
Abstract
Down syndrome predisposes individuals to haematological abnormalities, such as increased number of erythrocytes and leukaemia in a process that is initiated before birth and is not entirely understood1,2,3. Here, to understand dysregulated haematopoiesis in Down syndrome, we integrated single-cell transcriptomics of over 1.1 million cells with chromatin accessibility and spatial transcriptomics datasets using human fetal liver and bone marrow samples from 3 fetuses with disomy and 15 fetuses with trisomy. We found that differences in gene expression in Down syndrome were dependent on both cell type and environment. Furthermore, we found multiple lines of evidence that haematopoietic stem cells (HSCs) in Down syndrome are ‘primed’ to differentiate. We subsequently established a Down syndrome-specific map linking non-coding elements to genes in disomic and trisomic HSCs using 10X multiome data. By integrating this map with genetic variants associated with blood cell counts, we discovered that trisomy restructured regulatory interactions to dysregulate enhancer activity and gene expression critical to erythroid lineage differentiation. Furthermore, as mutations in Down syndrome display a signature of oxidative stress4,5, we validated both increased mitochondrial mass and oxidative stress in Down syndrome, and observed that these mutations preferentially fell into regulatory regions of expressed genes in HSCs. Together, our single-cell, multi-omic resource provides a high-resolution molecular map of fetal haematopoiesis in Down syndrome and indicates significant regulatory restructuring giving rise to co-occurring haematological conditions.
Similar content being viewed by others


Main
Children with trisomy of chromosome 21 (Ts21), known as Down syndrome, have a 150-fold higher risk of developing myeloid leukaemia in the first 5 years of life than the general population1,2. The current leukaemogenesis model in Down syndrome proposes that specific somatic mutations in the transcription factor GATA1 occur in Ts21 fetal liver HSCs, leading to pre-leukaemia6. Pre-leukaemia spontaneously resolves in most newborn babies, but can progress to leukaemia when persistent GATA1-mutant clones acquire additional mutations post-birth, often in chromatin regulators and signalling factors7,8. However, this model does not explain further numerous haematological abnormalities present in Ts21 fetal liver and newborn babies7. Neonates with Down syndrome frequently exhibit defects in red blood cell (RBC) production, such as polycythaemia (increased erythrocyte numbers) or macrocytosis (large erythrocytes)3, independent of leukaemia-inducing mutations. These abnormalities vary in severity and penetrance, suggesting contributions from epigenetics, microenvironment composition and gene–environment interactions9,10,11. For example, during fetal development, haematopoiesis occurs simultaneously in both the fetal liver and the bone marrow. Previous research in Down syndrome has shown an increase in megakaryocyte–erythroid progenitors in the fetal liver but not in the bone marrow, along with a reduction in granulocyte–monocyte progenitors, emphasizing the relevance of the microenvironment3.
Ts21 fetal liver haematopoietic stem and progenitor cells exhibit a unique mutational signature related to oxidative stress4, a potent driver of mutagenesis and a potential cause for the GATA1 mutations observed in Ts21 pre-leukaemia and acute megakaryocytic leukaemia4,5. However, given the well-known and substantial effects of chromatin organization on mutation patterns12,13, and the effect of Ts21 on reconfiguring the entire 3D genome14, it remains to be evaluated whether Ts21-induced genome-wide reshaping of chromatin organization in a cell-type-specific manner underlies distinct haematological phenotypes observed in Ts21, such as an increase in erythroid cells but an impairment in the lymphoid lineage. Furthermore, it remains to be determined how altered chromatin organization and heightened oxidative stress in Ts21 might create an environment conducive to acquiring mutations that contribute to the development of pre-leukaemia and leukaemia.
Here we combined single-cell RNA sequencing (scRNA-seq) from 1.1 million cells, 10X multiome, spatial transcriptomics and single-cell in vitro differentiation assays to examine how fetal haematopoietic cells with an altered dosage of chromosome 21 genes interact with particular niches (fetal liver versus bone marrow) and how these interactions differ from those in fetuses with disomy. We further assessed the regulatory landscape of haematopoietic cells in Down syndrome and its effect on gene expression, cellular differentiation and the regional distribution of somatic mutations.
Multi-omics of fetal haematopoiesis
To capture a full diversity of blood and niche cell types present in the fetal liver and bone marrow (isolated from the femur), we implemented various sorting and column-enrichment strategies. We isolated cells from 15 fetuses with Ts21 and 3 fetuses with disomy (median age of 14 post-conception weeks), from each of the following 3 populations: CD235− (niche and haematopoietic cells depleted of erythrocytes), CD34+Lin− (haematopoietic progenitors) and CD45+ (haematopoietic marker) and sequenced their transcriptomes using 10X Genomics (Fig. 1a and Supplementary Figs. 1 and 2). In addition, we collected part of the liver from 11 fetuses with Ts21 and 5 fetuses with disomy for spatial transcriptomics analysis using 10X Genomics Visium. Finally, for three Ts21 and three disomic liver samples, we performed 10X multiome analysis of CD45+ cells, combining a single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) and single-nucleus RNA sequencing (snRNA-seq) from the same cells (Fig. 1a).

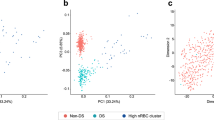
a, Experimental workflow. Cells (CD45+, CD34+Lin− and CD235a−) from fetuses with disomy and Ts21 were isolated from the bone marrow (femur) and the liver and processed for scRNA-seq. CD45+ cells from the fetal liver (number of fetus (k) = 3) were processed for multiome, and tissue sections from fetuses with disomy (k = 5) and Ts21 (k = 11) were used for spatial transcriptomics. Illustrations adapted from ref. 50, Springer Nature Limited. b, 3D uniform manifold approximation and projection (UMAP) visualization of cells from Ts21 liver (number of cells (n) = 780,299, k = 15), Ts21 femur (n = 162,775, k = 12), disomic liver (n = 110,671, k = 3) and disomic femur (n = 53,807, k = 3) coloured by broad cell-type categories. NK, natural killer. c, Stacked barplot of relative abundances of different broad cell types in fetuses with Ts21 and disomy. The arrows indicate a significant increase or decrease in cell number in the Ts21 liver compared with the disomic liver. The difference in cell proportion was tested by a two-sided Wilcoxon rank-sum test with significance determined at FDR < 0.1.
Cell composition differs in Ts21
Following quality control (Methods), our single-cell RNA sequencing (scRNA-seq) dataset comprised 943,074 cells from fetuses with Ts21 and 164,478 cells from fetuses with disomy (Supplementary Tables 1 and 2). We examined putative GATA1 somatic mutations from the scRNA-seq data; however, none was likely to impact GATA1 function (Supplementary Note 1). We identified eight major blood and niche cell types in both the Ts21 and the disomic fetal liver and femur (Fig. 1b and Supplementary Fig. 2), which were further resolved into 37 cell types in the femur and 25 in the liver (Methods; Extended Data Fig. 1, Supplementary Tables 3–7, Supplementary Figs. 1c–f and 2–5 and Supplementary Notes 2–5). In the Ts21 liver, we detected a significant increase in the proportion of HSC/multipotent progenitors (MPPs; as previously described15), erythroid cells, mast cells and megakaryocytes, and a decrease in the proportion of myeloid cells (false discovery rate (FDR) < 0.1; Fig. 1c, Supplementary Table 8 and Supplementary Fig. 6). Furthermore, we identified an additional population of highly cycling HSCs in the Ts21 liver, but did not detect a distinct cluster of mature B cells, in line with previously reported impairment in B cell development15 (Supplementary Fig. 4).
Using flow cytometry, we further compared the immunophenotypic stem and progenitor compartments of the Ts21 and disomic fetal liver, confirming an increase in the number of HSCs and megakaryocyte–erythroid–mast progenitors and a decrease in B cell populations in Ts21 (P < 0.05; Supplementary Fig. 7). We confirmed the robustness of our cell-type annotations and differences in frequency of cell types between Ts21 and disomic liver samples after integration using Harmony16 and scVI17, as well as reference-query mapping from a published disomic fetal liver atlas18 and our Ts21 liver cells to disomic liver cells (Supplementary Figs. 8–10 and Supplementary Note 6). Finally, we assessed differences in cell abundances using 10X Visium (Supplementary Tables 9 and 10, Supplementary Figs. 11 and 12 and Supplementary Note 7) and which ligand–receptor pairs are involved in HSC/MPPs interactions with the fetal liver niche (Supplementary Fig. 12 and Supplementary Note 8). Overall, the Ts21 liver showed greater haematopoietic perturbation than the bone marrow, where cycling HSCs and dysregulated B cell maturation were predominantly observed.
Gene–environment interactions in Ts21
An increase in somatic mutation rate in Ts21 fetal haematopoietic stem and progenitor cells (an estimated 1.3-fold increase4) cannot sufficiently explain the increased leukaemic development rate in children with Down syndrome (approximately 150-fold1,2). As HSCs are believed to be a cell of origin in Ts21 leukaemia, and the genetic background (trisomic versus disomic) and haematopoietic environment (liver versus femur) are significant factors4, we analysed gene–environment interactions specifically in HSCs7.
We contrasted the gene expression of HSC/MPPs collected from fetuses with disomy and Ts21 between the liver and bone marrow environments. By subsampling liver HSCs to balance the number of cells and donors across genotypes and environments, we discerned consistent gene expression changes between the liver and bone marrow that are present independent of Ts21 genotype (environment driven) versus those differences that are only present in Ts21 cells (Ts21 induced; see the Methods; Fig. 2a, Extended Data Fig. 2, Supplementary Tables 11–14, Supplementary Figs. 13–15 and Supplementary Notes 9–14).

