
Abstract
Crimean-Congo hemorrhagic fever virus (CCHFV) is a tick-borne virus of the Orthonairovirus genus, Nairoviridae family, that causes severe febrile hemorrhagic disease in humans with a case fatality rate ranging from approximately 3-30%. This zoonotic pathogen is distributed across a broad geographic area spanning Asia, Europe, and Africa. Despite its significant public health threat and outbreak potential, no licensed vaccines are available. In this study, we developed and systematically assessed the immunogenicity and protective efficacy of mRNA vaccines encoding either CCHFV nucleoprotein (NP) or glycoprotein precursor (GPC) in mouse models. Vaccination with the NP-encoding mRNA alone provided complete protection against lethal cross-genotype CCHFV challenge. Moreover, combined vaccination with both the NP and GPC mRNAs elicited robust immune responses and conferred protection against CCHFV infection. Notably, a single-dose immunization with 2 μg mRNA-NP was sufficient to confer protection against lethal challenge. Furthermore, the passive transfer of NP-immune serum provided partial protection, supporting the role of NP-specific antibodies in mediating protection. Overall, these mRNA vaccines demonstrate protective efficacy against CCHFV, with combined antigenic protection and dose-sparing potential, highlighting their potential for outbreak preparedness and further clinical development.
Similar content being viewed by others


Introduction
Crimean-Congo hemorrhagic fever (CCHF) is a tick-borne disease with high fatality rates in humans ranging from approximately 3–30%, and it is endemic across Asia, Europe, Africa, and the Middle East1,2. The causative agent, Crimean-Congo hemorrhagic fever virus (CCHFV), is a negative-sense, enveloped RNA virus that is classified within the genus Orthonairovirus, family Nairoviridae in the family genus Orthonairovirus, family Nairoviridae3,4,5,6 and has a tri-segmented genome. CCHFV is transmitted primarily by ticks of the genus Hyalomma within the Ixodidae family3. In humans, CCHF infection occurs most commonly in agricultural workers through tick bites, as well as in slaughterhouse employees and healthcare workers via exposure to blood, tissues, or bodily fluids from infected animals or patients, respectively3. The disease manifests with highly variable clinical outcomes, spanning from silent infections to critical and potentially lethal illness, progressing through incubation, pre-hemorrhagic, hemorrhagic, and convalescent phases7. Initial symptoms include a nonspecific febrile syndrome with acute onset of fever, headache, dizziness, and myalgia8,9. The hemorrhagic stage is characterized by extensive ecchymoses and uncontrolled multiorgan hemorrhage1. Owing to its endemicity, annual case reports, and a lack of licensed vaccines or effective therapies, CCHFV has been recognized by the World Health Organization (WHO) as a high-priority pathogen requiring the development of effective antiviral strategies10.
The genome of CCHFV consists of small (S), medium (M), and large (L) RNA segments encoding the viral nucleoprotein (NP), glycoprotein precursor (GPC), and RNA-dependent RNA polymerase (RdRp), respectively11. Regarding vaccine development, NP and GPC have been prioritized as key targets because of their essential roles in viral replication and entry. Several CCHFV vaccine candidates targeting these antigens have been evaluated in rodent and nonhuman primate models, with reported protective efficacies ranging from 0% to 100%12,13,14,15. While GPC serves as a primary immunogen targeted by both neutralizing and non-neutralizing antibodies, NP is also capable of eliciting humoral responses. In addition, both antigens can stimulate T-cell immunity, and the relatively high conservation of NP among various CCHFV strains makes it particularly suitable for development of vaccines that can mediate protection in against multiple genotypes. These immunogenic and structural characteristics make NP and GPC promising targets for vaccine development. Compared with traditional vaccines, mRNA vaccines provide several advantages, including a robust immune response, rapid development, design flexibility, and improved safety, as they do not involve live pathogens or genomic integration risks16. Currently, various mRNA vaccines targeting infectious diseases, including SARS-CoV-217, influenza virus18, respiratory syncytial virus (RSV)19,20, rabies virus21,22, mpox virus23, and Zika virus24,25, have either been approved for use or are being studied in clinical trials. This highlights the promise of mRNA vaccines in combating emerging epidemics and acute outbreaks.
In this study, we developed two mRNA vaccines encoding the full-length NP (mRNA-NP) and GPC (mRNA-GPC) of CCHFV, each of which are encapsulated separately in lipid nanoparticles (LNPs) to increase stability and delivery. We evaluated the immunogenicity of mRNA-NP, mRNA-GPC, and mRNA-NP + mRNA-GPC by assessing CCHFV-specific antibody and T-cell responses and subsequently evaluated their protective efficacy in a lethal Ifnar1−/− (C57BL/6JGpt background) mouse challenge model. Our results revealed that a two-dose regimen of mRNA-NP or mRNA-NP + mRNA-GPC conferred complete protection against lethal cross-genotype CCHFV challenge, and even a single dose of as low as 2 μg mRNA-NP resulted in 100% survival. This protection was attributed primarily to NP-specific immunity, as mRNA-NP alone provides complete survival. However, combined immunization with mRNA-NP and mRNA-GPC reduced the proportion of viral RNA positivity in tissues compared with mRNA-NP alone, especially at low dose, highlighting a complementary role for GPC in enhancing protective efficacy. Additionally, passive transfer of serum from mRNA-NP-immunized mice conferred partial protection (33.33%) to naïve mice, indicating a role for NP-specific antibodies. These findings support the potential of mRNA vaccines targeting NP and GPC to elicit protective immune responses and significantly improve survival in a lethal model of CCHFV infection.
Results
The mRNA vaccination elicits robust B- and T-cell responses in mice
To systematically evaluate the CCHFV-specific immunogenicity of the mRNA vaccine candidates, C57BL/6 J or Ifnar1−/− mice (n = 6 per group, aged 6‒8 weeks) were divided into seven groups and immunized (Fig. 1A‒B). The mice were subcutaneously (S.C.) injected with either mRNA-NP (1 or 5 μg), mRNA-GPC (1 or 5 μg), or mRNA-NP + mRNA-GPC (1 μg of each, totaling 2 μg; or 5 μg of each, totaling 10 μg), with empty LNPs serving as controls. All the groups were subjected to a prime-boost immunization regimen, receiving the second vaccination with the same dose 21 days later.

A Experimental scheme. C57BL/6 J and Ifnar1−/− mice received a prime dose followed by a booster three weeks later. Blood and spleens were collected at different time points to assess humoral and cellular immune responses. B Study design. A two-dose regimen specifying antigen composition, dosage, and administration route. C, D CCHFV-specific IgG responses. NP-specific (C) and Gc-specific (D) IgG levels in C57BL/6 J mice were quantified by ELISA. E, F Pseudovirus neutralization. Serum collected two weeks post-boost from C57BL/6 J (E) and Ifnar1−/− (F) mice was tested for neutralization against pseudotyped CCHFV (*GPC-VSVΔG/GFP). Neutralization efficacy was determined by quantifying GFP-expressing cells, and IC50 values were calculated from serum dilution curves and presented on a logarithmic scale. G, H Live virus neutralization. Serum from C57BL/6 J (G) and Ifnar1−/− (H) mice was tested for neutralization against live CCHFV YL16070 in Vero E6 cells. IC50 values were calculated from serum dilution curves based on viral inhibition, expressed on a logarithmic scale. I–K T-cell responses. IFN-γ-secreting splenocytes in C57BL/6 J mice immunized with mRNA-NP (I), mRNA-GPC (J), or mRNA-NP + mRNA-GPC (K) were quantified by ELISpot. Data represent cumulative spot-forming cells (SFCs) against the NP (I), GPC (J), and NP + GPC (K) peptide pools after background subtraction (DMSO-only controls), normalized to 106 splenocytes. Data are shown as mean ± SEM. Statistical significance for (I) was determined by one-way ANOVA with Tukey’s multiple comparisons test. Statistical significance for (E–H, J, and K) was determined by Kruskal–Wallis test followed by Dunn’s multiple comparisons test. Exact p values are shown.
