
Abstract
Advances in electrochemical devices have been primarily driven by the discovery and development of electrolyte materials. Yet the development of high-performance and chemically stable proton-conducting oxide electrolytes remains a challenge due to proton trapping and the resulting trade-offs between ionic carrier concentration and conductivity in doped oxides. Here we demonstrate that cubic perovskite oxides with heavy Sc doping can overcome these limitations. BaSn0.3Sc0.7O3–δ and BaTi0.2Sc0.8O3–δ are found to exceed the technological threshold of a total proton conductivity of 0.01 S cm−1 for fuel cell electrolytes at 300 °C. The structural stability of BaSn0.3Sc0.7O3–δ is further validated under harsh chemical and fuel cell conditions. Molecular dynamics simulations using a machine learning force field illustrate rapid proton diffusion pathways along the ScO6 octahedral network, effectively mitigating proton trapping, while protons are preferentially associated with Sc. Lattice softness is proposed as a primary design descriptor for increasing Sc content in perovskite oxides and developing high-performance electrolytes for electrochemical devices.
Similar content being viewed by others
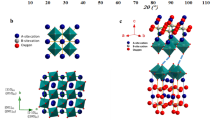

Main
The discovery and development of novel electrolyte materials has historically led to advancements in electrochemical devices1,2,3,4,5. Examples include lithium-ion conduction in the Li–Ge–P–S system4 and proton conduction in SrCe0.95Yb0.05O3−δ perovskite oxide1, which have paved the way for all-solid-state batteries5, intermediate-temperature proton ceramic fuel cells6 and electrolysers for fuel production7. An ideal electrolyte should exhibit both high ionic conductivity and enduring chemical and electrochemical stability during device operation. However, meeting these criteria remains a challenge. Liquid electrolytes8 offer high ionic conductivity but can compromise device integrity owing to potential leakage. By contrast, a solid electrolyte eliminates such risks but requires overcoming substantial activation barriers for ionic carrier migration. Hydride-conducting compounds9 offer a conductivity of 0.01 S cm−1 between room temperature and 400 °C but are unstable under oxidizing conditions. Proton-conducting CsH2PO4 fulfils the requirements within only a narrow operating temperature and water solubility window of 233–254 °C at a water partial pressure of 0.4 atm (ref. 10), limiting its broader application. Currently, no stable solid-state and/or protonic electrolytes exist that can operate under fuel cell and electrolyser conditions at 300 °C.
Proton-conducting perovskite oxides11,12 exhibit a high proton conductivity of 0.01 S cm−1 at 450 °C with strong chemical stability, as reported for Y-doped barium zirconate2,13. Perovskite oxides with a cubic or pseudo-cubic structure represented by the general chemical formula ABO3, where A and B are cations, are characterized by a network of corner-sharing BO6 octahedra with A-cation sublattices. Upon doping with cations of lower valency (that is, acceptor doping), usually on the B site, oxygen vacancies are generated to maintain charge neutrality, as represented using the Kröger–Vink notation:
These oxygen vacancies are filled with hydroxyl groups when exposed to moisture (hydration reaction):
The incorporated protons are often strongly associated with acceptor dopants14 that reside on the nearest or second nearest neighbour oxygen to the acceptor dopant14,15,16,17:
Further details of this general defect chemistry for acceptor-doped perovskite oxides can be found in Supplementary Section 1. This association effect acts to stabilize the hydrated state16, where the maximum proton content equals the amount of acceptor dopant in the case of a trivalent cation substituting at a tetravalent cation site. The protons can diffuse throughout the material via a combination of rotation and hopping to neighbouring proton sites (Grotthuss mechanism11) on the three-dimensional network of BO6 octahedra.
The development of proton-conducting perovskite oxides has followed a design principle aimed at enhancing proton conductivity by increasing lattice volume through the incorporation of larger host cations and acceptor dopants within the perovskite structure11 (Fig. 1a). However, the proton conductivity in barium zirconate peaks at a Y-doping level of 20 at.% (refs. 2,18), substantially below its solubility limit of approximately 50 at.% (ref. 19), and any further increase in Y doping reduces its conductivity20, which implies that carrier diffusivity also decreases (Fig. 1b,d). This has conventionally been explained as the detrimental effect of the proton–dopant association leading to proton trapping14,16. More recent theoretical works have pointed out the formation of deeper proton traps surrounded by Y clusters as the underlying mechanism for the decrease in conductivity at high dopant concentrations21. This trade-off is inherent in proton-conducting oxides, where the proton–dopant association stabilizes protons in oxides, enabling the hydration of oxides but impeding proton diffusion in the lattice (proton trapping), as found for proton-conducting barium zirconates2,14,18,20,22. A similar phenomenon is also observed for oxide-ion conduction in doped zirconia and ceria23.

a, Proton conductivity in bulk (σH.bulk) plotted against lattice volume. b, Proton conductivity in bulk versus dopant content (x). c, Proton concentration (CH) versus x. d, Proton diffusivity in bulk (DH.bulk) versus x. e, Apparent activation energy for proton diffusivity (Qapp). f, Proton diffusivity in bulk plotted against proton concentration. The lattice volumes of BSS70 and BTS80 in a are derived from Supplementary Table 1. The apparent activation energies in e were determined in the temperature range between 200 °C and 300 °C. The yellow region in a shows materials that meet the technological threshold26 of electrolyte for solid-state electrochemical devices. The vertical dashed lines in b–e show the solubility limits for Sc in each system. The diagonal dashed line in c shows the theoretical maximum proton content. The proton diffusivity for Y-doped barium zirconates in d and f was obtained using the Nernst–Einstein relationship and reported values for proton conductivity2,18, proton concentration39 and lattice volume39. Error bars in e represent the standard error of the slope obtained from the least-squares fits of proton diffusivities in the Arrhenius plots, using 9, 8, 6, 8, 6, 8, 11, 8 and 10 data points for BaSn0.3Sc0.7O3−δ, BaSn0.8Sc0.2O3−δ, BaTi0.2Sc0.8O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ, BaHf0.8Sc0.2O3−δ, BaZr0.4Sc0.6O3−δ, BaZr0.8Sc0.2O3−δ and BaZr0.8Y0.2O3−δ, respectively. Diffusivity datasets for BaZr0.4Sc0.6O3−δ (ref. 3), BaZr0.8Sc0.2O3−δ (ref. 3) and BaZr0.8Y0.2O3−δ (ref. 14) were reproduced from the respective publications. Bulk proton conductivities (σH) in f are presented with a corresponding colour bar; the solid contour lines show proton conductivities of 3.6 × 10−3 S cm−1 and 3.6 × 10−4 S cm−1. Abbreviations are as follows: BSS70 for BaSn0.3Sc0.7O3−δ, BSS20 for BaSn0.8Sc0.2O3−δ, BTS80 for BaTi0.2Sc0.8O3−δ, BTS60 for BaTi0.4Sc0.6O3−δ, BZS60 for BaZr0.4Sc0.6O3−δ (ref. 3), BZS20 for BaZr0.8Sc0.2O3−δ (ref. 3), BZY20 for BaZr0.8Y0.2O3−δ (refs. 2,39), BZY10 for BaZr0.9Y0.1O3−δ (refs. 18,39), BZY30 for BaZr0.7Y0.3O3−δ (refs. 18,39), SCYb5 for SrCe0.95Yb0.05O3−δ (refs. 40,41), BCY10 for BaCe0.9Y0.1O3−δ (refs. 42,43), BCY20 for BaCe0.8Y0.2O3−δ (refs. 42,43) and BCZYYb7111 for BaCe0.7Zr0.1Y0.1Yb0.1O3−δ (refs. 44,45). The proton contents given for the compounds are determined using thermogravimetry.
