity)26. Moreover, we defined the cognitive impairment as severe with MoCA ≤ 18. The malignant phenotype was defined as the concomitant presence of RBD, hyposmia, visual hallucinations, and cognitive impairment.
Genetic analysis
Genetic screening for pathogenic variants in the genes GBA1, LRRK2, Parkin and PINK1 was performed as previously described29. Due to the limited sample size, patients carrying heterozygous or bi-allelic mutations in LRRK2, Parkin, PINK1, and SNCA were excluded from the study. Of the 333 included participants, 48 carried a variant in GBA1.
Endpoint dilution α-syn SAA
Samples were centrifuged within 60 min and frozen at −80 °C within 90 min after collection. SAA experiments were performed at the Institute of Neurological Science of Bologna (ISNB). We performed purification of recombinant wild-type human α-syn as previously described1. The CSF SAA real-time quaking-induced conversion (RT-QuIC) was conducted according to an established protocol, with minor modifications concerning the number of loaded sample replicates at first screening and the duration of the assay (see below)1,30. CSF samples of all participants were initially analyzed in octuplicate. We loaded one negative and three positive controls on each plate to verify the correct functioning of the assay. As a negative control, we loaded the CSF pool we used to dilute the CSF samples from PD patients (see below). As positive controls, we used brain homogenates (10% in PBS 1X) from three well-characterized LBD patients, showing 4 out of 4 positive replicates at a screening SAA run. These were then diluted 10-5 in a pool of α-syn SAA-negative CSF samples collected from patients diagnosed with normal pressure hydrocephalus.
Participants’ samples were deemed positive when at least 3 replicates out of 8 loaded wells exceeded the arbitrarily set positivity threshold, defined as 30% of the median of the maximum fluorescence intensity (Imax) reached by the positive control replicates. We repeated the analysis in quadruplicate for 60 samples that initially showed unclear results (i.e., 1 or 2 positive replicates). In the case of repeated analyses, samples yielding 3 positive replicates out of 12 and showing at least one 2/4 run were considered as “low positives” and subsequently subjected to ED SAA analysis to evaluate their seeding behavior across serial dilutions. As a minor modification from previous studies, we established a duration of 40 h for the run. This allowed the inclusion of curves with slower kinetics, overcoming the positivity threshold near or slightly after the canonical 30-h incubation period. Notably, this extension did not affect the interpretation of the binary test results, as the outcome of the negative plate controls remained unchanged (i.e., they were deemed negative at both 30 h and 40 h).
CSF samples showing positive results were then analyzed in octuplicate by the ED SAA RT-QuIC to quantify pathological seeds in CSF samples. Two-fold serial dilutions were prepared and analyzed. Diluted samples were considered positive when at least 3 out of 8 replicates showed a positive response. For each sample, initial dilutions (typically up to 1:4 or 1:8) were run on the same plate, with the number of dilutions selected based on the proportion of positive replicates obtained in the neat sample during the screening assay. When additional dilutions were required, they were performed in separate experiments, considering that a limited number of freeze–thaw cycles of the same sample has no significant impact on assay performance, as previously shown12. The serial dilution series was tested until the assay yielded ≤ 2 out of 8 positive replicates. The dilutions were prepared using a pool of CSF samples that tested negative by α-syn SAA assay. The negative pool was extensively validated before use and was systematically included as a plate control in quadruplicate. Specifically, each CSF sample included in the negative pool was individually tested multiple times to confirm the absence of seeding activity, and the final pooled sample was also repeatedly assessed for consistent negativity. For each new pool batch, additional validation was performed by diluting highly positive CSF sample with a well-characterized dilution profile to exclude the presence of matrix inhibitory effect. Plates showing more than one positive replicate in the negative pool were excluded from analysis, and the assay was repeated.
Then, we utilised the midSIN mathematical algorithm to estimate the SD50, namely the seeding dose that resulted in 50% of positive replicate wells for each sample. The SD50 value (expressed in log10SD50/ml) was calculated by a computing open-source tool (https://midsin.physics.ryerson.ca) based on the number of dilutions, the dilution factor (i.e., 0.5) and the Nrep at each loaded dilution14,31.
Other SAA parameters analysed included the number of positive seeds, defined as the Nrep in each neat CSF, the total Nrep across the dilution series, defined as LFP score, and the TTT, which refers to the time required for the kinetic curve to reach the positivity threshold. Specifically, the calculated TTT value represented the mean of the three shortest TTT values among the positive replicates at baseline (i.e., best3-TTT). To account for potential inter-plate variability, the TTT for each plate was normalized to the corresponding positive controls included in the same plate. All CSF measurements were done, and results were reported blinded to the clinical diagnosis and genetic status.
