
Abstract
Spectrometers based on high-finesse optical cavities have proven to be powerful tools for applied and fundamental studies. Extending this technology to the deep cryogenic regime reduces Doppler broadening, enhances peak absorption, narrows the Boltzmann distribution of rotational states and ensures that all unwanted molecular species disturbing the spectra are frozen out. Moreover, the dense spectra of complex polyatomic molecules become easier to assign. Here we demonstrate a cavity-enhanced spectrometer fully operating down to 4 K. This was enabled by uniformly cooling, not only the sample, but the entire cavity. Our approach isolates the cavity from external noise and cryocooler vibrations. We demonstrate the capabilities of our cavity-enhanced spectrometer by performing measurements in the deep cryogenic regime: an accurate test of quantum electrodynamics for molecules; the realization of the primary standards of the International System of Units for temperature, concentration and pressure; a measurement of the dihydrogen phase diagram; and the determination of the ortho-to-para spin-isomer conversion rate.
Similar content being viewed by others
Main
Cavity-enhanced spectroscopy has revolutionized various fields of research by providing unprecedented sensitivity and precision1. It has been instrumental in atmospheric studies, as it enables the detection of pollutants and greenhouse gases2 at parts-per-trillion levels and subpermille accuracies3. This technique has enabled the realization of standards for ultralow humidity4 and carbon isotope ratios5 and has been used to test quantum electrodynamics (QED)6. Even more sensitive, noise-immune cavity-enhanced optical heterodyne molecular spectroscopy7 has become invaluable in fundamental physics8. New developments have extended cavity-enhanced spectroscopy to study fast processes, large molecules and multispecies samples by combining it with optical frequency combs9. Accuracy has further been improved with rapid scanning10 and purely frequency-based methods11.
Molecular spectroscopy benefits in many respects from operating in the deep cryogenic regime, often far below 77 K. In this regime, Doppler broadening is reduced, peak absorption is enhanced, the Boltzmann distribution of rotational states is narrowed, all unwanted molecular species disturbing the spectra12 are frozen out and the dense spectra of complex polyatomic molecules become easier to assign13. Accurate spectroscopy at low temperatures is also important for testing state-of-the-art ab initio quantum-chemical calculations, especially the interactions between atoms and molecules for both bound14 and scattering15 states. The quantum-scattering phenomena (collision-induced line-shape effects) are particularly interesting in the deep cryogenic regime (below 50 K), where fundamental discrepancies between experimental results and ab initio calculations have been reported for many molecular species16 but without any way to resolve the discrepancies. Cryogenic spectroscopy is also important for providing reference molecular data for astrophysical studies, particularly for studying the atmospheres of planets and exoplanets17.
A typical approach to optical spectroscopy at cryogenic temperatures is based on Fourier spectrometers18,19. The Grenoble group developed a cavity ring-down spectrometer with a gas sample kept at temperatures down to 77 K (ref. 20). A phase-shift cavity ring-down spectrometer operating at 105 K was demonstrated in ref. 21. Optical cavities have also been combined with buffer-gas-cooled molecular samples13,22,23 and molecular beams24,25. Recently, high-resolution cryogenic cavity-enhanced spectrometers have been developed by the Hefei26 and Amsterdam8 groups. In the Hefei spectrometer, the cavity mirrors are kept at room temperature while cryogenic gas flows through the cell26. In the Amsterdam spectrometer, the entire optical cavity is uniformly cooled to 72 K (ref. 8). Cryogenic optical cavities (operating at T = 1.7 K to 10 K) are also used as state-of-the-art short-term frequency references27,28,29,30.
In this work, we demonstrate a cavity-enhanced spectrometer fully operating in the deep cryogenic regime down to 4 K. Not only the sample but also the entire cavity, including the mirrors and cavity-length actuator, are uniformly cooled, which ensures the thermodynamic equilibrium of a gas sample. The cavity is well isolated from external noise and cryocooler vibrations. This instrument enables a variety of fundamental and practical applications. We demonstrate a few examples based on the accurate spectroscopy of cryogenic hydrogen molecules: an accurate test of QED for molecules, optical realization of three SI units in the deep cryogenic regime, measurement of the H2 phase diagram and measurement of the ortho-to-para spin-isomer conversion rate.
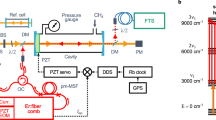
The cryogenic spectrometer is shown in Fig. 1. The greatest advantage of this spectrometer is that it allows nearly perfect thermalization of the entire gas sample down to 4 K. We uniformly cool not only the H2 molecules but also the entire inner cryogenic vacuum chamber, cavity spacer, mirrors and mirror position actuator. This feature is critical for accurate H2-based optical metrology, as temperature non-uniformity distorts the shape of an optical molecular resonance and, hence, introduces systematic uncertainties. The second important feature of this set-up is the system of heavy copper blocks and layers suspended on thin titanium rods and thermally linked by flexible copper connectors. This structure strongly attenuates vibrations and noise from the cryocooler and vacuum pumps as well as from external sources. Additionally, it suppresses the temperature fluctuations typical when operating the second stage of the cryocooler, such that the fluctuations at the cryogenic vacuum chamber are below 1 mK.

Left: illustration of the cryogenic part of the set-up. The entire cavity, including the mirror position actuator, is placed inside a cryogenic copper vacuum chamber (yellow) that can be cooled to 4 K to ensure the highly homogeneous thermodynamic equilibrium of the entire gas sample. The outer copper layers (orange), kept at approximately 40 K, block room-temperature thermal radiation. The entire system is placed inside a room-temperature vacuum system (grey) to maintain cryogenic conditions. The inner cryogenic and outer room-temperature vacuum chambers are independent; hence, the cryogenic chamber can be independently filled with H2 gas. The inner cryogenic vacuum chamber is suspended on thin titanium rods (not shown here) to eliminate thermal links and attenuate vibrations. The cryogenic mirror position actuator31 is shown in detail in the small inset below the vacuum chamber. Right: simplified scheme of the experimental set-up. We use an OPO to access the H2 fundamental-band transition with an idler beam operating at 2.2 μm. The idler beam is tightly locked to the cryogenic high-finesse cavity via a Pound–Drever–Hall lock33. The absolute frequency after idler frequency-doubling is determined by referring to an optical frequency comb at 1.1 μm. The length of the cryogenic cavity is actively stabilized by referring to the optical frequency comb. Both the comb repetition rate and the offset frequency are stabilized to the radio frequency from the hydrogen maser (H maser), which is integrated with the local representation of UTC at the Astrogeodynamic Observatory in Borowiec43. The plot in the right bottom corner shows a typical ring-down event acquired at 4 K, where τ and \({\mathcal{F}}\) are the photon lifetime and cavity finesse, respectively. AOS, Astrogeodynamic Observatory; PBS, polarizing beam splitter; PD, photodetector; PDH, Pound–Drever–Hall; SHG, second-harmonic generation; ECDL, external-cavity diode laser; AOM, acousto-optic modulator; EOM, electro-optic modulator.