a, Differential expression analysis (DEA) between the liver and femur within Ts21 and disomic cells. HSC/MPP genes were characterized as environment driven (detected in Ts21 and disomic cells), Ts21 induced (discovered in Ts21 cells) or Ts21 reverted (discovered in disomic cells) by downsampling Ts21 liver and femur data to the same size as disomic liver and femur data. Owing to reduced statistical power, differentially expressed genes (DEGs) were found in the full Ts21 dataset, but not in the downsampled Ts21 or disomic data, making it inconclusive whether they were Ts21 induced or environment driven. A volcano plot of DEGs between fetal bone marrow and liver HSC/MPPs from fetuses with Ts21 (bottom) or disomy (top) is also shown. The x axis indicates the log2 fold change (LFC), and the y axis denotes −log10 FDR. ** indicates significance in DEA of the specified genetic background. Illustrations adapted from ref. 50, Springer Nature Limited, and created using BioRender (https://biorender.com). b, Gene set enrichment analysis of upregulated Ts21-induced genes using EnrichR. The scatter plots show Gene Ontology (GO) terms (molecular function (MF) and biological process (BP)) or ENCODE and ChEA transcription factors (TFs). Significance is displayed using FDR values. c, Pie chart of cells in G1, G2M and S phase in cycling Ts21 fetal liver HSC/MPPs. d, Volcano plot between cycling and normal Ts21 HSC/MPPs. The blue dots represent genes with FDR < 0.05 and LFC > 1. The genes of interest are coloured by Gene Ontology terms. Analyses in panels a–d include the Ts21 liver (n = 780,299, k = 15), Ts21 femur (n = 162,775, k = 12), disomic liver (n = 110,671, k = 3) and disomic femur (n = 53,807, k = 3). e,f, Boxplots displaying per-sample MitoTracker (e) and MitoSOX (f) staining in disomic (n = 3 samples) and Ts21 (n = 3 samples) haematopoietic populations: HSCs (Lin−CD34+CD38−CD90+CD45RA− population), CD38− (Lin−CD45+CD34+CD38− population), CD38+ (Lin−CD45+CD34+CD38+ population) and Lin+ (CD3/CD8/CD11b/CD56/CD14/CD19, lineage-positive population). Nominal significance is shown using a single-cell Gaussian mixed model.
Compared with HSC/MPPs in the bone marrow, we found that both disomic and Ts21 HSC/MPPs in the liver upregulated genes involved in the cell cycle such as TOP2A, CDC20 and BIRC5, and downregulated genes involved in the cytokine response such as CXCR4 and IL6R (Fig. 2a). Subsequent cell-cycle analysis confirmed that HSC/MPPs, in both the Ts21 (G2/M + S = 29.8%) and the disomic (21.8%) liver, were cycling more than their counterparts in the bone marrow (26.9% in Ts21 and 18.3% in disomic)19 (Extended Data Fig. 2g).
In addition, we identified Ts21-induced gene expression changes that were specifically present in fetuses with Ts21 and upregulated in liver HSC/MPPs compared with the bone marrow. These included transcription factor and chromatin-binding genes such as GATA1, TAL1 and FOXO3, as well as genes involved in erythrocyte differentiation (KLF1 and AHSP) and haemostasis (ITGA2B and SERPINE2; Fig. 2a,b). Of note, genes involved in response to reactive oxygen species (ROS; FOXO3, SOD1, DUSP1, JUN and FOS) were upregulated in HSC/MPPs in the Ts21 fetal liver compared with the bone marrow (Fig. 2a,b), and not differentially expressed between disomy liver and bone marrow HSCs. Oxidative stress is a proposed cause of GATA1 mutations observed in Ts21 pre-leukaemia and acute megakaryocytic leukaemia, as suggested by previous mutational signature analysis5. Furthermore, oxidative stress in HSC/MPPs can stimulate cycling and cause their premature exhaustion20,21. This is consistent with the presence of cycling HSC/MPPs (G0/G1 = 1%) in the Ts21 liver (Fig. 2c). Besides increased expression of cell-cycle genes (MKI67, CCNB1, PLK1, CDK1 and NEK2), cycling HSCs in the Ts21 liver showed upregulation of genes involved in oxidative stress-induced intrinsic apoptotic signalling (SFPQ22, UBQLN1 (ref. 23) and SOD2 (ref. 24)), mitochondrial dysfunction (MGME1 (ref. 25), DNA2 (ref. 26) and LONP1 (ref. 27)), macroautophagy (ATG3/13 (ref. 28), VDAC1 (ref. 29) and YOD1 (ref. 30)) and type I interferon signalling pathway (RNF26 (ref. 31), POLA1 (ref. 32), MRE11 (ref. 33) and PIN1 (ref. 34)) compared with Ts21 liver HSC/MPPs (Fig. 2d). It has been demonstrated that mitochondrial dysfunction contributes to the number of Down syndrome-associated pathologies and severity of the phenotypes35,36. In addition, reduced mitochondrial activity, through increased autophagy of mitochondria, can lead to increased asymmetrical division of HSCs, an enlarged pool of progenitors and lineage bias37. Overall, we identified gene expression differences specific to trisomy and the environment in HSCs by including both liver and bone marrow samples. Critically, the increase in the levels of ROS, a potent mutagen4, was evident in Ts21 HSCs in the liver but not in the femur, and was further exacerbated in the subset of highly cycling HSCs.
Oxidative stress in Ts21 haematopoiesis
As oxidative stress is a potential cause of mutagenesis in Ts21 (refs. 4,5), we sought to validate higher oxidative stress and altered mitochondrial mass in Ts21 fetal haematopoietic cells than in disomic cells (Fig. 2e,f and Supplementary Fig. 16a). We measured mitochondrial ROS (mtROS) levels with MitoSOX probe and mitochondrial mass using MitoTracker green FM dye38, in the presence of verapamil39, in Ts21 (n = 3) and disomic (n = 3) fetal liver samples (Methods; Supplementary Fig. 16). We observed that Ts21 CD34+CD38+ cells had higher mitochondrial mass (FDR = 0.014) and mtROS levels (FDR = 2.5 × 10−7) than disomic progenitors, whereas CD34+CD38− and HSCs were comparable between the two conditions (Fig. 2e,f). Previous studies have shown that disomic HSCs exhibit very low electron transport chain activity and minimal mtROS production as they mostly rely on glycolysis40. By contrast, committed progenitors have much higher oxygen consumption, mitochondrial ATP production and maximal respiratory capacity. These results support the hypothesis that as the HSCs and immature CD34+CD38− cells start to differentiate to committed CD34+CD38+ progenitors, they ramp-up oxidative phosphorylation. This leads to higher mtROS production in Ts21 cells than in disomic cells, possibly due to their higher mitochondrial mass and dysfunction in the electron transport chain activity, which has been implicated in oxidative DNA damage, mutagenesis and leukaemia development41.
Erythroid priming in Ts21 HSCs
Our previous work has demonstrated that changes in motif accessibility in disomic HSCs precede activation of lineage-specific transcriptional programs and cell-fate choice19. To further investigate whether changes in the regulatory landscape between Ts21 and disomic haematopoietic cells contribute to differences in cell-type abundances, we performed 10X multiome sequencing on haematopoietic CD45+ cells from Ts21 (n = 3) and disomic (n = 3) fetal livers. After quality control (Methods), we obtained high-quality open chromatin profiles for 35,633 nuclei from Ts21 (a median of 13,476 unique open chromatin fragments per nucleus) and 21,257 nuclei from disomic samples (a median of 16,186 fragments per nucleus). Next, we identified cell populations within Ts21 and disomic samples separately by clustering the snRNA-seq multiome data (Methods). Overall, we found cell-type clusters that represent all broader cell types identified in the larger scRNA-seq atlas, with transcriptionally distinct cell populations appearing epigenetically distinct within the paired scATAC-seq uniform manifold approximation and projection (Supplementary Table 15 and Supplementary Fig. 17).
In line with our scRNA-seq analysis, we detected higher erythroid cell proportions in Ts21 (Supplementary Fig. 18 and Supplementary Note 15). Following integration of disomic and Ts21 datasets (Fig. 3a), we inferred trajectories of myeloid lineage cell populations (Extended Data Fig. 3a,b) and estimated the likelihood of each individual HSC to transition towards each lineage (Methods). HSCs demonstrated multipotent lineage commitment; however, Ts21 HSCs showed much greater bias towards the erythroid and megakaryocytic lineages than disomic HSCs (Fig. 3b), with greater GATA1 (which encodes a transcription factor essential for erythroid lineage development) gene body and promoter region accessibility than disomic HSCs (19.3% versus 6.7%). This provided evidence that the erythroid lineage skewing starts already at the HSC level.

a, Integration, UMAP and cell populations of 10X multiome ATAC profiles from the Ts21 and disomic datasets. cDC, classical dendritic cell; MEMP, megakaryocyte–erythroid–mast progenitor; pDC, plasmacytoid dendritic cell. b, Circular projection of absorption probabilities for every HSC towards terminal states (using CellRank; left). Disomic HSCs are in blue, and Ts21 HSCs are in red. The proportion of HSCs differentiating towards the erythroid and megakaryocytic lineage (absorption probability > 0.5) with binomial test P values is also shown (right). c, UMAP with cells coloured according to RNA and ATAC similarity in disomic (left) and Ts21 (middle) HSCs, as estimated by the mutual information of RNA and ATAC topics in a single cell. The HSC latent UMAP space is characterized by a joint representation of both RNA and ATAC topic models (using MIRA). The UMAP space of all disomy and trisomy HSCs (n = 3,784 Ts21 and n = 2,431 disomic HSCs) is in grey. The boxplots represent the 25th, 50th and 75th percentiles, and the whiskers represent 1.5× the interquartile range. Significance was determined using a two-sided Mann–Whitney U-test. d, UMAP with cells coloured according to pseudotime. The arrows denote vectors of the directed cell transition matrix. e, Branches of HSC differentiation in the UMAP space (using MIRA). Cells are coloured by their probability of belonging to a specific branch. f, Compositional STREAM graph indicating abundances of disomic and Ts21 HSCs in each branch along pseudotime. The dashed line denotes half of the pseudotime.
To assess how epigenetic changes lead to the observed erythroid biases, we examined the relationship between chromatin accessibility and gene expression in Ts21 compared with disomic HSCs. Although chromatin accessibility and gene expression are highly concordant in terminally differentiated cells, chromatin remodelling and gene expression are often out of sync in HSCs, such that the promoter and enhancers of a gene may be open but little to no transcription occurs19. To contrast RNA and ATAC profiles from the same cell, we described the transcriptional and chromatin accessibility state of single cells as a group of co-regulated genes (RNA topics) or cis-regulatory elements (ATAC topics), respectively42 (Methods; Supplementary Table 16). By quantifying the accessibility and expression of individual cells within topics, we found that Ts21 HSCs showed a greater concordance between RNA and ATAC topic compositions than disomic HSCs, suggesting they are ‘primed’ to differentiate19,42 (Fig. 3c and Extended Data Fig. 3c).
Heterogeneity of Ts21 HSCs
As a subset of Ts21 HSCs showed differentiation bias towards the erythroid lineage, we sought to further dissect their heterogeneity compared with disomic HSCs. We inferred a cell-state tree42 (see Methods), which revealed the organization of HSCs with three main branches that contained both Ts21 and disomic cells (Fig. 3d,e), but with different abundances (Fig. 3f). Branch 1 had a similar proportion of Ts21 and disomic HSCs, whereas branches 2 and 3 consisted predominantly of Ts21 HSCs, especially later in the pseudotime trajectory (Fig. 3f).