The recent discovery of superior proton conduction in heavily (60 at.%) Sc-doped barium zirconate3 offers a promising avenue for overcoming the trade-off in doped ionic conductors. In BaZr0.4Sc0.6O3–δ, the proton concentration, diffusivity and conductivity all increase continuously up to approximately the solubility limit (Fig. 1b–d). It is proposed that heavy acceptor doping facilitates proton diffusion in barium zirconates and analogous materials, based on computations24 and experiments3,25. These results suggest that heavy Sc doping promises to overcome the trade-off in doped ionic conductors.
In this study we performed systematic searches for Ba-based host perovskite oxides capable of dissolving substantial amounts of Sc. We found that heavy Sc doping, up to 70, 80 and 50 at.%, is possible in barium stannate, barium titanate and barium hafnate, respectively. The resultant total proton conductivities in the barium stannate and barium titanate exceed the technological threshold26 of 0.01 S cm−1 for fuel cell electrolytes at 300 °C (Fig. 1a), overcoming the well-known and problematic H+ concentration–conductivity trade-off in doped oxides. Doped barium stannates27,28 and barium titanates29 have long been overlooked due to their lower proton conductivities and less favourable hydration behaviour at dopant contents up to ~20 at.%, and proton conductivity investigations under heavy doping close to the solubility limit had not been attempted. Molecular dynamics simulations employing state-of-the-art machine learning force fields reveal that the heavy Sc doping creates proton diffusion pathways along the ScO6 octahedral network that mitigate the influence of proton trapping on diffusion. A correlation between reduced bulk modulus and increased ScO6 content in the perovskite framework provides a design guideline for developing such electrolytes.
Heavy Sc doping of BaSnO3, BaTiO3 and BaHfO3 perovskites
The 20 at.% and 70 at.% Sc-doped barium stannates, 60 at.% and 80 at.% Sc-doped barium titanates and 20 at.% and 50 at.% Sc-doped barium hafnates were synthesized using solid-state reactions. Powder X-ray diffraction analysis and Rietveld refinement for dehydrated BaSn0.3Sc0.7O3−δ, BaTi0.2Sc0.8O3−δ, BaSn0.8Sc0.2O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ and BaHf0.8Sc0.2O3−δ (Supplementary Fig. 1) show that these compounds primarily exhibit cubic perovskite phases in the space group \({Pm}\bar{3}m\) (structural parameters are provided in Supplementary Table 1). BaSn0.3Sc0.7O3−δ, BaTi0.2Sc0.8O3−δ and BaHf0.5Sc0.5O3−δ contain 5.1 wt%, 2.3 wt% and 5.3 wt%, respectively, of secondary BaSc2O4. By using inductively coupled plasma optical emission spectrometry (ICP-OES) and considering the secondary phase, the chemical composition of BaSn0.3Sc0.7O3−δ perovskite was determined to be Ba1.00Sn0.311Sc0.695O3−δ (Supplementary Table 2). The substitutional acceptor Sc doping and vacancy formation for barium stannates, titanates and hafnates, described in equation (1), are validated by the monotonic increase or decrease of lattice volume upon Sc doping (Supplementary Fig. 2).
Proton conductivities from a.c. impedance
The a.c. impedance spectroscopy, conducted under a humidified Ar atmosphere, reveals that heavy Sc doping at levels of 70 at.% and 80 at.% in stannate and titanate, respectively, results in fast proton conduction at 300 °C. For 20 at.% Sc-doped barium stannate and 60 at.% Sc-doped barium titanate, the proton conductivity is only 0.001 S cm−1 at 300 °C (Fig. 1b and Supplementary Figs. 3–8). However, increasing the Sc content to 70 at.% and 80 at.% enhances the proton conductivity by a factor of ten, reaching 0.01 and 0.016 S cm−1, respectively (Fig. 1b). This tenfold increase represents a substantial improvement compared to the Y-doped barium zirconate system, which peaks at a dopant level of 20 at.% Y (refs. 2,18). Within the Sc-doped systems, barium stannate outperforms barium hafnate and barium zirconate3 in proton conductivity for all Sc contents by a factor of 2–30 and 2–4, respectively (Fig. 1b). The temperature dependence of the proton conductivity in the stannates, titanates and hafnates (Supplementary Fig. 9) also highlights the advantage of heavy Sc doping regardless of operating temperature.
BaSn0.3Sc0.7O3–δ and BaTi0.2Sc0.8O3–δ exhibit high total proton conductivities, exceeding 0.01 S cm−1, at and above 300 °C under a water partial pressure of 0.02 atm (Fig. 2a and Supplementary Fig. 15). The total conductivity comprises both bulk and grain boundary resistivities in the polycrystalline pellet. Large grains with a mean grain size of 16.7 μm for BaSn0.3Sc0.7O3–δ (Supplementary Fig. 16) minimize the resistive contribution across grain boundaries to the overall proton resistivity in the polycrystalline pellet.

a, Comparison of total ionic conductivity (σion.total) with those of conventional electrolytes10,46,47 used in fuel cells. T, temperature. b, H–D isotope effect at 300 °C, shown by total conductivity (σtotal) differences. c, Total conductivity plotted against the log of oxygen partial pressure, \(p_{{\rm{o}}_2}\), at 300 °C. d, Proton diffusivity. The dashed line in a shows the technological threshold26 of electrolytes for solid-state electrochemical devices. The partial pressure of light and heavy water in b was 0.02 atm. For the ab initio molecular dynamics calculations in d, deuterium was used instead of hydrogen. The data for 8YSZ (yttria-stabilized zirconia with a mole fraction of 8 mol% Y2O3 (ref. 48)), BaCe0.7Zr0.1Y0.1Yb0.1O3−δ (ref. 47), CsH2PO4 (ref. 10), Nafion 117 (ref. 46), BaZr0.4Sc0.6O3−δ (ref. 3) and BaZr0.8Y0.2O3−δ (ref. 14) were taken from the literature.
Proton concentrations from thermogravimetry
The thermogravimetry analysis (Supplementary Fig. 10) provides compelling evidence that the defect chemistry and hydration processes of these materials adhere to the principles governing acceptor-doped perovskite oxide proton conductors, as described by equations (1) and (2). Proton concentration at 300 °C nearly equals the Sc content (Fig. 1c) for all perovskite oxides examined excluding barium titanates, indicating almost complete hydration in those systems. The hydration enthalpies for BaSn0.3Sc0.7O3−δ, BaSn0.8Sc0.2O3−δ, BaTi0.2Sc0.8O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ and BaHf0.8Sc0.2O3−δ are determined by van’t Hoff plots as −125 ± 1, −131 ± 3, −99 ± 5, −71 ± 2, −112 ± 2 and −80 ± 2 kJ mol−1, respectively (Supplementary Fig. 11 and Supplementary Table 3), which are in the range of reported values for acceptor-doped perovskite oxides30,12. When comparing among heavily Sc-doped samples, barium titanates exhibit a much lower proton concentration (Fig. 1c) and less negative hydration enthalpy (Supplementary Fig. 12) than barium stannate and barium zirconate. In situ Fourier transform infrared (FT-IR) spectroscopy for Sc-doped barium stannates and barium titanates (Supplementary Fig. 13) also provides direct evidence of the hydration reaction of equation (2).