Humoral immune responses were assessed using enzyme-linked immunosorbent assays (ELISAs) and virus neutralization assays. The ELISA results revealed that all vaccine regimens effectively elicited antigen-specific IgG responses that targeted their respective immunogens (NP or GPC). Booster immunization significantly increased antibody titers, which peaked between 5–7 weeks post-priming and remained detectable for up to 38 weeks (Fig. 1C‒D). High-dose (5 μg) regimens induced significantly more robust and sustained antibody responses than low-dose (1 μg) regimens, as evidenced by higher NP-specific IgG titers at weeks 3, 6, 7, 23, and 38, and higher Gc-specific IgG titers at weeks 3, 4, 11, and 23 (Fig. 1C, D; Supplementary Data 1). Neutralizing antibody responses were assessed using both pseudovirus and live virus neutralization assays in C57BL/6 and Ifnar1−/− mice. Pseudovirus neutralization assays demonstrated that mRNA-GPC and mRNA-GPC + mRNA-NP vaccination elicited neutralizing antibodies in a dose-dependent manner, with higher titers in the 5 μg-vaccinated groups than in the 1 μg-receiving groups (Fig. 1E, F). Live virus neutralization assays against cross-genotype CCHFV strains (YL16070 and IbAr10200) revealed enhanced neutralization at the higher vaccine dose (5 μg), despite the vaccine being designed on the basis of the Turkey strain (Fig. 1G, H; Supplementary Fig. 2A, B). These findings indicate that mRNA-GPC vaccination elicits potent neutralizing antibody responses capable of cross-reacting with genetically distinct CCHFV strains that exhibit significant glycoprotein sequence divergence from the vaccine strain. Based on S-segment phylogeny, the vaccine sequence from strain Turkey (Europe 1 lineage) and neutralization test using strains YL16070 (Asia 2 lineage) and IbAr10200 (Africa 3 lineage) represent distinct CCHFV lineages, with GPC amino acid identities of approximately 74.9% (Turkey vs. YL16070) and 84.5% (Turkey vs. IbAr10200), respectively26,27.
The T-cell responses to vaccination were assessed via interferon-γ (IFN-γ) ELISpot assays using splenocytes collected 7 days after booster immunization in each group (Fig. 1A). Compared with the LNP control mice, a statistically significant increase in spot-forming cells (SFCs) was observed in the mRNA-NP (1 μg or 5 μg) and mRNA-GPC (1 μg or 5 μg) groups in response to the NP- or GPC-specific peptide pools, respectively (Fig. 1I‒K). In the mRNA-NP + mRNA-GPC groups, both the 1 μg and 5 μg dose groups presented significantly increased IFN-γ-positive SFCs compared with the NP, GPC, and NP + GPC peptide pools, except for the 1 μg dose group, which did not show a statistically significant increase in response to the NP peptide pools (Fig. 1K). Taken together, these data demonstrate that mRNA vaccination induces a robust antigen-specific cellular immune response.
Two-dose mRNA vaccination protects Ifnar1−/− mice from cross-genotype CCHFV infection
We next evaluated the protective efficacy of the vaccine candidates against lethal cross-genotype challenge with the CCHFV strain YL16070 in vivo. Comparative sequence analysis revealed antigenic divergence between the vaccine reference strain (Turkey) and the challenge isolate YL16070, with 96.06% amino acid identity in the NP and 74.93% in the GPC. To assess cross-protection, Ifnar1−/− mice (n = 12 per group) were subjected to a subcutaneous prime-boost immunization regimen with a 21-day interval between the two doses (Fig. 1B). Two weeks after the booster dose, the mice were intraperitoneally inoculated with a lethal dose of CCHFV YL16070 (3,000 TCID50). Of the twelve mice in each group, six were monitored daily for survival and body weight changes over a 14-day period, while the remaining six were sacrificed at 5 days post infection (d.p.i.) for viral load quantification and histopathology analysis (Fig. 2A). Following infection, the LNP control mice began losing body weight at 2 dpi and reached humane endpoints between 4–5 dpi (Fig. 2B–G). In contrast, mice subjected to either the high-dose or low-dose regimen of mRNA-NP or mRNA-NP + mRNA-GPC resulted in complete (100%) survival throughout the 14-day observation period (Fig. 2B, D), with vaccinated mice exhibiting no obvious clinical signs and maintaining relatively stable body weights after CCHFV infection (Fig. 2E, G). In the high-dose (5 μg) mRNA-GPC group, 33.33% (2/6) of the mice died from infection between 5–7 dpi, whereas the remaining 66.67% (4/6) survived. The surviving mice exhibited mild, transient weight loss (2.35–4.53%) between 4–7 dpi and fully recovered by 8–14 dpi (Fig. 2F). In contrast, the low-dose (1 μg) mRNA-GPC group presented reduced protective efficacy, with 83.33% (5/6) mortality occurring at 5–7 dpi (Fig. 2C). While the survival difference between dose groups was not statistically significant, both doses conferred significant protection compared to the LNP control (p = 0.0178 and p = 0.0034, respectively), and the higher dose achieved greater survival. Collectively, these findings demonstrate that mRNA-NP immunization elicits protective immunity against clinical disease progression in response to lethal cross-genotype CCHFV challenge.

A Study schedule. Ifnar1−/− mice (n = 12 per vaccination group) received a subcutaneous prime-boost immunization with a 21-day interval. Two weeks after the post-booster, the mice were intraperitoneally challenged with 3000 TCID50 of CCHFV and divided into two cohorts (n = 6 each). One cohort was monitored for 14-day survival and body weight, while the rest was euthanized at 5 dpi for viral load and pathogenesis analysis. B–D Survival analysis of mice immunized with mRNA-NP (B), mRNA-GPC (C), and mRNA-NP + mRNA-GPC (D). Statistical significance was determined using a Log-rank (Mantel-Cox) test. E–G Body weight changes in mice receiving mRNA-NP (E), mRNA-GPC (F), or mRNA-NP + mRNA-GPC (G). H-J. Viremia. Viral RNA loads in blood were quantified by qRT-PCR at 5 dpi in mice immunized with mRNA-NP (H), mRNA-GPC (I), or mRNA-NP + mRNA-GPC (J). K–M. Tissue viral loads. Viral RNA loads in the liver and spleen were measured by qRT-PCR at 5 dpi in mice receiving mRNA-NP (K), mRNA-GPC (L), or mRNA-NP + mRNA-GPC (M). Data are presented as the mean ± SEM. The dashed line indicates the limit of detection. Statistical significance for (I) was determined by one-way ANOVA with Tukey’s multiple comparisons test. Statistical significance for (H, J, K–M) was determined by Kruskal–Wallis test followed by Dunn’s multiple comparisons test. Exact p values are shown.
