
Abstract
Infinite-layer transition metal oxides with two-dimensional oxygen coordination exhibit intriguing electronic and magnetic properties due to strong in-plane orbital hybridization. The synthesis of this distinctive structure has primarily relied on kinetically controlled reduction of oxygen-rich phases featuring three-dimensional polyhedral oxygen coordination. Here, using in situ atomic-resolution electron microscopy, we scrutinize the intricate atomic-scale mechanisms of oxygen conduction leading to the transformation of SrFeO2.5 to infinite-layer SrFeO2. The oxygen release is highly anisotropic and governed by the lattice reorientation aligning the fast diffusion channels towards the outlet, which is facilitated by cooperative yet shuffle displacements of iron and oxygen ions. Accompanied with the oxygen release, the three-dimensional to two-dimensional reconfiguration of oxygen is facilitated by the lattice flexibility of FeOx polyhedral layers, adopting multiple discrete transient states following the sequence determined by the least energy-costing pathways. Similar transformation mechanism may operate in cuprate and nickelate superconductors, which are isostructural with SrFeO2.

Similar content being viewed by others
Main
SrFeO2 crystallizes in an infinite-layer structure, where divalent iron (Fe2+) is stabilized in a two-dimensional (2D) FeO4 layer with corner-shared, square-planar oxygen coordination1,2,3,4,5. This unique oxygen coordination of iron has garnered substantial attention, as the irons in most oxides are coordinated three-dimensionally by oxygen6,7. The absence of apical oxygen in SrFeO2 imparts highly anisotropic properties, such as 2D magnetism, implying strong Fe 3d to O 2p hybridization leading to in-layer magnetic interactions1,3,5,8. In recent decades, except for some cuprates9, the synthesis of infinite-layer transition metal oxides, including nickelates10,11, cobaltates12 and ferrites1, has been achieved through kinetically controlled reduction of oxygen-rich precursor phases, such as perovskite or brownmillerite, using a hydride13 or metallic14 reductant. During the deintercalation of apical oxygen, a small amount of hydrogen might be unintentionally introduced as donors and impact any unconventional physical properties observed thus far. The origin of nickelate superconductivity has recently come under scrutiny due to the possible hydrogen incorporation15; however, concerns have been allayed through the synthesis of superconducting nickelates without the use of hydrides14. Obtaining a pristine SrFeO2 structure devoid of hydrogen could be achieved by high-temperature annealing of oxygen-rich precursor phases in a hydrogen-free reducing environment. However, considering the relative instability of SrFeO2 compared with its thermodynamically stable end phases—iron and strontium oxide2,16—capturing the rapid transformation to metastable SrFeO2 with atomic resolution presents a challenge.
The transformation from three-dimensional (3D) polyhedral to 2D square-planar oxygen coordination involves not only the removal of some oxygen but also the reconfiguration of the remaining ones. Conversely, the strontium and iron in SrFeO2 experience minimal positional changes, largely maintaining their original locations. This transformation is critically important, yet its atomic-scale mechanism, particularly the conversion from 3D polyhedral to 2D square-planar oxygen coordination, has remained largely elusive. This is primarily due to the challenges in directly observing the rapid oxygen conduction process. The model proposed by Inoue et al. has provided substantial insights into the fundamental aspects of this atomic reconfiguration, particularly highlighting the exchange between apical and equatorial oxygen ions4,17,18. However, direct atomic-scale observation is still required to fully comprehend the energy cost at each stage of oxygen release and the subsequent reorganization, along with the associated structural modifications of the host lattice.
Here, we successfully captured the fast dynamic motion of individual atoms during the phase transformation of SrFeO2.5 to SrFeO2 using high-resolution transmission electron microscopy (HRTEM) under negative spherical aberration (Cs) imaging (NCSI) condition19,20,21,22. This transformation was observed during the annealing of ultrathin transmission electron microscopy (TEM) specimens (typically sub-10 nm in thickness) in a hydrogen-free vacuum environment of TEM. Through in situ atomic-resolution observations and quantitative image analysis supported by atomic simulations, we uncover a previously unknown oxygen conduction mechanism. This mechanism is facilitated by the lattice dynamics, characterized by the cooperative yet shuffle displacement of iron and oxygen ions, as well as the lattice flexibility of FeOx polyhedral layers in accommodating the oxygen conduction by adopting multiple polyhedral configurations.
Results and discussion
Nucleation and growth of infinite-layer SrFeO2
A 30-nm-thick brownmillerite SrFeO2.5 (001) film grown epitaxially on SrTiO3 (001) substrate serves as the starting material23,24 (Extended Data Fig. 1 and Supplementary Fig. 1a,b). The in-plane lattice of SrFeO2.5 film is elastically strained to match that of SrTiO3 substrate with establishing a coherent interface (Supplementary Fig. 1c–e). SrFeO2.5 comprises alternating rows of corner-shared FeO6 octahedral and FeO4 tetrahedral layers. The key distinction between these two iron oxide layers lies in the presence of one-dimensional oxygen vacancy (VO) channels within the FeO4 tetrahedral layer along the [010] direction. These channels, composed of vacant atomic sites, provide a rapid diffusion pathway for oxygen ions25, a characteristic common to other transition metal oxides sharing similar structures (Supplementary Table 1). Due to the one-dimensionality of the VO channels, the oxygen conduction and associated phase transition exhibit high anisotropy depending on the orientation of these channels with respect to the oxygen removal and supply environment17,26. Considering this, we prepared cross-sectional TEM specimens for heating experiments along the two orthogonal [100] and the [010] zone axes of the SrFeO2.5 (001) film (Extended Data Fig. 1). In the [100] sample, the VO channels run parallel to the plane surfaces of TEM specimen, while in the [010] sample, the channels run perpendicular to the plane surfaces of TEM specimen.
The phase transformation of SrFeO2.5 to SrFeO2 begins at approximately 450 °C under the high-vacuum condition of TEM column (~10−5 Pa). Once initiated, the transformation completes within just 30 s (Supplementary Video 1). This rapid process is in stark contrast to the hydro-reduction process, which typically takes several hours at similar temperatures1,17. The nucleation of SrFeO2 phase occurs at various locations in the SrFeO2.5 film, primarily at its surface (Supplementary Video 1), occasionally at the SrTiO3 interface (Supplementary Video 2) or within the film’s interior (Supplementary Video 3). Notably, the surface of the SrFeO2.5 film emerges as the favoured site for nucleation, due to the ease of oxygen release at the initial stages of nucleation. Before the nucleation of SrFeO2 phase, the oxygen configuration within the tetrahedral layers fluctuates, occasionally exhibiting the contrast similar to the octahedral layers (Extended Data Fig. 2). The fluctuation in the oxygen configuration is attributable to the dynamic motion of oxygen through the VO channels, as it migrates and escapes into the environment. After nucleation, a nanometre-sized SrFeO2 phase grows rapidly with the propagation of multiple ledges on adjacent layers (Supplementary Video 3). We confirmed that the final product of phase transformation is SrFeO2 through integrated differential phase contrast (iDPC) imaging and electron energy-loss spectroscopy (EELS) analysis, supported by theoretical calculations (Methods and Extended Data Fig. 3).
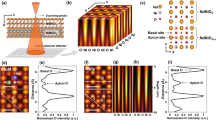
In the phase transformation recorded in real time under a NCSI HRTEM imaging condition, all constituent atoms, including oxygen, are clearly resolved (Fig. 1a). Across the moving phase boundary, the cation lattice frameworks of SrFeO2.5 and SrFeO2 remain highly commensurate, as strontium and iron atoms require only slight positional adjustments during the transformation. Given that the interlayer spacing of strontium layers (dSr–Sr) contracts by ~0.7 Å in SrFeO2 from SrFeO2.5 (Extended Data Fig. 4a,b), by mapping the dSr–Sr over a sequence of HRTEM images we were able to visualize the evolution of SrFeO2 phase and the morphology of the phase boundary with SrFeO2.5 (Fig. 1b,c and Extended Data Fig. 5). The phase boundary advances via step flow of multiple ledges on adjacent layers that effectively squeezes out oxygen from the SrFeO2.5 in front of the boundary (Extended Data Fig. 5, Supplementary Fig. 2 and Supplementary Videos 1 and 2). The multiple ledges arrange into a shear-band-like slanted stack, roughly along the {011} plane (Extended Data Fig. 5). The volume change (contraction) resulting from the release of oxygen ions at the moving front of ledge is accommodated by gradual lattice bending over several nanometres (~5 nm).

a, HRTEM images 6.84 s apart showing the phase transformation of SrFeO2.5 to SrFeO2 and their boundary migration at 450 °C (Supplementary Video 1). The dashed white line marks the phase boundary. In the SrFeO2.5 [100] zone axis sample, the VO channels are aligned laterally. Scale bars, 2 nm. b, Phase boundary outlined in a. c, Interplanar spacing map of the strontium layers, dSr–Sr, used for phase identification. Alternating red (tetrahedral layers) and blue (octahedral layer) stripes belong to SrFeO2.5, while uniform blue region corresponds to the SrFeO2 phase (refer to Extended Data Fig. 4). d, Intralayer iron–iron distance map, δFe–Fe, visualizing the evolution of normally aligned VO channels ahead of the moving phase boundary. The chequerboard pattern appearing in the SrFeO2.5 [100] specimen indicates 90° reconfiguration, which brings the laterally aligned VO channels to the normally aligned ones (yellow rectangles). Uniform green colour far below the phase boundary indicates the pristine SrFeO2.5 [100] without reconfiguration. The structural evolution of atomic structure during the phase transformation is labelled in sequence of (i) initial SrFeO2.5 [100], (ii) 90°-reconfigured to SrFeO2.5 [010], (iii) SrFeO2.5 [010]-SrFeO2 phase boundary and (iv) SrFeO2 [110] end phase. e,f, Atomic models (e) and HRTEM images (f) corresponding to each stage from (i) to (iv). The normally aligned VO channels are indicated by yellow rectangles. The whole phase transformation process can be described as (i) to (ii), 90° reconfiguration of the VO channels ahead of the moving front of phase boundary; (ii) to (iii), progress of the topotactic reduction with phase boundary migration; and (iii) to (iv), completion of the topotactic phase transformation to SrFeO2. Scale bars, 5 Å.