Proton diffusivities
Sc-doped barium stannate displays high proton diffusivity across a wide range of Sc concentrations. By using the Nernst–Einstein relationship and measuring proton conductivities and concentrations (Fig. 1b,c), the proton diffusivities in barium stannates with 20 at.% and 70 at.% Sc at 300 °C were determined to be 1.9 × 10−7 and 3.5 × 10−7 cm2 s−1, respectively (Fig. 1d). These proton diffusivities are 2.3 to 3.4 times greater than those in Sc-doped zirconates3, depending on Sc content. By contrast, barium titanates display a steep Sc dependence in proton diffusivity, similar to that observed in barium zirconates. Sc-doped barium hafnate shows the steepest dependence of diffusivity on Sc content, but the Sc solubility of 50 at.% limits the proton diffusivity to 1 × 10−7 cm2 s−1. The temperature-dependent proton diffusivity (Supplementary Fig. 14) in the range between 200 °C and 300 °C provides apparent activation energies, plotted against Sc content in Fig. 1e. In these perovskites, Sc doping lowers the activation energy. The reduced activation energy (Fig. 1e) and enhanced proton diffusivity (Fig. 1d) appear to be a general advantage of heavy Sc doping in the perovskites. As a result, heavy Sc doping overcomes the trade-off between proton diffusivity and proton concentration reported in conventional systems, leading to a high conductivity of 0.01 S cm−1 at 300 °C in barium stannate and barium titanate (Fig. 1f).
Confirmation of proton conduction in BaSn0.3Sc0.7O3–δ
An isotope exchange experiment between D2O and H2O moisture at 301 °C, while maintaining a water partial pressure of 0.02 atm, yielded an isotope effect of 1.95 (Fig. 2b). This strongly suggests that protons are the primary ionic carriers. The higher value (~2) than the square root of the mass ratio between a deuterium and a proton, 1.41, is likely attributable to a more profound influence of the zero-point energy between them31 at 301 °C. Recent calculations that incorporate both nuclear quantum effects and anharmonicity report an isotope effect of 1.9 at 227 °C in barium zirconate32, which closely aligns with our experimental result in 70 at.% Sc-doped barium stannate at 300 °C. A continuous isotope exchange experiment over 261 h at 301 °C confirmed the chemical stability of proton insertion and extraction in BaSn0.3Sc0.7O3–δ (Fig. 2b).
A constant total conductivity across a wide range of oxygen partial pressures ranging from highly reducing to oxidizing conditions between 10−22 and 1 atm at 300 °C (Fig. 2c) indicates a proton transference number close to unity. As proof of ionic conduction, the open circuit voltage for a BaSn0.3Sc0.7O3–δ electrolyte-supported protonic ceramic fuel cell at 300 °C matches the theoretical value (1.194 V) calculated from the Nernst equation for the given conditions (Supplementary Fig. 17). An anode-supported fuel cell, consisting of a thin BaSn0.3Sc0.7O3 electrolyte that includes 5 at.% Ni (Supplementary Fig. 18), shows an open circuit voltage of approximately 1 V at 300 °C (Supplementary Fig. 19 and Supplementary Table 4). The equivalence of the X-ray photoelectron spectra for reduced BaSn0.3Sc0.7O3−δ and undoped BaSnO3 (ref. 33; Supplementary Fig. 20) offers further evidence that the Sn cation remains tetravalent. All the results, including the isotope effect of Fig. 2b, confirm that BaSn0.3Sc0.7O3–δ is purely proton conductive at 300 °C, fulfilling another crucial requirement for fuel cell electrolytes.
Across a broad temperature spectrum, BaSn0.3Sc0.7O3–δ exhibits a proton diffusivity that is twice as high as that found for state-of-the-art 60 at.% Sc-doped barium zirconate3 (Fig. 2d) and is equivalent to that for 20 at.% Y-doped barium zirconate14. The nonlinear Arrhenius plot of proton diffusivity for BaSn0.3Sc0.7O3–δ suggests the existence of some sort of proton trapping.
Proton transport examined by machine learning force field
Molecular dynamics simulations using a machine learning force field were performed with realistic dopant configurations determined by Monte Carlo simulations17. Diffusivities predicted by the simulations for 60 at.% Sc-doped barium stannate and barium zirconate between 227 °C and 527 °C are in good agreement with the experimental results, although the absolute values are slightly overestimated (Fig. 3a; the data for the zirconate are taken from the literature12). The observation of the trajectory of protons in the molecular dynamics simulations confirms the Grotthuss mechanism, a combination of proton rotation around the donor oxygen and hopping between the oxygens (an animation is provided as Supplementary Data 3). A clear tendency of protons to reside near dopant Sc compared with Sn, that is, a proton–dopant association, is evident from the analysis of H–Sc and H–Sn pair distribution functions for the stannate (Fig. 3b; similar results for the zirconate can be found in the literature12). This is consistent with the conventional understanding of proton–dopant association in acceptor-doped perovskites15,16,17. The H–Sn distribution increases slightly with increasing temperature, indicating that protons are slightly more randomly distributed, but a strong preference for H–Sc association remains at 527 °C. A high proton diffusivity is observed despite such a strong association, and this can be explained by the fast proton-conducting pathways formed along the three-dimensional network of ScO6 (Fig. 3c,d).

a, Proton diffusivity from molecular dynamics using neural network potential (NNP-MD) compared to experiment. b, Radial distribution function (g(r), where r is distance). c,d, Proton trajectory at 227 °C (c) and 527 °C (d). Purple and blue spheres in c and d denote Sc and Sn cations, respectively. The proton density isosurface, rendered in yellow in c and d, corresponds to the isosurface level of 1.4 × 10−2 Å−3. The two-dimensional density maps at the a–b, b–c and c–a cross-sections are also given with the associated colour bar. The experimental3 and calculated12 data for BaZr0.4Sc0.6O3−δ in a were taken from the literature.