We then used the topics to characterize HSC cell states. Along inferred pseudotime, RNA topics appeared to differ more between Ts21 and disomic HSCs than ATAC topics (Extended Data Fig. 4a), suggesting larger transcriptional than epigenetic differences in Ts21. Furthermore, the RNA topic composition of HSCs in branch 1 was distributed across multiple topics, suggesting the maintenance of a multipotent state (Extended Data Fig. 3d). By contrast, HSCs in branch 2 highly expressed RNA topic 9, which contains genes related to ‘haem metabolism’ (Extended Data Fig. 3d,e), and were cycling as evidenced by high MKI67 expression (Extended Data Fig. 3f). The main RNA topic associated with branch 3 was topic 8 (associated with ‘downregulated genes in blood’; Extended Data Fig. 3d,e). Our findings indicate that Ts21 HSCs encompass a diverse population of cells that share transcriptional and epigenetic similarities with disomic HSCs, but a unique subset of Ts21 HSCs displays an increased erythroid bias (branch 2). This was further evident after inferring transcription factor regulation of branch-specific genes (Supplementary Table 17), with additional signalling necessary to enact transcription of erythroid lineage-related genes (Extended Data Fig. 4c–g and Supplementary Note 17). Thus overall, we observed systemic evidence, on both chromatin accessibility and transcriptional levels, that a subpopulation of Ts21 HSCs is primed to differentiate towards the erythroid lineage.
Transcription factor usage in Ts21 and disomic HSCs
Given that epigenetic and transcriptional priming was prevalent in Ts21 HSCs, we aimed to identify regulatory elements, causal genes and transcription factors that potentially drive the observed lineage bias in Ts21. We first identified transcription factor motifs that are enriched in differentially accessible regions of Ts21 fetal liver HSCs. We found a greater number of motifs in peaks with higher accessibility in disomic HSCs than in peaks with higher accessibility in Ts21 HSCs. Lineage transcription factor motifs such as GATA, RUNX1 and MECOM were enriched in regions with higher accessibility in Ts21, whereas regions with reduced accessibility were enriched for a diversity of motifs (Extended Data Fig. 5a, Supplementary Table 18 and Supplementary Note 18).
We next aimed to identify which transcription factor motifs were enriched in either (1) promoters or (2) enhancers within Ts21 and disomic HSCs (Fig. 4a). We analysed transcription factor enrichment in promoters using the Eukaryotic Promoter Database (EPD)43. EPD promoters were well represented in our dataset (Extended Data Fig. 5b and Supplementary Note 18), and genes linked with differentially accessible promoters (between Ts21 and disomic HSCs) were significantly more likely to be differentially expressed in our scRNA-seq dataset (Extended Data Fig. 5c). The Gene Ontology term enrichment analysis of genes linked to promoters with greater accessibility in Ts21 revealed associations with IFNα and IFNβ, as well as interferon-mediated signalling pathways (Extended Data Fig. 5d). In addition, we found differential utilization of AP-1 motifs between Ts21 and disomic HSCs44 (Fig. 4b). Ts21 HSCs exhibited enrichment of cAMP-response element AP-1 motifs in differentially accessible promoters, which were linked to genes involved in mitochondrial dysfunction and autophagy. Disomic HSC differentially accessible promoters were enriched for TPA-response element AP-1 motifs, and were linked to genes associated with granulopoiesis (Extended Data Fig. 5e,f and Supplementary Note 18).

a,b, All motif enrichments (a) and AP-1 motif-related enrichments (b) in promoters and enhancers for elements with greater accessibility in Ts21 or disomic HSCs. Significance was determined using monaLisa. CRE, cAMP-response element; TRE, TRA-response element. c, Schematic of the SCENT peak–gene multiome analysis performed in n = 3,784 Ts21 and n = 2,431 disomic HSCs. P–G, peak–gene. Schematic was created using BioRender (https://biorender.com). d, Number of significant peak–gene links in Ts21 and disomic HSCs using SCENT (FDR < 0.2), including after downsampling Ts21 HSCs (top), and the proportion of significant peak–gene links with a significant accessibility-by-trisomy interaction term (FDR < 0.2; bottom). e, Proportion of catalogued Ts21 and disomic HSC mutations in the introns of disomy HSC-expressed genes (top), Ts21 HSC-expressed genes (middle) and Ts21 cycling HSC-expressed genes (bottom) that overlap candidate cis-regulatory elements from the ENCODE project. The histograms represent the empirical distribution of this overlap from 10,000 bootstrapped sets of somatic variants in disomic HSCs. P values were determined by the proportion of bootstrapped disomic values that are more extreme than the observed Ts21 value. f, Enrichment of fine-mapped GWAS SNPs from RBC counts in SCENT peak–gene links (termed active cis-regulatory regions) as identified in Ts21 HSCs (top), and enrichment of target GWAS genes within DEGs, as identified in the large scRNA-seq analysis of HSCs (bottom). Odds ratios (ORs) and standard errors were calculated by Fisher’s exact test using three sets of peak–gene links based on significance. The red dashed line indicates the null hypothesis of OR = 1. g–i, Colonies (n = 322) generated by single-cell-sorted HSC/MPPs from three independent disomic and Ts21 fetal livers. Total cell number (live and dead) in Ts21 and disomic HSC-derived colonies (g); lineage proportions in uni-lineage, bi-lineage or multi-lineage colonies (top) and only in uni-lineage colonies (bottom; h); and lineage output (defined as the lineage with the highest cell count by flow cytometry) of HSC/MPP-derived colonies (i) are shown. Significance was determined by Wilcoxon rank-sum test (g) and chi-squared test (h).
To examine transcription factor motif enrichment in enhancers in Ts21 and disomic HSCs, we used the activity-by-contact (ABC) model45 to prioritize functional enhancers. Around 80% of expressed genes in HSCs had at least one ABC enhancer (Extended Data Fig. 6a,b), and differentially accessible ABC enhancers were significantly more likely to be linked to differentially expressed genes (Extended Data Fig. 6c and Supplementary Note 18). Furthermore, ABC enhancers were significantly more likely to be differentially accessible (FDR < 0.1) than promoters (18.1% ABC enhancers versus 9.1% promoters were differentially accessible; binomial test P = 10−219); we identified 2.1-times the number of differentially accessible ABC enhancers (n = 2,650) compared with differentially accessible promoters (n = 1,281). Differential motif usage was also observed between Ts21 promoters and enhancers, such as the GATA family of transcription factors having a stronger effect at enhancers than at promoters (Extended Data Fig. 6, Supplementary Table 18 and Supplementary Note 18).
Ts21 restructures regulatory interactions
We next set out to link regulatory elements to genes in disomic and Ts21 HSCs and assess the effects of altered regulatory elements on downstream gene expression. We established maps of non-coding element–genes in disomic and Ts21 HSCs by correlating peak accessibility with gene expression (Methods). We identified 191 and 1,564 significant peak–gene links in disomic and Ts21 HSCs, respectively (FDR < 0.2), with a greater number of links being identified in Ts21 HSCs even after downsampling cells to the disomic HSC count (773 significant links; 4.1-times more). Of all identified peak–gene links, only 69 were shared between disomic and Ts21 HSCs (representing 36.1% of those found in disomic HSCs; Fig. 4c,d, Extended Data Fig. 7a and Supplementary Table 19).
Given stark differences in the peak–gene map for Ts21 HSCs, we directly tested whether Ts21 modifies the relationship between peak accessibility and gene expression by using an accessibility-by-trisomy interaction term in our statistical models (Methods). A significant interaction term indicates that the relationship between peak accessibility and gene expression is stronger or weaker in Ts21 HSCs than in disomic HSCs. Overall, we found a significant interaction term for 62.3% (76 of 122) of all significant peak–gene links that were specific to disomic HSCs. This finding shows that there is a widespread loss of disomic peak–gene links in the Ts21 background (Fig. 4d), such that peak accessibility has a weaker association with gene expression in Ts21.
By contrast, only 3.9% (59 of 1,495) of Ts21-specific peak–gene links had a significant interaction term that suggests that trisomy strengthens a peak–gene connection. Compared with all tested peaks and genes, the Ts21-specific peaks and genes (without a significant interaction) were significantly more accessible and upregulated in Ts21 than in disomy within differential accessibility analysis (1.9-fold enrichment; P = 5.6 × 10−12) and the large scRNA differential expression analysis (1.7-fold enrichment; P = 9.7 × 10−7). This suggests that the greater activation of those regulatory elements and more widespread transcription (that is, more genes expressed and higher expression) in Ts21 HSCs allowed these peak–gene links to be discovered. Finally, 21.7% (15 of 69) of peak–gene links detected in both disomic and Ts21 HSCs (FDR < 0.2) had a significant interaction, resulting in the dampening or strengthening of the peak–gene relationship in Ts21. Therefore, our results indicate that trisomy restructured the regulatory element–gene landscape by altering the correlation between peak accessibility and gene expression, which led to an increase in accessibility and upregulation of these elements in Ts21 compared with the control, overall indicating that a small number of lineage transcription factors (including GATA transcription factors and other regulatory transcription factors on and beyond chromosome 21) become more active in Ts21, increase coordination between chromatin accessibility and gene expression, and drive lineage bias (Supplementary Table 20).
Our findings offer a potential link between altered chromatin organization in Ts21 and oxidative stress with somatic mutations accrual, thus providing an alternative perspective on the aetiology of leukaemia in Down syndrome. Of note, chromatin organization is well known to impact regional variation of the mutational rate in different cancers46, resulting in preferential accumulation of mutations in distinct chromatin regions. To test whether Ts21-induced changes in chromatin accessibility impact mutagenesis in regulatory regions of genes, we examined patterns of somatic mutations identified in fetal Ts21 and disomic HSCs from Hasaart et al.4. We found that Ts21 mutations located in the introns of Ts21 HSC-expressed genes are 60% more likely to reside within matched ENCODE cis-regulatory elements than intronic mutations identified in disomic HSCs (P = 3 × 10−4). This enrichment was stronger at Ts21 cycling HSC-expressed genes, and there were no significant differences at disomic HSC-expressed genes (Fig. 4e, Extended Data Fig. 7b,c and Supplementary Note 19). These results are in line with the hypothesis that the altered chromatin accessibility landscape in Ts21 HSCs allows differential mutation acquisition in regulatory regions of genes, with the potential to increase the propensity of Ts21 HSCs to pre-leukaemia and leukaemia development.
GWAS SNPs pinpoint regulatory activity
Our peak–gene analysis identified active regulatory elements and their target genes in Ts21 and disomic HSCs. To pinpoint which regulatory elements are relevant for different blood cell types, and in particular for erythroid lineage bias in Ts21, we used published genome-wide association study (GWAS) loci relevant for blood cell traits and evaluated their activity in HSCs. We found that HSCs from branch 1 were significantly enriched for cells with increased accessibility of GWAS SNPs relevant to white blood cell count, whereas HSCs from branch 2 were significantly enriched for cells with increased accessibility of RBC count GWAS SNPs (FDR < 0.1; Extended Data Fig. 8, Supplementary Figs. 19 and 20 and Supplementary Note 20). Considering that branch 2 was mostly composed of Ts21 HSCs, our analysis suggests that RBC GWAS-harbouring enhancers were more accessible in a subset of Ts21 HSCs.