Viral RNA loads in the blood, liver, and spleen of the 5 dpi euthanized mice were subsequently quantified. The data revealed that CCHFV RNA was undetectable in the blood of all the mice vaccinated with mRNA-NP or mRNA-NP + mRNA-GPC, with significantly lower viral loads than those of the LNP controls (Fig. 2H, J). In contrast, viral RNA was detected in the blood of 5/6 mice in the low-dose mRNA-GPC group and 3/6 in the high-dose group, although both groups presented significantly lower viral loads than the control group (Fig. 2I). The results of tissue viral RNA quantification revealed a similar trend. In the high-dose mRNA-GPC group, all recipients (6/6) presented high viral loads in the liver, and viral RNA was detectable in the spleens of 50% (3/6) of these mice (Fig. 2L). The low-dose mRNA-GPC group presented relatively high viral loads in both the liver and spleen, indicating insufficient suppression of viral replication in these tissues. In contrast, mRNA-NP vaccination led to markedly reduced viral RNA levels in both dose groups (Fig. 2K). High-dose recipients presented no detectable viral RNA in either the liver or spleen, whereas low-dose recipients presented residual viral RNA in the liver (3/6) but undetectable levels in the spleen (0/6) (Fig. 2K). Notably, both the high- and low-dose mRNA-NP + mRNA-GPC regimens resulted in viral RNA levels reduced to below the detection limit in the liver and spleen (Fig. 2M). These findings suggest that while mRNA-GPC vaccination alone provides only partial control of CCHFV replication (Fig. 2L), mRNA-NP vaccination provides robust suppression of viral loads (Fig. 2K). Furthermore, the combination of mRNA-NP and mRNA-GPC further reduced the frequency of detectable viral RNA in liver tissue (3/6 in mRNA-NP vs. 0/6 in mRNA-NP + mRNA-GPC group; Fig. 2K, M), although this difference did not reach statistically significant.
To determine whether the observed reduction in viral loads correlated with decreased tissue damage, histopathological examinations and immunohistochemistry (IHC) staining for CCHFV NP antigen were performed on liver and spleen samples. Consistent with the viral load results, animals vaccinated with mRNA-NP or mRNA-NP + mRNA-GPC presented little to no histopathology signs of infection, with no detectable viral antigen. The only exception was mild inflammatory infiltration in the livers of the low-dose mRNA-NP group (Fig. 3A‒D). In contrast, LNP control and mRNA-GPC-vaccinated mice presented obvious liver and spleen pathology at 5 dpi. Liver histopathology revealed hepatocellular necrosis, inflammatory cell infiltration, steatosis, and edema (Fig. 3A), accompanied by strong viral antigen positivity via IHC staining (Fig. 3B). Similarly, spleen sections exhibited disrupted splenic nodule architecture, reduction of small lymphocytes within white pulp, infiltration of a small number of neutrophils, and hemosiderin deposition (Fig. 3C). Viral antigens were also detected in the spleens, with a lower antigen-positive rate identified in the high-dose mRNA-GPC group than in the low-dose and LNP control groups, which was consistent with the viral load data (Fig. 3D). These findings indicate that mRNA-NP + mRNA-GPC vaccination effectively suppresses viral replication, significantly alleviates liver and spleen pathology, and provides robust protection against lethal cross-genotype CCHFV challenge.

A Hematoxylin and eosin (HE) staining of the liver. Severe liver pathology was observed in LNP control and mRNA-GPC groups, while the mRNA-NP and mRNA-NP + mRNA-GPC groups exhibited mild inflammatory infiltration. Red arrows indicate inflammatory cell infiltration, black arrows indicate hepatocellular necrosis, blue arrows indicate steatosis, and yellow arrows indicate edema. B Immunohistochemistry (IHC) analysis of the liver. High levels of CCHFV antigen were detected in non-immunized control and mRNA-GPC group, whereas little or no antigen was detected in the livers from mRNA-NP or mRNA-NP + mRNA-GPC. C HE staining of spleen. Severe pathological damage was observed in LNP control and mRNA-GPC groups, whereas the mRNA-NP and mRNA-NP + mRNA-GPC groups exhibited minimal histopathological abnormalities. Orange arrows indicate neutrophil infiltration, green arrows indicate hemosiderin deposition, and white arrows indicate reduced small lymphocytes in the white pulp. D IHC analysis of the spleen. High levels of CCHFV antigen were detected in control mice, with moderate levels in the mRNA-GPC group. In contrast, little or no antigen was detected in the spleens from mRNA-NP or mRNA-NP + mRNA-GPC. Scale bars are shown at 100 μm for large lower images, and 20 μm scale bars (inset) for magnified rectangle higher power images.
Prime-only mRNA vaccination protects against cross-genotype CCHFV infection
To further explore the correlation between the immunization regimen, RNA dosage, and protective effectiveness against cross-genotype CCHFV infection, the mice received a single immunization with 10, 5, or 2 μg mRNA and were challenged with 3000 TCID50 CCHFV YL16070 five weeks later (Fig. 4A, B). Serum samples were collected up to 35 days postvaccination, and ELISA results revealed that mRNA-NP and mRNA-NP + mRNA-GPC vaccination induced NP-specific IgG antibody titers ranging from 104–105 (Fig. 4C). The challenge results revealed that all vaccinated mice achieved 100% survival (Fig. 4D). Although the 2 μg mRNA-NP group experienced slight weight loss from 4–8 dpi, body weight began to recover by 9 dpi (Fig. 4E). Viral RNA quantification at 5 dpi revealed significantly reduced viral loads in the blood, livers, and spleens of all immunized groups (Fig. 4F‒H), indicating that a single low-dose immunization effectively suppresses CCHFV replication. Among the regimens tested, 10 μg mRNA-NP + 10 μg mRNA-GPC resulted in the most effective viral suppression, with undetectable levels of viral RNA observed in both blood and tissues. The 10 μg mRNA-NP and 5 μg mRNA-NP + 5 μg mRNA-GPC groups also demonstrated low proportions of samples with detectable viral RNA (1/6 or 2/6, Fig. 4F‒H). In contrast, the lower-dose groups (2 μg mRNA-NP, 1 μg mRNA-NP + 1 μg mRNA-GPC, and 5 μg mRNA-NP) presented moderate reductions in viral load. Notably, viral RNA levels in the liver at 5 dpi were significantly lower in the 10 μg mRNA-NP + 10 μg mRNA-GPC group than in the 2 μg mRNA-NP group (p = 0.0218, Fig. 4F). By 14 dpi, viral RNA was nearly undetectable in all immunized groups, except for a residual presence in the 2 μg mRNA-NP group (Fig. 4F‒H). These findings highlight the superior efficacy of high-dose mRNA-NP vaccination for suppressing viral replication, with further enhancement observed when it is combined with mRNA-GPC.

A Study design. Ifnar1−/− mice (n = 12 per vaccination group) received a single subcutaneous immunization. Five weeks later, the mice intraperitoneally challenged with 3000 TCID50 of CCHFV. Six mice per group were monitored for survival and body weight over 14 days, and the remaining six were euthanized at 5 dpi to assess viral loads. B Vaccine design. Single-dose vaccination regimen detailing antigen composition, and administration route. C CCHFV-specific IgG responses. NP-specific IgG levels in Ifnar1−/− mice were measured by ELISA five weeks post-immunization. D Survival analysis. Statistical significance was determined using a Log-rank (Mantel-Cox) test. E Body weight changes. F Viremia. Viral RNA loads in blood were quantified by qRT-PCR at 5 and 14 dpi. G, H. Tissue viral loads in the liver (G) and spleen (H) at 5 and 14 dpi. Data are presented as the mean ± SEM. The dashed line indicates the limit of detection. Statistical significance for (F-H) was determined by Kruskal–Wallis test followed by Dunn’s multiple comparisons test. Exact p values are shown.