Atomistic structure of BaSn0.3Sc0.7O3−δ
An analysis of the atomistic structure and chemical composition using transmission electron microscopy confirmed that the BaSn0.3Sc0.7O3−δ electrolyte had a perovskite-type structure with a high Sc content. Electron diffraction patterns were acquired from one crystal grain of the dehydrated BaSn0.3Sc0.7O3−δ electrolyte with two different electron incidence angles approximately 90° apart (Supplementary Fig. 21). All the Bragg reflections were reasonably indexed assuming a perovskite-type structure with the space group \({Pm}\bar{3}m\). The lattice parameter of 0.42 nm deduced from the electron diffraction pattern was consistent with the X-ray diffraction result of 0.4143 nm (Supplementary Fig. 1a). Both high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM; Fig. 4a and Supplementary Fig. 22) and energy-dispersive X-ray spectroscopy (EDS; Supplementary Fig. 23) imaged the sublattices making up the perovskite-type structure: that is, A sites occupied by Ba and B sites occupied by Sn and Sc, although the sublattice of O was unclear from these observations. The results showed neither cation ordering, expected in the double perovskite-type structure, nor oxygen-vacancy ordering in the brownmillerite-type structure. An analysis of the HAADF-STEM images revealed a negligible change in the intensity of the B sites, suggesting that the distribution of Sn and Sc at the B sites is random. An EDS analysis determined the atomic ratio of Ba/Sn/Sc to be 1:0.30:0.67 (Supplementary Fig 22c,f and Supplementary Table 2), indicating that Sc constitutes 69 at.% of the B-site cations (Supplementary Table 5). The results are in good agreement with the chemical analysis using ICP-OES, considering the 5 wt% BaSc2O4 (Supplementary Table 2).

a, HAADF-STEM image of dehydrated BaSn0.3Sc0.7O3−δ electrolyte and digital diffractogram (inset at bottom right). b, Time course of in situ powder X-ray diffraction patterns for hydrated BaSn0.3Sc0.7O3−δ under a concentrated and humidified CO2 stream. 2θ, the angle between the transmitted beam and reflected beam; pCO2, CO2 partial pressure. c, Amount of secondary BaSc2O4 plotted against exposure time to humidified CO2. The green, pink and red circles in a indicate the positions of the A sites (occupied by Ba), B sites (occupied by Sn and Sc) and O sites (occupied by O) in the perovskite-type structure, respectively. The time in b and c refers to the duration of the CO2-containing gas flow. Prior to the humidified CO2 flow, the powder sample was hydrated in an X-ray diffraction chamber for 9 h under a flowing humidified N2 stream with a water partial pressure (pH2O) of 0.02 atm. θ is the incident angle of X-ray. Colour in b represents the X-ray diffraction intensity. X-ray diffraction peak at 31.3° in b and c corresponds to the main peak of the BaSc2O4 phase. Assuming the degradation rate is proportional to the frequency of CO2 impacts on the material, and considering that the CO2 concentration during testing was 2,450 times that of ambient air (400 ppm), this accelerated stability test in b and c simulates over 100 years of exposure to ambient atmosphere at 300 °C.
CO2 tolerance of BaSn0.3Sc0.7O3−δ at 300 °C
An accelerated chemical stability test, conducted under harsh, humidified CO2 conditions at 300 °C using in situ X-ray diffractometry, confirmed the chemical stability of hydrated BaSn0.3Sc0.7O3−δ. The diffraction patterns for hydrated BaSn0.3Sc0.7O3−δ powder did not vary at 300 °C under a flow of highly concentrated CO2 for 398 h (Fig. 4b and Supplementary Fig. 24). The amount of the secondary BaSc2O4 phase, shown by the peak intensity at 31.3°, did not increase during the test (Fig. 4c). These results show the chemical stability of the BaSn0.3Sc0.7O3−δ powder, in agreement with the stability expected for the perovskite phases34; it is yet to be determined whether these results indicate thermodynamic or kinetic factors or both.
Discussion
A distinct characteristic of Sc, particularly at higher concentrations, is its ability to create rapid diffusion pathways even when protons are strongly associated with acceptor dopants in the oxide. The measured proton diffusivity versus Sc content for various host cations (Figs. 1d and 5d) shows that the diffusivity at low Sc content (~20 at.%) is lower than that at higher Sc content (>50 at.%). This, in turn, indicates that proton trapping, which is unfavourable for proton diffusivity, is prevalent at lower Sc content but is mitigated at higher Sc content. Also notable is that the diffusivities exhibit a clear dependence on the host B-site cation at lower Sc content but converge to similar values at higher Sc content; this is a natural consequence of Sc becoming the major B-site cation under heavy doping. The microscopic picture responsible for this effect is evident from our molecular dynamics data for 60 at.% Sc-doped BaSnO3 (Supplementary Fig. 3c,d) and BaZrO3 (ref. 12). Sc doping increases the population of ScO6 octahedra, forming oxygen sites corner-shared by ScO6–BO6 and ScO6–ScO6 pairs. A further increase in Sc concentration creates a connected network of ScO6 octahedra, to which protons preferentially associate, as seen in our simulated data (Fig. 3c,d) and discussed in the literature15,16,17. Despite such a clear association, the calculated diffusivity is rather high as a result of proton migration along the ScO6 network without the need to completely detrap itself from the acceptor dopant.
To highlight the difference between Sc and Y dopants, we compare local migration barriers for Sc-doped and Y-doped BaZrO3, obtained from ab initio calculations on the dopant configuration shown in Fig. 5a,b. We find that associated protons along ScO6–ScO6 octahedra pairs from sites 5 to 8 (pink region in Fig. 5a) exhibit the lowest site energy (proton–dopant association), and the values are almost equal. Transitions between these sites encompass both proton rotation and hopping (Grotthuss mechanism) in the oxide. By contrast, Y-doped barium zirconate, which exhibits the concentration–conductivity trade-off2,18, shows a large difference of 0.18 eV in the site energies of protons at YO6–YO6 octahedra pairs (between sites 5 and 8 in Fig. 5b). Similar tendencies are observed in the ScO6–ZrO6 and YO6–ZrO6 pairs across sites 2 to 4 (grey regions in Fig. 5a,b). The overall activation energies for protons to move from one of the lowest energy sites to site 4 or 9 at the YO6–ZrO6 pair (0.42–0.51 eV) exceed those for Sc (approximately 0.3 eV). These results show the importance of forming an interconnected ScO6 octahedral network within the cubic perovskite structure for enhanced proton diffusion. Although heavy acceptor M-doping into perovskite oxides always leads to the formation of an MO6 octahedral network, creating a relatively flat energy landscape that mitigates proton trapping is a distinct benefit of using Sc as a dopant, while the cubic perovskite oxide retains structural stability under harsh chemical and fuel cell conditions.

a,b, Energy diagrams for proton conduction (shown at top) with heavy Sc (a) and Y (b) doping. c, Experimental maximum Sc content versus bulk modulus. d, Proton diffusivity versus Sc content. The motions between sites 2, 3 and 4 and between sites 6, 7 and 8 in a represent proton rotation about the Sc–O–Zr and Sc–O–Sc vectors, respectively, while the other motions represent proton hopping. The pink and blue regions in a and b show proton sites sandwiched with two Sc or Y dopants, respectively. The grey regions in a and b show proton sites sandwiched by one dopant and a Zr host, whereas the darker grey regions on the left correspond to a proton site sandwiched by two Zr host cations. The bulk moduli of the host oxides in c were adopted from the Materials Project49. Closed and open symbols in d denote data in the Sc solubility limit and solid solution, respectively.
Saito and Yashima25 have recently proposed starting from a hypothetical BaScO2.5 and adding Mo6+ or W6+ as donors to control the oxygen content for stabilization of the cubic state. BaScO2.5 exists in a brownmillerite structure at ambient pressure with limited proton conductivities35, but the cubic perovskite structure can be stabilized by applying 4 GPa (ref. 36) or by adding Mo6+ or W6+ into the Sc3+ site to introduce oxide ions; the resultant BaSc0.8Mo0.2O3–δ (ref. 25) and BaSc0.8W0.2O3–δ (ref. 37) adhere to the principle of an interconnected ScO6 octahedral network facilitating rapid proton diffusion in cubic perovskite.
