
Abstract
Electronic ground states are of central importance in chemical simulations, but have remained beyond the reach of efficient classical algorithms except in cases of weak electron correlation or one-dimensional spatial geometry. We introduce a hybrid quantum-classical eigenvalue solver that constructs a wavefunction ansatz from a linear combination of matrix product states in rotated orbital bases, enabling the characterization of strongly correlated ground states with arbitrary spatial geometry. The energy is converged via a gradient-free generalized sweep algorithm based on quantum subspace diagonalization, with a potentially exponential speedup in the off-diagonal matrix element contractions upon translation into compact quantum circuits of linear depth in the number of qubits. Chemical accuracy is attained in numerical experiments for both a stretched water molecule and an octahedral arrangement of hydrogen atoms, achieving substantially better correlation energies compared to a unitary coupled-cluster benchmark, with orders of magnitude reductions in quantum resource estimates and a surprisingly high tolerance to shot noise. This proof-of-concept study suggests a promising new avenue for scaling up simulations of strongly correlated chemical systems on near-term quantum hardware.
Similar content being viewed by others

Introduction
Following a century of developments in ab initio electronic structure theory since Hartree’s self-consistent field1, the chemical ground state problem remains intractable for large systems exhibiting strong electron correlation. In these cases a superposition of many Slater determinants may be necessary to characterize the ground state accurately, and the computation time and memory requirements on classical hardware can then scale exponentially with the system size, under a combinatorial explosion of the configuration space. In recent decades tensor network2,3,4 methods such as the density matrix renormalization group (DMRG)5,6,7,8,9 have emerged as powerful tools to partially tame, if not exactly break, this so-called ‘curse of dimensionality’. Tensor networks promise to efficiently represent some classes of many-body quantum states by exploiting the locality of correlations among the parts of the system. A necessary but not sufficient condition for a system to be efficiently described by a tensor network is that it obeys an area law of entanglement10, meaning that entropy measures between certain partitions of the system scale only with the number of sites lying on the partition boundaries, as compared to volume law entanglement that scales with the total number of sites in the bulk. Tensor networks have proven very powerful in simulating one-dimensional lattice systems, for which the matrix product state (MPS)11,12 ansatz offers attractive qualities such as flexibility, variationality, and systematic convergence with increasing bond dimension. While matrix product states have had some success in quantum chemical settings6,7,8,9,13,14, systems without a one-dimensional area law may require exponentially large bond dimensions, and even states that follow the law are not guaranteed to be efficiently simulable15. Extensions to two-dimensional connectivity known as projected-entangled pair states (PEPS)16 have been proposed, but these are generically hard to contract even in terms of average-case complexity17. This reveals a broader challenge within the field of tensor networks, which is that generalizations beyond one-dimensional connectivity tend to incur exponentially hard classical contraction bottlenecks unless approximations or restrictions are applied18.
Quantum computers19,20 present an alternative route to breaking the curse of dimensionality by encoding highly correlated ground states in entangled registers of qubits. An ideal quantum computer could in principle represent a state with arbitrarily large entanglement with no increase in the space requirement, and useful properties such as ground state energies may then be extracted via quantum phase estimation21,22,23,24. Given the significant technical challenges associated with preserving and manipulating an array of entangled qubits, algorithms designed for the current generation of noisy intermediate-scale quantum (NISQ) processors must utilize as little quantum resource as possible. Hybrid quantum-classical algorithms, such as the variational quantum eigensolver (VQE) and its contemporary variants25,26,27,28,29, aim to achieve practical ground state energy estimates with extremely low depth quantum circuits at the expense of a greater measurement cost. While VQE algorithms have gathered interest due to their ease of quantum circuit implementation, the most widely studied ansätze based on single-reference unitary coupled cluster with single and double excitations (UCCSD—we shall refer to related approaches as UCC-type) suffer from poor treatment of strong correlation, expensive gradient calculations, and barren plateaus in the training landscape26,30. The lack of strong correlation can be ameliorated by multi-reference methods such as the recently developed non-orthogonal quantum eigensolver (NOQE)31, which promises a quantum advantage for systems with finite numbers of radical sites exhibiting both strong and weak electron correlation. Furthermore, optimizing the UCCSD ansatz requires a two-qubit gate count per QPU call of O(N4), where N is the number of molecular orbitals, which may be prohibitively expensive for medium-sized or large systems. While tensorial decompositions offer lower order polynomial scaling in practice31,32, and there is numerical evidence that adaptive methods such as ADAPT-VQE27 can reduce gate counts considerably, these cost reductions are system dependent and the worst-case scaling for the most challenging systems may still be quartic in N.
Given that quantum circuits are tensor networks with unitary constraints, several recent works33,34,35,36,37,38,39,40,41,42,43,44 have suggested that classical tensor network methods may be adapted to generate more compact and robustly trainable quantum circuit ansätze, so that quantum computers may help to resolve the computational bottlenecks in the contraction of higher-dimensional tensor networks. Here we present a new hybrid quantum-classical algorithm, which we call a tensor network quantum eigensolver (TNQE), that solves for chemical ground state energies by constructing a highly compact wavefunction from a superposition of M matrix product states of fixed bond dimension χ in rotated orbital bases. We show that computing expectation values with such a construction by classical tensor network contraction is exponentially costly in the general case, but is however easily translated into quantum circuits of depth O(N). We emphasize that although the TNQE method employs a hybrid quantum-classical parameter optimization routine which satisfies the Rayleigh-Ritz variational principle, it is not a “VQE method” in the typical sense, in which a parameterized quantum circuit is optimized using energy gradients. In contrast, it is a gradient-free approach that iteratively sets up and solves a generalized eigenvalue problem in a basis of non-orthogonal matrix product states. It can therefore be regarded as a member of the emerging family of quantum subspace diagonalization (QSD) methods31,45,46,47,48,49, which use a QPU to evaluate the Hamiltonian and overlap matrix elements within a subspace of low-energy state vectors, and as such may also be extendable to the calculation of low-lying excited states.
We know of several existing proposals for MPS-based ansätze within the VQE paradigm34,50,51,52,53, which fall into one of two categories. The first of these uses a single parameterized quantum circuit ansatz that encodes a matrix product state of bond dimension χ, which can be efficiently prepared by quantum circuits with gate count at most O(Nχ2)34,50,51. In general, this requires the quantum many-body system under study to obey a one-dimensional area law of entanglement, unless some additional restriction on the form of the MPS tensors is enforced. It has therefore been argued that these single-reference MPS methods can at best achieve a polynomial advantage over classical optimizers34. The second category uses an MPS as a classically pre-optimized initial state which is then augmented by a conventional parameterized quantum circuit, in an attempt to “short-cut” the non-linear VQE optimization past barren plateaus and local minima52,53. There is no guarantee of successfully side-stepping local minima by this method, although it has been shown to lead to an improvement in the cases studied. The circuit depth required by a single parameterized circuit to characterize generic chemical ground states is not well established, but is expected to be at least super-linear if not super-polynomial in N in the general case.
Our method advances on these prior works in two important respects. First, the superposition of single-particle bases extends the reach of the TNQE ansatz beyond states obeying a one-dimensional area law of entanglement in a systematic manner. This has not been attempted previously and may now allow for circumvention of the simulability constraints of single MPS-based ansätze15. Second, our optimization routine is entirely gradient-free and can instead be considered as a natural generalization of the DMRG sweep algorithm to linear combinations of non-orthogonal matrix product states. Recent results have shown that the canonical form of the MPS, which is utilized by the sweep algorithm, can provide rigorous guarantees of the absence of barren plateaus in the training landscape54,55. This may significantly reduce the overall measurement cost of ground-state energy estimation compared to conventional gradient-based variational optimizers56. The results presented in this first study are consistent with this prediction. We show that for ground states the energy can be reliably converged via iterative quantum subspace diagonalizations, using O(NM2χ4) calls to a QPU per sweep, with a gate count for each QPU call that scales at worst as O(N2 + Nχ2) and with circuit depths linear in N. We achieve chemical accuracy in numerical tests on two small chemical systems—a stretched water molecule and an octahedral arrangement of six hydrogen atoms—empirically demonstrating both significantly better converged energy estimates and a far lower sensitivity to shot noise than VQE with a single UCCSD ansatz circuit, resulting in orders of magnitude reductions in the estimated quantum resources for the H6 cluster, with even greater reductions expected for larger systems.
Results
Preliminaries
Expressed in a basis of N molecular orbitals, the second-quantized electronic structure Hamiltonian takes the form
where the one- and two-body coefficients hpq and hpqrs encode one- and two-electron integrals over the single-particle molecular orbital basis, and the creation and annihilation operators act on the Fock space of η-electron Slater determinants \(\left\vert \overrightarrow{k}\right\rangle =\left\vert {k}_{1},\ldots ,{k}_{N}\right\rangle\)57. Each orbital occupancy index kp has d possible values, where d = 2 with occupancy values 0 or 1 when expressed in terms of spin-orbital sites, or d = 4 with binary occupancy values 00, 01, 10, or 11 for spatial orbital sites, corresponding to the local basis states \(\left\vert {\rm{vac}}\right\rangle\), \(\left\vert \uparrow \right\rangle\), \(\left\vert \downarrow \right\rangle\), or \(\left\vert \downarrow \uparrow \right\rangle\), respectively. The ground state problem consists in finding a superposition of Slater determinants \(\left\vert \Psi \right\rangle\) that minimizes the expectation value \({E}_{0}=\langle \Psi | \hat{H}| \Psi \rangle\). The full configuration interaction (FCI) coefficient tensor \({\Psi }^{{k}_{1}\ldots {k}_{N}}\) encodes the coefficients exactly describing the ground state of the system,
The FCI coefficient tensor can be decomposed via tensor-train factorization58 – a sequence of index groupings and singular value decompositions (SVDs)—which is equivalent to representing the wavefunction in a tensor network format known as a matrix product state (MPS) or a tensor train,
where the \({\Phi }^{{k}_{p}}\) are the site tensors, each with χ2d free parameters, so that the total number of tensor parameters is O(Nχ2d) (see Fig. 1). The generality of the SVD ensures that, before truncation of singular values, the MPS preserves all of the information in the FCI wavefunction and hence is an equivalent representation of the true ground state. The Schmidt decomposition allows quantification of the entanglement between any left block A = {1, …, p} and right block B = {p + 1, …, N} of the MPS59, which is written as
where σ1≥…≥σχ are the singular values, and the left and right Schmidt vectors have the important property \(\langle {\Phi }_{l}^{A}| {\Phi }_{{l}^{{\prime} }}^{A}\rangle =\langle {\Phi }_{l}^{B}| {\Phi }_{{l}^{{\prime} }}^{B}\rangle ={\delta }_{l{l}^{{\prime} }}\). The Schmidt rank is upper bounded by the bond dimension \(\chi \le \,\min ({d}^{{N}_{A}},{d}^{{N}_{B}})={d}^{N/2}\) at the central bond. The MPS can be placed into this so-called canonical form by gauge transformations of the tensors, such that the canonical center is the diagonal matrix of singular values, as depicted in Fig. 2a (see Supplementary Note 1). The von Neumann entropy across the partition is then obtained as
The memory cost can be reduced at the expense of accuracy by truncating χ at each bond, equivalent to projecting onto the first χ left and right Schmidt vector pairs. This truncation necessarily enforces a notion of locality to the ansatz; an MPS with fixed bond dimension χ ≪ dN/2 obeys a one-dimensional area law of entanglement between the molecular orbitals, with maximal entropy between the left and right blocks equal to \(\ln (\chi )\), seen by setting all singular values to be equal in Equations (4) and (5). In the MPS format expectation values can be extracted by efficient tensor network contraction with an analogous tensor-train factorization of the operator, known as a matrix product operator (MPO)8, so that the sum over dN Slater determinants need not be explicitly computed. This is the basis for efficient classical ground state solvers, whereby an MPS is initialized with random parameters and optimized variationally to minimize the expected energy. This is typically by the one- or two-site DMRG sweep algorithm, wherein the canonical center is contracted over a single site or a pair of sites (Fig. 2b, c), which are then locally optimized8.