We next assessed whether RBC GWAS-harbouring enhancers influence gene expression. We intersected fine-mapped RBC GWAS SNPs with peak–gene links and Ts21 differentially expressed genes from our large scRNA-seq atlas. We found that fine-mapped GWAS-harbouring peaks were enriched for association with gene expression compared with peaks that do not contain GWAS SNPs (P = 0.03). Furthermore, target genes of GWAS-harbouring peaks (as identified by peak–gene links) were more likely to be differentially expressed between disomic and Ts21 HSCs in scRNA-seq (P = 0.01), confirming the functional relevance of these peaks (Fig. 4f).
As one example, the intronic variant rs2075672 lies in a peak primarily accessible in Ts21 HSCs (P = 5.0 × 10−3) and its accessibility correlates with TFR2 expression (Extended Data Fig. 8a, left; P = 2 × 10−4), which has been implicated in the regulation of erythropoiesis47. Within our scRNA-seq dataset, TFR2 expression was significantly higher in Ts21 HSCs than in disomic HSCs (P = 1.8 × 10−3), with even higher expression found in Ts21 cycling HSCs (Extended Data Fig. 8b, left; P = 3.5 × 10−31). Overexpression of TFR2 in erythroid-like HUDEP-2 cells resulted in increased expression of the maturation marker CD235 (Extended Data Fig. 8c, Supplementary Fig. 20 and Supplementary Note 20). Although dysregulation of any individual gene would have only a minor effect on HSC differentiation, genome-wide reshaping of regulatory element–gene maps in Ts21 is likely to have a major effect on haematopoiesis.
Erythroid cell bias confirmed in vitro
To confirm that the observed erythroid lineage bias can be recapitulated in vitro, we examined the differentiation potential of individual HSC/MPPs (CD34+, CD38−, CD62L+ and CD52+) sorted (Supplementary Fig. 21a) from disomic (n = 3) and Ts21 (n = 3) fetal livers into 96-well plates without a feeder layer. After 2 weeks, 322 colonies were assessed for their size and the lineage output by flow cytometry (CD41a (megakaryocytic), CD235a (erythroid), CD3/CD56 (lymphoid) and CD11b (myeloid); Supplementary Fig. 21b). Although there was no significant difference in the proportion of colonies that consisted of one (uni-) or multiple lineages, the overall frequency of myeloid, lymphoid, megakaryocytic and erythroid lineages showed significant differences (P = 0.01) between trisomy and disomy (Fig. 4h, top). In the disomic samples, uni-lineage colonies exclusively contained myeloid cells; whereas in Ts21, megakaryocytic and erythroid colonies were additionally present (Fig. 4h).
To assess cell proliferation of fetal liver haematopoietic stem and progenitor cells, we counted the total number of cells in individual colonies derived from individually sorted HSCs. Compared with disomic colonies, colonies obtained from Ts21 fetal liver HSCs showed a significantly higher number of total cells than the controls (P = 1.3 × 10−3; Fig. 4g) and were biased towards the erythroid lineage (Fig. 4h). This was pronounced when comparing the largest colonies. The top 20% of Ts21 (‘big’) colonies were bigger than the 95th size percentile of disomic colonies, and, although 50% of the largest disomic colonies (top 20% in size) differentiated towards the erythroid lineage, 73.9% of the largest Ts21 colonies (top 20% in size) had erythroid lineage output (P = 0.035; Fig. 4i). These results support the notion that Ts21 HSCs show megakaryocytic and erythroid bias in vitro and that the observed lineage bias is more pronounced in cycling HSCs.
Discussion
Our study expands the single-cell multi-omics map of blood development in Down syndrome. We increased the number of cells over 50-fold to 1.1 million scRNA-seq cells48, collected from haematopoietic and niche cells from matched fetal liver and bone marrow. We explored the spatial and epigenomic landscapes using spatial transcriptomics and 10X multiome. This dataset enabled us to discover the molecular changes in haematopoiesis as cells migrate from the fetal liver to bone marrow, and elucidate how Ts21 affects this process, leading to haematological abnormalities and elevated leukaemic risk.
Our analyses revealed the key importance of cellular and genomic context for studying fetal haematopoiesis in Down syndrome. Expression differences between Ts21 and disomic cells were dependent on both cell type and environment. In-depth analysis of HSCs in both disomic and trisomic genetic backgrounds, within liver and femur environments, revealed that Ts21 liver HSCs displayed increased cell cycle activity, higher oxidative stress, and mitochondrial dysfunction compared to their counterparts. Furthermore, we experimentally validated increased mitochondrial mass and ROS as phenotypes of Ts21 haematopoietic cells.
Infants with Down syndrome often exhibit high counts of and defects in RBCs3. Our epigenomic and transcriptomic profiling revealed that Ts21 HSCs, although multipotent, showed an erythroid lineage bias. Ts21 HSCs had greater concordance between RNA and ATAC profiles, typical of mature cell states19,42, with broad molecular dysregulation in Ts21 that extended beyond chromosome 21. Ts21 increased the accessibility and expression of regulatory elements and their genes critical to RBC differentiation, implying a genome-wide effect on erythroid lineage differentiation. Thus, our results show that chromatin remodelling underlies the priming of Ts21 HSCs towards the erythroid lineage, a finding confirmed through in vitro differentiation assays.
Finally, our study provides evidence that the altered chromatin organization, gene expression dynamics and oxidative stress create an environment potentially conducive to pre-leukaemic and leukaemic changes. We discovered that mutations linked to Down syndrome4 are concentrated within regulatory regions49 of genes expressed by HSCs, indicating a plausible mechanistic link between chromatin alterations, oxidative stress and heightened leukaemic risk in Ts21. A future goal is to further refine these observations by pairing cell-type-specific and highly sensitive whole-genome sequencing with multi-omic data across cellular and tissue contexts in the same samples.
Methods
Ethics and tissue acquisition
Human fetal bone and liver samples were obtained from 15 fetuses with Ts21 12–20 post-conception weeks (PCW) of age and 5 disomic fetuses 11–19 PCW of age, following termination of pregnancy and informed written consent. The human fetal material was provided by the Joint MRC/Wellcome Trust (grant MR/R006237/1) Human Developmental Biology Resource (http://www.hdbr.org), with maternal informed consent, in accordance with ethical approval by the National Health Service (NHS) Research Health Authority, REC Ref: 18/LO/0822. HDBR is regulated by the UK Human Tissue Authority (HTA; www.hta.gov.uk) and operates in accordance with the relevant HTA Codes of Practice. Sample size was not predetermined but based on sample availability and limited by the time period. Randomization and blinding were not applicable because samples were collected based on their karyotype, and data were analysed following automated computational pipelines.
Dissociation of fetal tissues
Fetal livers and femurs were received in L15 media and processed within 3 h from dissection. Livers were cut in smaller pieces with a scalpel and transferred to a tube containing prewarmed digestion media: RPMI (Gibco) supplemented with 10% FBS (Gibco), penicillin–streptomycin (10 U ml−1 penicillin and 100 ng ml−1 streptomycin, Sigma-Aldrich), 2 mM l-glutamine (Thermo Scientific), 1× MEM NEAA (Gibco), 1 mM sodium pyruvate (Gibco) and 1.6 mg ml−1 collagenase IV (Sigma-Aldrich). The tube was vortexed for 10 s, then incubated at 37 °C for 30 min, and vortexed for 10 s every 15 min. The digested tissue was filtered through a 100-μm filter and diluted in cold D-PBS (Gibco). Cells were centrifuged at 300g for 5 min, then aliquoted and cryopreserved in KnockOut Serum Replacement (Gibco) + 5% DMSO (Sigma-Aldrich). For femurs, adherent material was removed, then the epiphyses were removed with a scalpel and the bone marrow flushed with D-PBS. The remaining bone was cut in small pieces, then ground with a mortar and pestle using digestion media and incubated at 37 °C for 30 min, vortexing every 15 min. The digested material and the bone marrow flush were mixed and filtered through a 100-μm filter. Cells were centrifuged at 300g for 5 min, then aliquoted and cryopreserved in KnockOut Serum Replacement (Gibco) + 5% DMSO (Sigma-Aldrich). Cells were stored in liquid nitrogen until further analysis.
The karyotype for each sample used in this study was determined by quantitative fluorescent PCR (QF-PCR). The analysis was performed by the tissue bank from which the samples were obtained. QF-PCR was performed using chromosome-specific microsatellite markers. The analysis showed normal results with an apparently normal diploid complement for chromosomes 13, 15, 16, 18, 21 and 22 and the sex chromosomes in disomic samples and trisomy for chromosome 21 in Ts21 samples. No mosaicisms were detected.
FACS sorting for scRNA-seq
On the day of FACS sorting, cells were rapidly thawed at 37 °C and transferred to complete RPMI media (RPMI (Gibco) supplemented with 10% FBS (Gibco), penicillin–streptomycin (10 U ml−1 penicillin and 100 ng ml−1 streptomycin, Sigma-Aldrich), 2 mM l-glutamine (Thermo Scientific), 1× MEM NEAA (Gibco) and 1 mM sodium pyruvate (Gibco)). Live-cell enrichment was performed using MACS Dead Cell Removal Kit (130-090-101, Miltenyi Biotec) following the manufacturer’s instructions. When depleting for CD235a+ cells, a magnetic negative selection was performed using CD235a Microbeads (130-050-501, Miltenyi Biotec) and MACS LS columns (130-042-401, Miltenyi Biotec) following the manufacturer’s instructions.
For FACS sorting, cells were stained with Zombie Aqua (Thermo Fisher) to exclude dead cells and the cocktail of antibodies (Supplementary Table 21, sc-sorting panel) for 30 min at 4 °C. Cells were centrifuged for 5 min at 300g at 4 °C, resuspended in a final volume of 500 μl of 5% FBS in PBS, subsequently filtered into polypropylene FACS tubes (352063, Thermo Fisher) and sorted on a BD FACSAria Fusion.
scRNA-seq
Each cell suspension was submitted for 3′ scRNA-seq using Single Cell G Chip Kit, chemistry v3.1 (10X Genomics), following the manufacturer’s instructions. Libraries were sequenced on an Illumina NovaSeq S4 targeting 50,000 reads per cell, and mapped to the GRCh38 human reference genome using the Cell Ranger toolkit (v3.0.0).