A remaining issue is to characterize B-site cations that combine with Sc to stabilize Ba-based perovskite oxides with high Sc content. Although it becomes increasingly ambiguous whether BaScO2.5 or BaBO3 should be identified as the ‘host’ material at such high Sc content, considering the latter as the host turns out to be useful for our purpose; we find that the lattice softness (low bulk modulus) of ‘host’ BaBO3 correlates very strongly with high Sc ‘solubility’ (Fig. 5c). This experimental observation is also consistent with our ab initio calculations: the computed solution energy for Sc in the host oxides aligns with their maximum Sc content (Supplementary Fig. 25a) and decreases as the bulk modulus decreases (Supplementary Fig. 25b). The soft nature of the host lattice is also confirmed on a microscopic level by the lattice compression upon the formation of \({{\mathrm{v}}}_{{\rm{O}}}^{\bullet \bullet }\), which is linearly correlated with the bulk modulus (Supplementary Fig. 25c). This intrinsic lattice softness may reduce the energy increase associated with structural distortions induced by point defects, thereby facilitating Sc incorporation. A similar decreasing trend in solution energy for lower bulk modulus is also observed for Y (Supplementary Fig. 25d), indicating the general applicability of lattice softness as a descriptor for enhanced dopant content. Y doping of up to 50 at.% (ref. 19) with lower proton conductivities20 has been reported in BaZrO3, but this is not inconsistent with the above discussion; the solution energy is calculated for B-site substitution while experiments report partial substitution on the A site38 and/or phase separation19, which would lower the connectivity of the dopant network. We also considered the correlation between maximum Sc content and the ionic radius of the B-site cation in six-coordinate species, including Mo6+ and W6+, but the trend is not as clear (Supplementary Fig. 26).
The favourable impact of extremely heavy doping had been overlooked for almost half a century, since the first report of proton conduction in perovskite oxides1. Our work suggests that such heavy doping, enabled by the choice of host materials with ‘soft’ lattices, could open new avenues for high-performance materials for various solid-state devices.
Methods
Synthesis
BaSn0.3Sc0.7O3−δ, BaTi0.2Sc0.8O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ, BaSn0.8Sc0.2O3−δ and BaHf0.8Sc0.2O3−δ were synthesized using a solid-state reaction method and sintered at 1,600 °C for 12–24 h in dry air. The resulting relative densities of BaSn0.3Sc0.7O3−δ, BaTi0.2Sc0.8O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ, BaSn0.8Sc0.2O3−δ and BaHf0.8Sc0.2O3−δ pellets were 97%, 97%, 99%, 90%, 68% and 50%, respectively.
Structural and chemical characterization
Crystal structure data were obtained from sintered pellets using an X-ray diffractometer (D2 Phaser, Bruker AXS) equipped with a Ni filter. The DIFFRAC.TOPAS software (v.5, Bruker AXS) was used for Rietveld refinements. The chemical composition of BaSn0.3Sc0.7O3−δ was determined using ICP-OES at the Center of Advanced Instrumental Analysis, Toshiba Nanoanalysis Corporation.
Thermogravimetry
The proton concentration of Sc-doped barium stannates, barium titanates and barium hafnates was evaluated using thermogravimetric analysis (STA449F3 Jupiter, NETZSCH). The proton concentration was determined in the temperature range 100–1,000 °C. The samples were first dehydrated at 1,000 °C for 2 h in dry Ar and subsequently exposed to a wet gas flow saturated with a water partial pressure of 0.02 atm. The proton concentration was calculated by assuming a weight gain from the dehydrated state to the hydrated state based on the hydration reaction \({{\mathrm{v}}}_{{\mathrm{O}}}^{\bullet \bullet }+{{\mathrm{O}}}_{{\mathrm{O}}}^{\times }+{{\mathrm{H}}}_{2}{\mathrm{O}}\leftrightarrow 2{\mathrm{O}}{{\mathrm{H}}}_{{\mathrm{O}}}^{\bullet }\). To reduce the buoyancy effect, each weight change measurement using thermogravimetric analysis was corrected by performing the measurement without the sample under the identical temperature and gas flow conditions.
Transmission electron microscopy
Thin-foil specimens of a dehydrated BaSn0.3Sc0.7O3−δ pellet, which were transparent to incident electrons, were prepared using a focused ion beam system with an acceleration voltage of 16 kV for shaping and 2 kV for thinning (at the final stage of polishing). In addition to polishing, electron backscatter diffraction and scanning electron microscopy analyses were carried out using a Helios 5 UX DualBeam (Thermo Fisher Scientific). Focused ion beam sample preparation was performed after dehydrating the BaSn0.3Sc0.7O3−δ pellet by ramping up the temperature to 1,000 °C at a rate of 20 °C min−1 under 1 × 10−10 atm and maintaining the temperature at 1,000 °C for 1.5 h. Transmission electron microscopy, electron diffraction, HAADF-STEM and EDS were carried out using a JEM-ARM300F2 (JEOL) microscope at an acceleration voltage of 300 kV.
Electrochemical characterization
Temperature-dependent electrical conductivities of sintered BaSn0.3Sc0.7O3−δ, BaSn0.8Sc0.2O3−δ, BaTi0.2Sc0.8O3−δ, BaTi0.4Sc0.6O3−δ, BaHf0.5Sc0.5O3−δ and BaHf0.8Sc0.2O3−δ pellets were measured by a.c. impedance spectroscopy. The pellets were polished to ~500 μm thickness and coated with 650 nm Ag films on both surfaces by d.c. sputtering (Sanyu Electron, SC-701HMC II). Silver meshes (Niraco Corporation) affixed with silver paste (Tanaka Kikinzoku Kogyo, TR-3025) acted as current collectors. Specimens were pre-annealed at 720 °C in dry Ar and then exposed to humidified Ar (pH2O = 0.02 atm). The impedance spectra were acquired with an electrochemical station (BioLogic VSP-300) in the temperature range of 35–631 °C under the humidified atmosphere over a range from 10−1 Hz to 3 × 106 Hz and fitted to an equivalent circuit of serial RC components, where R and C were in parallel, for the grain interior, grain boundary and electrode regions using EC-Lab software (v.11.33, Biologic Science Instruments). Here, R is the resistance from an ideal resistor with impedance ZR = R, and C is a constant phase element with ZC = (Y(jω)l)−1, where j is \(\sqrt{-1}\), ω is the frequency and Y and l are constants with 0 ≤ l ≤ 1. H/D isotope effects were evaluated for BaSn0.3Sc0.7O3−δ at 300 °C by switching between H2O-saturated and D2O-saturated Ar streams (pH2O = pD2O = 0.02 atm). The oxygen partial pressure dependence of proton conductivities in BaSn0.3Sc0.7O3−δ was determined at 300 °C under pH2O = 0.02 atm across the range from pO2 = 2.4 × 10−22 atm to 9.6 × 10−1 atm.