One of the cavity mirrors is installed on a position actuator (see the inset under the vacuum chamber in Fig. 1), which allows the cavity length to be tuned by a few wavelengths. The actuator is designed to operate over a wide temperature range from room temperature down to 4 K (ref. 31). A key feature of the actuator design is that it does not become misaligned when the temperature is cycled between 4 K and 300 K (we initially align the optical cavity at room temperature and it remains aligned after the temperature is tuned within this range). The cavity has a length of 69 cm (free spectral range of 217 MHz) and finesse of 35,000 (photon lifetime of 26 μs). It is composed of two dielectric concave mirrors with a radius of curvature of 1 m. The length of the cavity is actively stabilized by locking the frequency of one of the cavity resonances to an optical frequency comb; see the cavity-length feedback line in Fig. 1.
To fully demonstrate the capabilities of this experimental platform, we consider the hydrogen molecule, which is the molecule with the highest equilibrium vapour pressure at deep cryogenic temperatures. To access the strongest band of H2 (the fundamental band at 2.2–2.4 μm), we developed a laser system (like the one recently reported in ref. 32) based on a continuous-wave (CW) optical parametric oscillator (OPO); see the right-hand panel in Fig. 1. A spectrally narrow (10 kHz) CW seed laser operating at 1,064 nm after amplification to 10 W pumps an OPO cavity. In this work, we focus on the 1 - 0 S(0) transition at 2.22 μm, which is covered by the tuning range of the OPO idler beam. In contrast to the signal beam, whose frequency is fixed by the OPO cavity, the idler beam inherits the frequency modulation of the seed laser. This allows us to tightly lock the idler beam to the high-finesse cryogenic cavity using the Pound–Drever–Hall technique33 with the Pound–Drever–Hall feedback signal being directly sent to the seed laser (Fig. 1). The absolute frequency of the idler beam is determined by referring to the optical frequency comb after doubling the idler frequency in a second-harmonic-generation crystal. This also allows stabilization of the cavity length.
In the bottom right corner of Fig. 1, we show the cavity photon lifetime recorded at 4 K. The high finesse is preserved when the temperature of the entire cavity is cycled between 4 K and room temperature. Molecular hydrogen ice is deposited at deep cryogenic temperatures on all inner surfaces (including mirrors); however, we did not observe any deterioration of the mirror reflectivity (the finesse remains the same). Figure 2 shows an example of the cavity ring-down spectra recorded with our instrument, the 1 - 0 S(0) line of the H2 molecule at 2.22 μm. The wide dynamic range of the absorption coefficient (four orders of magnitude has already been demonstrated in this work, with much room for improvement) allows us to record a clear spectrum even below 5 K, far below the H2 boiling and melting points. To our knowledge, this is the lowest temperature for high-resolution cavity-enhanced spectroscopy at thermal equilibrium. In the following four sections, we present several examples of the applications of this instrument that address scientific and technological challenges in different fields.

The 1 - 0 S(0) line of the H2 molecule at 2.22 μm is recorded at several temperatures from 4.9 K to 7.8 K, which are far below the H2 melting and boiling points where the vapour pressure strongly depends on temperature. The horizontal axis is shifted to the unperturbed position of the transition, ν0. The red lines are the fitted line-shape models that include both Doppler broadening and relevant collisional effects (we used the SDHC profile55,56 with β correction57 included). Note the log scale on the vertical axis. The pressures and temperatures indicated in this figure are the values determined optically. See sections ‘Realization of the primary SI units at deep cryogenic temperatures’ and ‘Hydrogen phase diagram’ and Table 1.
Precision tests of QED for molecules
Accurate spectroscopy of simple atomic and molecular systems, whose structure can be calculated from first principles with quantum theory, allows one not only to test the QED sector of the standard model at high precision6 but also to search for new physics beyond the standard model34. An important example of such systems is molecular hydrogen, for which the accuracy of ab initio calculations of the energies of the ro-vibrational transitions reaches 10 significant digits35. The present accuracy of the leading (non-relativistic) contribution reaches 13 significant digits36, which paves the way for further substantial progress.
In this section, we present complementary experimental results for the 1 - 0 S(0) transition of the H2 molecule. The experiment was carried out at T = 7.8 K, at which the vapour pressure was high enough to reach a high signal-to-noise ratio. We benefited from operating under deep cryogenic conditions in several ways. Compared with room temperature, at T = 7.8 K, the Doppler broadening is 6.2 times smaller and the peak absorption is 47.7 = 6.2 × 7.7 times larger, with the 6.2 factor being due to the smaller Doppler broadening and the 7.7 factor being due to the collapse of the entire Boltzmann distribution to the ground rotational state; compare the blue and red curves in the left-hand panel in Fig. 3a. Overall, the higher and narrower resonance potentially increases the accuracy of the line position determination at T = 7.8 K by a factor of nearly 300 compared with room temperature. Furthermore, at low temperatures, all unwanted molecular species with strong dipole transitions that would disturb the spectrum12 are frozen out.

a, Accurate test of QED for molecules. Left: difference Δν0 between the position of the 1 - 0 S(0) line of H2 as a function of pressure popt (black points) and its extrapolation to the zero-pressure limit (grey line). The red and blue lines are the simulations of the line at the same H2 concentration but different temperatures, which illustrate the metrological gain due to operation in the deep cryogenic regime. Inset: the α and ν axes set the coordinate frame for the red and blue curves. Right: difference Δν0 between ν0 determined in this work (marked with the horizontal dashed line) compared with previous experimental results37,58,59,60,61 and the most recent theoretical value35. The better than 1-MHz agreement of Fink61 with our result is incidental; ref. 61 does not report uncertainty, but based on our knowledge of the experimental techniques available at that time, we estimate that their accuracy is not better than that of Bragg37. b, Optical realization of SI units in the deep cryogenic regime. Topt and nopt are the optically determined temperature (left-hand panel) and amount of substance per volume (right-hand panel), respectively, whereas Tref and nref are the same quantities simultaneously measured with independent techniques. The dashed lines indicate the expected ideal equivalences between the optical and reference measurements. Insets: ΓD and A are the Doppler broadening and line area, respectively. c, Measured H2 phase diagram. The black points are the results obtained in this work with the fully optical approach. The red lines are from ref. 45. d, Ortho-to-para H2 spin-isomer conversion on copper surfaces. The blue and red colours correspond, respectively, to the ortho and para symmetries of the nuclear wavefunction Ψnuc. The ortho-to-para conversion time measured in this work (the 1/e decay time) is denoted as τ. The blue line comes directly from our measurement, whereas the red line is determined based on the condition that the total number of molecules inside the cavity is fixed. The error bars in all the panels represent the 1σ standard combined uncertainties containing both statistical and systematic contributions. The lack of error bars in all the panels means that the uncertainties are too small to be visible at this scale, except for the first three points in the right-hand plot in a, for which the uncertainties are large but are not reported in refs. 58,59,60,61.