Statistical analysis
Statistical analysis was performed using IBM SPSS 26.0, GraphPad Prism V. 7 (GraphPad, La Jolla, CA, USA) and R (version 4.4.3) software. The normality of continuous variables was assessed using Shapiro-Wilk and Kolmogorov-Smirnov tests. For normally distributed variables, a t-test was used; for non-normally distributed variables, comparisons were made using the Mann-Whitney U test. Spearman’s Rho coefficients were used to assess the correlation between SD50 values and scores on clinical scales. P values < 0.05 were considered statistically significant.
Univariable and multivariable linear regression models were used to assess the association between log10SD50 values (independent variables) and baseline clinical scale scores (dependent variables). In the multivariable models, adjustments were made for age at sampling and disease duration at baseline. Results are reported as β coefficients with 95% confidence intervals (95% CI). Univariable and multivariable logistic regression models were used to evaluate the association between log10-transformed SD50 values and the presence/absence of clinical features at baseline. Multivariable models were adjusted for age at sampling and disease duration at baseline. Results are expressed as odds ratios (OR) with 95% CI.
For the prognostic analysis, Kaplan-Meier estimates were used to calculate the cumulative time-dependent probability of developing specific disease milestones. The time of entry into the analysis was the date of the baseline visit, and the endpoint was the date of milestone occurrence or the last available follow-up, whichever came first. The milestones evaluated included: severe cognitive impairment (defined as MoCA score ≤ 18), Hoehn and Yahr stage ≥ 3, hallucinations, motor wearing-off, repeated falls. Univariable and multivariable Cox regression models were used to assess the prognostic role of SD50 values. Multivariable models were adjusted for age at sampling and disease duration at baseline. The results are reported as hazard ratios (HR) with 95% CI. The assumption of proportional hazard was assessed using Schoenfeld residuals. Participants who had already reached the milestone at the time of the baseline and those with no follow-up data available after the baseline were excluded from the prognostic analyses.
Participants were also stratified into tertiles based on their individual CSF α-syn SD50 and the kinetic parameter TTT (best3-TTT). Group comparisons of dichotomous data were analysed using the likelihood-ratio chi-square test or Fisher’s exact test, as appropriate. Group comparisons between the highest and lowest tertiles of SD50 with respect to demographics, clinical characteristics, and other CSF parameters were performed using unpaired t-tests for normally distributed variables, and the Mann-Whitney U test for non-normally distributed variables.
Finally, to analyse longitudinal changes in SD50 values, linear mixed-effects modelling analysis with random intercept was used. The log10SD50 values were the dependent variable, and time (in months) was the independent variable.
Ethics approval and consent to participate
The study was conducted in accordance with the revised Declaration of Helsinki for the protection of human participants and Good Clinical Practice guidelines approved by the Ethics Committee of the University of Tuebingen (26/2007BO1, 404/2010BO1, 199/2011BO1, 702/2013BO1). All participants gave written informed consent.
Data availability
Pseudonymised data are available upon reasonable request to Kathrin Brockmann (mailto:kathrin.brockmann@uni-tuebingen.de) or Piero Parchi (mailto:piero.parchi@unibo.it).
References
Rossi, M. et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 140, 49–62 (2020).
Iranzo, A. et al. Misfolded α-synuclein assessment in the skin and CSF by RT-QuIC in isolated REM sleep behavior disorder. Neurology 100, e1944–e1954 (2023).
Palmqvist, S. et al. Cognitive effects of Lewy body pathology in clinically unimpaired individuals. Nat. Med. 29, 1971–1978 (2023).
Siderowf, A. et al. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol. 22, 407–417 (2023).
Wang, Z. et al. Skin α-synuclein aggregation seeding activity as a novel biomarker for Parkinson's disease. JAMA Neurol. 78, 1–11 (2020).
Simuni, T. et al. A biological definition of neuronal α-synuclein disease: towards an integrated staging system for research. Lancet Neurol. 23, 178–190 (2024).
Höglinger, G. U. et al. A biological classification of Parkinson’s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol. 23, 191–204 (2024).
Bentivenga, G. M. et al. Performance of a seed amplification assay for misfolded alpha-synuclein in cerebrospinal fluid and brain tissue in relation to Lewy body disease stage and pathology burden. Acta Neuropathol. 147, 18 (2024).