In situ X-ray diffraction measurements under concentrated and humidified CO2 streams
To confirm the stability of BaSn0.3Sc0.7O3−δ against CO2, high-temperature X-ray diffraction (D8 DISCOVER, Bruker AXS) was performed in a high-temperature chamber (XRK900, Anton Paar). Cu Kα radiation was used in the measurement under humidified CO2 (pH2O = 0.02 atm, pCO2 = 0.98 atm) at 300 °C. Prior to exposure to humidified CO2, protons were introduced while maintaining the temperature at 300 °C in wet N2 (pH2O = 0.02 atm) for 9 h. Note that the BaCO3 phase did not exist before exposure to the CO2 atmosphere. The diffraction patterns were continuously recorded in θ–2θ mode for 398 h.
In situ FT-IR in a humidified atmosphere
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was conducted on a Nicolet iS50 (Thermo Fisher Scientific) at 300 °C. Powders of BaSn0.3Sc0.7O3−δ, BaTi0.2Sc0.8O3−δ, BaSn0.8Sc0.2O3−δ and BaTi0.4Sc0.6O3−δ were first dehydrated at 1,000 °C for 1 h and subsequently introduced into the in situ chamber and heated to 300 °C under dry Ar. Spectra obtained in this dry atmosphere served as the reference. The gas flow was then switched to wet Ar (pH2O = 0.02 atm), and spectra were collected up to 9 h.
X-ray photoelectron spectroscopy
The Sn valence state in reduced BaSn0.3Sc0.7O3−δ was analysed by X-ray photoelectron spectroscopy (AXIS-ULTRA, KRATOS Analytical). Measurements employed a monochromatic Al Kα X-ray source. Prior to measurement, the powder was reduced at 300 °C in flowing humidified H2 (partial pressure of H2 pH2 = 0.98 atm; pH2O = 0.02 atm), corresponding to pO2 = 2 × 10−47 atm. Surface charging was done with an electron flood gun, and binding energy values were referenced to the C 1s peak at 285.0 eV.
Electrolyte-supported protonic ceramic cells
A BaSn0.3Sc0.7O3−δ disc pellet with a thickness of 0.3 mm was used as the electrolyte. Platinum paste (Tanaka Precious Metal, TR-7907) was applied on both sides of the disc as cathode and anode electrodes in a circular pattern with a diameter of 8 mm. The discs were then baked at 700 °C for 2 h in dry air, followed by a temperature reduction to 100 °C for 100 h in moist air. As the reference electrode, a Pt wire was attached to the side of the disc, while a Pt mesh attached to both electrodes using Ag paste (TR-3025) served as the current collector. After the Ag paste dried, 60 μl of 2 mol% cerium nitrate solution was dropped onto both electrodes. The samples were then fired at 300 °C for 2 h in humid air.
Anode-supported protonic ceramic cells
Anode-supported proton ceramic single cells were fabricated using BaSn0.3Sc0.7O3−δ electrolyte through cold-pressing and dip-coating. A mixture of 70 wt% Nano NiO (Kceracell), 30 wt% BaCe0.7Zr0.1Y0.1Yb0.1O3−δ and 20 wt% starch was pressed at 100 MPa and pre-sintered at 900 °C for 2 h. The electrolyte film was dip-coated using 100 μl of a BaSn0.3Sc0.7O3−δ slurry and then sintered at 1,300 °C for 5 h. PrBaCo2O5+δ mixed with BaCe0.7Zr0.1Y0.1Yb0.1O3−δ was brush-painted onto the electrolyte and sintered at 950 °C for 2 h. Ag and Pd were applied to the cathode and anode, respectively, as current collectors. The single cells were sealed in alumina tubes using glass sealant. The electrochemical measurements were made using a BioLogic VSP-300 with 2% H2O/H2 (20 sccm) at the anode and 2% H2O/air (100 sccm) at the cathode. The microstructure of single cells was observed using a micro-calorimeter field emission scanning electron microscope (TES + ULTRA55, Zeiss).
Molecular dynamics simulations using a machine learning force field
The calculations were all performed in a 3 × 3 × 3 cubic perovskite supercell corresponding to 60 at.% Sc doping in BaSnO3. First, cation configurations corresponding to equilibrium at 1,900 K, which corresponds roughly to the sintering temperature, were obtained by replica exchange Monte Carlo calculations combined with a neural-network configuration energy model using our abICS framework50,51. The dry sintering conditions were simulated with a supercell containing 27 Ba, 16 Sc, 11 Sn and 73 O. Details of the calculation procedure and the neural-network model can be found in our previous work that studied 22 at.% Sc-doped BaZrO3 (ref. 17). The cation configuration is fixed to that determined by these Monte Carlo calculations, in subsequent molecular dynamics calculations using LAMMPS code52. We employed the Allegro model53 as the machine learning force field, and its training was performed in the fully hydrated system containing 27 Ba, 16 Sc, 11 Sn, 81 O and 16 H. The Supplementary Information contains details of the training procedure. In the final production run, deuterium was used instead of hydrogen to slow the local vibrations and enable accurate equation-of-motion integration with a time step of 1 fs. The diffusivities were calculated from 10 ns trajectories at 600–800 K and 100 ns trajectories at 500 K and 550 K. The calculated values are multiplied by \(\sqrt{2}\) to compare the simulations using deuterium with experiments using protons.
Proton migration barriers
The proton migration (hopping and rotation) barriers were calculated using the Vienna Ab initio Simulation Package for a 4 × 4 × 4 cubic perovskite supercell with 64 Ba, 61 Zr, 3 B (B = Sc, Y), 192 O and 1 H, where the B site dopants Sc and Y were placed as shown in Fig. 5a. Only the gamma point was used for k-point sampling. The plane-wave cut-off was set at 500 eV. A homogeneous background charge was introduced to satisfy charge neutrality considering formal ionic charges. The climbing-image nudged elastic band method54 was used to calculate minimum energy paths for the proton migration processes shown in Fig. 5a and obtain energy barriers.
Dopant solution
The solution energy of the Sc and Y dopants was calculated for cubic BaSnO3, BaTiO3, BaZrO3 and BaHfO3. The trigonal (R3m), orthorhombic (Amm2) and tetragonal (P4mm) phases were also calculated for BaTiO3, considering its phase stability. The solution energy of dopant M for B-site cations was calculated using the following equation55:
where \({E}_{{\rm{def}}}^{\sup}({{\rm{M}}}_{{\rm{B}}}^{{\prime}})\) and \({E}_{{\rm{def}}}^{\sup}({{\mathrm{v}}}_{{\rm{O}}}^{\bullet \bullet})\) are the energies of a charged supercell of a perovskite compound containing a dopant M (Sc or Y) and an oxide-ion vacancy, respectively. The lattice constants for these supercells were fixed during structural optimization. \({E}_{{\rm{per}}}^{\sup }\) is the energy of a perfect perovskite supercell, and μB, μO and μM are the chemical potentials for B, O and M, respectively. The Supplementary Information contains details of the computational conditions.
Data availability
All data supporting the findings of this study are available in the article and its Supplementary Information except for the molecular dynamics trajectories and training data/hyperparameters for the machine learning potential, which are available via the ISSP Data Repository at https://datarepo.mdcl.issp.u-tokyo.ac.jp/repo/54. Source data are provided with this paper.