Nuclei preparation and multiome sequencing
Nuclei preparation was performed following 10X genomics recommendations. Live, CD45+-sorted cells were centrifuged at 300g for 10 min at 4 °C. Pellets were resuspended in 45 µl chilled lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 substitute, 0.01% digitonin, 1% BSA, 1 mM dithiothreitol and 1 U ml−1 RNase inhibitor in nuclease-free water) and incubated on ice for 5 min. Of chilled wash buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Tween-20, 1 mM dithiothreitol and 1 U ml−1 RNase inhibitor in nuclease-free water), 50 µl was added and nuclei were centrifuged at 500g for 7 min at 4 °C. After removing the supernatant, the nuclear pellets were washed with 45 µl chilled diluted nuclei buffer (1× nuclei buffer, 1 mM dithiothreitol and 1 U ml−1 RNase inhibitor in nuclease-free water) without pipetting. Nuclei were centrifuged at 500g for 10 min at 4 °C. Nuclei were resuspended in 7 µl of chilled diluted nuclei buffer and counted. Fifteen thousand nuclei were targeted for library preparation. Each nuclei suspension was submitted for library preparation using the Chromium Next GEM Chip J Single Cell Kit (10X Genomics), following the manufacturer’s instructions. Libraries were sequenced on an Illumina NovaSeq S4 targeting 50,000 reads per nucleus, and mapped to the GRCh38 human reference genome using the Cell Ranger Arc toolkit (v1.0.1).
Processing of tissues for 10X Visium spatial transcriptomics
Tissues were frozen in dry-ice-cooled isopentane and stored in air-tight tissue cryovials at −80 °C. Before undertaking any spatial transcriptomics protocol, the tissues were embedded in an optimal cutting temperature compound (OCT) and tested for RNA quality with RNA integrity number (RIN). Tissues with RIN values > 7 were cryosectioned in a pre-cooled cryostat at 10 μm thickness. Two consecutive sections were cryosectioned at 10 μm thickness in a pre-cooled cryostat and transferred to the four 6.5 mm × 6.5 mm capture areas of the gene expression slide. Slides were fixed in methanol for 30 min before staining with haematoxylin and eosin and then imaged using the Nanozoomer slide scanner. The tissues underwent permeabilization for 6 min. Reverse transcription and second-strand synthesis was performed on the slide with cDNA quantification using quantitative PCR with reverse transcription (qRT–PCR) using the KAPA SYBR FAST-qPCR kit (KAPA Biosystems) and analysed on the QuantStudio (Thermo Fisher). Following library construction, these were quantified and pooled at 2.25 nM concentration. Pooled libraries from each slide were sequenced on NovaSeq SP (Illumina) using 150-bp paired-end dual-indexed setup to obtain a sequencing depth of approximately 50,000 reads as per the 10X Genomics recommendations.
Single-cell in vitro culture
Single-cell colony-forming unit (sc-CFU) was performed on fetal HSCs, as previously described51. Single, live, Lin−, CD34+, CD38−, CD62L+, CD52+ cells isolated from the fetal liver of three different fetuses with Ts21 (median of 13 PCW) and disomy (median of 12 PCW) were index-sorted into 96-well plates (Supplementary Table 21, HSC sc-CFU panel) containing StemSpan SFEM (Stemcell Technologies) supplemented with penicillin–streptomycin (10 U ml−1 penicillin and 100 ng ml−1 streptomycin, Sigma-Aldrich), 2 mM l-glutamine (Thermo Scientific), 20 ng ml−1 G-CSF (Peprotech), 20 ng ml−1 SCF (Peprotech), 20 ng ml−1 Flt3-L (Peprotech), 50 ng ml−1 TPO (Peprotech), 20 ng ml−1 IL-3 (Peprotech), 20 ng ml−1 IL-6 (Peprotech), 20 ng ml−1 IL-5 (Peprotech), 20 ng ml−1 M-CSF (Peprotech), 20 ng ml−1 GM-CSF (Peprotech) and 20 U ml−1 EPO (RnD). Cells were cultured for 15 days at 37 °C at 5% CO2. At the end of the culture, colonies were assessed for their lineage output by the expression of CD41a (megakaryocytic), CD235a (erythroid), CD3/CD56 (lymphoid) and CD11b (myeloid) by flow cytometry using a BD LSR-Fortessa analyser (Supplementary Table 21, sc-CFU lineage panel). Colonies were considered positive for a lineage if 30 or more cells were detected in the relative gate. The total number of cells in the colony was determined by Trypan blue exclusion using a Countess II cell counter (Thermo Fisher). To assess differences in the colony output between Ts21 and disomy, we performed a chi-squared test using Ts21 as the observed distribution and disomy as the expected distribution.
To compare Ts21 erythroid lineage output to disomic erythroid lineage output in the largest colonies, we first subset the Ts21 colonies with a cell count higher than 95% of all disomic colonies (equivalent to 20% of all Ts21 colonies), and the top 20% size colonies in disomic colonies. The output of multi-lineage colonies was binarized to the lineage with the highest number of cells in the relative gate. We then performed a binomial test with n = 17 observed Ts21 erythroid lineages, k = 23 total Ts21 lineages, and P = 0.5 (the proportion of the disomic lineages that are erythroid).
Fetal liver phenotyping by flow cytometry
Cells were rapidly thawed in complete RPMI media at 37 °C, then centrifuged at 300g for 5 min and washed again with D-PBS. Cells were then resuspended in D-PBS and LIVE/DEAD blue was added at a 1:800 final concentration. Cells were incubated for 15 min in the dark at room temperature, then washed with D-PBS. Cells were then stained for 30 min in the dark at room temperature with the antibody cocktail (Supplementary Table 21, phenotype panel), in the presence of BD Horizon Brilliant Stain Buffer (final dilution 1:4) and Miltenyi FcR blocking reagent (final dilution 1:5) in a final volume of 200 µl. Cells were washed with D-PBS and immediately acquired on a Cytek Aurora (five lasers setup). Data were analysed on FlowJo (v10.8.2).
MitoTracker and MitoSOX staining
MitoSOX is a specific fluorogenic dye for live-cell mitochondria, producing bright green fluorescence upon oxidation by mitochondrial superoxide. MitoTracker green FM accumulates in mitochondria independent of membrane potential and oxidative stress, serving as a reliable tool for mitochondrial mass measurement38. Considering dye efflux bias in HSCs and progenitor cells39, we used verapamil treatment to block xenobiotic efflux pumps and mitigate preferential dye efflux (Supplementary Fig. 16c,e), ensuring a more accurate representation of mitochondrial mass and mtROS levels.
Cells were rapidly thawed in complete RPMI media at 37 °C, then centrifuged at 300g for 5 min and washed again with D-PBS. Immediately before the incubation with the dyes, MitoTracker Green FM reagent was dissolved in DMSO, and MitoSOX green was dissolved in anhydrous N,N-dimethylformamide at a concentration of 1 mM. Cells were resuspended in 1 ml D-PBS and incubated with MitoTracker green FM (final dilution 1:1,000) or 2 µM MitoSOX green for 30 min at 37 °C in the presence of 50 µM verapamil (diluted from an aqueous 10 mM solution). Cells were then washed with D-PBS and stained for 30 min in the dark on ice with the antibody cocktail (Supplementary Table 21, mito panel) in the presence of BD Horizon Brilliant Stain Buffer (final dilution 1:4), Miltenyi FcR blocking reagent (final dilution 1:5) and 50 µM verapamil, in a final volume of 100 µl. Cells were washed again in D-PBS and immediately acquired on a Cytek Aurora (five lasers setup). Data were analysed on FlowJo (v10.8.2). The populations of interest (Lin+, CD38+, CD38−, HSCs) were exported as a fsc file and imported in R via flowCore 2.2 to obtain the fluorescence data of each mitochondrial probe for each cell.
MitoTracker and MitoSOX data analysis
To test whether Ts21 cells have significantly different mitochondrial mass or mtROS from disomic cells, we fit a Gaussian generalized linear mixed model. At single-cell resolution, we transformed the MitoSOX and mitochondrial mass values using a rank inverse normal transformation and used the transformed values as the response variable. We accounted for age as a fixed effect and sample as a random intercept. We tested for the effect of disease status (a fixed effect) within our model and determined significance using FDR across the eight fitted models (four cell types by two response variables).
TFR2 overexpression in HUDEP-2 cells
For the preparation of lentiviruses, HEK293T cells were co-transfected with lentiviral TFR2 expression plasmid or an empty vector (purchased from vectorbuilder) together with psPAX2 packing plasmid and pMD2.G envelope plasmid using Lipofectamine 3000 (Thermo Fisher). The viral supernatants were harvested at 48 h post-transfection and 72 h post-transfection, pooled and concentrated by ultracentrifugation (for 90 min at 90,000g). The pellet was resuspended in StemSpan SFEM II medium (Stemcell Technologies), aliquoted and stored at −80 °C. Functionality and titre of the lentiviruses were determined by serial dilutions on 293T cells and assessing GFP/TFR2 expression 48 h after transduction. HUDEP-2 cells were cultured and differentiated as previously described52. In brief, the cultivation medium of HUDEP-2 is based on StemSpan SFEM II medium (Stemcell Technologies) supplemented with 2% penicillin–streptomycin (20 U ml−1 penicillin and 200 ng ml−1 streptomycin, Sigma-Aldrich), 2 mM l-glutamine (Thermo Scientific), 50 ng ml−1 SCF (Peprotech), 3 U ml−1 EPO (R&D), 1 µM dexamethasone (Sigma) and 1 μg ml−1 doxycycline (Sigma). HUDEP-2 cells were incubated with concentrated lentiviruses (multiplicity of infection of 1) for 6 h, spun down and resuspended in fresh cultivation medium. After 2 days, cells were spun down and resuspended in differentiation medium, which is composed of IMDM (Thermo Fisher), supplemented with 2% penicillin–streptomycin (20 U ml−1 penicillin and 200 ng ml−1 streptomycin, Sigma-Aldrich), 2 mM l-glutamine (Thermo Scientific), 50 ng ml−1 SCF (Peprotech), 3 U ml−1 EPO (R&D), 5% human AB serum (Sigma), 10 μg ml−1 insulin (Sigma), 330 μg ml−1 holo-transferrin (Sigma), 2 U ml−1 heparin (Sigma) and 1 μg ml−1 doxycycline (Sigma). Cells were cultured for 4 days in differentiation medium at 37 °C at 5% CO2. Cells were assessed for TFR2 and erythroid lineage marker expression (CD235a, CD71 and CD36) by flow cytometry using a BD LSR-Fortessa analyzer (Supplementary Table 21, HUDEP panel). HUDEP-2 and HEK293T cells were gifted by the Grønbæk and Issazadeh-Navikas laboratories, respectively (BRIC, University of Copenhagen), and routinely tested for mycoplasma contamination.