Reconfiguration of VO channels by atomic shuffle displacements
The oxygen release and rearrangement processes at the moving front of the phase boundary were observed to be highly anisotropic (Extended Data Fig. 6). Tracking the phase boundary motion on the in situ HRTEM movies revealed that the boundary moves approximately two to three times faster in the [010] sample than that in the [100] sample. The anisotropy in the oxygen release and rearrangement originates from the way how the VO channels are aligned with respect to the oxygen sink, determining the kinetics of oxygen conduction, as schematically illustrated in Extended Data Fig. 1.
In the SrFeO2.5 [100] sample where the VO channels run parallel to the plane surface of TEM specimen, we observed that the crystallographic reorientation occurs in the vicinity of the phase boundary before the transformation to SrFeO2 phase26 (Supplementary Video 4). Specifically, the crystallographic reorientation results in the 90° reconfiguration of the SrFeO2.5 [100] to [010] direction, aligning the VO channels perpendicular to the top and bottom surfaces of TEM lamella specimen as in the [010] sample, shortening the total length of VO channels to the oxygen sink and facilitating the subsequent phase transformation (Fig. 1d–f). These normally aligned VO channels are visualized by mapping the intralayer iron–iron distance (δFe–Fe) projected in the plane of visualization (Fig. 1d and Extended Data Fig. 4c). In the pristine [100] sample where the VO channels are aligned horizontally, the measured δFe–Fe is a uniform 2.7 Å (green). In contrast, in the pristine [010] or the 90°-reconfigured [100] sample where the VO channels are aligned normally, the δFe–Fe exhibits a chequerboard pattern of 2 Å (navy) and 3.9 Å (yellow), with the latter corresponding to the VO channels. The 90° reconfiguration of VO channels is confirmed by the appearance of a chequerboard pattern near the phase boundary in the δFe–Fe map (Fig. 1d and Extended Data Fig. 7). Once the 90° reconfiguration takes place ahead of boundary, the phase boundary advances rapidly (Extended Data Fig. 6b, red), indicating that the alignment of VO channels with respect to the environment governs the overall transformation rate.
The in situ atomic observation and simulation revealed that the 90° reconfiguration of VO channels towards the shortest dimension of system is facilitated by collective yet shuffle atomic displacements. The HRTEM video was analysed frame by frame, capturing the atomic process leading to the 90° reconfiguration of the SrFeO2.5 [100] to the [010] orientation (Fig. 2a and Supplementary Video 5). At the completion of the reconfiguration, the structural features of the SrFeO2.5 [010] are perfectly restored, including the anti-ferrodistortive (AFD) rotation (β ≈ ±7°) of FeO6 octahedral layers and the appearance of VO channels (δFe–Fe = 3.9 Å) within the tetrahedral layers (Extended Data Fig. 1d). To track the progression of 90° reconfiguration, the δFe–Fe between two adjacent unit cells was monitored until the reconfiguration was fully accomplished (Fig. 2b and Supplementary Video 6). While the overall process completes quickly within seconds, a transient state, referred to as ‘(ii)’ in Fig. 2, was observed to exist between the two end states, ‘(i)’ for SrFeO2.5 [100] and ‘(iii)’ for SrFeO2.5 [010]. This transient state persists for an extended period, exhibiting intermediate values of δFe–Fe and β values that were halfway between those of SrFeO2.5 [100] and SrFeO2.5 [010].

a, Time sequence HRTEM images showing the 90°-reconfiguration process of SrFeO2.5 [100] to [010] (Supplementary Video 5). Images (i) and (iii) represent the two end states, while image (ii) is a transient state that spans a relatively long time. The white line connecting iron and oxygen in the octahedral layers depicts the evolution of AFD rotation during the reorientation as indicated by angle β. b, Intralayer iron–iron distance, δFe–Fe, of the two adjacent unit cells defined in a measured as a function of time (Supplementary Video 6). The δFe–Fe values in the plot are the average of measurements from three-consecutive two-adjacent unit cells, with error bars indicating the standard deviation. The uniform δFe–Fe values are split into one smaller (δFe–Fe_1) and one larger (δFe–Fe_2) values via a transient state (ii), with the latter being the vertically aligned VO channel in SrFeO2.5 [010]. c, Structural models from DFT simulations corresponding to states (i), (ii) and (iii) in a, which correspond to (1), (6) and (11), respectively, in a series of structural models calculated by DFT (Extended Data Fig. 8a). d, Top view of the structural models (i), (ii) and (iii) in c. The yellow arrows indicate the direction of VO channels lying in the tetrahedral layers. e, Visualization of atomic shuffle displacement vectors by overlapping the two atomic arrangements from tetrahedral layers corresponding to (i) and (iii) in d. Superposition of the tetrahedral layers before and after the 90° reconfiguration and connecting the positions of each atom define the displacement vector.
The process leading to the 90° reconfiguration of VO channels is facilitated by a shuffle-like displacement of iron and oxygen ions within the FeO4 tetrahedral layer, akin to the atomic shuffle mechanism proposed for deformation twinning27,28,29. It appears that there is no way to reposition all iron and oxygen ions to achieve the necessary 90° reconfiguration by a simple shear displacement process. To delve deeper into this phenomenon, we performed first principles calculations, taking into account the shuffle displacements for the 90° reconfiguration (Fig. 2c,d). In this process, each iron and oxygen atom is subject to a unique displacement vector (Fig. 2e and Supplementary Video 7). This shuffle displacement can be visualized by overlaying the two atomic arrangements before and after the 90° reconfiguration and connecting the position of each atom to define the corresponding displacement vector (Fig. 2e).
Twelve successive configurations, generated by the first principles nudged elastic band method, depicting the shuffle displacement process, are shown in Extended Data Fig. 8a and Supplementary Video 8. The initial and final configurations with the reaction coordinate (1) and (12) correspond to the SrFeO2.5 [100] and the [010] zone axis orientation, respectively. To enable a direct comparison between experimental observations and the theoretical calculations, we simulated the HRTEM images using the atomic configuration files corresponding to each reaction coordinate (Extended Data Fig. 8b and Supplementary Video 8) and conducted cross-correlation analysis to assign proper reaction coordinates to the experimental HRTEM images (Extended Data Fig. 8c). Notably, a relatively long-lived transient state is observed at the midpoint of the reconfiguration process, corresponding to the reaction coordinates (6) and (7) (or state (ii)).
The energy landscape calculated (Extended Data Fig. 8d, blue) along the reaction coordinate of shuffle displacement reveals the presence of a local energy minimum at the coordinates numbered (6) and (7). This energy minimum arises from the reduced Coulomb repulsion between oxygen ions due to their relocations. This particular reaction coordinate appears as a subtle yet distinct local maximum in the probability plot (Extended Data Fig. 8d, orange) assessed by Boltzmann statistics of each reaction coordinate (Methods), manifesting a relatively long-lived transient state compared with the rest of the reaction coordinates in the course of otherwise fast shuffle displacement. The atomic configuration of this transient state predicted by the calculation matches well with the experimentally observed one from the midpoint of the reconfiguration (Extended Data Fig. 8c, (ii)). The shuffle displacement leading to 90° reconfiguration of SrFeO2.5 is facilitated by the free space available for the iron and oxygen ions around the VO channels.
Given that the energy states before and after the 90° reconfiguration are equivalent, as demonstrated in Extended Data Fig. 8d, there appears to be no inherent driving force for this process. However, the 90° reconfiguration of the VO channels can reduce the energy required for the subsequent oxygen release process. When the VO channels are oriented perpendicular to the plane surfaces of the TEM specimen, as depicted in Extended Data Fig. 1f, there is a 103-fold increase in the direct exposure of the VO channels to the vacuum, compared with the configurations where the channels run parallel to the longest dimension of the specimen, as shown in Extended Data Fig. 1e. This anisotropy in oxygen conduction, where the conduction is much more efficient along the VO channels, suggests a preference for the 90° reconfiguration of VO channels to facilitate easier oxygen removal.
Atomic-scale mechanism of oxygen release and rearrangement
The 90°-reconfigured VO channels expedite the release of oxygen. The subsequent oxygen rearrangement is facilitated by the lattice flexibility of iron oxide layers that dynamically accommodates the change in oxygen coordination by adopting various FeOx polyhedral configurations. This raises fundamental questions as to which oxygen ion is removed first and which one later, and how the remaining ones are rearranged into the square-planar configuration. Time-resolved tracking of the atomic position and intensity change from real-time NCSI HRTEM recordings (Supplementary Video 9) provides valuable clues to the underlying atomic-scale mechanism. A series of transient states identified during the one-unit event of layer-by-layer phase transformation unveils the sequence of oxygen release and rearrangement, as well as the associated multiple polyhedral configurations, with each configuration being corroborated by density functional theory (DFT) calculations (Fig. 3).