Change history
18 March 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41563-026-02580-z
References
Iwahara, H., Esaka, T., Uchida, H. & Maeda, N. Proton conduction in sintered oxides and its application to steam electrolysis for hydrogen production. Solid State Ion. 3–4, 359–363 (1981).
Yamazaki, Y., Hernandez-Sanchez, R. & Haile, S. M. High total proton conductivity in large-grained yttrium-doped barium zirconate. Chem. Mater. 21, 2755–2762 (2009).
Hyodo, J., Kitabayashi, K., Hoshino, K., Okuyama, Y. & Yamazaki, Y. Fast and stable proton conduction in heavily scandium-doped polycrystalline barium zirconate at intermediate temperatures. Adv. Energy Mater. 10, 2000213 (2020).
Kanno, R. & Murayama, M. Lithium ionic conductor thio-LISICON. J. Electrochem. Soc. 148, A742–A746 (2001).
Kato, Y. et al. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 16030 (2016).
Duan, C. et al. Readily processed protonic ceramic fuel cells with high performance at low temperatures. Science 349, 1321–1326 (2015).
Morejudo, S. H. et al. Direct conversion of methane to aromatics in a catalytic co-ionic membrane reactor. Science 353, 563–566 (2016).
Wang, J., Wasmu, S. & Savinel, R. F. Evaluation of ethanol, 1-propanol, and 2-propanol in a direct oxidation polymer-electrolyte fuel cell. J. Electrochem. Soc. 142, 4218–4224 (1995).
Fukui, K., Iimura, S., Iskandarov, A., Tada, T. & Hosono, H. Room-temperature fast H− conduction in oxygen-substituted lanthanum hydride. J. Am. Chem. Soc. 144, 1523–1527 (2022).
Haile, S. M., Chisholm, C. R. I., Sasaki, K., Boysen, D. A. & Uda, T. Solid acid proton conductors: from laboratory curiosities to fuel cell electrolytes. Faraday Discuss. 134, 17–39 (2007).
Kreuer, K. D. Proton-conducting oxides. Annu. Rev. Mater. Res. 33, 333–359 (2003).
Fujii, S. et al. Emerging computational and machine learning methodologies for proton-conducting oxides: materials discovery and fundamental understanding. Sci. Technol. Adv. Mater. 25, 2416383 (2024).
Kreuer, K. D. Aspects of the formation and mobility of protonic charge carriers and the stability of perovskite-type oxides. Solid State Ion. 125, 285–302 (1999).
Yamazaki, Y. et al. Proton trapping in yttrium-doped barium zirconate. Nat. Mater. 12, 647–651 (2013).
Björketun, M. E., Sundell, P. G. & Wahnström, G. Structure and thermodynamic stability of hydrogen interstitials in BaZrO3 perovskite oxide from density functional calculations. Faraday Discuss. 134, 247–265 (2007).
Yamazaki, Y. et al. Oxygen affinity: the missing link enabling prediction of proton conductivities in doped barium zirconates. Chem. Mater. 32, 7292–7300 (2020).
Hoshino, K. et al. Probing local environments of oxygen vacancies responsible for hydration in Sc-doped barium zirconates at elevated temperatures: in situ X-ray absorption spectroscopy, thermogravimetry, and active learning ab initio replica exchange Monte Carlo simulations. Chem. Mater. 35, 2289–2301 (2023).
Iguchi, F., Tsurui, T., Sata, N., Nagao, Y. & Yugami, H. The relationship between chemical composition distributions and specific grain boundary conductivity in Y-doped BaZrO3 proton conductors. Solid State Ion. 180, 563–568 (2009).
Imashuku, S., Uda, T., Nose, Y. & Awakura, Y. To Journal of Phase Equilibria and Diffusion phase relationship of the BaO-ZrO2-YO1.5 system at 1500 and 1600 °C. J. Phase Equilibria Diffus. 31, 348–356 (2010).
Fabbri, E., Pergolesi, D., Licoccia, S. & Traversa, E. Does the increase in Y-dopant concentration improve the proton conductivity of BaZr1−xYxO3−δ fuel cell electrolytes? Solid State Ion. 181, 1043–1051 (2010).
Toyoura, K., Meng, G., Han, D. & Uda, T. Preferential proton conduction along a three-dimensional dopant network in yttrium-doped barium zirconate: a first-principles study. J. Mater. Chem. A 6, 22721–22730 (2018).
Imashuku, S., Uda, T., Ichitsubo, T., Matsubara, E. & Awakura, Y. A pseudoternary phase diagram of the BaO–ZrO2–ScO1.5 system at 1600 °C and solubility of scandia into barium zirconate. J. Phase Equilibria Diffus. 28, 517–522 (2007).
Koichi, E. Ceramic materials containing rare earth oxides for solid oxide fuel cell. J. Alloy. Compd. 250, 486–491 (1997).
Draber, F. M. et al. Nanoscale percolation in doped BaZrO3 for high proton mobility. Nat. Mater. 19, 338–346 (2020).
Saito, K. & Yashima, M. High proton conductivity within the ‘Norby gap’ by stabilizing a perovskite with disordered intrinsic oxygen vacancies. Nat. Commun. 14, 7466 (2023).
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Li, L. & Nino, J. C. Proton-conducting barium stannates: doping strategies and transport properties. Int. J. Hydrog. Energy 38, 1598–1606 (2013).
Kinyanjui, F. G. et al. Crystal structure and proton conductivity of BaSn0.6Sc0.4O3–δ: insights from neutron powder diffraction and solid-state NMR spectroscopy. J. Mater. Chem. A 4, 5088–5101 (2016).
Kreuer, K. D., Münch, W., Fuchs, A., Klock, U. & Maier, J. Proton conducting alkaline earth zirconates and titanates for high drain electrochemical applications. Solid State Ion. 145, 295–306 (2001).
Hyodo, J., Tsujikawa, K., Shiga, M., Okuyama, Y. & Yamazaki, Y. Accelerated discovery of proton-conducting perovskite oxide by capturing physicochemical fundamentals of hydration. ACS Energy Lett. 6, 2985–2992 (2021).
Nowick, A. S. & Vaysleyb, A. V. Isotope effect and proton hopping in high-temperature protonic conductors. Solid State Ion. 97, 17–26 (1997).
Yamada, S., Kanayama, K. & Toyoura, K. Nuclear quantum effects on proton diffusivity in perovskite oxides. Phys. Rev. B 111, 064117 (2025).
Roy, T., Sahani, S., Madhu, D. & Chandra Sharma, Y. A clean approach of biodiesel production from waste cooking oil by using single phase BaSnO3 as solid base catalyst: mechanism, kinetics & E-study. J. Clean. Prod. 265, 121440 (2020).
Haile, S. M., Staneff, G. & Ryu, K. H. Non-stoichiometry, grain boundary transport and chemical stability of proton conducting perovskites. J. Mater. Sci. 36, 1149–1160 (2001).
Omata, T., Fuke, T., Otsuka, Y. & Matsuo, S. Hydration behavior of Ba2Sc2O5 with an oxygen-deficient perovskite structure. Solid State Ion. 177, 2447–2451 (2006).