Analysis of the scRNA-seq data
For each CellRanger output corresponding to a specific technical and biological replicate, we identified low-quality cells or empty droplets by applying the barcodeRanks and emptyDrops functions using the R package DropletUtils53. We then merged all CellRanger outputs into a single Scanpy object54. Following per-sample droplets removal, quality control was applied based on three parameters: the total unique molecular identifier count (lower-upper threshold (750, 110,000)), the number of detected genes (lower-upper threshold (250, 8,500)), and the proportion of mitochondrial gene count per cell (an upper bound of 20%). We further applied Scrublet55 to remove potential doublets.
Next, we subsetted to samples of the same organ and Ts21 status and merged into a single dataset (for example, a dataset containing only Ts21 liver samples). We reasoned that Ts21 and disomic cell transcriptomes would be influenced heavily by an extra copy of chromosome 21 (for example, 50% higher expression within Ts21 cells that would not be present in disomic cells). As a result, the highly variable genes will be impacted by disomic versus Ts21 differences and insufficiently capture the genes relevant to liver-residing or femur-residing cells. Therefore, we chose to create disomic liver, Ts21 liver, disomic femur and Ts21 femur datasets to most accurately annotate the population of the individual cells in the data. Within each of the four merged datasets, we applied log-normalization, using the scaling factor 10,000 to correct for between-sample differences in library size, and calculated highly variable genes, using the Seurat (v5.0.3) implementation56. We performed principal component analysis (PCA) on the highly variable genes for dimensionality reduction, retaining the top 15 components using the Scree plot elbow rule. Data were batch-corrected using Harmony16 to account for additional technical variations arising between samples that are non-biological in origin.
We then performed an iterative clustering procedure to identify clusters in the single-cell data. Broadly, our iterative clustering procedure first found initial clusters using the Leiden algorithm, then merged clusters from seemingly identical cell populations, and finally subclustered into further refined populations using K-means clustering. Thus, the iterative clustering allowed us to further refine initial clustering, such that initial clusters containing multiple cell types can be further split into lower-level cell types. This is particularly useful for broad cell types such as erythroid cells that were further split into early and late erythroid cells. Following the between-sample batch correction above, we computed a neighbourhood graph using the uniform manifold approximation and projection (UMAP) approach implemented in Scanpy and subsequently clustered with the Leiden algorithm. For visualization purposes, we used UMAP manifold embedding to capture the global features in two and three dimensions. We identified marker genes for each cluster by performing a Wilcoxon signed-rank test with FDR correction, and we annotated clusters using these marker genes and canonical marker genes. We performed further clustering, by manually choosing clusters to subcluster using K-means clustering, merging clusters of the same cell type and performing marker gene detection. With this approach, we generated four separate annotated scRNA-seq datasets, together with associated marker genes, for the Ts21 (liver and femur) and disomic (liver and femur) datasets.
Contrasting cell-type abundances between Ts21 and disomic samples
To compare cell-type abundances, we calculated the proportion of each major cell type group in each sample. We contrasted cell-type proportions between developmental stage-matched Ts21 and disomic samples with the same sorting strategy using a Mann–Whitney U-test. Finally, we corrected for multiple testing using FDR and assessed significance at FDR < 0.1.
Ligand–receptor analysis using CellPhoneDB
We inferred statistically significant ligand–receptors and their corresponding cell types using CellPhoneDB on a subsampled Ts21 liver dataset, such that the proportion of cells in the reduced sample recapitulated the proportion in the full Ts21 dataset and corresponded to the number of cells in the disomic dataset. We repeated the same analysis on the full disomic dataset (which now has an identical cell size). We kept any pairs that did not involve HLA or a protein complex, and kept only those that involved a single receptor. Among the significant ligand–receptors (P < 0.001), we selected ligands or receptors identified in HSC/MPPs and used to communicate with vascular endothelial cells, and performed gene set enrichment analysis (GSEA) on those using EnrichR.
Analysing differential trajectory of the osteo-lineage
For the disomic and Ts21 femur, we computed partition-based graph abstraction (PAGA) using all annotated stromal cells (with a PAGA threshold of 0.05). We also computed a force-directed diffusion graph using Pegasus57, and overlaid the Pegasus and PAGA outputs.
Next, we focused on two different cases of osteo-linage transitioning: (1) within CAR cells, LepR+ CAR cells, osteoprogenitors and osteoblasts, and (2) within arterial endothelial cells, transitioning endothelial cells and osteoblasts. We computed pseudotime using Scanpy, and used the pseudotimes as input into PseudotimeKernel in CellRank58 (without usage of RNA velocity information) to obtain generalized Perron cluster cluster analysis (GPCCA) estimators for identifying macrostates and computing transition probabilities among them. We set the terminal state number according to the shape of the force-directed graph. For case (1), we chose two states in disomic cells based on the observation that there are two clear branches splitting between CAR cells and osteoblasts, and we chose one state in Ts21 because we observed a single branch leading to osteoprogenitors. For case (2), we chose two states considering osteoblasts as one end and some transitioning endothelial cells as the other. Next, we plotted the STREAM plot using the scVelo package59 to visualize the cell-type transition matrix. Finally, we correlated gene expression with estimated absorption probabilities (Pearson correlation, as implemented in the CellRank package). We identified the positively or negatively correlated genes at a significance level of FDR < 0.05 separately in Ts21 and the disomic femur. We checked the Gene Ontology terms of the top 500 genes that were most positively or negatively correlated to absorption probabilities using the clusterProfiler R package60.
Performing differential expression analysis in scRNA-seq
Within each cell type, four distinct differential expression analyses were performed to identify differentially expressed genes (DEGs) due to disease status (Ts21 or disomic) or the microenvironment (liver or femur).
-
(1)
Liver versus femur in disomic samples
-
(2)
Liver versus femur in Ts21 samples
-
(3)
Ts21 versus disomic in liver samples
-
(4)
Ts21 versus disomic in femur samples
Previous literature has shown that pseudobulk differential expression methods have improved FDRs compared with single-cell differential expression methods61. As a result, our analyses were performed by first computing cell-type-specific pseudobulk profiles for each sample and then analysing pseudobulk RNA-seq profiles using limma62.
To calculate sample-level pseudobulk profiles, we aggregated the read counts across cells of the same type. We kept samples for analysis that contained at least ten cells, and we used the filterByExpr() function in the edgeR package with default settings to retain genes for differential expression analysis and reduce the burden of multiple test correction, by removing genes with low expression across samples63.
Next, limma-voom was used to perform a statistical analysis for differential expression. In brief, sample-level weights were calculated by computing normalization factors for transforming count data into log2 counts per million and deriving weights based on a mean-variance relationship (using the calcNormFactors() function in edgeR and the voom() function in limma in R). Log fold changes for each gene were estimated using a linear model with sorting strategy as a covariate. P values were estimated after empirical Bayes shrinkage (lmFit and eBayes() functions in limma). A Benjamini–Hochberg FDR correction was applied across all gene P values, and significance was assessed at FDR < 0.05.
Analysis of Ts21 versus disomic DEGs
As we observed an exponential cross-dependency between the proportion of DEGs on chromosome 21 and other chromosomes, we investigated additional factors that could be relevant. We first tested whether cell-type-specific overexpression of a particular gene on chromosome 21 can lead to greater dysregulation on either chromosome 21 or on other chromosomes. As the probability of a gene being differentially expressed is linked to the number of cells tested in the differentially expressed analysis, we tested this using log fold change values. For each chromosome 21 gene and across cell types, we tested the Pearson correlation between log2 fold change of gene expression (from Ts21 versus disomic samples) and (1) chromosome 21 DEG (%) or (2) non-chromosome 21 DEG (%), and determined the significance of the correlation using FDR < 0.1. Second, we reasoned that overall expression of an important chromosome 21 gene could lead to greater dysregulation, as a highly expressed chromatin modifier or transcription factor might have a consistent 50% overexpression in Ts21 across cell types, but the overexpression might matter more in the cell types where the gene is expressed. We tested this for each chromosome 21 gene. Across cell types, we tested the Pearson correlation between average cell-type-specific gene expression (from Ts21 and disomic samples) and (1) chromosome 21 DEG (%) or (2) non-chromosome 21 DEG (%).
Identification of context-specific DEGs in HSC/MPPs
It is difficult to ascertain whether a gene is commonly or uniquely upregulated in single-cell data (for example, a gene upregulated in Ts21 liver HSCs compared with Ts21 femur HSCs, but not disomic liver HSCs compared with disomic femur HSCs). The presence of a DEG in one cell type and the absence in another may be a result of differences in population size, and thus purely statistical.
As there are sample and cell count differences between datasets, we could not directly take the Ts21 liver versus femur DEGs as the Ts21 population was much larger than the disomic datasets. Instead, we identified DEGs specific to disease status (in liver versus femur analyses) and the microenvironment (in Ts21 versus disomic analyses) in HSC/MPPs by using a subsampling procedure. Downsampling allows the ability to compare two analyses from distinct datasets that are confounded by differences in size. We did not repeat the same procedure across additional cell populations to conclude whether genes are differentially expressed in specific cell populations, as this would require to downsample to the smallest population sizes. This would erode statistical power and be computationally expensive.
Within liver versus femur analyses, we downsampled the Ts21 liver and Ts21 femur dataset to have the same number of fetuses contributing the same number of samples with the same number of HSC/MPPs as the disomic liver and disomic femur data. As a result, the Ts21 data matched the disomic data in terms of fetus sample cell counts. Similarly, within Ts21 versus disomic analyses, we downsampled the Ts21 and disomic liver data based on fetus sample cell counts in the Ts21 and disomic femur data. As an additional restriction in our downsampling, we ensured that fetuses present in both liver and femur data, with equal or greater number of cells and samples in the liver than in the femur, would still be selected in the downsample. The downsampling routine was repeated 100 times, such that 100 new datasets were created that match the smaller dataset. Differential expression analysis was performed identically to the full data using sample-level pseudobulks and limma-voom. The median nominal P value for each DEG was calculated across 100 iterations. We verified the robustness of this choice of 100 iterations by visualizing the variability of the median P value across iterations to assess its stability.
Next, we used differential expression analyses in the full data and in the downsampled data to categorize the context dependence of DEGs. In the liver versus femur analysis, we implicate environment-driven DEGs, Ts21-induced DEGs and Ts21-reverted DEGs.
-
(1)
Environment-driven DEGs are genes with an adjusted FDR < 0.05 in either Ts21 or disomic samples, and nominal P < 0.05 in the other dataset.
-
(2)
Ts21-induced DEGs were discovered by identifying genes with adjusted FDR < 0.05 and median P value across 100 subsamples of P < 0.05 in the Ts21 data and a nominal P > 0.05 in the disomic data.