a, Time sequence HRTEM images showing the phase transformation of the SrFeO2.5 [010] after 90° reconfiguration (Supplementary Video 9). The four distinct states (i)–(iv) were identified during the transformation of octahedral and tetrahedral layers to square-planar layers. AFD rotation and polar distortion in states (i) and (ii), respectively, are indicated by arrows and lines. The dashed arrows in states (iii) and (iv) correlate to the concurrent intensity change of the two oxygen, the apical oxygen of FeO6 octahedral and the equatorial oxygen of FeO4 tetrahedral, indicating the oxygen migration from the former to the latter. Colour bar indicates intensity level after normalization. Octa., octahedral; tetra., tetrahedral. b, SrO column intensity of the four SrO layers defined in a (Exp., yellow marker). The same measurement on the simulated images (Extended Data Fig. 9) was added for comparison (Sim., grey). The upper (SrO with all oxygen) and lower (only strontium without oxygen) bounds are marked by the dashed line. The intensity values in the plot represent the average of measurements from the lateral four SrO columns, with error bars indicating the standard deviation. Green dashed boxes and white arrows highlight the intensity change. c, Intensity of the VO channel (red) and FeO column (blue) along the tetrahedral layer defined in a. The correlated intensity changes of the apical oxygen in SrO and the oxygen occupying VO channels are indicated by the dashed arrows pointing from b to c. d, Atomic models, corresponding to states (i) to (iv) in a, proposed by DFT calculation.
The FeO4 tetrahedral and FeO6 octahedral layers of SrFeO2.5 undergo transformation to the FeO4 square-planar layer following distinct yet highly correlated pathways, as summarized below.
-
1.
The transformation of the tetrahedral layer to the square-planar layer begins with the release of the oxygen shared between two neighbouring tetrahedra (as they are the easiest to remove), followed by the sequential incorporation of the apical oxygen released from both the top and bottom adjacent octahedral layers. The experimental evidence supporting the release of oxygen from the tetrahedral layer is provided by the noticeable intensity trail across the VO channels, together with the reduced intensity of FeO columns (Supplementary Video 9).
-
2.
The transformation of the octahedral layer to the square-planar layer occurs through the sequential release of the two apical oxygen while retaining the strongly bound equatorial oxygen. Initially, the release of one apical oxygen results in a polar distortion arises in the resulting FeO5 square-pyramidal layer. This polar distortion disappears as the FeO5 unit further transitions to the FeO4 square-planar layer by releasing the remaining apical oxygen. The released apical oxygen then moves towards the nearest tetrahedral layers and participate in subsequent rearrangement within the layer.
The analysis of HRTEM images and complementary DFT calculations, elucidating the transformation sequence of each layer and their interplay, are summarized in Fig. 3. In the FeO6 octahedral layer, the initial yet most prominent change is observed as a decrease in the intensity of the apical SrO (SrO (1)) column, accompanied by the evolution of polar distortion in the equatorial oxygen and iron ions (from state (i) to (ii) in Fig. 3a–c), overriding the intrinsic AFD rotation. The reduction in the intensity of the SrO column can be attributed to the release of apical oxygen from the corresponding FeO6 octahedra. As verified by DFT calculation (Fig. 3d), the release of apical oxygen causes a structural change of the octahedral layer, that is, the AFD rotation to polar distortion; the removal of the top apical oxygen truncates the FeO6 octahedra into a FeO5 square pyramid, causing the equatorial oxygen ions to move upwards and the iron ions to move downwards, counterbalancing the emerging dipole moment and resulting in a buckling with an amplitude of 24 pm. The released apical oxygen subsequently migrates towards the (upper) nearest tetrahedral layer.
The first noticeable change in the tetrahedral layer is the rise of the intensity at the vacant sites (VO channels) beyond the background level and the simultaneous decrease of the intensity of the FeO columns (from state (i) to (ii) in Fig. 3a–c). These intensity changes indicate the release of the oxygen from the FeO columns, which is corner-shared between two neighbouring FeO4 tetrahedra, and subsequent migration through the VO channels to the vacuum outlet. As the oxygen release and evacuation continues in the tetrahedral layer, the oxygen coordination of iron atom reduces gradually from FeO4. At some point in the course of the oxygen evacuation, the tetrahedral layer transitions to a completely new atomic arrangement, where the VO channels are filled with oxygen (Fig. 3a, state (iii)). We note that this change happens simultaneously with the release of apical oxygen from the FeO5 square pyramid in the upper layer (Fig. 3b,c, state (iii)). The transition layer is likely to adopt a FeO3 trigonal-planar configuration by receiving the apical oxygen released from the FeO5 square pyramid in the upper layer (Fig. 3d, state (iii)). The trigonal-planar configuration then transitions into FeO4 square-planar configuration by completely filling up the vacant sites with the oxygen delivered from the FeO6 in the lower octahedral layer (Fig. 3a–d, state (iv)). This unit event converts one octahedral and tetrahedral layers into two corner-shared FeO4 square-planar layers and continues in the subsequent layers.
The proposed model, derived from the analysis of real-time HRTEM data, is validated by DFT simulations, confirming the sequence of oxygen release and rearrangement (Fig. 3d, Extended Data Fig. 9 and Supplementary Video 10). For instance, by tracking the variation of dSr–Sr (Extended Data Fig. 9f and Supplementary Video 11) during the phase transformation, the stepwise evolution of distinct transient states can be identified. These transient states exhibit different spacing, one measuring 4.16 Å and the other measuring 3.79 Å, between the two end phases (4.21 Å for SrFeO2.5 and 3.49 Å for SrFeO2). Each transient state persists for a specific duration within a second-level time span. The presence of such distinct, stepwise structural changes suggests that the phase transformation occurs discretely through a series of short-lived transient states. As validation, we carried out the same analysis on the simulated HRTEM images using the DFT results corresponding to each step shown in Fig. 3. The intensity measured from the simulated images is included as grey bars in Fig. 3, and the full results are summarized in Extended Data Fig. 9. The intensity changes of each atomic column associated with oxygen loss and redistribution measured from the simulated HRTEM images agree well with the experimental measurements.
Energy landscape along the observed oxygen release and rearrangement pathway
According to the DFT calculations, the equatorial oxygen in FeO6 (O1) exhibits the highest VO formation energy (4.11 eV), indicating that these oxygen ions are the least likely to be removed (Fig. 4a–c). Conversely, the corner-shared oxygen between two neighbouring FeO4 within the tetrahedral layer (O3) exhibits the lowest VO formation energy (3.25 eV). Given the substantial amount of oxygen that needs to be removed in SrFeO2.5, it is evident that this type of oxygen is the most easily removable one. Notably, the VO formation energy is substantially reduced for all types of oxygen at the phase boundary (Fig. 4b,c). Once the corner-shared oxygen is removed, the FeO4 unit adopts a FeO3 trigonal-planar configuration by receiving an apical oxygen from the upper octahedral layers and, subsequently, another one from the lower octahedral layers, achieving the square-planar configuration (Fig. 4d). Regarding the apical oxygen (O2) in perfect SrFeO2.5 (Fig. 4a,c), each oxygen atom is shared between the FeO6 octahedral and FeO4 tetrahedral layers at its vertices. The energy required to remove this apical oxygen is high (3.76 eV) when both connecting polyhedra maintain their regular oxygen coordination. However, when the oxygen coordination of the polyhedra is disrupted by losing oxygen, such as from the FeO6 octahedra to a FeO5 square pyramid (as in pathway 1 in Fig. 4d), or from the FeO4 tetrahedra to a FeO3 trigonal-planar (as in pathway 3), our DFT calculations show that the activation energy required to migrate the apical oxygen is lowered substantionally (Fig. 4e), facilitating the release and migration towards the tetrahedral layers. This energy difference in displacing the apical oxygen, depending on the local oxygen coordination, determines the energetically favoured pathway and controls the sequence of oxygen removal and rearrangement processes (Fig. 4d,e). Consequently, the phase boundary propagates in a layer-by-layer fashion, akin to falling dominoes, following the reaction pathway with the least energy cost in the energy landscape.

a,b, Atomic model of perfect SrFeO2.5 (a) and SrFeO2.5/SrFeO2 boundary (b). Oxygen in different iron coordination is classified for the calculation of VO formation energy; equatorial oxygen in FeO6 (O1 in SrFeO2.5, O1′ in SrFeO2.5/SrFeO2 boundary), apical oxygen in FeO6 (O2, O2′) and equatorial oxygen in FeO4 (O3, O3′). c, VO formation energy calculated for the oxygen in different Fe coordination of SrFeO2.5 (green) and SrFeO2.5/SrFeO2 boundary (orange). d, Oxygen migration pathways for apical oxygen in different iron coordination to the tetrahedral layer: (1) FeO5 square pyramid; (2) ordinary FeO6; (3) FeO3 trigonal planar. e, Energy landscape along the different migration paths defined in d. Pathway passing through (1) to (3) is energetically favoured over (2) to (3), explaining the observed sequence of oxygen removal and rearrangement processes.
The oxygen conduction mechanism identified in this study differs from the typical high-temperature hopping process. Instead, it is facilitated by the lattice flexibility of FeOx polyhedral layers, which accommodate oxygen conduction by adopting multiple polyhedral configurations. To illustrate this, we tracked the trajectory of atomic movements, highlighting their discrete nature in response to changes in oxygen coordination (Extended Data Fig. 10). These discrete movements result from changes in the oxygen coordination around Fe atoms within FeOx polyhedral layers, differing from conventional vacancy-driven oxygen conduction that typically leaves a diffusive trace as result of random walks. Additionally, we analysed the moving rate of the phase boundary to assess its time (t) dependency, determining whether it follows diffusion control, indicated by a t1/2 dependency, or interface reaction control, marked by a linear t dependency. The results, shown in Supplementary Fig. 3, reveal a linear t dependency in the boundary motion, suggesting that the process is driven by an interface reaction controlled type process. Specifically, this interface reaction involves oxygen rearrangement through discrete configurations of FeOx polyhedral layers at the phase boundary, in contrast to conventional vacancy-driven diffusion process. Remarkably, upon cooling in the vacuum of TEM column, the reverse reaction takes place spontaneously (Supplementary Fig. 4). This suggests not only that oxygen is sufficiently mobile at low temperatures but also that the cation framework can adjust itself to form a new topotactic structure.