Brockmann, K. et al. CSF α-synuclein seed amplification kinetic profiles are associated with cognitive decline in Parkinson’s disease. NPJ Park Dis. 10, 24 (2024).
Mastrangelo, A. et al. Alpha-synuclein seed amplification assay longitudinal outcomes in Lewy body disease spectrum. Brain 148, 2038–2048 (2025).
Coughlin, D. G. et al. α-synuclein seed amplification assay, amplification parameters and the risk of progression in prodromal Parkinson disease. Neurology 104, e210279 (2025).
Mammana, A. et al. Improving protocols for α-synuclein seed amplification assays: analysis of preanalytical and analytical variables and identification of candidate parameters for seed quantification. Clin. Chem. Lab Med. 62, 2001–2010 (2024).
Bellomo, G. et al. Cerebrospinal fluid lipoproteins inhibit α-synuclein aggregation by interacting with oligomeric species in seed amplification assays. Mol. Neurodegener. 18, 20 (2023).
Srivastava, A. et al. Enhanced quantitation of pathological α-synuclein in patient biospecimens by RT-QuIC seed amplification assays. PLoS Pathog. 20, e1012554 (2024).
Bernhardt, A. M. et al. A quantitative Lewy-fold-specific alpha-synuclein seed amplification assay as a progression marker for Parkinson’s disease. Acta Neuropathol. 149, 20 (2025).
Mastrangelo, A. et al. Quantification of Lewy body pathology by cerebrospinal fluid endpoint dilution RT-QuIC in a neuropathological autopsy cohort of clinically heterogeneous participants. Acta Neuropathol. 149, 67 (2025).
Leuzy, A. et al. Considerations in the clinical use of amyloid PET and CSF biomarkers for Alzheimer’s disease. Alzheimers Dement 21, e14528 (2025).
Mattsson-Carlgren, N. et al. Cerebrospinal fluid biomarkers in autopsy-confirmed Alzheimer disease and frontotemporal lobar degeneration. Neurology 98, e1137–e1150 (2022).
Poggiolini, I. et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain 145, 584–595 (2022).
Russo, M. J. et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol. Commun. 9, 179 (2021).
Kang, U. J. et al. Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov. Disord. 34, 536–544 (2019).
Litvan, I. et al. Movement disorders society scientific issues committee report: SIC task force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov. Disord. 18, 467–486 (2003).
Goetz, C. G. et al. Movement disorder society task force report on the Hoehn and Yahr staging scale: status and recommendations. Mov. Disord. 19, 1020–1028 (2004).
Goetz, C. G. et al. Movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170 (2008).
Chaudhuri, K. R. et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov. Disord. 21, 916–923 (2006).
Hoops, S. et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 73, 1738–1745 (2009).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Bergeron, D. et al. Multicenter validation of an MMSE-MoCA conversion table. J. Am. Geriatr. Soc. 65, 1067–1072 (2017).
Lerche, S. et al. Parkinson’s disease: glucocerebrosidase 1 mutation severity is associated with CSF alpha-synuclein profiles. Mov. Disord. 35, 495–499 (2020).
Quadalti, C. et al. Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat. Med. 29, 1964–1970 (2023).
Cresta, D. et al. Time to revisit the endpoint dilution assay and to replace the TCID50 as a measure of a virus sample’s infection concentration. PLoS Comput. Biol. 17, e1009480 (2021).
Acknowledgements
Supported by the Italian Ministry of Health (grants RF-2021-12374386 and “Ricerca Corrente”), the #NextGenerationEU (NGEU) funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006), BMBF (PDdementia), DZNE (PD research; MIGAP study), the PD-Strat project (FKZ 031L0137B) supported by the German Federal Ministry of Education and Research (BMBF) in the frame of ERACoSysMed2.
Author information
Authors and Affiliations
Contributions
P.P., K.B. and A.T. contributed to the conception and design of the study. A.T., K.B., S.L., A.Mam., A.Mas., P.P., E.V., B.R., A.H., C.D., S.B., I.W. and T.G. contributed to the acquisition and analysis of the data. A.T., A.Mas., S.L., K.B. and P.P. contributed to drafting a significant portion of the manuscript and preparing the figures. All authors contributed to copy editing and approval of the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brockmann, K., Ticca, A., Lerche, S. et al. Quantification of cerebrospinal fluid α-synuclein seeds by endpoint dilution seed amplification assay in Parkinson’s disease. npj Parkinsons Dis. 12, 5 (2026). https://doi.org/10.1038/s41531-025-01221-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01221-7