We acquired the spectra at three pressures, 0.98 Pa, 3.8 Pa and 10.8 Pa, and extrapolated the pressure-perturbed line position to the zero-pressure limit; see the black points and grey line in the left-hand panel in Fig. 3a. The details of the experimental conditions, spectral analysis and uncertainty budget are given in Methods. Our ultimate determination of the 1 - 0 S(0) transition frequency is
This value includes the recoil shift. The measured photons are blueshifted; hence, we subtracted the recoil shift, which is 20.02 kHz in this case, from the measured zero-pressure value. We improved the previous best measurement of this line37 by three orders of magnitude; see the right-hand panel in Fig. 3a. The most recent theoretical value35 is
The deviation between our measurement and this theoretical value is as small as 88 kHz, with a total combined uncertainty of 380 kHz, which corresponds to validating the quantum theory for molecules at the tenth significant digit, among the most accurate validations of QED for four-body systems35.
Our accuracy of ν0 is higher than those of previous Doppler-limited measurements of homonuclear isotopologues of molecular hydrogen6,38,39 and that for Doppler-limited measurements of the HD isotopologue20,40, the dipole line intensities of which are much larger than those of the quadrupole line considered here.
Realization of the primary SI units at deep cryogenic temperatures
The redefinition of the SI unit system in 201941 revolutionized metrology at the fundamental level. Instead of referring to unit artefacts, the SI units are defined based on several recently fixed constants (such as the Boltzmann and Planck constants), and the only remaining measurable reference is the hyperfine transition frequency of caesium atoms. This opens exciting possibilities for accurate metrology and the open-access dissemination of SI unit primary standards but also poses new scientific and technological challenges regarding the actual physical realization of the redefined units, especially under difficult-to-access conditions such as the deep cryogenic regime. We address this problem for the SI units of temperature (kelvin), amount of substance per unit volume (moles per metre cubed) and pressure (pascal). We demonstrate that optical methods can be used to realize these SI units in the difficult-to-access 4-K to 20-K temperature regime.
The thermodynamic temperature T can be spectroscopically determined by measuring the Doppler component ΓD of the broadening of a molecular line42
where c, kB and \({m}_{{{\rm{H}}}_{2}}\) are the speed of light, Boltzmann constant and mass of H2, respectively. ΓD is the half-width at half-maximum of the Doppler broadening. The first parentheses contain fixed constants defining the SI units. The second parentheses contain quantities determined experimentally, but their combined relative uncertainty is less than 10−9; hence, they can also be treated as an exact number. The only quantity that needs to be measured is the frequency span ΓD, which we do using the primary time standard (the local representation of the Coordinated Universal Time (UTC) at the Astrogeodynamic Observatory in Borowiec43). This method provides the kelvin unit based only on the primary time standard and physical constants; hence, it can be regarded as a primary temperature standard41 (it does not require any calibration to other temperature standards).
We demonstrate the optical realization of the kelvin unit at T = 4.9 K to 7.8 K based on the spectra shown in Fig. 2. In the left-hand panel of Fig. 3b, we show the optically determined temperature Topt against a reference temperature Tref measured with a commercial silicon diode temperature sensor; see Table 1 for exact values of Topt and Tref and their uncertainties. The uncertainty in Topt depends on temperature. For the two lowest temperatures, the H2 vapour pressure decreases to a level at which it clearly limits the signal-to-noise ratio of the spectra; hence, the statistical contribution dominates the uncertainty in Topt, which reaches 180 mK at the lowest temperature and decreases to below 20 mK at T > 6 K. The optically determined temperature agrees with the reference diode-sensor measurements within the estimated total combined uncertainty σ for all the data points except the highest temperature, at which the deviation is 4σ. We do not know whether the 4σ discrepancy is caused by our optical method or the reference diode sensor.
The amount of substance per unit volume n (mol m−3) is proportional to the line area A (which has a unit of m−1s−1; see the areas under the red curves in Fig. 2),
where NA, ℏ, α and me are the Avogadro constant, reduced Planck constant. fine-structure constant and electron mass, respectively. This relation has the same structure as equation (1), in that it links the SI unit (mol m−3) to a spectroscopically determined quantity A, which is based only on the time unit (the m−1 s−1 unit is linked to the s−2 unit via the fixed value of the speed of light). Like equation (1), the first and second parentheses contain, respectively, the fixed constants defining the SI units and quantities determined experimentally with negligibly small uncertainties. Equation (2) also includes a dimensionless factor ξ for the transition moment. It comes from ab initio calculations (as H2 is the simplest neutral molecule, the corresponding ξ factor can be calculated with high accuracy, ξ = 6.124(6) × 10−3; ref. 44). Equation (2) should also contain the Boltzmann distribution; however, the populations of H2 rotational excited states are completely negligible. In the right-hand panel of Fig. 3b, we show the optically determined amount of gas per volume nopt against its reference value nref (see Methods for nref). The optical method allows us to reach, in the difficult-to-access cryogenic regime, an exceptionally small uncertainty of the mol m−3 unit that is far below 1%, which is two orders of magnitude better than the values obtained from the previous best p(T) dependence in ref. 45. Root-mean-square temperature fluctuations in our experimental cryogenic chamber are at the level of 0.5 mK, which at the vapour pressure condition translates to concentration fluctuations of 0.08% at T = 7.8 K.
Hydrogen phase diagram
The simultaneous optical realization of the SI units of temperature and amount of substance per volume unit, Topt and nopt, as described in ‘Realization of the primary SI units at deep cryogenic temperatures’, also automatically gives the SI unit of pressure via the equation of state of a gas, which in the relevant low-pressure regime is popt = kBNAnoptTopt; based on the second virial coefficient from ref. 46, we estimated the correction to the ideal gas (for 11 Pa and 7.8 K) as 10−4, which is negligible in our case. Note that Topt and nopt are multiplied only by the exact SI defining constants; hence, this optical realization of the pascal unit can be regarded as a primary SI standard (it does not require any calibration to other intermediate pressure standards). This allowed us to measure a part of the phase diagram of molecular hydrogen with the purely optical method (Fig. 3c). This experimental demonstration covers more than three orders of magnitude of pressure and considerably improves the accuracy of the previous best p(T) curve45 in this range (Methods). This measurement was done for purely para H2 many days after cooling the sample. The phase diagram for a normal para–ortho distribution can be obtained with the same methodology but with frequent replacing of the H2 sample; see ‘Conversion between H2 spin isomers’ for details. One of the highly promising applications that could be realized with this technology is an accurate purely optical determination of the H2 triple point, which not only is an important anchor point for calibrating cryogenic temperature sensors but also has recently gained interest from the perspective of its first-principles determination47.