Conclusion
The atomic-scale mechanism underlying one of the most intriguing topotactic phase transformations in transition metal oxides, from 3D polyhedral to 2D square-planar configuration, has been revealed through real-time tracking of atomic motions during the phase transformation. The phase transformation was assessed by heating an ultrathin SrFeO2.5 sample in the vacuum of TEM column, eliminating the need for a controversial hydride reductant. Due to the pronounced anisotropy in oxygen conduction, the rate-limiting step is governed by the atomic reconfiguration process, aligning the Vo channels along the shortest dimension of the sample. This reorientation is facilitated by the cooperative shuffle displacement of iron and oxygen ions within the lattice. The release and rearrangement of oxygen occur in a well-defined sequential pathway via transient states, as the energy required for oxygen release varies depending on the evolving local oxygen coordination. In contrast to conventional vacancy-driven oxygen conduction at high temperatures, this conduction mechanism is enabled by the lattice flexibility of FeOx polyhedral layers, which can accommodate the removal or addition of oxygen through the adoption of multiple polyhedral configurations with varying degrees of iron–oxygen bond valence and covalence, as well as polar distortion or anti-polar rotation.
The insights gained from this observation not only shed light on the specific transformation mechanism in SrFeO2 but also provide a foundation for understanding similar processes in other materials, such as cuprate and nickelate superconductors that share similar structural characteristics. By elucidating the intricate atomic-scale dynamics involved in the transformation of 3D polyhedral to 2D square-planar oxygen coordination, our research paves the way for further exploration and potential advancements in the field of infinite-layer transition metal oxides.
Methods
SrFeO2.5 thin film growth and TEM specimen preparation
Epitaxial SrFeO2.5 thin film was grown on atomically flat (001)-oriented single-crystalline SrTiO3 substrates using pulsed laser epitaxy at 700 °C under 1 mTorr of oxygen partial pressure. An excimer (KrF) laser of 248 nm wavelength (IPEX 864, Lightmachinery) with an energy fluence of 1.3 J cm−2 and a repetition rate of 4 Hz was used. The atomic structures and the crystallinity of the epitaxial thin films were characterized by high-resolution X-ray diffraction. The thickness of the thin films was determined as 30 nm, using X-ray reflectivity.
Cross-sectional TEM specimens were prepared along the [100] and [010] zone axis of the epitaxial SrFeO2.5 thin films by using focused ion beam (FIB) milling. The prepared SrFeO2.5 lamella by FIB were attached to silicon micro-electromechanical systems chip (Wildfire, DENSsolutions) for in situ heating experiments. At the final stage of FIB milling, a low energy Ga+ ion beam at 2 kV was used to reduce the beam damages.
As shown in Extended Data Fig. 1, the geometry of the SrFeO2.5 thin film region of the TEM specimen is approximately 5.6 nm (lamella thickness) × 30 nm (as-grown thin film thickness) × 104 nm (lamella length). If the VO channels are aligned perpendicular to the plane surfaces of the [010] TEM specimen (Extended Data Fig. 1f), it would lead 103 times more direct exposure of the VO channels towards the vacuum, compared with the [010] specimen with VO channels running along the longest dimension (Extended Data Fig. 1e).
In situ NCSI HRTEM imaging
A Cs-corrected (S)TEM (Grand ARM300F, JEOL) operated at 300 kV was used for in situ heating experiments. The in situ heating was performed using a heating holder (Wildfire, DENSsolutions). After initially ramping up to 350 °C, the annealing temperature was subsequently increased to 500 °C in increments of 50 °C, with the sample maintained for 1 h at each temperature step. We observed that the temperature window for phase transition was notably narrow; specifically, the phase transition did not occur at temperatures below 450 °C. Conversely, at temperatures exceeding 450 °C, the phase transition occurred concurrently with thermal decomposition. This limited temperature range posed a challenge in varying the temperature within the narrow operational window for observing the phase transformation. Real-time in situ movies were acquired at 450 °C to investigate the phase transformation using a 4 K × 4 K charge-coupled detector (CCD) camera (OneView, Gatan) at 25 frames per second (fps). The NCSI condition, which gives enhanced contrast (bright atomic columns on dark background) and minimal delocalization (~±0.03 nm), was used to resolve all atoms (including oxygen columns) under spherical aberration coefficient Cs = −13 μm combined with Δf = +6.2 nm Scherzer defocus20,21. The electron dose rate was on the order of 103–104 e− Å−2 s−1.
For the TEM specimen with the thickness of 5.6 nm (Supplementary Fig. 5), all atomic columns in the NCSI HRTEM images show linear intensity–thickness relationships that justify the use of the column intensity to trace the change in the number of atoms in the column (Supplementary Fig. 6). This is key to the direct interpretation of the intensity change associated with the migration and loss of oxygen.
HAADF-STEM imaging
The detector collection angle in the range of 70 to 175 mrad was set for high-angle annular dark-field (HAADF) imaging mode. The convergence semi-angle of a focused electron probe was 25 mrad.
Electronic structure analysis by STEM EELS
Atomic-resolution EELS has been carried out using an EEL spectrometer (GIF Quantum ER, Gatan) with an energy resolution of 0.6 eV at the acceleration voltage of 300 kV under STEM imaging mode. The probe convergence angle and collection angle were 23 and 36 mrad, respectively. To determine the valence state of iron ions before and after phase transformation, atomic-resolution EELS has been carried out with focusing the electron probe at individual oxygen (for O K edge) and iron (for Fe L2,3 edge) column sites. Zero-loss peak was acquired simultaneously for the energy calibration. The electron dose rate was about 104 e− Å−2 s−1. The energy dispersion and dwell time per pixel were 0.1 eV and 0.2 s, respectively. There is no noticeable electron beam damages during data acquisition.
The presence of divalent Fe2+ ion in the square-planar oxygen coordination was confirmed by the chemical shift of EELS Fe-L2,3 edge towards a lower energy, compared with the Fe-L2,3 edge of tetrahedral or octahedral oxygen coordination of Fe3+ ions in SrFeO2.5 (refs. 30,31) (Extended Data Fig. 3) Moreover, the increased hybridization strength between Fe 3d to O 2p in SrFeO2, compared with SrFeO2.5, was confirmed by a shift in the EELS O-K edge to a higher energy—a key characteristic of the square-planar FeO4 layers in SrFeO2. This finding has been further supported by the DFT calculations on the density of state (DOS) and EELS O K edge of SrFeO2 and SrFeO2.5.
Oxygen configuration characterization by iDPC-STEM imaging
Differential phase contrast (DPC) signal was obtained by using a segmented field detector (SAAF Octa. JEOL) under STEM mode. The beam deflection was measured and converted to iDPC images using a commercial software (qDPC, HREM Research Ltd.). iDPC is a suitable imaging technique to visualize light elements such as oxygen when the sample thickness is not ideal for NCSI HRTEM. The convergence angle of electron probe for DPC was 25 mrad. The angular ranges for the inner and outer detectors were set to 0–16.7 and 16.7–33.3 mrad, respectively.
Viewing along the [100] zone axis, the apical oxygen of octahedra or tetrahedra is overlapped with strontium while the equatorial oxygen is visible without overlap with other elements (Extended Data Fig. 1). This image projection thus hinders unambiguous interpretation of the oxygen configuration in the newly formed phase. To avoid confusion, we performed a heating experiment along the [110] zone axis under the same temperature and characterized the atomic structure by iDPC imaging under STEM mode (Extended Data Fig. 3). Along this zone axis, the apical oxygen is isolated without overlap with strontium or iron. After transformation, compared with pristine SrFeO2.5 structure, all the apical oxygen in the newly formed phase was lost, indicating that the newly formed structure is SrFeO2 with square-planar coordinated infinite-layer structure.
Geometric phase analysis
Geometric phase analysis (HREM Research) software was used for the strain analysis of HRTEM images and HAADF-STEM images. For the strain maps shown in Extended Data Fig. 5 and Supplementary Figs. 1 and 2, the fractional change of the in-plane and out-of-plane lattice parameters from the reference SrFeO2.5 phase, εxx and εzz, respectively, was used to differentiate SrFeO2 from SrFeO2.5 and identify the phase boundary. The lattice rotation map (Rxz) was used to visualize the lattice bending over ledge(s) in the phase boundary, which arises to accommodate the large mismatch in the out-of-plane lattice parameters between SrFeO2.5 and SrFeO2. For the epitaxial misfit strain analysis applied on HAADF-STEM images in Supplementary Fig. 1, the reference area was set on SrTiO3 substrate region. Given that the SrFeO2.5 film used in this study is compressively strained on SrTiO3 substrate (misfit strain of −1.4%), the effect of lattice strain on the oxygen reconfiguration and conduction mechanisms needs further exploration.
Energy-filtered TEM for thickness estimation of TEM specimen
For the determination of TEM specimen thickness, energy-filtered TEM was conducted using an energy filter (Quantum spectrometer ER965, Gatan) combined with a 2 K × 2 K CCD camera (UltraScanXP 100FT, Gatan). An objective aperture with a diameter of 20 μm was used to select the transmitted beam for thickness map. Thickness maps were recorded using the imaging filter to remove inelastically scattered electrons outside an energy window of 0 ± 5.0 eV with the exposure time of 4 s. The mean free path for inelastic scattering of 94.6 nm was used for SrFeO2.5.