The black tensor denotes the canonical center in each case. The arrows indicate contraction (a.k.a. `bubbling') of the highlighted tensors. a The canonical center (diamond-shaped tensor) is a diagonal matrix of singular values corresponding to the Schmidt decomposition over partitions A (top) and B (bottom) as in Eq. (4). b The canonical center is contracted into the single-site tensor at site p (in A for this example). c The canonical center is contracted into the two-site tensor at sites p and p + 1.
A more general tensor network state can be depicted by a graph with N nodes representing the sites, in this case the molecular orbitals, and up to \(\left(\begin{array}{c}N\\ 2\end{array}\right)\) edges representing the bonds, as in Fig. 3. An MPS is then described by a path visiting each node via N − 1 unique edges, a PEPS corresponds to a graph with \(2N-2\sqrt{N}\) edges, and so on, up to a tensor network with full connectivity between all pairs of nodes. A fully connected tensor network is not a classically efficient description of a quantum state, requiring O(NχN−1) tensor parameters. The set of all fully connected states also contains that of all PEPS, the exact contraction of which is #P-hard60, and is thus believed to be exponentially hard even for a quantum computer up to arbitrary precision. In practice, however, we are often more interested in approximate contraction up to an additive error, which is BQP-complete for tensor networks defined on graphs of bounded degree61.

The solid lines denote the bond indices of a matrix product state as defined in Eq. (3). The restricted Hartree-Fock orbitals of the H\({}_{3}^{+}\) cation in the STO-3G basis set are provided as a minimal example of a closed-shell molecule with six spin-orbital sites.
Operators in the fermionic Fock space can be mapped onto the state space of a quantum register under the Jordan-Wigner transformation62, establishing a one-to-one correspondence between the spin-orbitals and the qubits, both of which are local Hilbert spaces of dimension d = 2. The fermion operators are then represented as a linear combination of unitary Pauli strings,
where \({\hat{X}}_{p}\) is short-hand for \({\hat{{\mathbb{1}}}}^{\otimes p-1}\otimes \hat{X}\otimes {\hat{{\mathbb{1}}}}^{\otimes N-p}\). The string of Pauli \(\hat{Z}\) gates on the qubits 1, …, p − 1 enforces the fermionic anticommutation relations. The electronic structure Hamiltonian in Eq. (1) can then be fully specified in any orbital basis with O(N4) Pauli strings (\({\hat{P}}_{\alpha }\)), written as
Under this representation the Slater determinants \(\left\vert \overrightarrow{k}\right\rangle\) are equivalent to the computational basis vectors of the qubit register (also known as ‘bitstring’ states). Near-term variational quantum eigenvalue solvers (VQEs) involve optimizing the parameters of a polynomial-size quantum circuit \(\hat{U}\), which prepares some ansatz state \(\left\vert \phi \right\rangle =\hat{U}\left\vert 0\right\rangle\), to minimize the expectation value \(\langle \phi | \hat{H}| \phi \rangle\) as computed from the Pauli decomposition in Eq. (7). A common choice of ansatz circuit for quantum chemistry is fermionic unitary coupled cluster with single and double excitations (UCCSD)26.
Alternatively, quantum multi-reference methods use a wavefunction ansatz constructed from a linear combination of efficiently preparable reference states,
subject to the constraint that \(\left\vert \psi \right\rangle\) is normalized and thus strictly variational. For example, the NOVQE method46 uses UCC-type reference states, where the gate rotation angles are computed using a hybrid quantum-classical variational gradient-based optimizer, while the NOQE method31 also UCC-type reference states, but instead selects the circuit parameters using classical chemistry heuristics, avoiding all hybrid variational parameter optimization. Given a set of M arbitrary reference states \({\{\vert {\phi }_{j}\rangle \}}_{j = 1}^{M}\), the optimal coefficients {cj} to approximate the ground state of the Hamiltonian by their linear combination are obtained by solving the generalized eigenvalue problem
where the elements of the matrix pencil (H,S) are given by
The matrix E is a diagonal matrix of generalized eigenvalues E1≤ ⋯ ≤EM. The optimal coefficients corresponding to the ground state estimate E1 are then given by the first column of C, i.e., cj = Cj1. This technique of solving Eq. (9) on a classical computer with matrix elements obtained by a quantum computer has come to be known more broadly as quantum susbpace diagonalization49.
In the following sections we will make frequent use of the direct equivalence between tensors subject to unitary constraints and quantum gates. In the figures we will use circles and rounded shapes to denote classical tensors in tensor network diagrams, and we will use squares and rectangles to denote quantum gates in circuit diagrams. For an in-depth introduction to tensor network notation and the relation to quantum circuits, see e.g.3,4.
TNQE ansatz
The TNQE ansatz is a quantum multi-reference state, with the form of \(\left\vert \psi \right\rangle\) in Equation (8), where the M non-orthogonal reference states \({\{\vert {\phi }_{j}\rangle \}}_{j = 1}^{M}\) are matrix product states of fixed bond dimension χ, as defined in Eq. (3), expressed in different orbital bases. Thus the TNQE wavefunction is specified by the O(MNχ2) MPS tensor parameters, the choice of single-particle basis for each MPS, and the coefficients {cj}, which are subject to the constraint that \(\left\vert \psi \right\rangle\) is normalized. This wavefunction form is strictly variational, and inherits size consistency from its closest classical analogues, namely the non-orthogonal configuration interaction63,64 and orbital-optimized DMRG65,66,67. While each MPS obeys a one-dimensional area law within its own single-particle basis, their superposition allows for the efficient description of a broader class of wavefunctions. This additional flexibility also incurs an exponential separation in complexity between the classical and quantum evaluations of the off-diagonal matrix elements in Eqs. (10), as we shall now demonstrate.
To gain some intuition we shall first introduce the restricted case of orbital permutations, i.e., the same set of orbitals but with different site orderings, before generalizing to arbitrary rotations of the single-particle basis. In this first case, the superposition in Eq. (8) allows for the combination of matrix product states with different site connectivity, as shown in Fig. 4a. The set of all states that can be described by Eq. (8) using both M and χ of size polynomial in N may then be expected to lie somewhere in-between that of a single matrix product state and that of a fully connected tensor network. If the orbital sets were identical, then the off-diagonal matrix element computations in Equations (10) would be tractable by classical tensor network contraction, assuming an efficient MPO representation of \(\hat{H}\). However when the orbital sets differ additional tensors must be inserted to transform between the representations. An arbitrary permutation of the orbitals is achieved with the correct treatment of fermionic antisymmetry via a nearest-neighbor fermionic-SWAP (FSWAP) network68. The sequence of FSWAP operations \({\hat{F}}_{ij}\) to rearrange the orbital ordering of \(\left\vert {\phi }_{i}\right\rangle\) to that of \(\left\vert {\phi }_{j}\right\rangle\) is illustrated in Fig. 4b. Representing these operations with classical tensors results in the tensor network in Fig. 5a to compute the matrix element \(\langle {\phi }_{i}| \hat{H}| {\phi }_{j}\rangle\), which has a cost of contraction that rises steeply with the depth of the FSWAP network. Any pair of orderings can be connected by a path-restricted sorting network of depth lower bounded by O(N)69,70, e.g., a bubble sort71. The application of each FSWAP gate to the MPS on the left can grow the bond dimension by up to a factor of d, so χ will rise exponentially with N unless severe truncations are applied. This implies that a large amount of entanglement can be generated in the ansatz through the site rearrangement, even though each matrix product state is individually of low bond dimension. We note that while at first glance these tensor network contractions appear to be classically hard, the matrix elements can in principle be efficiently approximated by randomly sampling computational basis vectors from the matrix product state (see Supplementary Note 3). This suggests the potential for a powerful, although limited, new class of quantum-inspired classical tensor network methods via random sampling. Because FSWAP networks belong to the Clifford subgroup, this is somewhat comparable to a multi-reference generalization of the recently proposed Clifford-augmented matrix product states72,73. We may say that this permutation-only variant conveys the basic intuition behind TNQE but does not fulfill its potential as a hybrid quantum-classical method.

a MPSs \(\left\vert {\phi }_{j}\right\rangle\) and \(\left\vert {\phi }_{i}\right\rangle\) with different site orderings over the same set of N = 6 molecular orbitals, and the superposition state \({c}_{i}\left\vert {\phi }_{i}\right\rangle +{c}_{j}\left\vert {\phi }_{j}\right\rangle\). b A sequence of FSWAP operations \({\hat{F}}_{ij}\) transforms the MPS \(\left\vert {\phi }_{i}\right\rangle\) into the orbital ordering of \(\left\vert {\phi }_{j}\right\rangle\), giving rise to the tensor network in Fig. 5a.