Conversion between H2 spin isomers
Molecular hydrogen exists in two distinct isomeric states, referred to as para- and ortho-H2, which differ by the symmetry of the nuclear wavefunction and are related to the total nuclear spin I (I = 0 and 1 for para- and ortho-H2, respectively). Although the energy of the interaction between nuclear spins is extremely small (far below 1 mK), the Pauli exclusion principle, which leads to a relation between the symmetries of the nuclear and rotational wavefunctions, means that an even rotational quantum number j is allowed only for para-H2 and an odd j is allowed only for ortho-H2, resulting in a large energy difference between the para and ortho isomers (above 170 K). On the one hand, this makes the ortho-to-para H2 conversion a highly exothermic process (the heat released exceeds the evaporation heat). On the other hand, the ortho-to-para transition requires a nuclear spin flip, making the conversion process inherently slow. These features have important technological consequences, especially for liquid H2 energy storage technologies48 and imply the necessity of developing efficient ortho-to-para catalysts. Research on the ortho-to-para conversion is also important from a basic physics perspective, as it enables studies of the magnetic properties and structural composition of solid surfaces49, processes on the non-magnetic surfaces of noble metals50, and various physical properties of solid hydrogen51. Moreover, the ortho-to-para conversion is crucial for astrophysics52 and ab initio quantum calculations53.
In this work, we demonstrate that using the experimental platform shown in Fig. 1, we can precisely track (with fully optical methods) the composition of the H2 spin-isomer mixture. We use that the optical determination of the H2 concentration n, discussed in ‘Realization of the primary SI units at deep cryogenic temperatures’, is spin-isomer selective. We start the measurement process by filling the experimental cryogenic chamber with a new portion of H2 (taken directly from the room-temperature chamber), which immediately thermalizes with the cryogenic surfaces, but the rotational distribution remains separated between the para and ortho states, thus preserving the normal (1:3 ratio) para:ortho distribution. This sets the initial conditions for our measurement, that is, 0.25 and 0.75 fractions of H2 occupy the j = 0 (para-H2) and j = 1 (ortho-H2) states, respectively; see the blue and red curves at t = 0 in Fig. 3d. The blue curve comes directly from our measurement of the spectral line area, and the red curve comes from the condition that the total number of H2 molecules in the cavity is fixed. The temperature at which the conversion was measured (6.7 K) is much lower than the energy (divided by kB) of the j = 1 state (170.5 K); hence, the remaining ortho population (after isomer thermalization) is negligible, and the para and ortho fractions converge asymptomatically to 1 and 0, respectively. The ortho-to-para conversion time measured in this work (the 1/e decay time) is 32.4(1.1) h. The conversion time of approximately 50 h from the literature54 indicates that the residual magnetic properties of the components used to construct the cryogenic vacuum chamber and the optical cavity slightly enhanced the ortho-to-para conversion. Determining the ortho-to-para conversion rate for the specific combination of the geometry and material composition of our chamber is necessary for the optical metrology of pressure and concentration and for measuring the phase diagram demonstrated in ‘Realization of the primary SI units at deep cryogenic temperatures’ and ‘Hydrogen phase diagram’. To determine the ortho-to-para conversion rate for a given material, one would need to introduce into the cryogenic chamber a material sample with a well-defined geometry and measure the deviation from the nominal decay time.
Future perspectives
There are many directions for further development of the instrument. One direction is the implementation of the frequency-agile, rapid-scanning spectroscopy technique10, which will improve the signal-to-noise ratio of the measured spectra and further reduce systematic instrumental errors6. Another direction is to implement purely frequency-based spectroscopic methods11, which would further reduce systematic instrumental errors and extend the dynamic range to higher pressures, which would be particularly beneficial for accessing the H2 triple point. We also plan to benefit from the high power of the laser (the 3-W idler beam), which, together with the high finesse of the cavity, makes saturation of the H2 ro-vibrational lines and working in the Doppler-free regime feasible, as the cryogenic environment is beneficial (compared with room temperature), owing to the larger peak absorption, longer time of flight through the laser beam and better control of the H2 pressure down to below millipascals, with all the other species perfectly frozen out.
This experimental platform opens a way to address many other challenges in various fields, such as accurate measurements of weakly bound molecular complexes, spectroscopy of buffer-cooled complex molecules (whose spectra are difficult to resolve at elevated temperatures), acquisition of reference spectroscopic data for astrophysical applications and studies of the collisional effects at few-wavenumber collision energies.
Methods
Cryogenic cavity design and the laser system
The large cryogenic vacuum chamber (with a length of 79 cm), whose temperature can be controlled down to 4 K, lies at the heart of the set-up; see the thick yellow layer in Fig. 1. The cryogenic vacuum chamber is independent of the outer room-temperature vacuum chamber (the grey layers in Fig. 1), which allows us to fill the cryogenic chamber with a gas sample without introducing convection heat leaks between the cryogenic layers and the outer room-temperature chamber. The cryogenic high-finesse optical cavity (the thick brown cylinder with white mirrors at the ends in Fig. 1) is installed inside the cryogenic vacuum chamber. Both the cavity spacer and the cryogenic vacuum chamber are made of high-purity copper and annealed after machining to ensure high thermal conductivity and, hence, excellent temperature uniformity. Based on the thermal properties of the materials used in our set-up and the known heat loads and sinks, we estimated the temperature inhomogeneity inside the cavity spacer to be less than 1 mK. The exceptions are the central spots on the surfaces of the mirrors, which in the present configuration are heated by the laser to approximately 13 mK above the other surfaces. This inhomogeneity influences only approximately 1% of the molecules. Hence, its influence on the optically determined temperature is smaller than 1 mK, which is negligible compared with the total combined uncertainty for Topt; see the Topt row in Table 1. For future developments, the temperature inhomogeneity along the mirror surfaces will be further reduced (by many orders of magnitude) by using sapphire mirror substrates whose thermal conductivity is many orders of magnitude larger than those of fused silica.
The system can operate at any temperature in the range from 4 K to 300 K. For the first measurement campaign (presented in this work), we operated in the most challenging temperature range, which is from 4 K to 20 K. The set-up can be switched to higher temperatures by using thermal links (between the cryocooler and the cryogenic vacuum chamber) with lower thermal conductivity. We used a low-vibration two-stage cryocooler (PT420-RM, Cryomech) with 1.8 W of cooling power at 4.2 K.
The large masses of the cryogenic vacuum chamber and the thermal shields (130 kg and 200 kg, respectively), which are suspended on thin titanium and stainless steel rods, respectively, together with the high flexibility of the thermal connectors considerably attenuate the noise and vibrations. We did not observe any negative influence of the cryocooler vibrations on the operation of the optical cavity. Furthermore, we observed that the large mechanical inertia of the heavy components of the vacuum and cryogenic systems made the optical cavity much more immune to external perturbations than typical room-temperature cavity-enhanced spectrometers.
In contrast to the previous design of the mirror position actuator reported in ref. 31, we did not glue the mirrors to the copper holders but used stainless steel springs that mechanically press the mirrors against the holders (Fig. 1). When the mirrors are glued, the mismatch of the thermal expansion between the copper mirror holder and the mirror substrate material leads to misalignment of the cavity at low temperatures, which is not the case when springs are used. The mirrors (FiveNine Optics) have dielectric coatings deposited on Corning 7979 (infrared grade) fused silica substrates. The CW seed laser operating at 1,064 nm and CW OPO are the TOPTICA CTL and TOPTICA TOPO, respectively. We removed all unwanted reflections from the optical elements that form (together with the cavity mirror) unwanted etalons, as these would perturb the spectral baseline with a structure of sine fringes.