Control experiments I—electron beam effects
Evaluating the effects of the electron beam is crucial in in situ TEM heating experiments. In this study, we carefully maintained the electron dose rate at approximately 1 × 104 e− Å−2 s−1. This rate was chosen to optimize the signal-to-noise ratio while minimizing potential electron beam-induced damages. Fortunately, the benefit of using HRTEM for atomic-scale imaging over STEM is its relatively lower electron dose rate, which is typically in the order of 103–104 e− Å−2 s−1. We also performed the heating experiment at a reduced dose rate of ~103 e− Å−2 s−1 and obtained similar results. To further assess the impact of the electron beam, we conducted controlled experiments, the results of which are detailed in Supplementary Figs. 4 (~20-nm-thick TEM specimen) and 7 (~180-nm-thick TEM specimen). In these experiments, we activated the electron beam only for capturing images or obtaining diffraction patterns necessary for phase identification. By maintaining identical heating–cooling conditions but without electron beam exposure, we observed that the SrFeO2.5 film in the 20-nm-thick TEM specimen transformed into SrFeO2 at a temperature of 450 °C and then reverted to SrFeO2.5 reversibly upon cooling to room temperature, consistent with the behaviour of the ultrathin TEM specimen. This confirms that the reversible transformation between SrFeO2.5 and SrFeO2 is primarily driven by temperature changes, rather than by stimulation from the electron beam.
Control experiments II—sample size effects
The synthesis of infinite-layer SrFeO2 with the square-planar oxygen configuration is limited to only a few kinetically controlled processes such as hydro-reduction as SrFeO2 is not a thermodynamically stable phase in most experimental conditions. Considering the relative instability of SrFeO2, thermal annealing of the initial phase SrFeO2.5 for a sufficiently long time is likely to induce the decomposition to the stable end phases. Whether kinetically driven oxygen deintercalation topotactic transition dominates thermodynamically driven decomposition depends on the annealing and sample conditions. In this context, providing a favourable condition for the fast deintercalation of oxygen can result in the dominance of SrFeO2 formation over phase decomposition. This can be achieved by lowering the oxygen partial pressure in the annealing atmosphere, which enhances the escaping tendency of oxygen (chemical potential of oxygen). Not only the driving force but also the physical area through which oxygen escapes should be increased and the energy barrier for oxygen migration should be reduced to promote the fast deintercalation of oxygen. This can be accomplished by increasing the surface-to-volume ratio by optimizing the geometry, crystallographic orientation and sample size.
Note that the pressure of TEM column is in the level of 10−5 Pa, which is several orders of magnitude lower than the pressure adopted for most hydro-reduction processes1,13. More importantly, while the size of powder samples prepared by mechanical grinding in all reported studies was in the range of 1–10 μm, the thickness of TEM specimens used in the present study was in the order of nanometre, resulting in a high surface-to-volume ratio, favourable for oxygen release. All these sample and environmental conditions can promote the fast deintercalation of oxygen before decomposition. In fact, in situ heating of the ultrathin TEM sample (5.6 nm in thickness) prepared for NCSI HRTEM imaging showed the fast transformation of SrFeO2.5 to SrFeO2. Upon cooling, the SrFeO2 phase transforms back to SrFeO2.5 reversibly, demonstrating that, for the ultrathin sample, kinetically driven oxygen deintercalation or intercalation completes before thermodynamically driven process sets in.
As demonstrated in Extended Data Fig. 6, the activation of VO channel reconfiguration plays a pivotal role in determining the overall rate of phase transformation, thus acting as a critical rate-limiting step in this kinetically controlled process. We note that the reconfiguration of the VO channels incurs a substantial energy cost, necessitated by the collective and cooperative shuffle displacements of all involved atoms, as depicted in the energy curve in Extended Data Fig. 8d. With increasing sample size, the energy required for this reconfiguration escalates due to the greater number of atoms needing displacement. Consequently, effective reconfiguration is primarily observable in exceedingly thin samples, such as those prepared for TEM, or only in the near-surface area of bulk sample where the VO channels are not aligned towards the largest exposed surface.
We note that, as the phase transformation proceeds, the SrFeO2/SrFeO2.5 phase boundary moves downwards from the film surface, transforming the volume of the SrFeO2.5 film swept by the phase boundary into SrFeO2, leaving no residual SrFeO2.5 phase in its wake. The released oxygen moves through the VO channels and eventually escapes into the vacuum at the (010) edge of the film. Therefore, in this specific TEM sample scenario, the released oxygen is directly transported to the vacuum via the VO channels without passing through SrFeO2 phase. However, in the transformation of bulk SrFeO2.5, oxygen diffusion through the SrFeO2 phase probably plays a critical role. If this step is slow and becomes rate limiting, it might compete with the thermal decomposition process.
As a control experiment to investigate the effects of TEM specimen thickness on the oxygen deintercalation topotactic transition, we prepared thicker TEM samples with thickness of ~20 nm and ~180 nm. The rate of transformation to SrFeO2 via oxygen deintercalation depends critically on the sample thickness. For the TEM specimen with a thickness of ~20 nm (Supplementary Fig. 4), the reversible transformation completes within 5 min in a heating–cooling cycle between 450 °C and 20 °C. However, reversibility diminishes with increasing TEM specimen thickness, and thermodynamics comes into play. For the ~180-nm-thick TEM specimen (Supplementary Fig. 7), the transformation from SrFeO2.5 to SrFeO2 was slower and more prolonged. Additionally, we observed the formation of a Fe-rich (Sr-deficient) phase on the film surface, indicating decomposition of the SrFeO2.5 or SrFeO2 ternary oxide into Fe–O and Sr–O binary oxides, with SrO being volatile; therefore, the chemical composition deviates from exact stoichiometry (due to the preferential loss of strontium). Intriguingly, the SrFeO2 phase persisted without reverting to SrFeO2.5 even after extended post-cooling air exposure at room temperature. This behaviour aligns with previous studies using micrometre-sized powder samples, presenting challenges in synthesizing a single phase SrFeO2 through thermal reduction; such attempts either lead to the loss of reversibility or generate byproducts. Despite these challenges, our findings offer a novel avenue for achieving reversible phase transformation in infinite-layer transition metal oxides, potentially in ultrathin membrane forms. Further, it excludes the potential incorporation of hydrogen, which has been speculated as a mechanism that stabilizes metastable infinite-layer phase32.
As seen in the light-element sensitive iDPC-STEM image in Extended Data Fig. 3, no signature indicative of hydrogen incorporation was observed at the apical oxygen sites of the resulting SrFeO2 phase. As a comparison, the hydride reduction process, involving hydrogen ions reacting with oxygen ions to form H2O (ref. 33) or metallic hydroxide13, probably operates via a different atomic-scale mechanism than what we observed in this study. The hydrogen intercalation into the brownmillerite lattice destabilizes and extracts lattice oxygen during annealing, significantly impacting the energetics of diffusion pathways33. However, the reduction process, operating at low temperature, inherently involves oxygen ion conduction across an energy barrier within the brownmillerite lattice, controlled by reaction kinetics. Consequently, despite these differences, the kinetic pathway for oxygen conduction may still bear similarities.
HRTEM image simulations
The HRTEM image simulation was performed by using the abTEM Python package34 working with a multi-slice algorithm. The NCSI imaging condition used for the simulation is as follows: accelerating voltage 300 kV, spherical aberration coefficient Cs = −13 μm, chromatical aberration coefficient Cc = 1.6 mm, defocus Δf = 6.2 nm, convergence angle α = 0.5 mrad and focus spread Δfs = 2.4 nm. All simulated images have been calibrated by the modulation transfer function of the CCD camera (OneView, Gatan), which is the major factor causing the Stobbs-factor problem on simulated and experimental image matching35.
To determine the thickness of the TEM specimen used for NCSI HRTEM imaging, image simulation was carried out for SrFeO2.5 structure as a function of sample thickness (Supplementary Figs. 5 and 6). As a quantitative evaluation, the cross-correlation factor (XCF) was calculated for each simulated and experimental image. An XCF of ‘1’ indicates a perfect match. Judging from both the image contrast and the XCF value, the sample thickness of 10-unit-cell (5.6 nm) yields the best match with the experimental image as shown in Supplementary Fig. 5. Once the sample thickness was determined, the image simulation was conducted for various transient states predicted by DFT calculations using the same simulation setup.
Theoretical calculations
All calculations were performed using DFT with the plane-wave-based Vienna Ab initio Simulation Package (VASP)36,37,38. For the exchange-correlation function, the revised Perdew, Burke and Ernzerhof generalized gradient approximation (PBEsol) is adapted39. We used the projector-augmented wave potentials that include ten valence electrons for strontium (4s2, 4p6 and 5s2), eight for iron (3d7 and 4s1) and six for oxygen (2s2 and 2p4)40.
To investigate the dynamics of ions, we designed 1 × 2 × 6 SrFeO2.5 supercell. Energy barriers for the atomic shuffling and phase transformation to SrFeO2 were computed using the nudged elastic band method, implemented in the VASP code41. A plane-wave cut-off energy of 500 eV with a Monkhorst–Pack grid with 6 × 6 × 6 and 2 × 2 × 2 k-point meshes were used for the structural relaxations and supercell calculations, respectively42. We applied the onsite Hubbard U correction of 5 eV using the Dudarev formalism for the correlated iron d electrons43. We considered the G-type antiferromagnetic collinear spin ordering on the iron ions. All calculations were converged in energy to 10−5 eV per unit cell, and the structures were fully optimized with the force convergence of 10−1 eV Å−1.