a The MPSs are related by a permutation over the same set of orbitals. The FSWAPs, denoted by pairs of crossed tensors, are selected by a bubble sort comparator network. Each comparator applies an FSWAP to rearrange neighboring orbitals if their order does not match the order in which they appear on the right. The dashed outlines indicate the positions of non-swapping comparators. b The MPSs are related by an arbitrary orbital rotation \({\hat{G}}_{ij}\), which is decomposed into a pyramidal structure of Givens rotation tensors (see Eq. (11)).
We now show that the restriction to orbital permutations can be lifted to create a far more expressive ansatz under arbitrary rotations of the single-particle basis. Kivlichan et al.74 have shown how to decompose an arbitrary rotation of the molecular orbitals \({\hat{G}}_{ij}\) into a sequence of nearest-neighbor Givens rotation gates of linear depth in N,
where Θij is a set of \(\left(\begin{array}{c}N\\ 2\end{array}\right)\) pairs (p, θ) of site positions and rotation angles, and a Givens rotation between orbitals p and q with rotation angle θ is written as
This decomposition follows from a QR factorization of the N × N orbital rotation matrix, and is completely general as a consequence of the Thouless theorem75. As shown in Fig. 5, there is an obvious resemblance between the sequence of Givens rotations to perform an arbitrary orbital rotation and the comparator network that implements an arbitrary orbital permutation. The connection becomes clear when one considers that the FSWAP gate is a special case of a Givens rotation gate with rotation angle \(\theta =\frac{\pi }{2}\), up to a change of phase on one of the orbitals (see Supplementary Note 1.C), so that in some sense the transformation \({\hat{G}}_{ij}\) can be thought of as a ‘quantum sorting network’. Although there is no longer an adequate orbital graph representation for the transformed state \({\hat{G}}_{ij}\left\vert {\phi }_{i}\right\rangle\), it is clear that any path rearrangement can be recovered with the appropriate set of discrete angles \(\theta \in \{0,\frac{\pi }{2}\}\). The generalization to arbitrary continuous angles then implies a kind of smooth distribution over exponentially many possible paths.
In quantum mechanics, the entanglement of a system is dependent on the global basis in which it is measured; a system of low entanglement in one basis may have very high entanglement when expressed in a different basis. For instance, the tensor network in Fig. 6 depicts a linear-depth sequence of orbital rotations that transforms a completely unentangled bitstring state, which corresponds to a d = 2, χ = 1 MPS, to a state with maximal Schmidt rank at the central partition, i.e. χ = 2N/2 (this transformation is explained in Supplementary Note 4). This appears to suggest that one may write down an MPS superposition ansatz with no classically efficient representation in any common orbital basis. It is argued in Supplementary Note 3 that in contrast to the restricted case of orbital permutations, the matrix elements between matrix product states differing by arbitrary orbital rotations cannot be efficiently evaluated via random sampling, so there is no known classical algorithm to efficiently compute Equations (10) in this case.

A sequence of \(\frac{\pi }{2}\) and \(\frac{\pi }{4}\) orbital rotations transforms the unentangled state \({\left\vert 1\right\rangle }^{\otimes {N}_{A}}\otimes {\left\vert 0\right\rangle }^{\otimes {N}_{B}}\) to a state with maximal Schmidt rank and von Neumann entropy across the A/B partition, where NA = NB = N/2, shown above for N = 6 and explained in Supplementary Note 4.
TNQE algorithm
Converging the expected energy of the TNQE asnatz requires an efficient optimization routine for both the tensor parameters and the orbital rotation angles. We introduce a generalized sweep algorithm (Algorithm 1) to co-optimize both of these quantities. The intuition behind this approach is that each MPS should “learn” those specific entanglement features which are most efficiently represented within its own single-particle basis. This means that the tensor parameters should be informed by the choice of orbital bases, and vice-versa.
First, we show how the DMRG sweep algorithm can be naturally extended to optimize the TNQE ansatz parameters via quantum subspace diagonalization (lines 4–7 of Algorithm 1), resulting in a gradient-free optimization strategy that converges reliably even in the presence of relatively large noise perturbations to the matrix elements. A key step in this procedure is the decomposition of the local two-site tensor (T) into a basis of “one-hot” tensors (line 4), which are tensors of the same dimension as T that have a single entry equal to 1 and all other entries 0 (see the subsection on “Parameter optimization” and Supplementary Note 1.A). These span the space of all states that could be obtained by locally updating the MPS parameters. A QPU is needed to compute the expanded subspace matrix elements in line 5, with all of the additional co-processing steps being executed entirely on classical hardware. Second, it follows from Equation (11) that any orbital rotation can be built up from pairwise orbital rotations, and in this work we explore this “bottom-up” perspective to manipulate the orbital entanglement during the MPS optimization. We show how the rotation angles can be classically co-optimized (line 8), with no additional QPU calls, to reduce the bond entanglement during the parameter sweep, enacting a ‘transfer’ of entanglement between the bonds of the MPS and the orbital rotations. We note that similar strategies have been proposed to update the site ordering76 or the orbital basis65 of a single MPS ansatz for use in classical methods.
Each sweep of Algorithm 1 requires O(NM2χ4) matrix element evaluations to optimize all the states simultaneously, which scales favorably provided that χ is kept small and the resources of the ansatz are increased through M. In numerical experiments we have found it to be an effective strategy to increase M in stages until convergence in the energy estimate is achieved. The first MPS (M = 1) is constructed in orbitals obtained by a restricted Hartree-Fock calculation, and initialized with classical DMRG. At the next stage (M = 2), a new MPS is added in the same set of orbitals, with random tensor parameters. The entire subspace is then optimized by Algorithm 1 for some number of sweeps. The same procedure is then repeated at each stage (M = 3, 4, …), with each new MPS initialized with random tensor parameters, starting in the same set of orbitals as the previous MPS. With this iterative subspace construction, the TNQE algorithm is essentially a “black-box” routine, where in principle the system information is entirely supplied by the Hamiltonian coefficients, and the only free parameter is the fixed bond dimension cutoff χ. In practice, however, some additional fine-tuning of the optimizer and system dependent parameter initializations may improve convergence, the exploration of which we leave to future work. A more detailed explanation of the TNQE algorithm with further derivations, intermediate steps, and lower-level pseudocode, is provided in Supplementary Note 1.
Algorithm 1
Generalized sweep algorithm (high level)
1: for p = 1, …, N − 1 do
2: Put each MPS in canonical form centered at site p
3: Contract MPS two-site tensors (T) over sites p, p + 1
4: Decompose each T into ‘one-hot’ tensors
5: Query QPU for expanded subspace matrices \({{\bf{H}}}^{{\prime} }\), \({{\bf{S}}}^{{\prime} }\)
6: Solve generalized eigenvalue problem on CPU
7: Update T parameters from solution matrix \({{\bf{C}}}^{{\prime} }\)
8: Apply local two-site FSWAPs or orbital rotations
9: SVD of T and truncate to χ singular values
10: end for
Matrix elements
Figure 7 shows a quantum circuit used to efficiently evaluate the off-diagonal matrix elements in Equation (10), equivalent to the tensor network contraction in Fig. 5b, given linear qubit connectivity on the system register and all-to-one connectivity to a single ancilla qubit. By measuring this ancilla in the z-basis, one obtains the matrix elements required in line 5 of Algorithm 1 up to standard error δ with O(δ−2) repetitions (see Methods). This circuit requires a controlled state preparation unitary for each MPS. There are several proposals for MPS quantum circuit encodings77,78,79,80, the simplest exact approach being the direct synthesis of unitary encodings of each MPS tensor77,78, with a total gate count that scales at worst as O(Nχ2), following standard asymptotic results for generic unitary synthesis81,82. For near-term hardware we present here a cost analysis based on the approximate disentangler technique pioneered by Ran79 and developed by others83,84, but in principle any encoding scheme may be substituted. This technique is briefly explained in Methods, wherein we show how to cheaply add controls to the disentanglers, then decompose the entire circuit into CNOT gates and single-qubit rotations to derive an exact two-qubit gate count for each circuit in terms of the number of qubits N and the disentangler depth D, resulting in
We have omitted the cost of the controlled Pauli string, which will be constant in N for any k − local fermionic observable since the Pauli \(\hat{Z}\) strings of the Jordan-Wigner mapping act trivially on the vacuum state. The scaling of D with χ will vary depending on the MPS parameters, but is not expected to be more expensive than O(χ2). The intuition behind the TNQE ansatz, which may be tested empirically, is that χ can be made constant in N, provided that the non-local correlations are recovered through the inclusion of multiple references in different orbital bases. This comes at the cost of a greater number of MPS reference states, M, which appears as a quadratic factor in the number of matrix elements, and hence in the overall measurement cost. With this choice of fixed-bond ansatz, Eq. (13) presents a substantive advantage over existing ansätze, such as the UCC-type ansätze, that are characterized by a two-qubit gate count per circuit that scales as O(N4)26.

This circuit requires controlled unitaries \({\hat{U}}_{i}\) and \({\hat{U}}_{j}\) to prepare \(\left\vert {\phi }_{i}\right\rangle\) and \(\left\vert {\phi }_{j}\right\rangle\) from the all-zero state in their respective orbital bases, an efficient Pauli string decomposition of \(\hat{H}\) as in Eq. (7), and an orbital rotation \({\hat{G}}_{ij}\) decomposed into Givens rotation gates as defined in Eq. (11). The details of the Hadamard test evaluation and the circuit construction are provided in Methods and Supplementary Note 2.A.
With a single ancilla qubit, the layer depth in terms of CNOT gates and single-qubit rotations is given by
(see Methods). This results from having to apply each controlled disentangler in sequence, introducing an unwanted scaling of O(ND). If, however, the hardware is of sufficient fidelity to prepare and maintain a coherent superposition of the all-zero and all-one states over N/2 qubits, known as a Greenberger-Horne-Zeilinger (GHZ) state, then this limitation can be circumvented at the cost of using N/2 ancillas. With this modification up to N/2 controlled rotations can be applied in parallel with control from different ancilla qubits. The layer depth is then given by
which may be worth the additional overhead of N/2 ancillas and preparation of the GHZ state. In either case, the depth of the circuit grows only linearly in the number of qubits for a fixed bond dimension χ.
Parameter optimization
Matrix product states are commonly optimized to lower the expected energy by a two-site sweep algorithm, whereby the MPS is put into canonical form centered on a two-site tensor over the sites p and p + 1 (Fig. 2c), which are then contracted, optimized, and decomposed back into site tensors. Here, we explain how to generalize the conventional two-site algorithm to a linear combination of non-orthogonal matrix product states. The key idea behind the generalized sweep algorithm is to expand each MPS into an orthonormal basis that represents the local degrees of freedom at sites p and p + 1. This follows from an expansion of the local two-site tensor as a sum over a set of orthonormal tensors, together with the canonical form of the MPS. By solving a generalized eigenvalue problem, like that in Eq. (9), in the expanded subspace, a new set of optimal parameters are obtained for each two-site tensor to minimize the new ground state energy estimate \({E}_{1}^{{\prime} }\le {E}_{1}\).
More concretely, each two-site tensor T is decomposed as a sum over the set of all unique tensors with the same indices as T and a single element equal to 1, with all other elements equal to 0. We will refer to these as ‘one-hot’ tensors, analogous to one-hot vectors that are common in computer science85. This sum over the one-hot tensors, indexed by m, is initially weighted by the two-site tensor parameters {tm} (see Fig. 8a). We will further define the one-hot state, \(\vert {\varphi }_{m}^{[j]}\rangle\), obtained by replacing T in \(\vert {\phi }_{j}\rangle\) with the unique one-hot tensor indexed by m. Because the MPS is in canonical form centered at the sites p and p + 1, the one-hot states represent a decomposition of \(\vert {\phi }_{j}\rangle\) into an orthonormal basis,
which spans the space of all possible matrix product states that could be obtained by locally updating the tensor parameters at sites p and p + 1 with any values satisfying the normalization condition. The optimal values of the tensor parameters can then be found by diagonalizing in this expanded basis. Each MPS can be decomposed simultaneously from two-site tensors located at the same or different sites, resulting in a new matrix pencil \(({{\bf{H}}}^{{\prime} },{{\bf{S}}}^{{\prime} })\) in the expanded subspace of dimension Md2χ2, with matrix elements given by
Equation (17) describes the simultaneous expansion of all reference states, such that im denotes the combined row or column index (i − 1)d2χ2 + m, as in Fig. 8b. One may also choose to optimize only a single MPS, or any subset thereof. After solving the generalized eigenvalue problem in the expanded subspace,
the new optimal coefficients \(\{{t}_{m}^{{\prime} [j]}\}\) for each two-site tensor are obtained by slicing and normalizing from the first column of the expanded solution matrix \({{\bf{C}}}^{{\prime} }\),
Further details and derivations of Eqs. (16), (17), and (19) are provided in Supplementary Note 1.A. Optimizing all states simultaneously requires O(M2χ4) matrix element evaluations per site decomposition, so a full sweep over all sites requires O(NM2χ4) matrix elements, each of which may be efficiently obtained by a quantum circuit as in Fig. 7. In practice, the number of one-hot tensors can be reduced by enforcing particle number and z-spin conservation through an internal block-sparse structure of the tensors86,87. The effect of ill-conditioning of the generalized eigenvalue problem has been rigorously studied in ref. 88. In practice the condition number of \({{\bf{S}}}^{{\prime} }\) can be effectively regulated by discarding subspace vectors with a high degree of linear dependence (see Methods).