-
(3)
Ts21-reverted DEGs are those discovered in only the disomic dataset: an adjusted FDR < 0.05 in disomic samples but nominal P > 0.05 in the Ts21 dataset.
-
(4)
Environment-driven or Ts21-induced DEGs are those genes that have FDR < 0.05 in the full Ts21 dataset, but have a median nominal P value across 100 Ts21 subsamples of P > 0.05 and FDR > 0.05 in the disomic data. For these genes, we do not have sufficient evidence to claim context dependence nor reject context dependence, as the discovery of these DEGs might be highly dependent on sample size.
To visualize gene–environment interactions, we examined the expression of environment-driven DEGs and Ts21-induced DEGs across Ts21 and disomic liver and femur HSC/MPPs. We scaled expression across cells for each DEG to mean = 0 and variance = 1. We averaged the scaled expression across genes within the environment-driven and Ts21-induced gene sets, such that each cell has its own value for each gene set. We visualized the mean and standard error of these values across cells in Fig. 2c.
GSEA of upregulated Ts21-induced genes was performed by inputting the list of genes into EnrichR. Scatterplots show the top Gene Ontology terms (molecular function and biological process) or ENCODE and ChEA transcription factors.
Identifying differences in cell cycling across cell types, environment and Ts21
We assigned a cell cycle using the score_genes_cell_cycle() function in Scanpy with the standard list of cycling genes from Tirosh et al.64, as applied to all cells from samples of the same environment and disease type. We determined cycling by the predicted cycling phase being equal to ‘G1’ or not (either ‘G2M’ or ‘S’). We compared the proportions of cycling cells using a Mann–Whitney U-test.
Evaluating the regional distribution of catalogued somatic mutations
Using publicly available data, we evaluated the hypothesis that the regulatory landscape is affected in Down syndrome to influence where somatic mutations occur. First, we downloaded somatic mutation data from Hasaart et al. in fetal Ts21 and disomic HSCs4, and converted the mutation positions from hg19 to hg38 using liftOver. Second, we identified genes expressed in Ts21 HSCs. We used the filtered set from the Ts21 cycling versus less-cycling HSC differential expression analyses, which were identified by the filterByExpr() function. Next, we narrowed down the Ts21 and disomic sets of mutations within the HSC-expressed genes to the set of non-coding intronic mutations. Finally, we downloaded candidate cis-regulatory elements (cCREs) in hg38 from ENCODE; for Ts21 and disomic mutation sets, we calculated the proportion of intronic somatics in Ts21 HSC-expressed genes that overlap with ENCODE cCREs. To determine significant differences, we bootstrapped the disomic intronic mutation set for 1,000 times and compared the observed Ts21 proportion to the disomic distribution. We calculated P values as the proportion of bootstrapped disomic mutation sets with larger values than the Ts21 value.
Analysis of the 10X Visium data
For disomic and Ts21 liver datasets, we used our annotated cell types from the disomic and Ts21 large scRNA-seq datasets as our input reference data for Cell2location65. Next, we merged all SpaceRanger outputs of tissue sections for the disomic liver and then separately for the Ts21 liver to create two Scanpy objects56. We removed mitochondrial genes and spots with the total expressed gene count of less than 800 (the remaining spots were of sufficient good quality for downstream analysis).
We then estimated cell-type abundances for each spatial spot. Using Cell2location, we trained a negative binomial regression model on the input reference data. We applied our model to the Scanpy formatted data, considering tissue section as a covariate to account for distinct batches. We used the estimated posterior mean value of each cell type (from Cell2location) as the local abundances. For each section, we computed the section-level relative abundance of each cell type as the proportion of its estimated abundance across all spots over the total estimated abundance of all cell types across all spots. We compared relative abundances between disomic and Ts21 using a Wilcoxon rank-sum test, and corrected P values by using Benjamini–Hochberg.
To evaluate cell-type colocalization, we computed spot-level relative abundance of each cell type, dividing the abundance of each cell type on an individual spot by the total abundance of all cell types on the same spot. We then computed a Pearson distance matrix among cell types, based on these spot-level relative abundances across sufficient-quality spots of tissue sections in the same disease status, respectively, for the Ts21 liver and disomic liver. We next performed hierarchical clustering, with inter-cluster distance estimated by the Ward variance minimization algorithm.
Processing the multiome data
We performed the initial processing of multiome data using Seurat (v5.0.3) and Signac (v1.13)66. After Cellranger processing, we identified high-quality multiome cells for downstream analysis if they satisfied the following criteria: more than 750 RNA unique molecular identifiers, more than 250 expressed genes, less than 40% mitochondrial read fraction, transcription start site (TSS) enrichment score of more than 3 and more than 1,000 ATAC fragments in peaks. We next identified and annotated transcriptionally distinct clusters within Ts21 and disomic samples using Seurat. We created Ts21-specific and disomic-specific expression matrices by merging the matrices across Ts21 or disomic samples, respectively. Within the separate Ts21 and disomic expression datasets, we log-normalized with a scaling factor of 10,000, identified 2,000 highly variable genes, scaled and centred the data, performed PCA, and used Harmony with lambda = 1 to batch correct for sample-specific variation. We then constructed a k-nearest neighbours graph based on the Euclidean distance in PCA space (using the first 30 components), and identified transcriptionally distinct clusters using the Leiden algorithm. We nominated marker genes for each cluster by performing a Wilcoxon signed-rank test that compares cells within one cluster to all other cells. We performed further clustering by performing K-means clustering and we merged clusters of the same cell type. With this approach, we annotated each cell in two separate multiome datasets (Ts21 and disomic). The Harmony-corrected datasets were visualized in two dimensions using UMAP.
Our overarching process for creating the chromatin accessibility matrix was to call peaks within each sample before calling a final set of peaks from cell-type-specific ATAC profiles. We first called peaks within each sample using macs2, as implemented with default parameters in the callPeaks() function in Signac. We created a unified set of peaks across all samples by combining any intersecting peaks into a single peak, and removing the combined peaks that were less than 20 bp or more than 10 kb wide. This set of peaks was used to compute a cell × peaks matrix for each sample from the ATAC fragments file. Using all peaks present in at least ten cells, we ran latent semantic indexing (a two-step procedure of first using term frequency-inverse document frequency normalization and then singular-value decomposition) to project the ATAC matrix into a reduced dimension representation. We performed batch correction over all samples using Harmony before constructing a k-nearest neighbours graph across the first 30 components (except omitting the first component, which correlates with sequencing depth). This graph was used to perform clustering using the Leiden algorithm with resolution = 1; within each cluster, a new set of peaks were called using macs2. This set of peaks was combined into a unified set of peaks, which was used to form the final cell × peaks matrix, which contained all final Ts21 and disomic cells.
For downstream analyses involving a single combined dataset, the two datasets were merged into a single matrix. Log-normalization, scaling, PCA, Harmony batch correction and UMAP were applied to the combined dataset.
Myeloid trajectory analysis of snRNA using CellRank
Processed snRNA data from multiome was subset to cells of the myeloid lineage. Both disomic and Ts21 cohorts were downsampled to 15,000 cells. A trajectory graph was calculated using the force-directed layout (FLE) function in Pegasus. Instead of the UMAP space, the cell trajectories were plotted in the FLE space. Trajectory analysis of the RNA expression data closely followed the CellRank tutorial ‘CellRank beyond RNA velocity’. Moments of connectivity were calculated using scVelo with 30 principal components and 15 neighbours. The root cell was manually selected according to the diffusion map of HSCs, selecting the cell with the greatest Euclidean distance in FLE space from the centre of the cluster, indicating a cell with a divergent transcriptome. The pseudotimeKernel was used to calculate pseudotime with default parameters. The CytoTRACEKernel was used to compute the transition matrix with the parameters used in the tutorial (threshold_scheme = ‘soft’, nu = 0.5). To compute terminal states and the probability of each cell differentiating towards each terminal state, the GPCCA estimator was utilized with default parameters. Schur decomposition was performed, and five terminal states were automatically selected according to an eigengap in the real part of the eigenvalues. Terminal states were labelled according to the cell type with the closest association (late erythroid, monocytes, mast cells, pDCs and megakaryocytes) and absorption probability was calculated. Significant differences in predicted terminal states between Ts21 and disomic HSCs were calculated using a binomial test that used disomic proportions as the background probabilities.
Topic modelling of the HSC multiome using MIRA
We used MIRA to perform topic modelling of HSCs in the 10X multiome dataset42. Only HSCs were included for all downstream MIRA analysis. Out of the 6,215 HSCs, 3,784 were Ts21 and 2,431 were disomic. All MIRA analysis closely followed the online tutorials.
To generate the latent topics for RNA, the variational autoencoder (VAE) framework uses raw expression counts as input. Rare genes were removed by filtering genes only expressed in 15 or fewer cells. Exogenous genes (n = 7,905) were selected using the highly variable gene function in Scanpy, selecting for all genes with a minimum mean dispersion of 0.1. Exogenous genes are genes that will be captured in topics but will not be used as VAE features. Endogenous genes (n = 4,359) were selected by filtering the exogenous genes for those with a normalized dispersion greater than 0.5. Endogenous genes were used as features for the VAE network. The ExpressionTopicModel was instantiated with the default parameters. The learning rate bounds were manually tuned to cover the portion of the learning rate versus loss curve with the steepest slope. The model was then tuned using TopicModelTuner with default iterations, a minimum number of topics set to 2, a maximum number of topics set to 15, a batch size of 32, threefold cross-validation and a training size of 0.8.
To generate the latent topics for ATAC, the VAE framework used binarized peak counts. Peaks were filtered according to the epiScanpy tutorial. Using the same process for RNA, 72,541 exogenous peaks and 45,095 endogenous peaks were selected according to a minimum mean dispersion of 0.05 and a normalized dispersion greater than 0.5. The AccessibilityTopicModel was instantiated with the default parameters and ‘dataset_loader_workers’ set to 3. The learning rate bounds were manually tuned to cover the portion of the learning rate versus loss curve with the steepest slope. The model was then tuned using TopicModelTuner with default iterations, a minimum number of topics set to 2, a maximum number of topics set to 15, a batch size of 8, onefold cross-validation and a training size of 0.8.
GSEA was performed on the expression topics using wrapper of enrichr in MIRA and the top 200 genes associated with each topic. For the accessibility topics, transcription factor-binding sites were annotated in peaks using the motif scanning in MIRA and the hg38 reference from the UCSC repository. Each peak was scanned according to the JASPAR 2020 vertebrate collection of transcription factor-binding motifs. Transcription factors that were not expressed in the RNA data were removed. For comparing RNA and ATAC topics, the joint data were split into a disomic and Ts21 dataset and the ‘get_topic_cross_correlation’ function in MIRA was performed.