Using the calculated energy for each shuffle step during the 90° reconfiguration of SrFeO2.5, we estimated the relative probability of observing each step (Extended Data Fig. 8a) given by Boltzmann distribution44
where Pi is the relative probability of observing step i, T is the temperature, kB is the Boltzmann constant and Ei is the enthalpy in each step. Relatively higher observation possibility can be recognized at the locations between configuration (6) and (7). We then carried out HRTEM image simulation for each configuration (Extended Data Fig. 8b) and cross-correlated them with all experimental HRTEM frame images from the in situ video (Supplementary Video 5) during the reconfiguration to find out the most likely configuration for each frame by estimating the XCF value.
The VO formation energy (\({E}_{{{\mathrm{vac}}}}^{\;f}\)) was calculated using
where Edefective is the energy of a structure with one VO, Eperfect is the energy of the perfect crystal structure and \({E}_{{{\mathrm{O}}}_{2}}\) is the energy of an oxygen molecule in the gas phase. The calculated VO formation energy has been compared with other oxides and summarized in Supplementary Table 2.
To investigate the effect of the structural phase transformation on the electronic and magnetic properties, we calculated the DOS at each reaction coordinate along the pathway from (1) to (3) shown in Fig. 4e. This analysis segmented the atomic model for each stage into three zones: SrFeO2, SrFeO2/SrFeO2.5 phase boundary and SrFeO2.5 (Supplementary Fig. 8). For these zones, the DOS calculations were performed at each stage. The results, detailed in Supplementary Figs. 9–12, reveal that only the SrFeO2/SrFeO2.5 phase boundary exhibits minor ferromagnetic metallic characteristics. In contrast, the regions of SrFeO2 and SrFeO2.5 predominantly exhibit G-type antiferromagnetic insulating properties, with the exception of tiny mid-gap states. Additionally, the DOS calculations indicate the persistence of G-type antiferromagnetic insulating properties throughout the atomic shuffling displacement leading to the 90° reconfiguration of VO channels (Supplementary Fig. 13).
To obtain EELS O-K edge spectra of SrFeO2.5 and SrFeO2, the dielectric function was calculated using the supercell core-hole method. The following imaginary part of the dielectric function (\({\epsilon }_{\alpha \beta }^{\left(2\right)}\left(\omega \right)\)) in the long wavelength limit (q = 0) is used for calculating the loss spectrum:
where \(\varOmega ,\,\varepsilon\) and \(M\) denote the unit cell volume, the orbital energies and the momentum matrix elements, respectively. \(\alpha\) and \(\beta\) denote the Cartesian indices of dielectric tensor. ω denotes the fequency, me, denotes the mass of electron, ẟ is the delta function, ħ is the reduced Planck’s constant, c is the speed of light and k is the wave vector. Here, we only consider in-plane components of the dielectric tensor due to the sample geometry of TEM sample. We applied a constant Gaussian broadening of 0.6 eV to the simulated EELS data to replicate the experimental data.
Data availability
The authors declare that all the theoretical atomic structures generated in this article have been included in Source data. All the raw HRTEM data supporting the major results are included in Supplementary Videos 1–3. Due to the heavy size, the raw format HRTEM videos are available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
The commercially available VASP software is used for the DFT calculations. The simulation settings are presented in Methods. Customized Python code for image simulation is available upon reasonable request.
Change history
07 April 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41557-025-01816-w
References
Tsujimoto, Y. et al. Infinite-layer iron oxide with a square-planar coordination. Nature 450, 1062–1065 (2007).
Hayward, M. A. & Rosseinsky, M. J. Materials chemistry: cool conditions for mobile ions. Nature 450, 960–961 (2007).
Kawakami, T. et al. Spin transition in a four-coordinate iron oxide. Nat. Chem. 1, 371–376 (2009).
Tassel, C. & Kageyama, H. Square planar coordinate iron oxides. Chem. Soc. Rev. 41, 2025–2035 (2012).
Köhler, J. Square-planar coordinated iron in the layered oxoferrate(II) SrFeO2. Angew. Chem. Int. Ed. 47, 4470–4472 (2008).
Hodges, J. P. et al. Evolution of oxygen-vacancy ordered crystal structures in the perovskite series SrnFenO3n−1 (n=2, 4, 8, and ∞), and the relationship to electronic and magnetic properties. J. Solid State Chem. 151, 190–209 (2000).
Takeda, Y. et al. Phase relation in the oxygen non-stoichiometric system, SrFeOx (2.5 ≤ x ≤ 3.0). J. Solid State Chem. 63, 237–249 (1986).
Rahman, M., Nie, Y. Z. & Guo, G. H. Electronic structures and magnetism of SrFeO2 under pressure: a first-principles study. Inorg. Chem. 52, 12529–12534 (2013).
Kanai, M., Kawai, T. & Kawai, S. Atomic layer and unit cell layer growth of (Ca,Sr)CuO2 thin film by laser molecular beam epitaxy. Appl. Phys. Lett. 58, 771–773 (1991).
Li, D. et al. Superconductivity in an infinite-layer nickelate. Nature 572, 624–627 (2019).
Puphal, P. et al. Topotactic transformation of single crystals: from perovskite to infinite-layer nickelates. Sci. Adv. 7, eabl8091 (2021).
Kim, W. J. et al. Geometric frustration of Jahn–Teller order in the infinite-layer lattice. Nature 615, 237–243 (2023).
Yamamoto, T. & Kageyama, H. Hydride reductions of transition metal oxides. Chem. Lett. 42, 946–953 (2013).
Wei, W., Vu, D., Zhang, Z., Walker, F. J. & Ahn, C. H. Superconducting Nd1−xEuxNiO2 thin films using in situ synthesis. Sci. Adv. 9, eadh3327 (2023).
Ding, X. et al. Critical role of hydrogen for superconductivity in nickelates. Nature 615, 50–55 (2023).
Chroneos, A., Yildiz, B., Tarancón, A., Parfitt, D. & Kilner, J. A. Oxygen diffusion in solid oxide fuel cell cathode and electrolyte materials: mechanistic insights from atomistic simulations. Energy Environ. Sci. 4, 2774–2789 (2011).
Inoue, S. et al. Anisotropic oxygen diffusion at low temperature in perovskite-structure iron oxides. Nat. Chem. 2, 213–217 (2010).
Inoue, S. et al. Single-crystal epitaxial thin films of SrFeO2 with FeO2 ‘infinite layers’. Appl. Phys. Lett. 92, 4–6 (2008).
Jia, C. L., Lentzen, M. & Urban, K. Atomic-resolution imaging of oxygen in perovskite ceramics. Science 299, 870–873 (2003).
Jia, C. L., Houben, L., Thust, A. & Barthel, J. On the benefit of the negative-spherical-aberration imaging technique for quantitative HRTEM. Ultramicroscopy 110, 500–505 (2010).
Du, H. et al. Multiple polarization orders in individual twinned colloidal nanocrystals of centrosymmetric HfO2. Matter 4, 986–1000 (2021).
Jia, C. L. et al. Determination of the 3D shape of a nanoscale crystal with atomic resolution from a single image. Nat. Mater. 13, 1044–1049 (2014).
Khare, A. et al. Topotactic metal-insulator transition in epitaxial SrFeOx thin films. Adv. Mater. 37, 1606566 (2017).
Kang, K. T. et al. A room‐temperature ferroelectric ferromagnet in a 1D tetrahedral chain network. Adv. Mater. 31, 1808104 (2019).
Mitra, C., Meyer, T., Lee, H. N. & Reboredo, F. A. Oxygen diffusion pathways in brownmillerite SrCoO2.5: influence of structure and chemical potential. J. Chem. Phys. 141, 4–9 (2014).
Xing, Y. et al. Atomic-scale operando observation of oxygen diffusion during topotactic phase transition of a perovskite oxide. Matter 5, 3009–3022 (2022).
Li, B. & Ma, E. Atomic shuffling dominated mechanism for deformation twinning in magnesium. Phys. Rev. Lett. 103, 035503 (2009).
Tochigi, E., Miao, B., Nakamura, A., Shibata, N. & Ikuhara, Y. Atomic-scale mechanism of rhombohedral twinning in sapphire. Acta Mater. 216, 117137 (2021).
He, Y., Li, B., Wang, C. & Mao, S. X. Direct observation of dual-step twinning nucleation in hexagonal close-packed crystals. Nat. Commun. 11, 2483 (2020).
Feldhoff, A. et al. Spin-state transition of iron in (Ba0.5Sr0.5)(Fe0.8Zn0.2)O3-δ perovskite. J. Solid State Chem. 182, 2961–2971 (2009).
Haruta, M. et al. Local electronic structure analysis for brownmillerite Ca(Sr)FeO2.5 using site-resolved energy-loss near-edge structures. J. Appl. Phys. 110, 033708 (2011).
Ferreira, T. et al. Unintended consequence of topochemical reduction of SrFeO3 to SrFeO2: design of infinite layered oxides. Phys. Rev. Mater. 5, 123401 (2021).
Li, H.-B. et al. Dehydration of electrochemically protonated oxide: SrCoO2 with square spin tubes. J. Am. Chem. Soc. 143, 17517–17525 (2021).
Madsen, J. & Susi, T. abTEM: ab initio transmission electron microscopy image simulation. Microsc. Microanal. 26, 448–450 (2020).
Thust, A. High-resolution transmission electron microscopy on an absolute contrast scale. Phys. Rev. Lett. 102, 5–8 (2009).