a Local tensor operations corresponding to the lines of Algorithm 1, omitting the orbital update step. The canonical center at site p (in black) is contracted into the two-site tensor T (line 3), which is decomposed over d2χ2 one-hot (dotted) tensors as in Equation (16) (line 4). After solving the generalized eigenvalue problem in the expanded subspace (lines 5, 6), the two-site tensor parameters are updated according to Eq. (19) (line 7). The two-site tensor is then decomposed back into single-site tensors by SVD (line 9). b The Hamiltonian subspace matrix H is expanded over one-hot tensor decompositions of the two-site tensors at sites p and p + 1, as in line 5 of Algorithm 1 and Eq. (17) (for legibility, the number of elements depicted in each matrix block is not to scale). The tensor networks on the right compute the matrix elements in the diagonal (i = j) and off-diagonal (i ≠ j) blocks. The expanded overlap matrix \({{\bf{S}}}^{{\prime} }\) is computed by similar tensor networks, but removing the Hamiltonian MPO. Each off-diagonal matrix element can be efficiently obtained using a quantum circuit encoding of the tensor network as in Fig. 7.
Orbital rotations
After each parameter update the optimized two-site tensors are decomposed into single-site tensors via SVD with a maximum Schmidt rank of dχ, which is then truncated to the χ largest singular values. At this step some information is discarded by projecting onto the space of the first χ left and right singular vector pairs of the Schmidt decomposition (see Equation (4)). Given that entanglement is a basis-dependent quantity, it can be reduced before the truncation step by a local rotation of the orbitals p and p + 1 without loss of information. Our pairwise orbital co-optimization strategy is similar to previous approaches that have been developed to reduce the bond truncation error of a single MPS reference state, either by swapping neighboring sites76, or by applying a local pairwise orbital rotation65. The procedure is outlined with tensor network diagrams in Fig. 9. Consider first the restricted case of a pairwise orbital permutation. The double application of an FSWAP gate is equivalent to the identity and so does not alter the state encoded by the two-site tensor. One of the FSWAPs is then contracted into the two-site tensor on the left prior to the SVD and the truncation error is computed as
The orbital rearrangement is accepted if the truncation error at the bond is reduced by the FSWAP contraction, in which case the second FSWAP is then merged into \({\hat{G}}_{ij}\) by updating the N × N orbital rotation matrix and re-computing the rotation angles. Otherwise, the FSWAP insertion is rejected, and the two-site tensor parameters are reverted to their original values. A similar method to reduce the bond entanglement by swapping neighboring site indices was studied in ref. 76 for dynamical simulations in a non-fermionic context, which is equivalent to substituting the generic SWAP gate in the procedure above.

a A schematic of the FSWAP insertion. The vertical arrow denotes the combined operations of contraction into the two-site tensor followed by SVD in tensor network diagrammatic notation3,4. The horizontal arrow indicates merging into \({\hat{G}}_{ij}\) if the update is accepted. b A schematic of the steps to compute the truncation error cost function ξ(θ) in Eq. (21) for the classical univariate optimization to determine θopt. The adjoint rotation with angle − θopt is then merged into \({\hat{G}}_{ij}\).
To apply an arbitrary pairwise orbital rotation, the unitary \(\hat{g}(\theta )\) is classically optimized to find the value of θopt that minimizes the truncation error,
This is a purely classical co-optimization step with a cost O(χ3) that derives solely from performing the SVD on the contracted two-site tensor, thus requiring no additional QPU calls or energy gradient calculations. The adjoint rotation \({\hat{g}}^{\dagger }({\theta }_{{\rm{opt}}})=\hat{g}(-{\theta }_{{\rm{opt}}})\) is then merged into \({\hat{G}}_{ij}\). In a sense this unitary ‘encodes’ a part of the information from the optimized two-site tensor, which is then merged into the basis rotation instead of being discarded by the singular value truncation.The key insight behind this approach is that switching the focus of the orbital optimization from directly minimizing the energy estimate to instead maximizing the fidelity of the wavefunction by reducing the truncation error, in combination with a DMRG-like optimization procedure for the MPS, can allow for more rapid and robust convergence of the orbital rotation angles. Essentially the same idea was proposed for orbital optimization in a fully classical context with a single MPS reference in ref. 65. In this classical setting only a single orbital basis must be updated, which amounts to a transformation of the fermionic Hamiltonian coefficients. By contrast, our multi-reference ansatz requires a set of distinct orbital rotation operators, \({\hat{G}}_{ij}\), to transform between every pair of orbital bases. Note that in line 8 of Algorithm 1 only a single Givens rotation angle is optimized for each MPS in order to minimize its local bond truncation error. However, when this new local orbital rotation is ‘merged’ into each of the \({\hat{G}}_{ij}\), every N × N orbital transformation matrix must be updated separately, after which, in order to factorize them back into Givens rotation circuits as in Eq. (11), all of the rotation angles need to be re-computed, using the technique of ref. 74.
In practice, it is observed that interleaving orbital permutation sweeps with orbital rotation sweeps is highly effective at breaking the optimizer out of local minima. This corresponds to alternating between rearranging the orbitals to different site positions via nearest neighbor swapping, and mixing of the orbitals through nearest neighbor quantum superpositions. After the rotation step, it is found to be beneficial to further mitigate the penalty in the expected energy incurred by singular value truncation using a sequence of single-site decompositions, with no additional matrix element computations required, as explained in Methods.
Numerical benchmarking
Here we evaluate the performance of the TNQE method on a stretched water molecule and on an octahedral arrangement of six hydrogen atoms. All calculations are performed in the STO-3G minimal basis set, corresponding to seven spatial molecular orbitals for H2O and to six spatial orbitals for octahedral H6. This would be equivalent to running quantum circuits on system registers of 14 and 12 qubits, respectively, under the Jordan-Wigner mapping. We have chosen to focus on spin-restricted rotations over the spatial orbitals; we suggest experimentation with spin-symmetry breaking as a topic of future work. We benchmark the method against classical DMRG calculations and against a simple sparse matrix implementation of VQE with a single UCCSD ansatz circuit (VQE-UCCSD) under a first-order Trotter decomposition. For details of the numerical calculations see Methods.
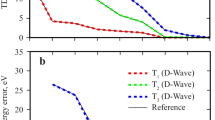
In Fig. 10a, we demonstrate that TNQE can be reliably converged to chemical accuracy for the stretched water molecule with O—H bond length between 2–3 Å, with bond dimension χ = 3 and subspace dimension M = 3. This dissociation region is known to be challenging for UCC-type ansätze29. The ease of convergence with low computational resources suggests that systems with weak to moderate amounts of electron correlation are not challenging for TNQE. In Fig. 10b, we show that TNQE also reliably attains chemical accuracy for the more strongly correlated octahedral H6 system with χ = 4 and M = 4 across a wide range of internuclear separations, demonstrating that TNQE can also handle cases of much stronger electron correlation with a very manageable increase in the computational resources. Figure 11 shows that the TNQE energy is drastically more accurate than the VQE-UCCSD energy estimate for this system across all bond lengths.

a TNQE (χ = 3, M = 3) energies for a stretched H2O molecule from r = 2 Å up to r = 3 Å. b TNQE (χ = 4, M = 4) energies for octahedral H6 from r = 1.13 Å up to r = 2.83 Å. The `kink' in the FCI curve at ~1.75 Å is an artefact of the minimal basis set. c Comparison of LC-MPS, TNQE-F (FSWAP), and TNQE-G (Givens) energies at χ = 3 with increasing M for octahedral H6 at r = 1.70 Å. d Comparison of DMRG, LC-MPS, TNQE-F (FSWAP), and TNQE-G (Givens) energies with increasing non-zero parameter count for octahedral H6 at r = 1.70 Å. The DMRG bond dimension is increased from χ = 8 to χ = 13, while the TNQE bond dimension is fixed at χ = 4, and M is increased from 2 to 4.