Reference temperature and pressure determination
The reference temperature Tref was measured with a commercial diode sensor (the DT-670 diode in the CU-HT package from Lake Shore Cryotronics, calibrated in the range from 1.4 K to 325 K) and read by a cryogenic temperature monitor (model 218S from the same manufacturer). The sensor is mounted directly on the copper cavity spacer (the innermost brown cylinder in Fig. 1). The uncertainty of Tref varied from 20 mK at 4.9 K to 8 mK at 7.8 K (Table 1).
The reference pressure pref was determined by combining Tref and the p(T) curve from ref. 45:
where p and T are given in pascals and kelvins, respectively. This approach works in the vapour pressure regime. Hence, pref is reported only for the highest pressure at 7.7 K in Table 1. The uncertainty of p(T) for the temperature range relevant here is not reported in ref. 45. Based on the uncertainties of the neighbouring temperature ranges, we estimated the uncertainty of pref as 50%. Having the reference temperature and pressure, we determined the reference amount of substance per unit volume nref and its uncertainty using the equation of state of a gas, which in the relevant low-pressure regime is the ideal gas equation.
Spectral simulations
Molecular hydrogen spectra are subjected to subtle collisional effects, such as the speed dependence of collisional broadening and shift and velocity-changing collisions that lead to notable Dicke narrowing62,63.
To account for these phenomena, all spectral simulations and analyses in this work use the speed-dependent hard-collision (SDHC) profile55,56:
where \({\widetilde{I}}_{\mathrm{SDV}}^{* }(\nu )\) is a complex speed-dependent Voigt profile:
Here vz is the z component of the velocity \({\bf{v}}\) of an active molecule and \({f}_{{\rm{m}}}({\boldsymbol{v}})={(\sqrt{{\rm{\pi }}}{v}_{{\rm{m}}})}^{-3}{{\rm{e}}}^{-{({\boldsymbol{v}}/{v}_{{m}})}^{2}}\) is the Maxwellian velocity distribution, with \({v}_{{\rm{m}}}=\sqrt{2{k}_{{\rm{B}}}T/m}\) being the most probable speed of the absorbing molecule with mass m at temperature T.
The SDHC profile accounts for Doppler broadening through νD = ν0vm/c, which is related to the half-width at half-maximum of the Doppler profile through \({\varGamma }_{{\rm{D}}}=\sqrt{ln2}{\nu }_{{\rm{D}}}\). The profile is further defined by four collisional parameters: the Lorentzian half-width at half-maximum and shift (Γ(v) and \(\Delta\) (v)), which depend on the speed of the absorbing molecule, and the real and imaginary parts of the complex Dicke parameter (\({\nu }_{\mathrm{opt}}^{{\rm{r}}}\) and \({\nu }_{\mathrm{opt}}^{{\rm{i}}}\)), which quantify the effects of velocity-changing collisions.
The ‘hard-collision’ component of SDHC assumes that any velocity-changing collision results in complete thermalization of the molecular velocity, regardless of its pre-collisional state64,65. Although sufficient for many systems, this approximation fails to capture the full dynamics of molecular hydrogen, which exhibits strong Dicke narrowing63. To account for this, we incorporated a simple analytical correction to SDHC that mimics the behaviour of a more physically justified description of the velocity-changing collisions57. The correction is made by replacing \({\nu }_{\mathrm{opt}}^{{\rm{r}}}\) in equations (4) and (5) with \({\beta }_{\alpha }(\chi ){\nu }_{\mathrm{opt}}^{{\rm{r}}}\), where βα(χ) is an analytical function of the absorber-to-perturber mass ratio, α (here α = 1), and the narrowing parameter, \(\chi ={\nu }_{\mathrm{opt}}^{{\rm{r}}}/{\varGamma }_{{\rm{D}}}\) (see ref. 57 for details). The final model used in the analysis is, therefore, the β-corrected SDHC.
A key advantage of this model is that its parameters can be determined from first principles by connecting them to energy-dependent generalized spectroscopic cross sections \({\sigma }_{\lambda }^{q}({E}_{{\rm{kin}}})\) (refs. 66,67). These relations are56,68
Here p is the pressure, vr is the relative speed of the colliding molecules, 〈vr〉 is its mean value at temperature T, \({\bar{v}}_{{\rm{p}}}\) is the most probable speed of the perturber distribution (in the self-perturbed case \({\bar{v}}_{{\rm{p}}}={v}_{{\rm{m}}}\)), \({E}_{\mathrm{kin}}=\mu {v}_{{\rm{r}}}^{2}/2\) is the relative collision energy, μ is the reduced mass of the colliding system and the integration variable is x = Ekin/kBT. The cross-sectional superscripts denote the rank of the transition (q = 2 for the S(0) quadrupole line), and the subscripts the type of cross section (λ = 0 for pressure broadening and shift and λ = 1 for the Dicke cross section). We determined the generalized cross sections from scattering S-matrix elements obtained from our quantum-scattering calculations66,67,69.
In this work, we computed the reference collisional line-shape parameters for the self-perturbed 1 - 0 S(0) transition in H2. We used the state-of-the-art six-dimensional (6D) H2–H2 potential energy surface (PES), which explicitly incorporates the dependence of the interaction energy on the bond lengths of the two molecules17. The 6D PES is expanded in bispherical harmonics and averaged over the wavefunctions of the isolated monomers (see equations (B.1)–(B.3) in ref. 17). Unlike our previous work on the room-temperature self-perturbed 2 - 0 P(3) transition11, which uses the same PES, here we perform full 6D quantum-scattering calculations, that is, we retain all possible combinations of coupling terms that enter equation (B.3) in ref. 17, including the full centrifugal distortion of the PES and vibrational coupling. Such a comprehensive treatment is crucial for realizing an accurate description of the collision dynamics at cryogenic conditions for two primary reasons. First, it correctly captures the resonant and near-resonant exchange of vibrational and rotational quanta between colliding partners70,71. Second, it allows for accurate modelling of quantum interference effects that arise from the indistinguishability of colliding molecules; these become particularly prominent at low energies, where only a few partial waves contribute to scattering72.
The explicit treatment of quantum indistinguishability in line-shape calculations has received little attention73,74,75. For self-perturbation, the colliding molecules are identical, meaning that resonant and interference contributions must be included along with the standard formulas for the generalized cross sections. For the 1 - 0 S(0) line considered in this work, S-matrix elements that couple the v = 0 and v = 1 vibrational levels are needed, making a full 6D calculation essential. This work fully accounts for quantum indistinguishability in the ab initio calculation of the spectral line shape.
The coupled-channel equations were solved using the renormalized Numerov method76 implemented in our in-house BIGOS code77,78. To ensure convergence of the thermal averages in equation (6), calculations were performed on a dense grid of collision energies spanning nine orders of magnitude (from 10−6 cm−1 to 3 × 103 cm−1). The radial propagation extended from 2.5a0 to 100a0, with a0 being the Bohr radius. The channel basis set involved rotational levels j = 0, 2 and 4 for both the v = 0 and v = 1 vibrational manifolds, which was sufficient to converge the cross sections to within 1% in the relevant energy range.