Tong, B. Y. & Sham, L. J. Application of a self-consistent scheme including exchange and correlation effects to atoms. Phys. Rev. 144, 1–4 (1966).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. J. Phys. Chem. A 54, 11169 (1996).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, 864 (1964).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 1–4 (2008).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Dudarev, S., Botton, G., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA + U study. Phys. Rev. B 57, 1505–1509 (1998).
Zheng, H. et al. Dynamics in a Cu2S nanorod. Science 333, 206–209 (2011).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (no. NRF-2020R1A2C2101735), Samsung Research Funding and Incubation Center of Samsung Electronics under project number SRFC-MA1702-01, Creative Materials Discovery Program (NRF-2019M3D1A1078296) and the KENTECH Research Grant (KRG2022-01-019). J.K.L. acknowledges the support of an NRF grant funded by the Korean government (NRF-2023R1A2C1003900). K.T.K. acknowledges the support of NRF grant funded by the Korean government (NRF-2022R1C1C1005168) and the Learning and Academic research institution for Master’s·PhD students, and Postdocs (LAMP) Program of the grant funded by the Ministry of Education (RS-2023-00301914). W.S.C. acknowledges the support of NRF grant funded by the Korean government (NRF-2021R1A2C2011340 and RS-2023-00220471). The TEM work at Korea Institute of Energy Technology (KENTECH) was supported by KENTECH Center for Shared Research Facilities.
Author information
Authors and Affiliations
Contributions
S.H.O. conceived the project; Y.X. conducted the TEM experiment and analysis under the supervision of S.H.O.; I.K. J.B. and J.L. performed DFT calculations; K.T.K. and W.S.C. conducted thin film growth; all authors contributed to the interpretation of data and visualization of results; Y.X. and S.H.O. prepared the paper, which was reviewed and edited by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Dangfeng Li, Yuichi Shimakawa and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Orientation of 1-D VO channels in the TEM samples of epitaxial SrFeO2.5 (001) film.
a, X-ray θ–2θ diffraction pattern of SrFeO2.5 epitaxial thin film grown on SrTiO3 (001) substrate. b, Schematic illustration of the two orthogonal sampling directions for TEM specimens, which are the [100] (green) and the [010] (purple). c and d, Crystal structure and HRTEM image of SrFeO2.5 along the [100] and the [010] zone axis, respectively. The VO channels seen in end-on projection are indicated by yellow rectangles in the [010] sample. e and f, Schematic illustration of the orientation of VO channels in the TEM specimens prepared along the [100] and the [010] zone axis, respectively. The VO channels are aligned laterally in the [100] and normally in the [010] samples. h is the film thickness of SrFeO2.5 (001) film. l and t is the width and the thickness of TEM specimen, respectively.
Extended Data Fig. 2 Nucleation of SrFeO2 phase within SrFeO2.5 matrix.
a, Atomic structure of SrFeO2.5. The alternate stack of octahedral and tetrahedral layers is indicated by yellow and red dotted lines, respectively. b, Fluctuation of the oxygen arrangement in the tetrahedral layers of SrFeO2.5 prior to nucleation of SrFeO2 as depicted by the interruption of the red lines by yellow lines. This temporal fluctuation in the oxygen arrangement is attributed to oxygen conduction along the VO channels and escape to the vacuum. c, Time-series HRTEM images showing the nucleation of SrFeO2 phase. The nanometer-sized nucleus is indicated by elliptical circle. d, GPA strain maps of the HRTEM images shown in c. SrFeO2 phase is distinguished from SrFeO2.5 matrix as the compressively strain (blue color) in the out-of-plane maps due to the smaller lattice parameter.
Extended Data Fig. 3 Atomic and Electronic structures of SrFeO2.5 and SrFeO2.
a and b, iDPC STEM image and intensity profile of pristine SrFeO2.5 ([110]) and transformed SrFeO2 ([010]) phase, respectively. Compared with SrFeO2.5, all the apical oxygen are missing in SrFeO2 as indicated by arrows. The apical oxygen absent in SrFeO2 is delineated by yellow dashed circles in the atomic structure. c, EELS Fe-L2,3 and d, O-K edge from SrFeO2 (green), SrFeO2.5 tetrahedral layer (orange), and SrFeO2.5 octahedral layer (blue). The chemical shift of the Fe-L2,3 edge of the SrFeO2 to a lower energy ( ~ 1.5 eV from that of SrFeO2.5 octahedral layer) indicates the reduction of the iron valence to 2+ . In the O-K edges the peak a of the SrFeO2, which originates from the transition to the O 2p-Fe 3d orbital hybridized state, shows the shift to a higher energy, resulting in a narrower separation with peak b. Besides, the peak b and c of the SrFeO2, which are attributed to the transition to O 2p-Sr 4d and O 2p-Fe 4p hybridized state, respectively, exhibit a reduced intensity level compared to those of SrFeO2.5. e, Total and projected density of states (DOS) of SrFeO2.5 and SrFeO2. From the DOS, it is apparent that the conduction band position of SrFeO2 is higher by about 2 eV than that of SrFeO2.5. The large band splitting of bonding and anti-bonding state is the common feature of strong hybridization. f, Simulated EELS O-K edges of SrFeO2.5 and SrFeO2. The vertical lines mark the energy of the first peak, guiding the chemical shift between each O-K edge.
Extended Data Fig. 4 Structural characterization of SrFeO2.5 and SrFeO2.
a, HRTEM images of SrFeO2.5 [100] (left), SrFeO2.5 [010] (middle), and SrFeO2 [110] (right). b, Interlayer strontium-strontium d-spacing, dSr-Sr, and c, intralayer iron-iron distance, δFe-Fe, measured directly on the HRTEM images. The dSr-Sr and δFe-Fe maps are displayed in the same order as in the HRTEM images. In the dSr-Sr maps of both SrFeO2.5 [010] and [100], the tetrahedral and octahedral layers are distinguished by red (4.2 Å) and blue (3.7 Å) stripe signals. The dSr-Sr map of SrFeO2 exhibit uniform blue signal corresponding to 3.5 Å. Only the δFe-Fe map of SrFeO2.5 [010] where the VO channels are aligned end-on shows a chequerboard pattern of 2 Å (navy) and 3.9 Å (yellow), where the latter corresponds to the VO channels. The δFe-Fe map of SrFeO2.5 [100] and SrFeO2 [110] show a uniform 2.7 Å (green).
Extended Data Fig. 5 HRTEM image analysis showing morphological development of SrFeO2.5-SrFeO2 phase boundary and strained lattice matching – SrFeO2.5 [100] sample.
a, Time series HRTEM images during phase transformation of SrFeO2.5 [100] sample at 450 °C (Supplementary Video 1). The phase boundary overlaid on each image was determined by the dSr-Sr measurement. b, Map of interplanar spacing of the strontium layers, dSr-Sr. The atomically smooth phase boundary advances via lateral migration of multiple ledges in a step-flow like manner. The phase transformation completes within 30 s. Geometrical phase analysis (GPA) of the HRTEM images displaying: c, In-plane strain maps (εxx); d, Out-of-plane strain maps (εzz); e, Lattice rotation maps (Rxz). The two phases maintain highly commensurate lattice across the phase boundary, as seen by the small but continuous in-plane strain distribution. The contraction of the out-of-plane lattice of SrFeO2 as oxygen is removed from SrFeO2.5 is clearly visible in the out-of-plane strain map. The lattice bending over the ledges accommodates the large mismatch in the out-of-plane lattice parameters between the two phases, resulting in the opposite sign of the lattice rotation in SrFeO2.5 and SrFeO2.
Extended Data Fig. 6 Anisotropy in the phase transformation rate of SrFeO2.5 depending on the orientation of VO channels.
a, Schematic model illustrating three different routes of phase transformation of SrFeO2.5 to SrFeO2 in terms of the orientation of VO channels; transformation of SrFeO2.5 [100] with or without 90°-reorientation, and direct transformation of SrFeO2.5 [010] to SrFeO2 [110]. b, dSr-Sr measurement during phase transformation of three different cases as a function of time. SrFeO2.5 has dSr-Sr of 4.2 Å (tetrahedral layers) and SrFeO2 has dSr-Sr value of 3.5 Å. Red and blue profiles are the measurement from SrFeO2.5 [100] sample undergoing the phase transformation with (red) or without (blue) 90°-reconfiguration, respectively. Orange profile is from SrFeO2.5 [010] sample. The dSr-Sr values in the plot represent the average of measurements from the nine unit cells, with error bars indicating the standard deviation. The SrFeO2.5 [010] sample (orange) shows the highest transformation rate of 1.78 u.c. s-1. The SrFeO2.5 [100] sample without 90°-reorientation shows the lowest transformation rate of 0.64 u.c. s-1. The SrFeO2.5 [100] sample with 90°-reorientation exhibits an intermediate transformation rate of 1.05 u.c. s-1. A distinct step locating at around 3.85 Å can be recognized at blue and red profiles, indicating the existence of transient state. This step is hardly detected in the SrFeO2.5 [010] sample due to the fast transformation.