The FCI curve appears underneath the TNQE curve at this energy scale. The `kink' in the UCCSD energy curve at ~1.75 Å is an artefact arising from the minimal basis set.
To assess the prospects for quantum advantage, the convergence of TNQE with increasing subspace dimension M is compared against two classically tractable variants of the method, the first of which removes all of the orbital rotations and permutations. We refer to this variant, which is tractable by classical tensor network contraction, as linear combinations of matrix product states (LC-MPS). The second variant restricts the orbital rotations to orbital permutations with FSWAP networks. As explained in Supplementary Note 3, this variant is in principle dequantizable by classical random sampling. We will refer to this variant in the text as TNQE-F. The TNQE ansatz using Givens rotations with arbitrary angles is not tractable using any known classical algorithm, and we refer to this as TNQE-G. Figure 10c, shows a clear advantage for TNQE-G over both LC-MPS and TNQE-F in the octahedral H6 system at 1.70 Å with χ = 3. While TNQE-G touches the threshold for chemical accuracy at M = 6, the dequantizable TNQE-F and classical LC-MPS sit at ~10 mHa and ~30 mHa, respectively, for M = 6, and have still not converged to chemical accuracy at M = 8.
In Fig. 10d, TNQE is benchmarked against classical DMRG by directly comparing the total number of tensor parameters in the non-zero blocks: this number scales as O(Nχ2) for DMRG and as O(MNχ2) for TNQE. For this example, we scale up the parameters in the TNQE variants at a fixed value of χ = 4 by increasing M from 2 to 4. For the DMRG curve, we have M = 1 by definition and we increase χ from 8 up to 13. For the octahedral H6 system at r = 1.70 Å we observe that TNQE achieves far better energies at low parameter counts. However we are careful not to claim this as proof that TNQE outperforms DMRG for this small system, and we mention several caveats to this comparison. First, at larger bond dimensions (χ ≥ 15) the DMRG curve converges almost exactly to the FCI energy, with a slightly decreased parameter count of ~180, roughly on par with the number of parameters for TNQE at χ = 4, M = 4. This may be due to a rearrangement of the internal block-sparse structure at larger bond dimensions. It may also be possible to improve the convergence of the DMRG curve with localized orbitals or other classical techniques. There is also some ambiguity regarding the inclusion of the Givens rotation angles in the total parameter count of TNQE. These are not ‘free’ parameters in the TNQE ansatz and are mutually interdependent, so it is not clear how or whether they should be counted. Nonetheless, the results at low parameter counts are encouraging for the prospect of observing a more decisive advantage in larger strongly correlated systems. Furthermore, these results have all been obtained with superpositions over spin-restricted orbital bases. This restriction can easily be lifted to include orbital bases of broken spin symmetry, which may reveal a yet stronger improvement in the energy compared to the standard spin-restricted variants of DMRG89.
Cost estimates
We provide detailed estimates of the exact CNOT gate count and circuit depth for TNQE (with both the one-ancilla and the GHZ-ancilla circuit variants) and VQE-UCCSD Trotterized to first order, using a CNOT-efficient circuit encoding of the UCC fermion operators90, for the octahedral H6 system (see Supplementary Note 2). We also estimate the total number of QPU calls to converge the energy estimate, where each QPU call corresponds to a single Hamiltonian or overlap evaluation, noting that the QPU calls in TNQE are highly parallelizable. We simulate shot noise in the matrix elements and expectation values with standard error δ, and we find that while VQE-UCCSD fails to converge with noise levels of δ = 10−7 and requires δ = 10−8 to match the noiseless result to within 3 mHa after 100 iterations, TNQE reliably converges to within chemical accuracy after 48 sweeps with δ = 10−4 in the Hamiltonian matrix elements and δ = 10−5 in the overlaps. We hypothesize that this may be due in part to effective regulation of the subspace condition number, as detailed in Supplementary Note 1.B. From this we derive estimates for the number of shots to resolve each QPU call to these levels of precision, and hence the total number of CNOT gate executions required for the computation. This is the total number of two-qubit entangling gate operations (ng), which gives a sense of the overall quantum compute requirement; with access to Nq qubits (in total across all available QPUs), one can implement at most Nq/2 two-qubit gates in parallel, so the number of entangling gate layers that must be executed in sequence is bounded from below by 2ng/Nq, and from above by ng. A greater overall two-qubit gate count necessitates either more sequential gate layers, raising the lower bound on the end-to-end execution time, or access to a greater number of qubits, thus increasing the hardware budget requirement. We summarize these findings in Table 1 for the octahedral H6 system at r = 1.70 Å in the STO-3G basis. We comment on the high prefactors in the number of shots for both methods in the Discussion. While the primary driver behind our cost reductions in the octahedral H6 system is the high noise tolerance of the TNQE optimizer, we show in Fig. 12 that the CNOT gate count per circuit also scales much more favorably for TNQE with increasing system size, observing a reduction in the CNOT count of nearly two orders of magnitude for 50 hydrogen atoms with disentangler depths as high as D = 100 (see “Methods” for an explanation of the disentangler depth).

Discussion
We have presented a new class of hybrid quantum-classical algorithm, which we call a tensor network quantum eigensolver, to evaluate chemical ground state energies on near-term quantum computers. We have combined techniques from classical tensor networks, variational quantum algorithms, and quantum subspace diagonalization to eliminate the reliance on costly and unreliable gradient-based optimizers. In the TNQE method, as in classical DMRG, it is the efficient reduction of truncation error that is the key to successful optimization of the ground state energy, which is now enabled through the superposition of matrix product states in different orbital bases. Although the TNQE ansatz is constructed using MPS of low bond dimension, the orbital rotations can mimic the entanglement structure of systems that do not follow a one dimensional area law, as is likely to be the case in many real-world instances of strong electron correlation such as cuprates91 or the oxygen-evolving complex of photosystem II92. We have demonstrated reliable convergence to chemical accuracy in small chemical systems, with low parameter counts that suggest a possible regime of practical quantum advantage over classical DMRG. Additionally we report a high tolerance to shot noise and efficient use of quantum resources, with per-circuit gate counts of O(N2) and circuit depths of O(N), signaling the potential of the method to scale well to larger systems in the NISQ era.
The argument for realizing an exponential quantum advantage with TNQE rests on the degree to which the orbital rotations are of practical utility in the energy calculations. This should now be rigorously tested on larger and more complex chemical problems. We note that the orbital rotation heuristics we have developed appear to be improvable from both a quantum information and a quantum chemistry perspective, for example using the quantum mutual information93, or initializing the orbitals from unrestricted Hartree-Fock calculations with broken spin symmetry64. Achieving practical results at scale will require further research into a number of open questions. First, the gate count, the number of measurements, and the classical cost of solving the generalized eigenvalue problem scale cheaply with the number of matrix product states, as O(1), O(M2), and O(M3) respectively, but somewhat less favorably with the bond dimension, at worst as O(χ2), O(χ4), and O(χ6). Establishing the optimal tradeoff between M and χ will be necessary to fully characterize the algorithmic complexity, as well as the scaling of both of these quantities with N, which is expected to be strongly system-dependent. The method also requires the efficient quantum circuit encoding of a large number of matrix product states to high fidelity. It is not known how this cost will scale using the disentangler method79, in terms of both the classical overhead and the circuit depth, and the optimal encoding of matrix product states is currently an ongoing topic of research. It is also unknown how the number of sweeps required for convergence will scale with the system size. Finally, a deeper understanding of the noise resilience and the identification of possible regimes where this breaks down will be important for implementations on real quantum hardware with both stochastic and coherent gate errors. As with any quantum algorithm, it is expected that the incorporation of some form of error mitigation or error correction will be vital for any successful practical implementation.
Compared with the closely related NOQE method31, TNQE offers more compact circuits, greater flexibility, and systematic convergence to the ground state. However, these advantages come at the cost of a greater number of measurements to converge the expected energy. While the estimate of ~1016 shots for a TNQE calculation on 12 system qubits is orders of magnitude cheaper than the estimate of ~1023 for VQE using UCCSD, the high measurement prefactor will need to be addressed in future work. Although the circuit repetitons are highly parallelizable, running calculations on ~1014 QPUs in parallel is prohibitive with current hardware availability. The shot noise limit, nS ∝ δ−2, is a generic problem facing near-term algorithms, and has been frequently discussed in the context of VQE26,94. It may be possible to use classical shadows to reduce the measurement prefactor by exploiting the operator structure of the chemical Hamiltonian95, although the comparative measurement scaling with increasing system size is unknown. Alternatively, improvements in hardware fidelity may enable amplitude amplification teqhniques96 to provide a quadratic reduction in the measurement scaling, to nS ∝ δ−1, for a tradeoff in the circuit depth.
Tensor networks have played a central role in the “dequantization” of quantum algorithms and simulations97,98, providing a richer understanding of the arguments for and against both near-term and fault-tolerant quantum advantage99. Here we have taken a complementary approach, identifying tensor network contractions that are thought to be classically hard and exploiting their efficient quantum evaluation as algorithmic components. This design philosophy yields quantum algorithms that are not limited by classical contraction constraints and are by construction, difficult to spoof with tensor networks. In the domain of quantum chemical simulation, this provides new tools to extend tensor network descriptions of chemical systems to higher connective geometries and to study highly entangled states of chemical interest with low-depth quantum circuits. We argue that far from being antithetical to quantum computing, the tensor network paradigm is compatible with and actively beneficial for emerging quantum algorithms, enabling a virtuous cycle between the development of both quantum-inspired classical methods and classical-inspired quantum methods. We anticipate the techniques developed in this work finding applications beyond quantum chemistry, in fields such as condensed matter physics and quantum machine learning.
Methods
Numerical details
We have presented above the results of numerical simulations using the ITensor Julia library100 for the classical tensor network parts of the algorithm, with conversion to a sparse matrix representation to evaluate the off-diagonal matrix elements. The code is made publicly available (see the section on “Code availability”). For numerical convenience, we have used spatial orbital sites with d = 4, which can be easily converted to the d = 2 representation for quantum circuit encoding by use of SVDs. The parameter counts were reduced by enforcing particle number and z-spin symmetry using a standard block-sparse representation86,87. An initial orbital basis was computed via restricted Hartree-Fock calculations in the PySCF Python package101,102,103, and an initial orbital ordering was selected using mutual information heuristics common in DMRG calculations93,104. The effect of shot noise on the quantum processor was emulated by adding random Gaussian perturbations to the elements of \({{\bf{H}}}^{{\prime} }\) and \({{\bf{S}}}^{{\prime} }\) with standard error δ. The algorithm was observed to reliably converge to chemical accuracy in the systems studied, almost always converging in the first run, and if not, then within two or three runs. We note that in rare cases, it was found to be beneficial to initialize the first RHF ordering at random instead of using the mutual information heuristic. The occurrence of these rare cases followed no discernible pattern, thus we attribute this to the inadequacy of the heuristic used, and we believe that a more robust starting guess for the RHF ordering could be developed. In these cases, when the random initial ordering was substituted, the algorithm would again typically converge in the first run, and if not, then within two or three runs.
The VQE-UCCSD benchmark was optimized via the L-BFGS algorithm105 with numerical gradients starting from zero amplitudes. While some reduction in the number of QPU calls for these benchmarking calculations may be possible by UCC parameter initialization with MP2 or projective coupled-cluster amplitudes106, and by use of analytic gradient calculations107, since our goal was to assess the feasibility and scalability of TNQE relative to a standard method, we have left a more detailed comparison against state-of-the-art VQE methods to future work.
Matrix element Hadamard test
Here, we assume the d = 2 representation so that N is equal to the number of system qubits under the Jordan-Wigner transformation. For the quantum evaluation of the matrix elements, we first require a unitary circuit to prepare each \(\left\vert {\phi }_{j}\right\rangle\) from the vacuum state in their respective orbital bases,
where \(\left\vert 0\right\rangle\) is understood to refer to the multi-qubit state \({\left\vert 0\right\rangle }^{\otimes N}\). We also require the Givens rotation circuits \({\hat{G}}_{ij}\) to rotate each state \(\left\vert {\phi }_{i}\right\rangle\) into the orbital basis of another state \(\left\vert {\phi }_{j}\right\rangle\). Finally we assume an efficient Pauli string decomposition for the observable \(\hat{H}\) in each orbital basis as in Equation (7). The matrix elements are expanded as a sum over expectation values of a set of unitary products with respect to the vacuum state,
which can be evaluated in parallel via standard Hadamard test108 circuits as shown in Fig. 7. This circuit makes a conditional application of \({\hat{U}}_{j}^{\dagger }{\hat{P}}_{\alpha }{\hat{G}}_{ij}{\hat{U}}_{i}\) with control from a single ancilla qubit. The particle number conserving operator \({\hat{G}}_{ij}\) acts trivially on the vacuum state, so does not require control. The ancilla qubit is measured in the z-basis at the end of the computation, and the real part of the expectation value is calculated from the probabilities
The imaginary component can also be obtained via a change of phase of the ancilla qubit, however, we note that the tensor elements in the TNQE ansatz can be restricted to real values without loss of generality, hence for real-valued observables there is no imaginary component of the expectation value. An overlap matrix element \(\langle {\phi }_{j}| {\hat{G}}_{ij}| {\phi }_{i}\rangle\) can be evaluated with a single Hadamard test circuit, omitting the Pauli string. For a number of shots nS the output of each Hadamard test circuit has variance ≤1/nS109. Consequently each overlap matrix element can be resolved to standard error δ with nS ≈ δ−2, and, following a standard optimal measurement allocation over the Pauli strings94, each Hamiltonian matrix element can be resolved up to standard error δ with
There are several approaches in the literature for reducing the number of Pauli terms, which will be important in the presence of gate noise110,111. We leave the incorporation of these methods to future work.
Circuit compilation
Here we break down the gate cost and layer depth for the disentangler-based implementation of the matrix element quantum circuits. The disentangler method first introduced by Ran79 works by truncating the target MPS to χ = 2 and then computing a staircase-like circuit of one- and two-qubit unitaries that map the truncated MPS to the all-zero state (the one-qubit gate can then be then merged into the last two-qubit gate). Applying these gates to the untruncated MPS results in a new MPS that is closer in fidelity to the all-zero state. Repeating this procedure D times results in a sequence of D disentangler layers that, when applied in reverse to the all-zero state, approximately prepares the original MPS up to some desired fidelity, as shown in Fig. 13. The fidelity can be further improved by numerically re-optimizing each unitary via a QR factorization of a black-box optimized matrix, similar to the strategies advocated in refs. 83,84.