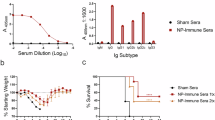
Passive transfer of anti-NP antibodies confers partial protection in Ifnar1−/− mice
To evaluate the protective efficacy of NP-specific and GPC-specific antibodies induced by vaccination, passive immunization experiments were conducted. Serum was collected from Ifnar1−/− mice vaccinated with mRNA-NP, mRNA-GPC, or mRNA-NP + mRNA-GPC, yielding antibody titers of approximately 106, as determined by ELISA (Fig. 5B, C). Serum-recipient mice received 300 μL heat-inactivated immune serum via intraperitoneal injection, and 18 h later, blood was collected before challenge with 3000 TCID50 CCHFV YL16070 (Fig. 5A). ELISA analysis revealed a substantial decrease in NP-specific IgG (Fig. 5D) and Gc-specific IgG (Fig. 5E) titers in the recipient mice. After challenge, the disease progression of the mice that received serum from mice in the mRNA-NP or mRNA-NP + mRNA-GPC groups was delayed, with the mean time to death (MTD) increased from 4 dpi in the LNP control animals to 5 dpi (Fig. 5F). Additionally, survival rates significantly increased from 0% in the control group to 33.33% (2/6) in the mRNA-NP and mRNA-NP + mRNA-GPC serum-treated groups (Fig. 5F). Two of the six mice in these groups experienced transient weight loss starting at 2 dpi but showed signs of recovery by 7–8 dpi (Fig. 5G). In contrast, all other mice either succumbed to infection or reached a humane endpoint (Fig. 5G). Although there were no statistically significant differences in viral loads among the blood, spleens, or livers between the groups, compared with the LNP control mice, the mice receiving mRNA-NP-immune serum presented 8.8-fold and 11.2-fold reductions in the viral loads in their livers and spleens, respectively (Fig. 5I). These data indicate that the passive transfer of mRNA-NP vaccination-induced serum anti-NP antibodies provides partial but detectable protection against CCHFV infection.

A Study design. Ifnar1−/− mice (n = 12 per vaccination group) received 300 μL of heat-inactivated immune sera by intraperitoneal injection. After 18 h, mice were challenged intraperitoneally with 3000 TCID50 of CCHFV. Six mice were monitored for 14-day survival and body weight, and six were euthanized at 5 dpi for viral load assessment. B, C CCHFV-specific IgG levels in donor sera. NP-specific (B) and Gc-specific (C) IgG levels were quantified by ELISA. D-E. CCHFV-specific IgG levels in recipient mice 18 h post-transfer, measured by ELISA for NP-specific (D) and Gc-specific (E) IgG. F Survival analysis. Statistical significance was determined using a Log-rank (Mantel-Cox) test. G Body weight changes. H Viremia. Viral RNA loads in blood were quantified by qRT-PCR at 5 dpi. I Tissue viral loads in the liver and spleen at 5 dpi. Data are presented as the mean ± SEM. The dashed line indicates the limit of detection.
Discussion
Despite recent progress in CCHFV vaccine development, no mRNA-based candidate has been approved to date28,29. Two previous studies explored CCHFV mRNA vaccines: one used naked mRNA without delivery systems28, and the other applied LNP-encapsulated mRNA encoding separate NP or Gn/Gc antigens. However, both studies were limited by high-dose regimens, and did not assess single-dose efficacy or cross-genotype protection. In our study, we developed LNP-formulated, nucleoside-modified mRNA vaccines encoding either NP, full-length GPC or both. Compared with previous approaches, our design incorporates several advancements. First, our GPC construct encodes the full glycoprotein precursor, which undergoes natural processing into Gn and Gc, potentially improving antigen authenticity and epitope presentation. Second, we demonstrate robust protection using as little as 1–2 μg total mRNA, revealing clear dose-sparing potential not evaluated in earlier work. Third, we report strong efficacy of single-dose immunization, even a prime-only with low-dose mRNA-NP + mRNA-GPC or mRNA-NP provided complete survival, underscoring the potency of this platform for dose-sparing and rapid-response strategies. Fourth, we evaluated cross-genotype protection by immunizing with a Europe 1 strain-derived vaccine and challenging with a genetically distant Asia 2 strain, thus demonstrating cross-genotype efficacy not addressed in prior studies. Additionally, the passive transfer of vaccination-induced anti-NP antibodies conferred partial protection, further highlighting the role of the NP in immune defense. These findings indicate that mRNA-NP, encoding the conserved NP antigen, plays a pivotal role in protective immunity against CCHFV. Moreover, coadministration of this formulation alongside mRNA-GPC further enhances vaccine efficacy, suggesting a complementary role for GPC in immune protection. Collectively, these findings provide valuable insight into the feasibility of mRNA-based strategies for CCHFV prevention and underscore the potential of NP-targeting mRNA vaccines as a foundation for future vaccine development.
Our data highlight the distinct protective roles of NP and GPC in mRNA vaccine-induced immunity against CCHFV. The GPC undergoes proteolytic processing to generate structural proteins (Gn and Gc), as well as non-structural proteins such as NSm, the mucin-like domain (MLD), and GP3830,31. The protein Gc, which is essential for viral attachment and entry32,33, is the primary target of neutralizing antibodies (nAbs)34, while GP38 has also been explored as a protective target in antibody-based therapies and vaccine development against CCHFV35,36,37,38. However, CCHFV GPC exhibits significant genetic diversity, with less than 75% amino acid conservation among strains3. This high level of genetic variability raises concerns about potential immune evasion. Consistent with the results of prior studies on DNA and replicating RNA (repRNA) vaccines14,38, our mRNA-GPC vaccine provides only partial protection against cross-genotype CCHFV challenge. In contrast, a vesicular stomatitis virus (VSV)-vector vaccine targeting GPC has demonstrated complete cross-genotype protection39. Moreover, modified vaccinia virus Ankara (MVA)- and adenovirus-vector vaccines have conferred full protection against homologous CCHFV challenge13,40, although their cross-protective potential remains unclear. Notably, even when DNA vaccines match the challenge strain, complete protection is not always achieved41, suggesting that GPC-based immunity may be influenced by platform-specific immune responses. Interestingly, a virus-like particle (VLP)-based vaccine induced higher neutralizing antibody titers than a DNA vaccine but exhibited a 60% lower survival rate37, whereas subunit vaccines targeting Gn or Gc ectodomains elicited strong neutralizing antibody responses but failed to protect against lethal challenge42. These findings indicate that protection does not directly correlate with neutralizing antibody titers and that humoral immunity alone is insufficient for effective CCHFV protection. Consistent with previous studies in mice, our data confirm that the mRNA-GPC vaccine alone induces a strong humoral response but fails to provide adequate protection against CCHFV challenge. To further assess the protective potential of vaccination-induced antibodies, heat-inactivated serum from mRNA-GPC-vaccinated mice was passively transferred to naive mice. However, no protective effect was observed post-challenge, which aligns with findings from MVA-GP vaccine studies43. Notably, despite donor serum exhibiting ELISA titers as high as 10⁶, recipient mice displayed a sharp decline in antibody levels post-transfer, with titers decreasing to 50–450. While this reduction likely results from antibody dilution and clearance, it complicates assessment regarding whether the lack of protection is solely due to the insufficiency of humoral immunity. Furthermore, studies regarding the MVA-GP vaccine have demonstrated that effective protection requires the transfer of both antibodies and T-cells from vaccinated mice43, indicating essential roles of both humoral and cellular immunity in vaccine-mediated protection.