Cervera, R. B. et al. Perovskite-structured BaScO2(OH) as a novel proton conductor: heavily hydrated phase obtained via low-temperature synthesis. Chem. Mater. 25, 1483–1489 (2013).
Saito, K., Umeda, K., Fujii, K., Mori, K. & Yashima, M. High proton conduction by full hydration in highly oxygen deficient perovskite. J. Mater. Chem. A 12, 13310–13319 (2024).
Yamazaki, Y., Yang, C.-K. & Haile, S. M. Unraveling the defect chemistry and proton uptake of yttrium-doped barium zirconate. Scr. Mater. 65, 102–107 (2011).
Han, D., Hatada, N. & Uda, T. Chemical expansion of yttrium‐doped barium zirconate and correlation with proton concentration and conductivity. J. Am. Ceram. Soc. 99, 3745–3753 (2016).
Knight, K. S. & Bonanos, N. The crystal structures of some doped and undoped alkaline earth cerate perovskites. Mater. Res. Bull. 30, 347–356 (1995).
Zheng, M. & Bo, Z. Proton conductivity in Yb-doped strontium cerates. Solid State Ion. 80, 59–65 (1995).
Loureiro, F. J. A., Shakel, Z., Graça, V. C. D., Holz, L. I. V. & Fagg, D. P. Benchmarking the yttrium content in the low temperature/low humidity electrical properties of yttrium-doped barium cerate. Ceram. Int. 49, 34303–34308 (2023).
Takeuchi, K. et al. The crystal structures and phase transitions in Y-doped BaCeO3: their dependence on Y concentration and hydrogen doping. Solid State Ion. 138, 63–77 (2000).
Clark, D. et al. Anomalous low-temperature proton conductivity enhancement in a novel protonic nanocomposite. Phys. Chem. Chem. Phys. 16, 5076–5080 (2014).
Nomura, K. et al. Phase transitions, thermal expansions, chemical expansions, and CO2 resistances of Ba(Ce0.8–xZrxY0.1Yb0.1)O3–δ (x = 0.1, 0.4) perovskite-type proton conductors. J. Electrochem. Soc. 169, 024516 (2022).
Sone, Y., Ekdunge, P. & Simonsson, D. Proton conductivity of Nafion 117 as measured by a four-electrode AC impedance method. J. Electrochem. Soc. 143, 1254–1259 (1996).
Nguyen, N. T. Q. & Yoon, H. H. Preparation and evaluation of BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) electrolyte and BZCYYb-based solid oxide fuel cells. J. Power Sources 231, 213–218 (2013).
Mori, M. et al. Cubic-stabilized zirconia and alumina composites as electrolytes in planar type solid oxide fuel cells. Solid State Ion. 15, 157–164 (1994).
Jain, A. et al. Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Kasamatsu, S., Motoyama, Y., Yoshimi, K. & Aoyama, T. Configuration sampling in multi-component multi-sublattice systems enabled by ab Initio Configuration Sampling Toolkit (abICS). Sci. Technol. Adv. Mater. Methods 3, 2284128 (2023).
Kasamatsu, S. et al. Facilitating ab initio configurational sampling of multicomponent solids using an on-lattice neural network model and active learning. J. Chem. Phys. 157, 104114 (2022).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2023).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Fujii, S., Shimizu, Y., Hyodo, J., Kuwabara, A. & Yamazaki, Y. Discovery of unconventional proton‐conducting inorganic solids via defect‐chemistry‐trained, interpretable machine learning. Adv. Energy Mater. 13, 2301892 (2023).
Acknowledgements
This work was supported by the Japan Science and Technology Agency (JST) CREST (JPMJCR18J3) and JST GteX (JPMJGX23H0 and JPMJGX23H7) programmes. S.F., S.K. and Y.Y. acknowledge financial support from the Japan Society for the Promotion of Science through KAKENHI (JP19H05786 and JP22H04914), from the JST through FOREST (JPMJFR2037 and JPMJFR235X) and from the Kyushu University Platform of Inter-/Transdisciplinary Energy Research (Q-PIT) through the Module-Research Program, respectively. The computational part of this work was supported by the Ministry of Education, Culture, Science, and Technology of Japan’s Program for Promoting Researches on the Supercomputer Fugaku (JPMXP1020230325), and computations were performed using supercomputer Fugaku at RIKEN Center for Computational Science (hp230205) and the Supercomputer Center at the Institute for Solid State Physics, the University of Tokyo. We thank T. Tamaoka for his advice on the transmission electron microscopy experiments and M. Yoshitake for her assistance with sample preparation.
Author information
Authors and Affiliations
Contributions
Y.Y. conceived and designed the project. K. Tsujikawa performed ex situ X-ray diffractometry, thermogravimetry, electrochemical measurements, X-ray photoelectron spectroscopy and in situ FT-IR as well as their analyses under the supervision of J.H. and Y.Y.; J.H. performed in situ X-ray diffractometry. Y.T. performed transmission electron microscopy experiments. Y.T. and Y.M. analysed the transmission electron microscopy data. K. Takahashi and S.K. performed the neural-network-potential molecular dynamics simulations. N.S. fabricated the anode-supported protonic ceramic fuel cell and performed electrochemical measurements. S.K. calculated the proton migration barriers, and S.F. calculated the dopant solution energies. K. Tsujikawa, S.F., Y.M., S.K. and Y.Y. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Materials thanks John Irvine, Truls Norby and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1 and 2, Tables 1–5 and Figs. 1–26.
Supplementary Data 1 (download ZIP )
Statistical source data for Supplementary Figs. 1–20 and 24–26.
Supplementary Data 2 (download ZIP )
Density functional theory structure data for Supplementary Fig. 25.
Supplementary Data 3 (download MP4 )
Animation of the proton hopping and rotation processes observed during molecular dynamics simulation.
Source data
Source Data Fig. 1 (download XLSX )
Proton conductivity data plotted against lattice volume in Fig. 1a, bulk proton conductivity data plotted in Fig. 1b, proton concentration data plotted in Fig. 1c, proton diffusivity data plotted in Fig. 1d, apparent activation energy data for proton diffusivity plotted in Fig. 1e and proton diffusivity data plotted against proton concentration in Fig. 1f.
Source Data Fig. 2 (download XLSX )
Ionic conductivity data plotted in Fig. 2a, H–D isotope effect data plotted in Fig. 2b, proton conductivity data plotted against oxygen partial pressure in Fig. 2c and proton diffusivity data plotted in Fig. 2d.
Source Data Fig. 3 (download XLSX )
Proton diffusivity data plotted in Fig. 3a and radial distribution function data plotted in Fig. 3b.
Source Data Fig. 4 (download XLSX )
Amount of secondary BaSc2O4 plotted against exposure time to humidified CO2 in Fig. 4c.
Source Data Fig. 5 (download XLSX )
Data of energy diagrams for proton conduction with heavy Sc and Y doping plotted in Fig. 5a,b, experimental maximum Sc content plotted against bulk modulus in Fig. 5c and proton diffusivity data plotted against Sc content in Fig. 5d.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsujikawa, K., Hyodo, J., Fujii, S. et al. Mitigating proton trapping in cubic perovskite oxides via ScO6 octahedral networks. Nat. Mater. 24, 1949–1956 (2025). https://doi.org/10.1038/s41563-025-02311-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41563-025-02311-w