The MPS on the left is approximately prepared on a quantum register via D layers of disentangler gates, shown here with D = 2.
Without loss of generality, the matrix product states used in the TNQE ansatz can be made entirely real-valued, from which it follows that each two-qubit disentangling gate U is a real-valued orthogonal transformation. We exploit this fact to reduce the prefactors in the CNOT gate count for each controlled disentangler. An orthogonal unitary U satisfies \(\det (U)=\pm 1\). Any orthogonal two-qubit gate with determinant +1 can be decomposed into two CNOT gates and six Ry gates via the Cartan decomposition112, depicted in Fig. 14a. Furthermore, for any orthogonal matrix with determinant +1 there exists an orthogonal matrix \(\sqrt{U}\) such that \({(\sqrt{U})}^{2}=U\) and \(\sqrt{U}{\sqrt{U}}^{T}={\sqrt{U}}^{T}\sqrt{U}={\mathbb{1}}\). By computing this matrix, we may apply controlled-U by a standard technique113, with one application each of \(\sqrt{U}\) and \({\sqrt{U}}^{T}\) and four additional CNOTs, as shown in Fig. 14b. Thus adding control to an arbitrary two-qubit orthogonal gate introduces a factor of two to the synthesis cost plus an additional four CNOT gates. Note in the case that \(\det (U)=-1\), we may instead compute \(\sqrt{-U}\), and after application of controlled-(−U) flip the phase of the ancilla \(\left\vert 1\right\rangle\) state by applying a Z gate to the ancilla qubit. We may then decompose each \(\sqrt{U}\) into six single-qubit Ry gates and two CNOT gates as before. We can do this regardless of the determinant of \(\sqrt{U}\) because we may equivalently decompose \(-\sqrt{U}\), as the global phase will cancel with that of \(-{\sqrt{U}}^{T}\). Therefore each controlled two-qubit rotation requires a total of eight CNOT gates with a layer depth of 14. Note that if one were to simply replace the CNOT gates in Fig. 14a with Toffoli gates, each would decompose into six CNOT gates114, for twelve CNOT gates in total. Therefore the implementation in Fig. 14b reduces the overall CNOT count by a third.

We can evaluate a matrix element with two controlled MPS preparation unitaries as in Fig. 7, each of which require (N − 1)D controlled two-qubit rotations, assuming one-all connectivity to the ancilla qubit. The \(\left(\begin{array}{c}N\\ 2\end{array}\right)\) Givens rotation gates in the orbital rotation circuit are also orthogonal and have determinant equal to + 1, and therefore can be implemented with two CNOT gates and five gate layers under the same two-qubit gate decomposition (Fig. 14a). Only three of the rotation unitaries contribute to the layer depth since the rest can be performed fully in parallel with the controlled disentanglers. We thus arrive at the Equations (13), (14), and (15).
Regulating the subspace condition number
When solving the generalized eigenvalue problem in Equation (18) (line 6 in Algorithm 1), the quantity of principal concern regarding numerical stability is the condition number of the expanded matrix \({{\bf{S}}}^{{\prime} }\). We can directly control this quantity by discarding one-hot states that have a high degree of linear dependence. Intuitively this representational flexibility is already present within the rest of the states in the subspace, so the discarded states are in some sense redundant. In fact the discarding step appears to be beneficial for convergence of the optimizer, possibly because it encourages each matrix product state to capture different features of the ground state. We formalize the linear dependence condition by measuring the squared norm of the projection of each of the one-hot states onto the subspace of the previous states. More concretely, let \({\hat{P}}^{\ddagger }\) be the projector onto the subset of the first jn − 1 one-hot states (the upper-left block of the expanded state space). Let S‡ denote the reduced overlap matrix consisting of the upper-left block of \({{\bf{S}}}^{{\prime} }\) over the rows and columns 1, …, jn − 1, and let \(\overrightarrow{s}\) be the reduced vector taken from the first 1, …, jn − 1 elements of the jn’th column of \({{\bf{S}}}^{{\prime} }\), i.e., \({s}_{im}\equiv \langle {\varphi }_{m}^{[i]}| {\varphi }_{n}^{[j]}\rangle\), for im = 1, …, jn − 1. Then the squared norm of the projection of \(\vert {\varphi }_{n}^{[j]}\rangle\) onto the upper-left subspace is given by
(see Supplementary Note 1.B). We discard the one-hot state \(\vert {\varphi }_{n}^{[j]}\rangle\) if one minus the squared norm is less than some set tolerance, e.g., 10−3. As a secondary check we also discard the one-hot state if the condition number of the new reduced overlap matrix including \(\vert {\varphi }_{n}^{[j]}\rangle\) is above a certain threshold. Since we have discarded the linearly dependent states in the upper-left block the matrix inverse should remain well-behaved (in practice we use the Moore-Penrose pseudoinverse with singular value tolerance \(\sqrt{\varepsilon }\parallel {{\bf{S}}}^{\ddagger }\parallel\) where ε is the machine epsilon and ∥ ⋅ ∥ is the operator 2-norm).
We can further control the solution of the generalized eigenvalue problem by thresholding the singular values of \({{\bf{S}}}^{{\prime} }\)88. We have implemented two common thresholding strategies known as projection and inversion. For both methods, we begin by computing
We filter the eigenvalues to keep only those greater than a chosen singular value tolerance, i.e. λ > ϵ. Then, by the projection method, we take the rectangular matrix of the remaining eigenvectors Uϵ and solve
Then the columns of the matrix product \({{\bf{U}}}_{\epsilon }^{\dagger }{{\bf{C}}}^{{\prime} }\) provide the approximate desired coefficients. By the inversion method, we remove the eigenvalues λ ≤ ϵ from Λ, and then solve
Letting \({\overrightarrow{c}}^{{\prime} }\) be the first column of \({{\bf{C}}}^{{\prime} }\), the approximate desired coefficients are then provided by \({\overrightarrow{c}}^{{\prime} }/\sqrt{{\overrightarrow{c}}^{{\prime} \dagger }{{\bf{S}}}^{{\prime} }{\overrightarrow{c}}^{{\prime} }}\). Using a combination of these measures we find that in practice the optimizer explores the space surrounding the ground state in a highly restricted manner, such that the more ill-conditioned regions of the non-orthogonal state space are largely avoided. This is backed up empirically by our finding that we can reliably converge the energy estimate with Gaussian noise in the overlap matrix elements on the order of 10−5.
Mitigating the energy penalty due to singular value truncation
In the final step of the generalized sweep algorithm (line 9 in Algorithm 1), after the orbital updates, the truncation error can still significantly and nontrivially affect the energy estimate of the ansatz. The optimal parameters of the two-site tensor after truncation are typically better than the previous parameters, however they may not be optimal for a pair of single-site tensors with a fixed singular value threshold. We mitigate this penalty due to truncation without growing the bond dimension by performing a sequence of alternating single-site decompositions at sites p and p + 1, starting from the two-site parameters following the initial truncation. This does not require any additional QPU calls, as all of the necessary information for this step is contained within the two-site expanded matrices \({{\bf{H}}}^{{\prime} }\) and \({{\bf{S}}}^{{\prime} }\). Instead, local classical tensor contractions are used to compute block transformation matrices (see Fig. 15), which are then applied to the blocks of \({{\bf{H}}}^{{\prime} }\) and \({{\bf{S}}}^{{\prime} }\) to give the subspace matrices for the single-site subspace expansions. This technique is explained in detail in Supplementary Note A.4.