Compared with GPC, CCHFV NP exhibits a greater degree of conservation3. Consistent with our findings and those of prior studies on DNA-based and repRNA vaccines, NP-based immunization alone is sufficient to confer complete protection in mice14,44. Notably, repNP + repGPC vaccination has been shown to provide full protection independent of CD8⁺ T-cells, whereas the survival rate decreases to 40% in B-cell-deficient (μMT) mice, suggesting a crucial role for humoral immunity14. In support of this, recent studies have demonstrated that a monoclonal antibody targeting the NP confers protection against lethal CCHFV challenge45, and the passive transfer of NP-immune sera achieves 40%–50% protection, with higher doses correlating with improved survival46. Unlike traditional viral neutralizing antibodies, NP-specific antibodies do not block viral entry but instead restrict viral replication intracellularly through Fc-dependent, TRIM21-mediated proteasomal degradation of NP-containing virus complexes46. The results of our passive transfer experiments align with these findings. Despite a significant decrease in NP antibody titers post-transfer (from 10⁶ in donor serum to 10³ in recipient serum), survival improved significantly (~33.33%). This finding suggests that a portion of the NP antibodies may have entered cells and contributed to intracellular antiviral effects. However, the precise mechanism of cellular entry remains unclear and requires further investigation.
Although our study provides important insights into mRNA vaccine-induced immunity against CCHFV, several limitations should be acknowledged. First, our experiments were conducted in Ifnar1−/− mice, which lack type I interferon signalling. While this model is widely used for CCHFV research because of its susceptibility to infection, it may not fully recapitulate human immune responses. Future studies should assess vaccine efficacy in more physiologically relevant models, such as nonhuman primates, to better predict human immunogenicity and protection. Second, although we evaluated the persistence of vaccination-induced IgG antibodies, we did not perform longitudinal challenge studies to assess the durability of protection. Protection against viral challenge is a critical factor in vaccine development, and further studies are necessary to determine whether long-term immunity requires booster doses. Finally, while our findings suggest that mRNA-NP confers robust protection, the exact mechanisms underlying NP-mediated immunity remain incompletely understood. Elucidating these mechanisms will be critical for optimizing CCHFV mRNA vaccine design and evaluating its importance for broad and durable protection.
Overall, our findings demonstrate that mRNA vaccines encoding NP and GPC elicit strong immune responses and protect against lethal CCHFV challenge. Both the two-dose and prime-only regimens were effective, and the passive transfer of anti-NP antibodies provided partial protection. Collectively, these results highlight the potential of mRNA vaccines for CCHFV prevention and establish a foundation for further preclinical and clinical development.
Methods
Ethics statement
All animal studies were reviewed and approved by the Animal Ethics Committee of the Wuhan Institute of Virology, Chinese Academy of Sciences (Ethics number: WIVA42202306). Experimental procedures involving CCHFV were conducted within the Animal Biosafety Level 3 (ABSL-3) facility at the National Biosafety Laboratory (Wuhan), Chinese Academy of Sciences. All procedures, including subcutaneous and intraperitoneal injections, retro-orbital blood collection, intracerebral injection in neonatal mice, and euthanasia, were performed under appropriate anesthesia. Adult mice were anesthetized with isoflurane before all procedures. Neonatal mice (1–2 days old) undergoing intracerebral injection were anesthetized via brief hypothermia, induced by placement on ice for 2–3 min, in accordance with institutional animal care protocols. Euthanasia of adult mice was conducted under deep isoflurane anesthesia followed by cervical dislocation, while neonatal mice were euthanized by decapitation following hypothermia-induced anesthesia. All procedures were performed in accordance with institutional ethical guidelines and the AVMA Guidelines for the Euthanasia of Animals (2020 Edition), ensuring animals were fully unconscious and insensible prior to death. All efforts were made to minimize animal suffering.
Cells, viruses, and antibody
HEK293T (ATCC: ACS-4500) and Vero E6 (ATCC: CRL-1586) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% Fetal bovine serum (FBS, Gibco), 100 units/mL penicillin, and 100 μg/mL streptomycin, at 37 °C in a 5% CO2. 293 Freestyle (293 F, a kind gift from Dr. Rui Gong; Wuhan Institute of Virology, CAS) cells were maintained in FreeStyle 293 expression medium supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin, in shaker incubators set at 150 rpm, 37 °C, and 8% CO₂. The CCHFV strain YL16070 (GenBank accession numbers KY354080, KY354081, KY354082) was propagated by intracranial injection into 1–2-day-old KM suckling mice. Once all symptomatic mice were identified, they were immediately euthanized. The brains were harvested and stored in 50% glycerol at −80 °C. Following the three washes with PBS, the brains were homogenized in 1 mL of PBS, and the virus was subsequently extracted and purified by centrifugation 6000 × g for 10 min at 4 °C. Viral titers (expressed as TCID50/mL) were quantified by an indirect immunofluorescence assay (IFA) on Vero E6 cells. Briefly, ten-fold serial dilution of the virus were inoculated onto Vero E6 monolayer cells in the 96-well plate and incubated at 37 °C with 5% CO₂ for 4 days. The cells were then fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% BSA for 30 min, followed by incubation with CCHFV-specific primary antibodies and subsequently with fluorescent dye–labeled secondary antibody. Fluorescent foci were visualized under an EVOS fluorescence microscope (Invitrogen), and TCID₅₀ values were calculated by the Reed–Muench method. The primary antibody against CCHFV NP was produced in-house by immunizing mice with mRNA-NP, followed by collection of blood three weeks after the booster dose; serum containing anti-NP polyclonal antibodies was obtained by centrifugation at 3000 × g for 10 min at 4 °C. The highly cross-adsorbed goat anti-mouse IgG (H + L) secondary antibody was obtained from Thermo Scientific (Cat# A-11001).
Synthesis and characterization of CCHFV mRNA
CCHFV mRNA vaccine was prepared as previously described47. In brief, the mRNAs encoded codon-optimized open reading frames of the CCHFV GPC (GenBank accession no. ARB18232) or NP (GenBank accession no. ARB18231), respectively. The constructs included 5’ untranslated region (UTR), 3’ UTR (see Supplementary Data 2 for sequences), and a 100-nucleotide poly(A) tail. mRNAs were synthesized via in vitro transcription (all reagents purchased individually from Hongene) using a linearized DNA template, with 100% substitution of uridine by N1-methylpseudouridine and incorporation of a co-transcriptional a cap structure (m7G (5’) ppp (5’) (2’OMeA) pG). The synthesized mRNA was then purified using lithium chloride precipitation and resuspended in RNase-free water for further analysis and application. The concentration and integrity of CCHFV mRNA were measured using a NanoDrop One (Thermo) and 5200 Bioanalyzer (Agilent), respectively.
Formulation and characterization of mRNA-LNP
mRNA-LNPs were prepared using a modified procedure. Lipid components were dissolved in ethanol at a molar ratio of 47:10:41.5:1.5 (ionizable lipid CS21001: DSPC: cholesterol: PEG-lipid). The lipids were rapidly mixed with mRNA dissolved in 25 mM sodium acetate buffer (pH4.0) at an N/P ratio of 6, using a 3:1 (aqueous: ethanol) volume ratio at a flow rate of 12 mL/min, via a microfluidic system (INanoTML from Micro&Nano Biologics). The resulting sample was diluted with 20 mM Tris, 10.7 mM sodium acetate buffer (pH7.5), then concentrated by 30 kDa Ultra Centrifugal Filters and sterile-filtered through a 0.2 µm filter. Dynamic light scattering (Malvern Panalytical Zetasizer Pro) was employed to measure particle size, polydispersity index (PDI), and zeta potential. Encapsulation efficiency was determined using the Quant-iT RiboGreen RNA assay kit (Invitrogen) as previously described47.