Spectral analysis
In this work, we acquired the H2 spectra at five temperatures from 4.9 K to 7.8 K in the vapour pressure regime, which correspond to pressures from 4.68 mPa to 10.8 Pa (Table 1). At the highest temperature, T = 7.8 K, we acquired the spectra also at two lower pressures, 1 Pa and 3.8 Pa, which we used to extrapolate the position of the 1 - 0 S(0) H2 line to the zero-pressure limit. To increase the signal-to-noise ratio, we repeated the scans many times at each pressure–temperature point (Table 1).
The black points in Fig. 3a demonstrate the dependence of the line position on pressure. They were determined by performing a single-pressure fit of the β-corrected SDHC profile to the experimental spectra. The grey line is a linear extrapolation to the zero-pressure limit, ν0. It turns out, however, that ν0 can be more accurately determined when all the spectra at all the pressures are fitted simultaneously, with the constraint that the collision-induced shift and line-shape parameters are linear in pressure. Furthermore, to reduce the systematic uncertainties originating from numerical correlations between ν0 and the line-shift and line-shape parameters, we fixed the line-shape parameters to their theoretical ab initio values11. Following ref. 6, to estimate the systematic uncertainties related to the description of the shape of the measured line, we repeated all the fits but changed the values of the ab initio line-shape parameters by a conservative amount of 10%, which resulted in a line-shape contribution to the ν0 uncertainty of 6 kHz. The statistical contribution (1σ) to the uncertainty of ν0 is 15 kHz. Other components of the uncertainty budget are negligible at this level. The standard combined uncertainty for ν0 is 16 kHz.
The amount of substance per unit volume
Equation (2) is valid for the quadrupole electric transitions, such as the case considered in this work, the 1 - 0 S(0) transition. In this case, the theoretical dimensionless ξ factor is given by \(\xi ={(| {Q}_{\mathrm{eg}}| /e{a}_{0}^{2})}^{2}\). Here Qeg denotes the quadrupole moment function of the ground electronic \({}^{1}\Sigma _{g}^{+}\) state of H2, Q(r) (ref. 44), averaged over the wavefunctions of the ground χg(r) and excited χe(r) ro-vibrational states of H2:
where r is the internuclear distance in H2, and e and a0 denote the elementary charge and Bohr radius, respectively.
For electric dipole lines (important for the HD isotopologue), equation (2) takes the form:
with the dimensionless theoretical factor \(\xi ={(| {d}_{\mathrm{eg}}| /e{a}_{0})}^{2}\). Here deg denotes the dipole moment function of the ground electronic 1Σ+ state of HD, d(r), averaged over the wavefunctions of the ground and excited ro-vibrational states of HD, as in equation (7).
H2 phase diagram
In this work, we improved the p(T) curve from ref. 45 by using our optically measured (p, T) points from the H2 phase diagram, the black points from Fig. 3c. We fitted the three numerical parameters from equation (3) to a combined set of the old (p, T) points used in ref. 45 and our much more accurate (p, T) points reported in this work, resulting in
This p(T) curve is considerably more accurate than the one from ref. 45, especially in the 4.9 K to 7.7 K temperature range, where the 50% estimated accuracy of equation (3) is improved to below 1% for equation (9).
Data availability
The data that support the plots within this paper and other findings of this study are publicly available in the Repository for Open Data79. Source data are available with this paper.
References
Gianfrani, L., Hu, S.-M. & Ubachs, W. Advances in cavity-enhanced methods for high precision molecular spectroscopy and test of fundamental physics. Riv. Nuovo Cimento 47, 229–298 (2024).
Gomez-Pelaez, A. J., Ramos, R., Cuevas, E., Gomez-Trueba, V. & Reyes, E. Atmospheric CO2, CH4, and CO with the CRDS technique at the Izaña Global GAW station: instrumental tests, developments, and first measurement results. Atmos. Meas. Tech. 12, 2043–2066 (2019).
Bielska, K. et al. Subpromille measurements and calculations of CO (3–0) overtone line intensities. Phys. Rev. Lett. 129, 043002 (2022).
Lisak, D., Havey, D. K. & Hodges, J. T. Spectroscopic line parameters of water vapor for rotation-vibration transitions near 7180 cm−1. Phys. Rev. A 79, 052507 (2009).
Fleisher, A. J. et al. Absolute 13C/12C isotope amount ratio for Vienna PeeDee Belemnite from infrared absorption spectroscopy. Nat. Phys. 17, 889–893 (2021).
Zaborowski, M. et al. Ultrahigh finesse cavity-enhanced spectroscopy for accurate tests of quantum electrodynamics for molecules. Opt. Lett. 45, 1603–1606 (2020).
Ye, J., Ma, L.-S. & Hall, J. L. Sub-Doppler optical frequency reference at 1.064 μm by means of ultrasensitive cavity-enhanced frequency modulation spectroscopy of a C2HD overtone transition. Opt. Lett. 21, 1000–1002 (1996).
Cozijn, F. M. J., Diouf, M. L. & Ubachs, W. Lamb dip of a quadrupole transition in H2. Phys. Rev. Lett. 131, 073001 (2023).
Bernhardt, B. et al. Cavity-enhanced dual-comb spectroscopy. Nat. Photonics 4, 55–57 (2010).
Truong, G.-W. et al. Frequency-agile, rapid scanning spectroscopy. Nat. Photonics 7, 532–534 (2013).
Cygan, A. et al. Dispersive heterodyne cavity ring-down spectroscopy exploiting eigenmode frequencies for high-fidelity measurements. Sci. Adv. 11, eadp8556 (2025).
Fasci, E. et al. Precision spectroscopy of HD at 1.38 μm. Phys. Rev. A 98, 022516 (2018).
Changala, P. B., Weichman, M. L., Lee, K. F., Fermann, M. E. & Ye, J. Rovibrational quantum state resolution of the C60 fullerene. Science 363, 49–54 (2019).
Jankowski, P., McKellar, A. R. W. & Szalewicz, K. Theory untangles the high-resolution infrared spectrum of the ortho-H2-CO van der Waals complex. Science 336, 1147–1150 (2012).
Słowiński, M. et al. H2-He collisions: ab initio theory meets cavity-enhanced spectra. Phys. Rev. A 101, 052705 (2020).
Thachuk, M., Chuaqui, C. E. & Le Roy, R. J. Linewidths and shifts of very low temperature CO in He: a challenge for theory or experiment? J. Chem. Phys. 105, 4005–4014 (1996).
Jóźwiak, H. et al. Accurate reference spectra of HD in an H2-He bath for planetary applications. Astron. Astrophys. 687, A69 (2024).
McKellar, A. R. W. Infrared spectra of hydrogen dimers. J. Chem. Phys. 92, 3261–3277 (1990).
Sung, K. et al. The rotational spectrum of HD broadened by H2 at temperatures between 100–296 K. J. Quant. Spectrosc. Radiat. Transf. 295, 108412 (2023).