a A tensor network to compute the elements of the d2χ2 × d2χ2 block transformation matrices {G[j]} corresponding to the local orbital rotation updates \(\hat{g}(\theta )\) in the basis of the two-site one-hot states of reference state j. b A tensor network to compute the elements of the dχ2 × d2χ2 isometries {T[j]} that, when applied to the blocks of the two-site expanded subspace matrices \({{\bf{H}}}^{{\prime} }\) and \({{\bf{S}}}^{{\prime} }\) (dim. Md2χ2), yield the blocks of the single-site expanded subspace matrices (dim. Mdχ2) (see Supplementary Note A.4 for a detailed explanation).
Finally, the parameter update is rejected if the new ground state energy estimate is higher than the previous estimate by a fixed tolerance, on the order of ~1 mHa. We find that in combination these measures can effectively mitigate any remaining convergence issues due to truncation.
Data availability
The numerical data presented in this study are available from the corresponding author upon reasonable request. The code used to generate all of the numerical data presented in this study is publicly available at https://github.com/oskar-leimkuhler/TNQE-Julia/.
Code availability
The code used to generate all of the numerical data presented in this study is publicly available at https://github.com/oskar-leimkuhler/TNQE-Julia/.
References
Hartree, D. R. The wave mechanics of an atom with a non-Coulomb central field. part I. theory and methods. Math. Proc. Camb. Philos. Soc. 24, 89–110 (1928).
Penrose, R. Applications of negative dimensional tensors. In Combinatorial Mathematics and its Applications (ed. Welsh, D. J. A.) 221–244 (Academic Press, 1971).
Biamonte, J. & Bergholm, V. Tensor networks in a nutshell. Preprint at. http://arxiv.org/abs/1708.00006 (2017).
Bridgeman, J. C. & Chubb, C. T. Hand-waving and interpretive dance: an introductory course on tensor networks. J. Phys. A Math. Theor. 50, 223001 (2017).
White, S. R. Density matrix formulation for quantum renormalization groups. Phys. Rev. Lett. 69, 2863–2866 (1992).
White, S. R. & Martin, R. L. Ab initio quantum chemistry using the density matrix renormalisation group. J. Chem. Phys. 110, 4127–4130 (1999).
Chan, G. K.-L. & Sharma, S. The density matrix renormalization group in quantum chemistry. Ann. Rev. Phys. Chem. 62, 465–481 (2011).
Chan, G. K.-L., Keselman, A., Nakatani, N., Li, Z. & White, S. R. Matrix product operators, matrix product states, and ab initio density matrix renormalization group algorithms. J. Chem. Phys. 145, 014102 (2016).
Baiardi, A. & Reiher, M. The density matrix renormalization group in chemistry and molecular physics: recent developments and new challenges. J. Chem. Phys. 152, 040903 (2020).
Eisert, J., Cramer, M. & Plenio, M. B. Colloquium: area laws for the entanglement entropy. Rev. Modern Phys. 82, 277–306 (2010).
Fannes, M., Nachtergaele, B. & Werner, R. F. Finitely correlated states on quantum spin chains. Commun. Math. Phys. 144, 443–490 (1992).
Klümper, A., Schadschneider, A. & Zittartz, J. Matrix product ground states for one-dimensional spin-1 quantum antiferromagnets. Europhys. Lett. 24, 293–297 (1993).
Sharma, S. & Chan, G. K.-L. Spin-adapted density matrix renormalization group algorithms for quantum chemistry. J. Chem. Phys. 136, 124121 (2012).
Sharma, S., Sivalingam, K., Neese, F. & Chan, G. K.-L. Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics. Nat. Chem. 6, 927–933 (2014).
Schuch, N., Wolf, M. M., Verstraete, F. & Cirac, J. I. Entropy scaling and simulability by matrix product states. Phys. Rev. Lett. 100, 030504 (2008).
Verstraete, F. & Cirac, J. I. Renormalization algorithms for quantum-many body systems in two and higher dimensions. Preprint at. https://arxiv.org/abs/cond-mat/0407066 (2004).
Haferkamp, J., Hangleiter, D., Eisert, J. & Gluza, M. Contracting projected entangled pair states is average-case hard. Phys. Rev. Res. 2, 013010 (2020).
Zaletel, M. P. & Pollmann, F. Isometric tensor network states in two dimensions. Phys. Rev. Lett. 124, 037201 (2020).
Feynman, R. P. Quantum mechanical computers. Found. Phys. 16, 507–531 (1986).
Nielsen, M. A. & Chuang, I. L.Quantum Computation and Quantum Information: 10th Anniversary Edition (Cambridge University Press, 2010).
Kitaev, A. Y. Quantum measurements and the abelian stabilizer problem. Preprint at https://arxiv.org/abs/quant-ph/9511026 (1995).
Aspuru-Guzik, A., Dutoi, A. D., Love, P. J. & Head-Gordon, M. Simulated quantum computation of molecular energies. Science 309, 1704–1707 (2005).
Su, Y., Berry, D. W., Wiebe, N., Rubin, N. & Babbush, R. Fault-tolerant quantum simulations of chemistry in first quantization. PRX Quantum 2, 040332 (2021).
Ding, Z. & Lin, L. Even shorter quantum circuit for phase estimation on early fault-tolerant quantum computers with applications to ground-state energy estimation. PRX Quantum 4, 020331 (2023).
Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 5, 4213 (2014).
Tilly, J. et al. The variational quantum eigensolver: a review of methods and best practices. Phys. Rep. 986, 1–128 (2022).
Grimsley, H. R., Economou, S. E., Barnes, E. & Mayhall, N. J. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nat. Commun. 10, 3007 (2019).
Ryabinkin, I. G., Yen, T.-C., Genin, S. N. & Izmaylov, A. F. Qubit coupled cluster method: a systematic approach to quantum chemistry on a quantum computer. J. Chem. Theory Comput. 14, 6317–6326 (2018).
Mizukami, W. et al. Orbital optimized unitary coupled cluster theory for quantum computer. Phys. Rev. Res. 2, 033421 (2020).
McClean, J. R., Boixo, S., Smelyanskiy, V. N., Babbush, R. & Neven, H. Barren plateaus in quantum neural network training landscapes. Nat. Commun. 9, 4812 (2018).
Baek, U. et al. Say no to optimization: a nonorthogonal quantum eigensolver. PRX Quantum 4, 030307 (2023).
Motta, M. et al. Low rank representations for quantum simulation of electronic structure. npj Quantum Inform. 7, 1–7 (2021).
Bauer, B., Bravyi, S., Motta, M. & Chan, G. K.-L. Quantum algorithms for quantum chemistry and quantum materials science. Chem. Rev. 120, 12685–12717 (2020).
Haghshenas, R., Gray, J., Potter, A. C. & Chan, G. K.-L. Variational power of quantum circuit tensor networks. Phys. Rev. X 12, 011047 (2022).
Kim, I. H. & Swingle, B. Robust entanglement renormalization on a noisy quantum computer. Preprint at http://arxiv.org/abs/1711.07500 (2017).
Huggins, W., Patel, P., Whaley, K. B. & Stoudenmire, E. M. Towards quantum machine learning with tensor networks. Quantum Sci. Technol. 4, 024001 (2019).
Borregaard, J., Christandl, M. & Stilck França, D. Noise-robust exploration of many-body quantum states on near-term quantum devices. npj Quantum Inf. 7, 1–6 (2021).
Jamet, F. et al. Anderson impurity solver integrating tensor network methods with quantum computing. APL Quantum 2, 016121 (2025).
Lubasch, M., Joo, J., Moinier, P., Kiffner, M. & Jaksch, D. Variational quantum algorithms for nonlinear problems. Phys. Rev. A 101, 010301 (2020).
Mc Keever, C. & Lubasch, M. Classically optimized hamiltonian simulation. Phys. Rev. Res. 5, 023146 (2023).
Huang, J. et al. Tensor-network-assisted variational quantum algorithm. Phys. Rev. A 108, 052407 (2023).
Watanabe, R., Fujii, K. & Ueda, H. Variational quantum eigensolver with embedded entanglement using a tensor-network ansatz. Phys. Rev. Res. 6, 023009 (2024).
Miao, Q. & Barthel, T. Quantum-classical eigensolver using multiscale entanglement renormalization. Phys. Rev. Res. 5, 033141 (2023).
Miao, Q. & Barthel, T. Convergence and quantum advantage of Trotterized MERA for strongly-correlated systems. Quantum 9, 1631 (2025).
McClean, J. R., Kimchi-Schwartz, M. E., Carter, J. & de Jong, W. A. Hybrid quantum-classical hierarchy for mitigation of decoherence and determination of excited states. Phys. Rev. A 95, 042308 (2017).
Huggins, W. J., Lee, J., Baek, U., O’Gorman, B. & Whaley, K. B. A non-orthogonal variational quantum eigensolver. New J. Phys. 22, 073009 (2020).
Cortes, C. L. & Gray, S. K. Quantum krylov subspace algorithms for ground- and excited-state energy estimation. Phys. Rev. A 105, 022417 (2022).
Francis, A., Agrawal, A. A., Howard, J. H., Kökcü, E. & Kemper, A. F. Subspace diagonalization on quantum computers using eigenvector continuation. Phys. Rev. Res 5, 023067 (2023).
Motta, M. et al. Subspace methods for electronic structure simulations on quantum computers. Electronic Struct. 6, 013001 (2024).
Fan, Y., Liu, J., Li, Z. & Yang, J. Quantum circuit matrix product state ansatz for large-scale simulations of molecules. J. Chem. Theory Comput. 19, 5407–5417 (2023).
Meth, M. et al. Probing phases of quantum matter with an ion-trap tensor-network quantum eigensolver. Phys. Rev. X 12, 041035 (2022).
Rudolph, M. S. et al. Synergistic pretraining of parametrized quantum circuits via tensor networks. Nat. Commun. 14, 8367 (2023).
Khan, A., Clark, B. K. & Tubman, N. M. Pre-optimizing variational quantum eigensolvers with tensor networks. New J. Phys. 25, 103027 (2023).
Miao, Q. & Barthel, T. Isometric tensor network optimization for extensive hamiltonians is free of barren plateaus. Phys. Rev. A 109, L050402 (2024).
Barthel, T. & Miao, Q. Absence of barren plateaus and scaling of gradients in the energy optimization of isometric tensor network states. Commun. Math. Phys. 406, 86 (2025).
Cervero Martín, E., Plekhanov, K. & Lubasch, M. Barren plateaus in quantum tensor network optimization. Quantum 7, 974 (2023).