Mice, vaccinations, and infection
C57BL6/J mice aged 6–8 weeks were purchased from Vital River Laboratories (Beijing, China). In both single-dose and double-dose groups, mice were inoculated subcutaneously with varying immunogens (NP, GPC, and NP + GPC) (n = 6 per group) at different doses of 1 μg, 5 μg, or empty LNP as a control. For double-dose groups, a booster of the same dose was given on day 21 post-initial immunization. To assess cellular immune response, a dedicated cohort of mice (n = 6 per group) were euthanized on day 7 after the boost, and spleens were collected for assessment of cellular immune responses by ELISpot (IFN-γ). To evaluate humoral immune responses, a separate cohort of immunized mice (n = 6 per group) was monitored longitudinally, with blood samples collected at designated timepoints for ELISA and neutralization assay.
Ifnar1−/− mice (C57BL/6 J background) were purchased from GemPharmatech Co., Ltd. (Nanjing, China). In the CCHFV challenge experiment, both single-dose and double-dose immunized, along with LNP control (n = 12) were bled and intraperitoneally infected with 3,000 TCID50 of the CCHFV YL16070 strain in a total volume of 100 μL. This challenge occurred four weeks after the first immunization in the single-dose groups and two weeks after the second immunization in the double-dose groups. Following infection, mice were randomly divided into two subgroups (n = 6): one subgroup was euthanized at 5 dpi for the collection of blood, liver, and spleen samples for viral loads quantification and pathological analysis, while the other subgroup was monitored for survival and body weight changes over a 14-day period. Mice were weighed daily and monitored for clinical symptoms. Humane endpoint criteria were defined as a ≥ 20% body weight loss from baseline or the presence of severe clinical signs such as lethargy, hunched posture, ruffled fur, or impaired mobility. Animals meeting these criteria were humanely euthanized in accordance with institutional animal welfare guidelines. In the single-dose groups, samples were also collected on day 14 post-infection from surviving animals.
Western blot
For protein expression analysis, HEK293T cells were seeded into 6-well plates and transfected with 2 μg of LNP-formulated mRNA-NP or mRNA-GPC by directing adding it to the medium. After 24 h of incubation at 37 °C in 5% CO2, cells were washed with PBS and lysed in 200 μL of RIPA buffer (Beyotime, China) supplemented with a protease inhibitor cocktail. The lysates were incubated on ice for 30 min and then clarified by centrifugation at 12,000 × g for 15 min at 4 °C. Equal amounts of total proteins were resolved on a 10% SDS-PAGE gel and electrotransferred onto PVDF membrane (Millipore) using a semi-dry transfer system. Membrane was blocked with 5% skim milk in TBST (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) at room temperature for 1 h, followed by incubation overnight at 4 °C with mouse anti-CCHFV NP (in-house generated by mRNA-NP immunization) or anti-Gc primary antibodies (a generous gift from Dr. Fei Deng, Wuhan Institute of Virology, CAS). After three washes in TBST, the membrane was incubated with HRP-conjugated goat anti-mouse IgG secondary antibody (Proteintech) for 1 h at room temperature. Signal was developed using an enhanced chemiluminescence (ECL) detection kit (Thermo Scientific) and imaged with a Tanon 5200 imaging system (Tanon, China). GAPDH (Abcam) was used as a loading control.
Expression and purification of CCHFV Gc and NP proteins
Codon-optimized coding sequences for the Gc ectodomain (residues 1053–1573 of the full-length M segment; GenBank accession no. KR864902.1) and full-length NP (GenBank accession no. KY362517.1) of the CCHFV Turkey strain were cloned into the mammalian expression vector pcDNA3.4, each bearing a C-terminal His-tag for affinity purification. To enable secretion of the normally intracellular NP protein, an N-terminal interleukin-10 (IL-10) signal peptide was added to the NP construct. Plasmid constructs were transiently transfected into 293 Freestyle (293 F) suspension cells using polyethyleneimine (PEI; Sigma-Aldrich) in accordance with the manufacturer’s instructions. The cells were cultured in FreeStyle™ 293 Expression Medium (Gibco) under conditions of 37 °C, 8% CO2, and 150 rpm shaking. Five days post-transfection, the supernatants were collected by centrifugation at 1000 rpm for 30 min to remove cellular debris. The clarified supernatants were passed through a 0.45 μm filter. Gc and NP proteins were purified via Ni Sepharose High Performance resin (Cytiva), following the manufacturer’s protocol for His-tagged protein purification.
ELISA
The binding levels of serum IgG antibodies specific to CCHFV Gc and NP proteins was assessed by ELISA. Briefly, 96-well EIA/RIA microplates (Corning) were coated with 200 ng per well of Gc or NP protein in 1× ELISA coating buffer (Solarbio, Cat# C1055), prepared by diluting the 10× stock with deionized water, and incubated overnight at 4 °C. Plates were blocked the next day with 100 μL of PBS containing 5% skim milk at 37 °C for 1 h. After washing with PBS supplemented with 0.1% Tween-20 (PBST), serial three-fold dilutions of mouse serum (starting at 1:50) were prepared in blocking buffer and added to the wells, followed by incubation at 37 C for 2 h. Subsequently, plates were washed four times with PBST and incubated with HRP-conjugated goat anti-mouse IgG secondary antibody (Abcam) at 37 °C for 1.5 h. After a final wash, 100 μL of TMB substrate solution (Beyotime) was added and incubated for 8 min in the dark for color development. The substrate reaction was stopped by adding 50 μL of stop solution (Beyotime), and absorbance was measured at 450 nm (OD450) using a microplate reader. The endpoint titer is defined as the reciprocal of the highest serum dilution at which the OD₄₅₀ value equals or exceeds the positivity threshold. The threshold was calculated as 2.1 times the OD₄₅₀ of blank wells (wells with no serum), consistent with several published studies48.
Enzyme-linked immunospot (ELISpot) assay
The Mouse IFN-γ ELISpot kit (Mabtech) was conducted to evaluate CCHFV-specific T-cell responses according to the manufacturer’s instructions. The CCHFV NP peptide pool (comprising eight peptides, each with a length of 18–20 amino acids, corresponding to NP pools #1 and #4) and the GPC peptide pool derived from the Gc region only (comprising four peptides, each with a length of 18–20 amino acids, corresponding to Gc pool #2), as described in the reference29, were obtained from Genscript and dissolved in DMSO (Sigma). Splenocytes were harvested from C57BL/6 J mice on day 7 after the booster immunization with mRNA vaccines. Freshly isolated splenocytes (5 × 105 cells per well) were plated into pre-coated ELISpot plate and stimulated with 250 ng of peptide pool. The plates were incubated at 37 °C with 5% CO2 for 20 h. For positive controls, cells were treated with phorbol 12-myristate 13-acetate (PMA) and ionomycin (Dakewe), while unstimulated cells served as negative controls. Spot-forming cells (SFCs) were analyzed using a CTL ImmunoSpot Analyzer and ImmunoSpot software (Cellular Technology Ltd). SFCs counts were normalized to 106 splenocytes.