Kassi, S., Lauzin, C., Chaillot, J. & Campargue, A. The (2-0) R(0) and R(1) transition frequencies of HD determined to a 10−10 relative accuracy by Doppler spectroscopy at 80 K. Phys. Chem. Chem. Phys. 24, 23164–23172 (2022).
Moehnke, C. J., Lewis, E. K., Lopez-Calvo, A. & Manzanares, C. E. Phase shift cavity ring down at low temperatures: vibration-rotation overtone absorption of H-D (Δν = 4) at 297 and 105 K. Chem. Phys. Lett. 418, 576–580 (2006).
Santamaria, L. et al. Comb-assisted cavity ring-down spectroscopy of a buffer-gas-cooled molecular beam. Phys. Chem. Chem. Phys. 18, 16715–16720 (2016).
Libert, A. et al. Assignment of the methanol OH-stretch overtone spectrum using the pattern recognition method. Phys. Chem. Chem. Phys. 26, 16505–16513 (2024).
Didriche, K., Lauzin, C., Földes, T., de Ghellinck D’Elseghem Vaernewijck, X. & Herman, M. The FANTASIO+ set-up to investigate jet-cooled molecules: focus on overtone bands of the acetylene dimer. Mol. Phys. 108, 2155–2163 (2010).
Perot, S. et al. Jet-cooled ethylene cavity ring-down spectroscopy between 5880 and 6200 cm−1. J. Quant. Spectrosc. Radiat. Transf. 324, 109065 (2024).
Liu, Q.-H., Lv, Y.-N., Zou, C.-L., Cheng, C.-F. & Hu, S.-M. Saturated absorption spectroscopy of HD at 76 K. Phys. Rev. A 106, 062805 (2022).
Robinson, J. M. et al. Crystalline optical cavity at 4 K with thermal-noise-limited instability and ultralow drift. Optica 6, 240–243 (2019).
Wiens, E. et al. Optical frequency reference based on a cryogenic silicon resonator. Opt. Express 31, 42059–42076 (2023).
He, L. et al. Ultra-stable cryogenic sapphire cavity laser with an instability reaching 2 × 10−16 based on a low vibration level cryostat. Opt. Lett. 48, 2519–2522 (2023).
Valencia, J., Iskander, G., Nardelli, N. V., Leibrandt, D. R. & Hume, D. B. Cryogenic sapphire optical reference cavity with crystalline coatings at 1 × 10−16 fractional frequency instability. Rev. Sci. Instrum. 95, 103002 (2024).
Słowiński, M. et al. Cryogenic mirror position actuator for spectroscopic applications. Rev. Sci. Instrum. 93, 115003 (2022).
Schenkel, M. R., Vogt, V. A. & Schiller, S. Metrology-grade spectroscopy source based on an optical parametric oscillator. Opt. Express 32, 43350–43365 (2024).
Drever, R. W. P. et al. Laser phase and frequency stabilization using an optical resonator. Appl. Phys. B 31, 97–105 (1983).
Salumbides, E. et al. Bounds on fifth forces from precision measurements on molecules. Phys. Rev. D 87, 112008 (2013).
Pachucki, K. & Komasa, J. From first-principles to quantum electrodynamics: pushing the limits of theory with the hydrogen molecule. J. Chem. Theory Comput. 21, 12664–12673 (2025).
Pachucki, K. & Komasa, J. Nonadiabatic rotational states of the hydrogen molecule. Phys. Chem. Chem. Phys. 20, 247–255 (2018).
Bragg, S. L., Brault, J. W. & Smith, W. H. Line positions and strengths in the H2 quadrupole spectrum. Astrophys. J. 263, 999–1004 (1982).
Lamperti, M. et al. Stimulated Raman scattering metrology of molecular hydrogen. Commun. Phys. 6, 67 (2023).
Fleurbaey, H., Koroleva, A. O., Kassi, S. & Campargue, A. The high-accuracy spectroscopy of H2 rovibrational transitions in the (2-0) band near 1.2 μm. Phys. Chem. Chem. Phys. 25, 14749–14756 (2023).
Castrillo, A., Fasci, E. & Gianfrani, L. Doppler-limited precision spectroscopy of HD at 1.4 μm: an improved determination of the R(1) center frequency. Phys. Rev. A 103, 022828 (2021).
Machin, G. The kelvin redefined. Meas. Sci. Technol. 29, 022001 (2018).
Daussy, C. et al. Direct determination of the Boltzmann constant by an optical method. Phys. Rev. Lett. 98, 250801 (2007).
Morzyński, P. et al. Absolute measurement of the 1S0 - 3P0 clock transition in neutral 88Sr over the 330 km-long stabilized fibre optic link. Sci. Rep. 5, 17495 (2015).
Roueff, E. et al. The full infrared spectrum of molecular hydrogen. Astron. Astrophys. 630, A58 (2019).
Souers, P. C. Hydrogen Properties for Fusion Energy (Univ. California Press, 1986).
Garberoglio, G. & Harvey, A. H. First-principles calculation of ortho-para effects in the second virial coefficients of H2 and D2 at low temperatures. J. Phys. Chem. A 129, 8453–8463 (2025).
Deiters, U. K. & Sadus, R. J. An intermolecular potential for hydrogen: classical molecular simulation of pressure-density-temperature behavior, vapor-liquid equilibria, and critical and triple point properties. J. Chem. Phys. 158, 194502 (2023).
Tzimas, E., Filiou, C., Peteves, S. D. & Veyret, J.-B. Hydrogen Storage: State-of-the-art and Future Perspective. Report No. EUR 20995EN (European Commission, 2003).
Ilisca, E. Ortho-para hydrogen conversion on metal surfaces. Mod. Phys. Lett. B 05, 1191–1198 (1991).
Ilisca, E. Ortho-para conversion of hydrogen molecules physisorbed on surfaces. Prog. Surf. Sci. 41, 217–335 (1992).
Driessen, A., van der Poll, E. & Silvera, I. F. Ortho-para conversion of solid hydrogen as a function of density. Phys. Rev. B 30, 2517–2526 (1984).
Lique, F., Honvault, P. & Faure, A. Ortho-para-H2 conversion processes in astrophysical media. Int. Rev. Phys. Chem. 33, 125–149 (2014).
Pachucki, K. & Komasa, J. Ortho-para transition in molecular hydrogen. Phys. Rev. A 77, 030501 (2008).
Strzhemechny, M. A., Hemley, R. J., Mao, H.-K., Goncharov, A. F. & Eggert, J. H. Ortho-para conversion of hydrogen at high pressures. Phys. Rev. B 66, 014103 (2002).
Lance, B., Blanquet, G., Walrand, J. & Bouanich, J.-P. On the speed-dependent hard collision lineshape models: application to C2H2 perturbed by Xe. J. Mol. Spectrosc. 185, 262–271 (1997).
Pine, A. S. Asymmetries and correlations in speed-dependent Dicke-narrowed line shapes of argon-broadened HF. J. Quant. Spectrosc. Radiat. Transf. 62, 397–423 (1999).