Helgaker, T., Jørgensen, P. & Olsen, J.Second Quantization, Ch. 1, 1–33 (John Wiley & Sons, Ltd, 2000).
Oseledets, I. V. Tensor-train decomposition. SIAM J. Sci. Comput. 33, 2295–2317 (2011).
Vidal, G. Efficient classical simulation of slightly entangled quantum computations. Phys. Rev. Lett. 91, 147902 (2003).
Schuch, N., Wolf, M. M., Verstraete, F. & Cirac, J. I. Computational complexity of projected entangled pair states. Phys. Rev. Lett. 98, 140506 (2007).
Arad, I. & Landau, Z. Quantum computation and the evaluation of tensor networks. SIAM J. Comput. 39, 3089–3121 (2010).
Jordan, P. & Wigner, E. Über das paulische Äquivalenzverbot. Z. Phys. 47, 631–651 (1928).
Sundstrom, E. J. & Head-Gordon, M. Non-orthogonal configuration interaction for the calculation of multielectron excited states. J. Chem. Phys. 140, 114103 (2014).
Thom, A. J. W. & Head-Gordon, M. Hartree–Fock solutions as a quasidiabatic basis for nonorthogonal configuration interaction. J. Chem. Phys. 131, 124113 (2009).
Krumnow, C., Veis, L., Legeza, O. & Eisert, J. Fermionic orbital optimization in tensor network states. Phys. Rev. Lett. 117, 210402 (2016).
Zgid, D. & Nooijen, M. The density matrix renormalization group self-consistent field method: orbital optimization with the density matrix renormalization group method in the active space. J. Chem. Phys. 128, 144116 (2008).
Ghosh, D., Hachmann, J., Yanai, T. & Chan, G. K.-L. Orbital optimization in the density matrix renormalization group, with applications to polyenes and β-carotene. J. Chem. Phys. 128, 144117 (2008).
Verstraete, F., Cirac, J. I. & Latorre, J. I. Quantum circuits for strongly correlated quantum systems. Phys. Rev. A 79, 032316 (2009).
Banerjee, I., Richards, D. & Shinkar, I. Sorting networks on restricted topologies. in SOFSEM 2019: Theory and Practice of Computer Science (eds. Catania, B., Královič, R., Nawrocki, J. & Pighizzini, G.) 54–66 (Springer International Publishing, 2019).
Knuth, D. E.The Art of Computer Programming, vol. 3: Sorting and Searching (Addison-Wesley, 1998).
Friend, E. H. Sorting on electronic computer systems. J. ACM 3, 134–168 (1956).
Qian, X., Huang, J. & Qin, M. Augmenting density matrix renormalization group with Clifford circuits. Phys. Rev. Lett. 133, 190402 (2024).
Lami, G., Haug, T. & Nardis, J. D. Quantum state designs with Clifford enhanced matrix product states. PRX Quantum 6, 010345 (2025).
Kivlichan, I. D. et al. Quantum simulation of electronic structure with linear depth and connectivity. Phys. Rev. Lett. 120, 110501 (2018).
Thouless, D. Stability conditions and nuclear rotations in the Hartree-Fock theory. Nuclear Phys. 21, 225–232 (1960).
Li, W., Ren, J., Yang, H. & Shuai, Z. On the fly swapping algorithm for ordering of degrees of freedom in density matrix renormalization group. J. Phys. Condens. Matter 34, 254003 (2022).
Schön, C., Solano, E., Verstraete, F., Cirac, J. I. & Wolf, M. M. Sequential generation of entangled multiqubit states. Phys. Rev. Lett. 95, 110503 (2005).
Fomichev, S. et al. Initial state preparation for quantum chemistry on quantum computers. PRX Quantum 5, 040339 (2024).
Ran, S.-J. Encoding of matrix product states into quantum circuits of one- and two-qubit gates. Phys. Rev. A 101, 032310 (2020).
Malz, D., Styliaris, G., Wei, Z.-Y. & Cirac, J. I. Preparation of matrix product states with log-depth quantum circuits. Phys. Rev. Lett. 132 (2024).
Shende, V., Bullock, S. & Markov, I. Synthesis of quantum-logic circuits. IEEE Trans. Comput.-Aided Des. Integr. Circuits Syst. 25, 1000–1010 (2006).
Low, G. H., Kliuchnikov, V. & Schaeffer, L. Trading t gates for dirty qubits in state preparation and unitary synthesis. Quantum 8, 1375 (2024).
Ben-Dov, M., Shnaiderov, D., Makmal, A. & Dalla Torre, E. G. Approximate encoding of quantum states using shallow circuits. npj Quantum Inf. 10 (2024).
Rudolph, M. S., Chen, J., Miller, J., Acharya, A. & Perdomo-Ortiz, A. Decomposition of matrix product states into shallow quantum circuits. Quantum Sci. Technol. 9, 015012 (2023).
Harris, D. & Harris, S.Digital Design and Computer Architecture (Morgan Kaufmann, 2010).
Singh, S., Pfeifer, R. N. C. & Vidal, G. Tensor network decompositions in the presence of a global symmetry. Phys. Rev. A 82, 050301 (2010).
Singh, S., Pfeifer, R. N. C. & Vidal, G. Tensor network states and algorithms in the presence of a global U (1) symmetry. Phys. Rev. B 83, 115125 (2011).
Epperly, E. N., Lin, L. & Nakatsukasa, Y. A theory of quantum subspace diagonalization. SIAM J. Matrix Anal. Appl. 43, 1263–1290 (2022).
Li, Z. & Chan, G. K.-L. Spin-projected matrix product states: versatile tool for strongly correlated systems. J. Chem. Theory Comput. 13, 2681–2695 (2017).
Yordanov, Y. S., Arvidsson-Shukur, D. R. M. & Barnes, C. H. W. Efficient quantum circuits for quantum computational chemistry. Phys. Rev. A 102, 062612 (2020).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Askerka, M., Brudvig, G. W. & Batista, V. S. The O2-evolving complex of photosystem II: recent insights from quantum mechanics/molecular mechanics (QM/MM), extended X-ray absorption fine structure (EXAFS), and femtosecond X-ray crystallography data. Acc. Chem. Res. 50, 41–48 (2017).
Rissler, J., Noack, R. M. & White, S. R. Measuring orbital interaction using quantum information theory. Chem. Phys. 323, 519–531 (2006).
Wecker, D., Hastings, M. B. & Troyer, M. Progress towards practical quantum variational algorithms. Phys. Rev. A 92, 042303 (2015).
Huang, H.-Y., Kueng, R. & Preskill, J. Predicting many properties of a quantum system from very few measurements. Nat. Phys. 16, 1050–1057 (2020).
Brassard, G., Høyer, P., Mosca, M. & Tapp, A. Quantum amplitude amplification and estimation. In Quantum Computation and Information (eds Lomonaco, S. J. Jr. & Brandt, H. E.) 53–74 (American Mathematical Society, 2002).
Begušić, T., Gray, J. & Chan, G. K.-L. Fast and converged classical simulations of evidence for the utility of quantum computing before fault tolerance. Sci. Adv. 10, eadk4321 (2024).
Shin, S., Teo, Y. S. & Jeong, H. Dequantizing quantum machine learning models using tensor networks. Phys. Rev. Res. 6, 023218 (2024).
Lee, S. et al. Evaluating the evidence for exponential quantum advantage in ground-state quantum chemistry. Nat. Commun. 14, 1952 (2023).
Fishman, M., White, S. R. & Stoudenmire, E. M. The ITensor Software library for tensor network calculations. SciPost Phys. Codebases 4 (2022).
Sun, Q. et al. Recent developments in the PySCF program package. J. Chem. Phys. 153, 024109 (2020).
Sun, Q. et al. Pyscf: the Python-based simulations of chemistry framework. WIREs Comput. Molecular Sci. 8, e1340 (2018).
Sun, Q. Libcint: an efficient general integral library for Gaussian basis functions. J. Comput. Chem. 36, 1664–1671 (2015).
Ali, M. On the ordering of sites in the density matrix renormalization group using quantum mutual information. Preprint at. http://arxiv.org/abs/2103.01111 (2021).
Liu, D. C. & Nocedal, J. On the limited memory BFGS method for large scale optimization. Math. Program. 45, 503–528 (1989).
Hirsbrunner, M. R., Chamaki, D., Mullinax, J. W. & Tubman, N. M. Beyond MP2 initialization for unitary coupled cluster quantum circuits. Quantum 8, 1538 (2024).
Izmaylov, A. F., Lang, R. A. & Yen, T.-C. Analytic gradients in variational quantum algorithms: algebraic extensions of the parameter-shift rule to general unitary transformations. Phys. Rev. A 104, 062443 (2021).
Cleve, R., Ekert, A., Macchiavello, C. & Mosca, M. Quantum algorithms revisited. Proc. R. Soc. Lond. A Math. Phys. Eng. Sci. 454, 339–354 (1998).
Polla, S., Anselmetti, G.-L. R. & O’Brien, T. E. Optimizing the information extracted by a single qubit measurement. Phys. Rev. A 108, 012403 (2023).
Huggins, W. J. et al. Efficient and noise resilient measurements for quantum chemistry on near-term quantum computers. npj Quantum Inf. 7, 23 (2021).
Inoue, W. et al. Almost optimal measurement scheduling of molecular Hamiltonian via finite projective plane. Phys. Rev. Res. 6, 013096 (2024).
Wei, H.-R. & Di, Y.-M. Decomposition of orthogonal matrix and synthesis of two-qubit and three-qubit orthogonal gates. Quantum Inf. Comput. 12, 262–270 (2012).
Zickert, F. Hands-on Quantum Machine Learning with Python: Volume 1: Get Started (Amazon Digital Services LLC—KDP Print US, 2021).
Shende, V. V. & Markov, I. L. On the CNOT-cost of TOFFOLI gates. Quantum Inf. Comput. 9, 461–486 (2009).
Acknowledgements
We thank Unpil Baek, William J. Huggins, and Torin F. Stetina for many invaluable discussions. This work was supported in part by the NSF QLCI program through grant number QMA-2016345, in part by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing Research under Award Number DE-SC0025526, and as part of a joint development agreement between UC Berkeley and Dow.
Author information
Authors and Affiliations
Contributions
O.L. proposed the project, developed the algorithm, implemented and performed the classical numerical simulations, worked out the resource estimates, and wrote the initial draft of the paper. K.B.W. participated in discussions, provided guidance throughout the project, and contributed to the writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leimkuhler, O., Whaley, K.B. A quantum eigenvalue solver based on tensor networks. npj Quantum Inf 11, 184 (2025). https://doi.org/10.1038/s41534-025-01128-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41534-025-01128-4