Neutralization assay based on pseudotyped CCHFV
CCHFV Hoti GPC (accession number EU037902.1), with a C-terminal deletion of 53 amino acids, was subcloned into the pcDNA3.1 expression vector to generate pcDNA3.1-Hoti-GPCdel53aa plasmid. HEK 293 T cells were plated in 6-well plate (NEST) in DMEM supplemented with 10% FBS and incubated at 37 °C with 5% CO2 for 18 h prior to transfection. Cells were then transfected with 3 μg of pcDNA3.1--Hoti-GPCdel53aa per well using Lipofectamine 3000 Transfection Reagent (Thermo Scientific). Following a 24-hour transfection period, cells were infected with *G-VSVΔG/GFP (a generous gift from Dr Manli Wang, Wuhan Institute of Virology, CAS) and incubated at 37 °C for 2 h. The monolayers were subsequently washed three times with serum-free DMEM and replenished with fresh complete DMEM (containing 10% FBS). After 48 h, Supernatants were collected, centrifuged, and filtered through a 0.22 μm filter to remove debris. The pseudotyped CCHFV (*GPC-VSVΔG/GFP) was stored at -80 °C and titrated using an end-point dilution assay. For neutralization assays, serum samples were heat-inactivated at 56 °C for 30 min, then serially diluted threefold in DMEM containing 2% FBS and incubated with an equal volume of pseudovirus at 37 °C for 1 h. After incubation, 100 μL of each serum-virus mixtures were added to confluent Vero E6 cell monolayers in black 96-well plates (Corning). Plates were incubated for 24 h at 37 °C with 5% CO2. Infection was assessed by quantifying GFP-positive cells using a fluorescence imaging system (EVOS M7000), and neutralizing antibody titers were determined accordingly.
Neutralization assay based on live CCHFV
Vero E6 cells were seeded into 24-well plates (NEST) with DMEM supplemented with 10% FBS and incubated at 37 °C with 5% CO2 for 18 h to allow monolayer formation. Serum samples were heat-inactivated at 56 °C for 30 min and prepared as a series of threefold dilutions (starting from a 1:50 dilution) in DMEM containing 2% FBS. Equal volumes of diluted sera were mixed with 100 PFU of CCHFV strain YL16070 and incubated at 37 °C for 1 h to enable neutralization. After incubation, 100 μL of each serum-virus mixture was added to Vero E6 monolayers and adsorbed for 1 h at 37 °C. Following this, the inoculum was removed, and wells were overlaid with DMEM containing 2% FBS and 0.8% carboxymethylcellulose sodium (Sigma) at 37 °C with 5% CO2 for 6 days. Cells were fixed with 8% formaldehyde (Sigma) for 30 min, permeabilized with PBS containing 0.1% Triton X-100 for 25 min, and then blocked in PBS containing 5% bovine serum albumin (BSA) for 30 min. Wells were then incubated with mouse anti-CCHFV NP antibody diluted in PBS with 1% BSA for 2 h at room temperature. After three washes with TBST, plates were incubated with HRP-conjugated goat anti-mouse IgG (Beyotime) in PBS with 1% BSA for 1 h. Following additional TBST washes, plaques were developed using DAB substrate and hydrogen peroxide from the Enhanced HRP-DAB Chromogenic Kit (TIANGEN), with incubation at room temperature in the dark for 5–30 min. Plaques were visualized and counted, and neutralization titers were determined accordingly.
CCHFV RNA detection
Whole blood, spleen, and liver samples were collected from infected mice for viral RNA quantification. Each tissue was weighed and mechanically homogenized in 1 mL of DMEM using a tissue grinder. The homogenates were clarified by centrifugation at 5000 × g for 20 min at 4 °C. A 140 μL aliquot of the supernatant was used for RNA isolation using the QIAamp Viral RNA Mini Kit (Qiagen), following the manufacturer’s instructions. For quantitative detection, 2 μL of RNA was used as a template in a one-step real-time reverse transcription PCR (qRT-PCR), performed using the HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme). Primers specific to the CCHFV S segment were used as follows: Forward primer: 5’-TCAAGTGGAGGAAGGACATAGG-3’; Reverse primer: 5’-TCCACATGTTCACGGCTCACTGGG’. Amplification reactions were run on a CFX96 Real-Time PCR System (Bio-Rad) under the following cycling conditions: reverse transcription at 50 °C for 15 min, initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. To quantify viral RNA copy numbers, a standard curve was generated using an RNA standard prepared by in vitro transcription of a known-length CCHFV NP fragment. The RNA standard was serially diluted 10-fold (10¹–10⁹ copies/μL) and used as the template in qRT-PCR to construct the standard curve. Sample Ct values were converted to RNA copy numbers based on this curve. Results were expressed as copies per μL of blood or copies per gram of tissue.
Histopathology and immunohistochemistry
Liver and spleen tissues were fixed in 8% formaldehyde (Sigma) for 7 days at room temperature, followed by routine dehydration and paraffin embedding. Tissue blocks were sectioned at a thickness of approximately 4 μm and mounted onto glass slides. Hematoxylin and eosin (H&E) staining was performed for histopathological evaluation under a light microscope. Multiple regions from each organ were examined to ensure representative sampling. For immunohistochemical (IHC) detection of viral antigen, tissue sections were deparaffinized and subjected to antigen retrieval prior to incubation with an in-house mouse monoclonal antibody against CCHFV NP. A biotinylated goat anti-mouse IgG (SeraCare) was applied as the secondary antibody. Visualization was achieved using DAB chromogen followed by hematoxylin counterstaining. Images were acquired using the Pannoramic MIDI digital slide scanner (3DHISTECH, Budapest, Hungary).
Statistical analysis
All statistics were done using GraphPad Prism 9.0 software. Data normality was assessed using the Shapiro–Wilk test, and homogeneity of variance was assessed using the Brown–Forsythe test. For datasets that satisfied both assumptions (normal distribution and equal variances), one-way ANOVA with Tukey’s multiple comparisons test was used. For non-normally distributed or heteroscedastic datasets, the Kruskal–Wallis test with Dunn’s multiple comparisons correction was applied. For longitudinal IgG antibody response comparisons between dose groups, two-way mixed-effects models (REML) with Geisser–Greenhouse correction and Tukey’s multiple comparisons test were employed. Survival data were analyzed using the Log-rank (Mantel–Cox) test. A p-value < 0.05 was considered statistically significant.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. Raw antibody titers and statistical analyses are provided in Supplementary Data 1. The nucleotide sequences of the 5′ and 3′ untranslated regions of the mRNA constructs are provided in Supplementary Data 2. Additional raw data are available from the corresponding author upon reasonable request.
References
Hawman, D. W. & Feldmann, H. Recent advances in understanding Crimean-Congo hemorrhagic fever virus. F1000Res 7, F1000 (2018).
Aslam, M., Abbas, R. Z. & Alsayeqh, A. Distribution pattern of Crimean-Congo Hemorrhagic Fever in Asia and the Middle East. Front Public Health 11, 1093817 (2023).
Bente, D. A. et al. Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 100, 159–189 (2013).
Whitehouse, C. A. Crimean-Congo hemorrhagic fever. Antivir. Res. 64, 145–160 (2004).
Ergönül, O. Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 6, 203–214 (2006).
Garrison, A. R. et al. ICTV virus taxonomy profile: Nairoviridae. J. Gen. Virol. 101, 798–799 (2020).
Hoogstraal, H. The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J. Med. Entomol. 15, 307–417 (1979).
Ergönül, O. et al. Characteristics of patients with Crimean-Congo hemorrhagic fever in a recent outbreak in Turkey and impact of oral ribavirin therapy. Clin. Infect. Dis. 39, 284–287 (2004).
Swanepoel, R. et al. The clinical pathology of Crimean-Congo hemorrhagic fever. Rev. Infect. Dis. 11, S794–S800 (1989).
WHO. Prioritizing diseases for research and development in emergency contexts. https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts. (WHO, 2018).
Overby, A. K., Pettersson, R. F., Grünewald, K. & Huiskonen, J. T. Insights into bunyavirus architecture from electron cryotomography of Uukuniemi virus. Proc. Natl. Acad. Sci. USA 105, 2375–2379 (2008).
Hawman, D. W. et al. Accelerated DNA vaccine regimen provides protection against Crimean-Congo hemorrhagic fever virus challenge in a macaque model. Mol. Ther. 31, 387–397 (2023).