Extended Data Fig. 7 90°-reconfiguration of SrFeO2.5 [100] ahead of advancing phase boundary.
a, Time sequence HRTEM images showing the phase transformation of SrFeO2.5 [100] to SrFeO2 [110]. White dashed line in each image indicates the phase boundary. b, Magnified images of the phase boundary outlined yellow in a. The structural features inherent to the SrFeO2.5 [010] appeared as a consequence of 90°-reconfiguration, that is, the AFD rotation (β ~±7°) of FeO6 in the octahedral layers and the VO channels (δFe-Fe = 3.9 Å) in the tetrahedral layers. c, Interlayer d-spacing map of the strontium layers, dSr-Sr, for phase identification. Alternating red (tetrahedral layers) and blue (octahedral layers) stripes correspond to SrFeO2.5 while uniform blue region corresponds to SrFeO2 phase. d, Intralayer Fe-Fe distance δFe-Fe map visualizing the normally aligned VO channels. The chequerboard pattern appearing in the SrFeO2.5 [100] ahead of moving phase boundary indicates 90°-reconfiguration which brings the laterally aligned VO channels to the normally aligned one. Uniform green color far below the phase boundary indicates the pristine SrFeO2.5 [100] without reorientation.
Extended Data Fig. 8 Atomic models for 90°-reconfiguration of SrFeO2.5 by atomic shuffle displacements.
a, Twelve successive atomic configurations in the course of 90°-reconfiguration of SrFeO2.5 [100] to [010] by atomic shuffle displacements (Supplementary Video 8). Each oxygen and iron are displaced by different magnitude of displacement vector rather than single shear displacement. The two end configurations with the reaction coordinate (1) and (12) correspond to the SrFeO2.5 [100] and [010], respectively. The atomic models were simulated using the first principles nudged elastic band method by referring to the in-situ HRTEM images. b, Simulated HRTEM images of the atomic models shown in a. The simulated HRTEM images were used to validate the atomic models by cross-correlating with the experimental HRTEM images. Among them, model (1), (6) and (12) were cross-correlated with state (i), (ii) and (iii) in Fig. 2a, respectively. The XCFs higher than 0.9 were obtained for all atomic model structures. c, DFT calculation on the duration time of the twelve structural configurations represented by the reaction coordinate (1) to (12). The structural models for the reaction coordinates were determined by cross-correlation of their simulated HRTEM images with experimental HRTEM images. The reaction coordinate (6) which corresponds to the transient state (ii) stays the longest time. d, Energy landscape (blue) along the reaction coordinates. The orange profile represents the probability of each coordinate in Boltzmann distribution (Methods).
Extended Data Fig. 9 Atomic-scale mechanism of oxygen release and rearrangement – Atomic models from DFT calculation.
a, Atomic structures obtained by DFT calculation (same as in Fig. 3d) corresponding to the transient states observed by in-situ HRTEM during one unit event of phase transformation. b, Simulated HRTEM images using the atomic structures in a. AFD rotation and polar distortion arises in (i) and (ii) state, respectively, indicated by arrows. c, SrO column intensity measurement of all four SrO layers in b. The intensity values in the plot represent the average of measurements from the lateral four SrO columns, with error bars indicating the standard deviation. d, Intensity measurement of each atomic site in tetrahedral layer. Red and blue data points represent the intensity measured at the VO channels and FeO columns, respectively. The intensity variation measured on the simulated HRTEM images match well the experimentally measured one in Fig. 3. e, Tracing of the intralayer Fe-Fe distance (δFe-Fe, defined in b and Fig. 3a) across VO channel with time in experimental data. f, Tracing of the strontium interlayer d-spacing (dSr-Sr, defined in b and Fig. 3a) with time in experimental data (Supplementary Video 11). The measured average values are indicated by black dashed lines, which distinguish the four distinct steps with different duration time. The dSr-Sr measured from the corresponding atomic models simulated by DFT (brown dashed lines) are added for comparison.
Extended Data Fig. 10 Trajectory analysis of atom movements during the phase transformation.
a, Atomic models obtained by DFT and b, HRTEM images captured during the phase boundary migration. These four sets of the atom configuration are the same as those presented in Fig. 3 of the main text. c, Atomic trajectory analysis of atoms of outlined region in b, during the phase transformation. d, Enlarged view of the atomic trajectory of outlined region in c. The “reference” atom marked in c is used to correct the drift, so that its motion trace is minimal. Trajectory was color-corded from the reference (t0) to 4.4 s. The trajectory map reveals the highly discrete nature of strontium, iron, oxygen atom movements during the phase boundary migration from (i) to (iv), characterized by sudden jump of atom position rather than diffusive motion. Such discrete atomic motions involving a large amount of sudden displacement along a specific direction are accounted for by the oxygen conduction via changes in the oxygen coordination around iron atoms within FeOx polyhedral layers.
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–13, Tables 1 and 2 and References.
Supplementary Video 1 (download MP4 )
HRTEM video showing the phase transformation from SrFeO2.5 [100] to SrFeO2 [110] at 450 °C. NCSI HRTEM video showing the migration of the phase boundary at 450 °C. The SrFeO2 phase initiates from the film surface and grows towards the interface with the SrTiO3 substrate as the phase boundary advances. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged.
Supplementary Video 2 (download MP4 )
HRTEM video showing the phase transformation from SrFeO2.5 [010] to SrFeO2 [110] at 450 °C. NCSI HRTEM video showing the migration of the phase boundary at 450 °C. The SrFeO2 phase initiates from the interface with the SrTiO3 substrate and grows towards the film surface as the phase boundary advances. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged.
Supplementary Video 3 (download MP4 )
HRTEM video showing the nucleation of SrFeO2 at 450 °C. NCSI HRTEM video showing the nucleation stage of the phase transformation from SrFeO2.5 [100] to SrFeO2 [110]. Before the nucleation of SrFeO2 phase, the oxygen configuration in the tetrahedral layers of SrFeO2.5 fluctuates, occasionally exhibiting the contrast similar to the octahedral layers. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged.
Supplementary Video 4 (download MP4 )
HRTEM video showing the lattice dynamics before the phase transformation. NCSI HRTEM video exhibiting the atomistic details of the phase transformation from SrFeO2.5 [100] to SrFeO2 [110]. Before the phase transformation, active lattice dynamics, such as lateral vibrations of atoms in tetrahedral layers and reorientation of Vo channels, are observed in SrFeO2.5. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged.
Supplementary Video 5 (download MP4 )
Magnified HRTEM video showing the 90° reorientation of VO channels. NCSI HRTEM video showing the 90° reorientation of VO channels before the phase transformation of SrFeO2.5 [100] sample. Initially, the iron and equatorial oxygen in octahedral layers are almost coplanar, and the iron in tetrahedral layers remains isometric. As the reorientation progresses, AFD rotation evolves in the octahedral layers, with a rotation angle β of approximately ±7˚, and the intralayer iron distance, δFe–Fe, in the tetrahedral layers alternates between 2.0 and 3.9 Å. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged.
Supplementary Video 6 (download MP4 )
Iron intralayer distance traced during 90° reorientation of VO channels. The intralayer iron–iron distance, δFe–Fe, is measured on the two adjacent unit cells in Supplementary Video 6. The initially uniform δFe–Fe values undergo a split into smaller (δFe–Fe_1) and larger (δFe–Fe_2) values through a transient state (ii), with the latter corresponding to the vertically aligned VO channel.
Supplementary Video 7 (download MP4 )
Dichromatic pattern showing 90° reorientation of VO channels by atomic shuffle displacement. Animated schematic illustrates the reorientation of VO channels situated between tetrahedral chains through atomic shuffle displacement, which is viewed along the [001] direction. Initially, the positions of iron and oxygen are represented by open circles and open squares, respectively, before the reorientation. After the reorientation, the positions of iron and oxygen are indicated by filled pink circles and squares, respectively. The VO channels are depicted by large open arrows. At the end of the animation, the shuffle displacement of each atom is defined by overlapping the two atomic arrangements and connecting the position of each atom.
Supplementary Video 8 (download MP4 )
DFT simulated sequential atomic models showing the 90° reorientation of VO channels by atomic shuffle displacement. On the left, animated atomic model shows each reaction coordinate during the atomic shuffle displacement for the 90° reorientation of VO channels, simulated using DFT. On the right, the corresponding HRTEM image simulations are presented.
Supplementary Video 9 (download MP4 )
High-magnification HRTEM video showing the atomic-scale mechanism of phase transformation. The HRTEM video captures the phase transformation starting from the reoriented SrFeO2.5 phase. To enhance the contrast of the original video, which was recorded at 25 fps, the three successive frames were averaged. Additionally, for clarity, the original video has been slowed down by 4 times.
Supplementary Video 10 (download MP4 )
DFT-simulated sequential atomic models showing atomic phase transformation mechanism. On the left, the animated atomic model shows the transient states with different polyhedral configurations observed by in situ HRTEM during one unit event of phase transformation, simulated by DFT. On the right, the corresponding HRTEM image simulations are presented.
Supplementary Video 11 (download MP4 )
Stepwise phase transformation visualized by tracing the strontium interlayer d-spacing during phase transformation. The interlayer strontium d-spacing, dSr–Sr, is traced throughout the phase transformation process. The dashed lines represent the measured average values, accompanied by a standard deviation of approximately ±0.001 Å. These lines delineate the four distinct steps, each lasting on the level of second. Additionally, the dSr–Sr values obtained from corresponding atomic models simulated by DFT are depicted as dashed brown lines.
Source data
Source Data Fig. 2 (download XLSX )
Source data of Fig. 2.
Source Data Fig. 3 (download XLSX )
Source data of Fig. 3.
Source Data Fig. 4 (download XLSX )
Source data of Fig. 4.
Source Data Extended Data Fig. 8 (download XLSX )
Source data of Extended Data Fig. 8.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xing, Y., Kim, I., Kang, K.T. et al. Monitoring the formation of infinite-layer transition metal oxides through in situ atomic-resolution electron microscopy. Nat. Chem. 17, 66–73 (2025). https://doi.org/10.1038/s41557-024-01617-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41557-024-01617-7