Konefał, M. et al. Analytical-function correction to the Hartmann-Tran profile for more reliable representation of the Dicke-narrowed molecular spectra. J. Quant. Spectrosc. Radiat. Transf. 242, 106784 (2020).
Rank, D. H., Rao, B. S., Slomba, A. F., Sitaram, P. & Wiggins, T. A. Fundamental band of the quadrupole spectrum of the hydrogen molecule. Nature 194, 1267–1268 (1962).
Rank, D. H., Rao, B. S., Sitaram, P., Slomba, A. F. & Wiggins, T. A. Quadrupole and induced dipole spectrum of molecular hydrogen. J. Opt. Soc. Am. 52, 1004–1009 (1962).
Rank, D. H. & Wiggins, T. A. Quadrupole spectrum of molecular hydrogen. J. Opt. Soc. Am. 53, 759–760 (1963).
Fink, U., Wiggins, T. & Rank, D. Frequency and intensity measurements on the quadrupole spectrum of molecular hydrogen. J. Mol. Spectrosc. 18, 384–395 (1965).
Wcisło, P., Thibault, F., Cybulski, H. & Ciuryło, R. Strong competition between velocity-changing and phase- or state-changing collisions in H2 spectra perturbed by Ar. Phys. Rev. A 91, 052505 (2015).
Wcisło, P. et al. The implementation of non-Voigt line profiles in the HITRAN database: H2 case study. J. Quant. Spectrosc. Radiat. Transf. 177, 75–91 (2016).
Nelkin, M. & Ghatak, A. Simple binary collision model for Van Hove’s Gs(r, t). Phys. Rev. 135, A4–A9 (1964).
Rautian, S. G. & Sobelman, I. I. The effect of collisions on the Doppler broadening of spectral lines. Sov. Phys. Uspekhi 9, 701 (1967).
Monchick, L. & Hunter, L. W. Diatomic-diatomic molecular collision integrals for pressure broadening and Dicke narrowing: a generalization of Hess’s theory. J. Chem. Phys. 85, 713–718 (1986).
Schaefer, J. & Monchick, L. Line shape cross sections of HD immersed in He and H2 gas. I. Pressure broadening cross sections. J. Chem. Phys. 87, 171–181 (1987).
Wcisło, P. et al. Accurate deuterium spectroscopy for fundamental studies. J. Quant. Spectrosc. Radiat. Transf. 213, 41–51 (2018).
Olejnik, A. et al. Ab initio quantum scattering calculations and a new potential energy surface for the HCl(X1Σ+)-O2(\({X}^{3}{\Sigma }_{g}^{-}\)) system: collision-induced line shape parameters for O2-perturbed R(0) 0-0 line in H35Cl. J. Chem. Phys. 159, 134301 (2023).
Quéméner, G., Balakrishnan, N. & Krems, R. V. Vibrational energy transfer in ultracold molecule-molecule collisions. Phys. Rev. A 77, 030704(R) (2008).
Fonseca dos Santos, S., Balakrishnan, N., Forrey, R. C. & Stancil, P. C. Vibration-vibration and vibration-translation energy transfer in H2-H2 collisions: a critical test of experiment with full-dimensional quantum dynamics. J. Chem. Phys. 138, 104302 (2013).
Huo, W. M. & Green, S. Quantum calculations for rotational energy transfer in nitrogen molecule collisions. J. Chem. Phys. 104, 7572–7589 (1996).
Ben Reuven, A. in Advances in Chemical Physics, Vol. 33 (eds Prigogine, I. et al.) 235–293 (Wiley, 1975).
Liu, W.-K. Symmetrized Liouville basis for indistinguishable particles. Application to spectral linewidth. J. Chem. Phys. 72, 4869–4872 (1980).
Monchick, L. Quantum kinetic equations incorporating the Fano collision operator: the generalized Hess method of describing line shapes. J. Chem. Phys. 101, 5566–5577 (1994).
Johnson, B. R. The renormalized Numerov method applied to calculating bound states of the coupled-channel Schroedinger equation. J. Chem. Phys. 69, 4678–4688 (1978).
Jóźwiak, H., Thibault, F., Viel, A., Wcisło, P. & Lique, F. Revisiting the rovibrational (de-)excitation of molecular hydrogen by helium. Astron. Astrophys. 685, A113 (2024).
Jóźwiak, H. The SCATTERING code adjusted for diatom-atom calculations. Zenodo http://zenodo.org/records/10776728 (2024).
Stankiewicz, K. et al. Data for manuscript entitled ‘Cavity-enhanced spectroscopy in the deep cryogenic regime for quantum sensing and metrology’. Repository for Open Data https://doi.org/10.18150/MRKBNY (2026).
Acknowledgements
We thank M. Drewsen and J. Glässel for discussions on cryogenic technologies. This research was funded by the European Union (Grant No. 101075678, ERC-2022-STG, H2TRAP). The views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. This work is a part of the 23FUN04 COMOMET project that has received funding from the European Partnership on Metrology, which is co-financed by the European Union’s Horizon Europe Research and Innovation Programme and by the participating states (Funder ID 10.13039/100019599). We gratefully acknowledge Polish high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support (Computational Grant Nos. PLG/2024/017252 and PLG/2025/018511). This work used resources provided by the Wroclaw Centre for Networking and Supercomputing (http://wcss.pl). The work was supported by the National Science Centre in Poland (Project Nos. 2019/35/B/ST2/01118 to K.S., M.M., K.L.S., B.B., H.J., N.S., M.P. and P.W., 2022/46/E/ST2/00282 to M.S. and D.K., 2023/51/B/ST2/00427 to D.L., 2021/42/E/ST2/00152 to S.W. and 2020/39/B/ST2/00719 to A.C.). The research was conducted using infrastructure from the National Laboratory of Photonic and Quantum Technologies and is part of the programme of the National Laboratory FAMO in Toruń, Poland.
Author information
Authors and Affiliations
Contributions
P.W. and D.L. conceived the research. K.S., M.M., M.S., K.L.S., B.B., S.W., A.C., G.K., P.M., M.P., D.L. and P.W. designed and constructed the experimental set-up. H.J.J. performed the theoretical calculations. K.S., M.M., M.S., K.L.S., B.B., M.N., D.K., G.K. and P.M. carried out the measurements and collected the experimental data. M.S., N.S., M.N., D.K., D.L. and P.W. analysed the experimental data. All authors discussed the results. K.S., H.J.J., D.L. and P.W. wrote the paper with input from all authors. The research work was supervised by P.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Physics thanks Cunfeng Cheng, Nikolai Klimov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Source Data Fig. 1 (download XLSX )
Source Data for Fig. 1.
Source Data Fig. 2 (download XLSX )
Source Data for Fig. 2.
Source Data Fig. 3 (download XLSX )
Source Data for Fig. 3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stankiewicz, K., Makowski, M., Słowiński, M. et al. Cavity-enhanced spectroscopy in the deep cryogenic regime for quantum sensing and metrology. Nat. Phys. (2026). https://doi.org/10.1038/s41567-026-03204-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41567-026-03204-8