Saunders, J. E. et al. Adenoviral vectored vaccination protects against Crimean-Congo Haemorrhagic Fever disease in a lethal challenge model. EBioMedicine 90, 104523 (2023).
Leventhal, S. S. et al. Replicating RNA vaccination elicits an unexpected immune response that efficiently protects mice against lethal Crimean-Congo hemorrhagic fever virus challenge. EBioMedicine 82, 104188 (2022).
Tipih, T. & Burt, F. J. Crimean-Congo hemorrhagic fever virus: advances in vaccine development. Biores Open Access 9, 137–150 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Chen, G. L. et al. Safety and immunogenicity of the SARS-CoV-2 ARCoV mRNA vaccine in Chinese adults: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Microbe 3, e193–e202 (2022).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
Wilson, E. et al. Efficacy and Safety of an mRNA-Based RSV PreF Vaccine in Older Adults. N. Engl. J. Med. 389, 2233–2244 (2023).
Qiu, X. et al. Development of mRNA vaccines against respiratory syncytial virus (RSV). Cytokine Growth Factor Rev. 68, 37–53 (2022).
Alberer, M. et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390, 1511–1520 (2017).
Aldrich, C. et al. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: a phase 1 trial. Vaccine 39, 1310–1318 (2021).
Mucker, E. M. et al. Comparison of protection against mpox following mRNA or modified vaccinia Ankara vaccination in nonhuman primates. Cell 187, 5540–5553 (2024).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Essink, B. et al. The safety and immunogenicity of two Zika virus mRNA vaccine candidates in healthy flavivirus baseline seropositive and seronegative adults: the results of two randomised, placebo-controlled, dose-ranging, phase 1 clinical trials. Lancet Infect. Dis. 23, 621–633 (2023).
D’Addiego, J. et al. Development of targeted whole genome sequencing approaches for Crimean-Congo haemorrhagic fever virus (CCHFV). Virus Res. 350, 199464 (2024).
Guo, R. et al. A new strain of Crimean-Congo hemorrhagic fever virus isolated from Xinjiang, China. Virol. Sin. 32, 80–88 (2017).
Aligholipour Farzani, T. et al. Immunological analysis of a CCHFV mRNA vaccine candidate in mouse models. Vaccines 7, 115 (2019).
Appelberg, S. et al. Nucleoside-modified mRNA vaccines protect IFNAR(-/-) mice against crimean-congo hemorrhagic fever virus infection. J. Virol. 96, e0156821 (2022).
Sanchez, A. J., Vincent, M. J., Erickson, B. R. & Nichol, S. T. Crimean-congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J. Virol. 80, 514–525 (2006).
Freitas, N. et al. The interplays between Crimean-Congo hemorrhagic fever virus (CCHFV) M segment-encoded accessory proteins and structural proteins promote virus assembly and infectivity. PLoS Pathog. 16, e1008850 (2020).
Hulswit, R. J. G., Paesen, G. C., Bowden, T. A. & Shi, X. Recent Advances in Bunyavirus Glycoprotein Research: Precursor Processing, Receptor Binding and Structure. Viruses 13, 353 (2021).
Sanchez, A. J., Vincent, M. J. & Nichol, S. T. Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol. 76, 7263–7275 (2002).
Bertolotti-Ciarlet, A. et al. Cellular localization and antigenic characterization of crimean-congo hemorrhagic fever virus glycoproteins. J. Virol. 79, 6152–6161 (2005).
Li, L. et al. Neutralizing monoclonal antibodies against the Gc fusion loop region of Crimean-Congo hemorrhagic fever virus. PLoS Pathog. 20, e1011948 (2024).
Golden, J. W. et al. GP38-targeting monoclonal antibodies protect adult mice against lethal Crimean-Congo hemorrhagic fever virus infection. Sci. Adv. 5, eaaw9535 (2019).
Hinkula, J. et al. Immunization with DNA plasmids coding for crimean-congo hemorrhagic fever virus capsid and envelope proteins and/or virus-like particles induces protection and survival in challenged mice. J. Virol. 91, e02076–16 (2017).
Suschak, J. J. et al. A CCHFV DNA vaccine protects against heterologous challenge and establishes GP38 as immunorelevant in mice. npj Vaccines 6, 31 (2021).
Rodriguez, S. E. et al. Vesicular stomatitis virus-based vaccine protects mice against Crimean-Congo hemorrhagic fever. Sci. Rep. 9, 7755 (2019).
Buttigieg, K. R. et al. A novel vaccine against Crimean-Congo Haemorrhagic Fever protects 100% of animals against lethal challenge in a mouse model. PLoS One 9, e91516 (2014).
Garrison, A. R. et al. A DNA vaccine for Crimean-Congo hemorrhagic fever protects against disease and death in two lethal mouse models. PLoS Negl. Trop. Dis. 11, e0005908 (2017).
Kortekaas, J. et al. Crimean-Congo hemorrhagic fever virus subunit vaccines induce high levels of neutralizing antibodies but no protection in STAT1 knockout mice. Vector Borne Zoonotic Dis. 15, 759–764 (2015).
Dowall, S. D. et al. Protective effects of a modified vaccinia ankara-based vaccine candidate against Crimean-Congo haemorrhagic fever virus require both cellular and humoral responses. PLoS One 11, e0156637 (2016).
Aligholipour Farzani, T. et al. Co-delivery effect of CD24 on the immunogenicity and lethal challenge protection of a DNA vector expressing nucleocapsid protein of Crimean Congo hemorrhagic fever virus. Viruses 11, 75 (2019).
Garrison, A. R. et al. Nucleocapsid protein-specific monoclonal antibodies protect mice against Crimean-Congo hemorrhagic fever virus. Nat. Commun. 15, 1722 (2024).
Leventhal, S. S. et al. Antibodies targeting the Crimean-Congo Hemorrhagic Fever Virus nucleoprotein protect via TRIM21. Nat. Commun. 15, 9236 (2024).
Wang, H. et al. mRNA based vaccines provide broad protection against different SARS-CoV-2 variants of concern. Emerg. Microbes Infect. 11, 1550–1553 (2022).
Lu, M. et al. Both chimpanzee adenovirus-vectored and DNA vaccines induced long-term immunity against Nipah virus infection. NPJ Vaccines 8, 170 (2023).
Acknowledgements
We sincerely thank Tao Du, Jin Xiong, Min Hou, Hao Tang, Jun Liu, and Jia Wu from the Wuhan Institute of Virology for managing the BSL-3 and ABSL-3 facilities for their invaluable support in conducting all authentic CCHFV experiments. We also extend our gratitude to He Zhao, Fan Zhang, and Li Li from the Animal Facility Center for their meticulous care of the animals. Special thanks to Researcher Manli Wang for generously providing *G-VSVΔG/GFP. Additionally, we appreciate the assistance of Liushuai Li, Yajie Liu, Ziyang Jiang, and Jia Liu in our ABSL-3 experiments.
Author information
Authors and Affiliations
Contributions
C.S. and H.M.W. designed the research. J.H.R., J.L., T.T.J., W.W.G., X.K.Z., Z.H.Z., J.L.S., M.Q.L., X.H., X.P.L., R.Q., T.Z., C.L.H., and K.P.L. performed research. J.H.R. analyzed the data. J.H.R. wrote the manuscript. C.S. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rao, J., Li, J., Jiang, T. et al. A single-dose mRNA vaccine protects mice from lethal Crimean-Congo hemorrhagic fever virus infection. npj Vaccines 10, 269 (2025). https://doi.org/10.1038/s41541-025-01310-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41541-025-01310-x