Trajectory analysis of the HSC multiome using MIRA
First, a joint embedding space was calculated using the ‘make_joint_representation’ function in MIRA to combine both modalities. A neighbourhood graph and UMAP embedding were performed on the joint representation (15 neighbours and a minimum distance of 0.1). The datasets were then batch corrected using Harmony on the joint UMAP features. The neighbourhood graph and UMAP embedding were re-calculated on the Harmony-adjusted feature space.
We next calculated several different HSC branches. First, the diffusion map was calculated using Scanpy ‘diffmap’ with default parameters and then normalized by MIRA to regularize distortions in magnitude of the eigenvectors. Schur decomposition was performed and the eigengap heuristic was used to automatically select the proper number of diffusion components within the data (3). The data were subsetted to three diffusion components and the neighbourhood graph was calculated on the diffusion map embedding and components were connected. Pseudotime was calculated using the ‘get_transport_map’ function in MIRA, which defines a transport map using a Markov chain model of forward differentiation. A root cell was selected as the maximum value of the third diffusion component based on the suggestion in the tutorial. This root cell was located in the centre of the highest density of HSCs. Terminal cells were identified using the ‘find_terminal_cells’ function in MIRA with 8 iterations and a threshold of 0.01. There were three distinct clusters of terminal cells, the cell in each cluster farthest away from the root cell was selected, and the probability of each cell differentiating towards those three branches was calculated. The lineage probabilities were parsed into a bifurcating tree structure using ‘get_tree_structure’ with the threshold set to 1. Expression and accessibility dynamics across time were plotted using a streamgraph that depicts this tree structure.
Regulatory potential modelling of the HSC multiome using MIRA
LITE modelling in MIRA was performed to link gene expression to nearby cis-regulatory elements. For every gene, MIRA learns a regulatory window describing a range in which changes in local accessibility appear to influence gene expression. The regulatory window decays exponentially both upstream and downstream according to the TSS of each gene. Consequently, each gene is associated with a unique TSS using non-redundant human TSS annotations (hg38 GENCODE VM39). The model was trained on the union of genes that were highly variable and were the top 5% most activated in each of the 10 expression topics (n = 5,367). The genes that did not have an annotated TSS were removed leaving 4,454 genes. The LITE model was instantiated with default parameters and the raw expression and accessibility data were then fitted (4 out of 4,454 genes failed to fit). Now that each gene contained a trained regulatory potential model, the expression of each gene was estimated by calculating the maximum a posteriori prediction given the accessibility state of each gene in each cell.
Next, we identified transcription factors that regulate the expression of genes specific to each of the three HSC branches using the probabilistic in silico depletion method in MIRA. MIRA simulates ‘computational knockouts’ of each transcription factor. MIRA uses regulatory potential modelling to predict gene expression based on local chromatin accessibility, and then masks cis-regulatory elements with specific motifs to define transcription factors where motif accessibility is important to gene expression prediction. This is measured by the changes in performance of the regulatory potential model of the gene to predict expression after computationally masking the binding sites of every transcription factor. In this manner, transcription factors that strongly regulate the expression of a gene will be prioritized because masking of their binding site will significantly decrease the accuracy of the LITE model prediction. The function ‘probabilistic_isd’ was run with default parameters across all modelled genes. To counteract the inefficiency of noisy transcription factor-binding site predictions, co-varying genes associated with individual topics were queried for a shared association across many transcription factors. The ‘driver_TF_test’ function was utilized to identify potential transcription factors regulating the expression of branch-specific topics. The top 150 topic-specific genes were included, and a Wilcoxon rank-sum test was performed over the association scores. After identifying transcription factors regulating topic expression, the top genes being regulated by these transcription factors were queried using ‘fetch_ISD_matrix’ and selected according to the ranked association score. We additionally performed this procedure across all RNA topics, which we reported in Supplementary Table 16.
Divergence in accessibility and expression of the HSC multiome using MIRA
To identify genes in which expression cannot be accurately predicted by local chromatin accessibility alone, MIRA NITE modelling was performed. In addition to cis-regulation, the NITE model expands on the scope of the LITE model by incorporating accessibility topics, which are genome wide. The NITE model was initialized using the same parameters, topics and genes as the LITE model using ‘spawn_NITE_model’. The model was fit and expression was predicted using default parameters. The difference between LITE and NITE model performance for every gene was calculated using ‘get_chromatin_differential’. The ‘get_NITE_score_genes’ function was used to calculate a cumulative metric per gene describing the divergence of local accessibility and expression across all cells. Genes with a high cumulative NITE score indicate genes that are regulated in part by non-local mechanisms. The top 500 genes with the highest NITE score were incorporated into the GSEA analysis in MIRA. To ascertain whether Ts21 HSCs expressed genes enriched for non-local regulation, pseudobulk differential expression was performed (DESeq2) between disomic and Ts21 HSCs. The distribution of cumulative gene NITE scores for the top 300 condition-specific genes (log2FC) was compared using Wilcoxon rank-sum test with Benjamini–Hochberg correction.
Defining differentially accessible promoters using the EPD
We downloaded a set of curated promoter annotations from the EPD43 available through EPDnew for build hg38 (https://epd.expasy.org/epd/human/human_database.php?db=human). We then intersected these annotations with peaks that were differentially accessible in Ts21 HSCs (as compared with disomy HSCs) using the genomicRanges R package. Differentially accessible promoters (FDR < 0.1) were then tested to determine whether they were more likely to be linked to a significantly DEG (FDR < 0.1) using a Fisher’s exact test.
Defining functional enhancers in ATAC data using ABC
We defined functional enhancers as those with a high potential to regulate gene expression. Following this definition, we used the ABC model to construct genome-wide maps of enhancer–gene connections45. The ABC scores generated for each enhancer reflect the chromatin activity and chromosome interaction between the enhancer and surrounding genes. We utilized merged ATAC-seq mapping results from disomy HSCs and Ts21 HSCs to identify potential enhancer locations and quantify their chromatin activity. We applied the averaged Hi-C contact data provided by the ABC authors to map contact interactions between chromosomal regions at 5-kb resolution. The default parameters and thresholds were then used to run the ABC model pipeline available on GitHub for implicating a list of relevant enhancers. For motif analyses, non-promoter enhancers were kept (those ABC enhancers that did not overlap with an EPD promoter).
Motif enrichment analysis in differentially accessible peaks using monaLisa
To identify transcription factor motifs that are responsible for differential accessibility, we utilized the JASPAR2022 (v0.99.7)67 and monaLisa (v0.63.0)68 R packages. First, liver HSC peaks with FDR < 0.1 in a Ts21 versus disomy HSC analysis were identified and stratified into separate bins based on the direction of differential accessibility. Next, to account for any size differences between groups, we resized all peaks to the median peak length across all groups. After accounting for differences in peak length, we used the ‘calcBinnedMotifEnrR’ function from monaLisa, which internally corrects for GC content and identifies motifs that are significantly enriched in either Ts21-biased or disomy-biased peaks. The motif enrichment analysis above was repeated using all differentially accessible peaks, only promoters and only enhancers.
Next, to assess differential accessibility of promoters, we downloaded a set of curated promoter annotations from the EPDnew database for hg38 (https://epd.expasy.org/epd/human/human_database.php?db=human). We then intersected these annotations with peaks that were differentially accessible using the genomicRanges R package. Differentially accessible promoters were then tested to determine whether they were more likely to be linked to a significantly DEG (FDR < 0.1) using a Fisher’s exact test. The motif enrichment analysis above was repeated using the EPD promoters.
To assess utilization of the AP-1 motif between Ts21 and disomy, we specifically used motifs for AP-1 identified in Isakova et al.44. We first identified which motifs belonged to the TRA-response element (TRE) or CRE family by calculating the similarity between motifs as the Pearson correlation between the position frequency matrices using the ‘MotifSimilarity’ function from monaLisa. We then identified two distinct clusters of motifs using hclust() and cutree() R functions with K = 2. These clusters corresponded to the TRE and the CRE motif families. All TRE and CRE motifs were then used in the pipeline described above to identify differential usage of AP-1 motifs. Gene Ontology terms were then assessed using enrichR.
Finally, we quantified the contribution of each motif to differential accessibility in Ts21 HSCs. We utilized motifmatchR (v1.20.0) to identify ABC enhancers or EPD promoters that contain a significant match to each motif. Then, for each motif, we fit a linear regression model logFC ~ match + peak_type + match:peak_type, where match indicates whether the peak contains a significant match of the motif, peak_type indicates whether the peak is an enhancer or promoter, and logFC represents the log fold change values of the peak. This model allows each motif to have a different effect on differential accessibility between enhancers and promoters. To quantify the contribution of the motif to overall differential accessibility, we computed the multiple R2 from the regression model.
Assessing enrichment of GWAS SNPs in single cells using SCAVENGE
Trait-relevant individual cells were calculated using SCAVENGE69, which combines network propagation and SNP enrichment analysis to map causal variants to their cellular context. First, variants with posterior inclusion probability (PIP) > 0.001 were downloaded in the format used by Yu et al.69, which were originally processed and described within Vuckovic et al.70. Second, the full multiome ATAC dataset (including all cell populations) was used as input to downstream tasks. A mutual k-nearest neighbour graph was computed to represent the relationship between neighbouring single cells, using k = 30. Next, g-chromVAR71 was used to calculate bias-corrected z scores for each tested trait and each single cell to estimate cell-trait relevance. The top 5% ranked cells served as seed cells for the SCAVENGE network propagation, which were further scaled and normalized to calculate the final SCAVENGE trait relevance score (TRS).
SCAVENGE TRS was contrasted between different lineage branches using a pseudobulk approach similar to the approach that we used for differential expression analysis. On a per-sample-and-branch level, we pseudobulked the SCAVENGE scores by summing SCAVENGE scores across all cells of the same branch and of the same sample. We then applied three linear models to assess the effect of each of the three branches on the SCAVENGE TRS. In each model, we accounted for the number of cells within the pseudobulk as a covariate. We determined significance by multiple test corrections at FDR < 0.1 across the 9 analyses (3 branches (1, 2, 3) and 3 traits (RBC, white blood cell and lymphocyte counts)).
Identification and analysis of peak–gene links using SCENT
SCENT72 was used to identify peak–gene links in disomic HSCs and Ts21 HSCs, by correlating peak accessibility (binarized) and gene expression (raw) counts. SCENT is a Poisson regression model that recomputes the standard errors in the model coefficients by using bootstrapping, which helps to maintain false-positive rates. We first identified all peak–gene combinations within 500 kb of each other. Separately within disomic and Ts21 HSCs, we retained all peak–gene combinations where more than 5% of cells were accessible and expressed the peak and the gene, retaining a total of 38,478 peak–gene combinations. Owing to biological differences between disomy and Ts21, this was an overlapping but not identical peak–gene set tested in disomic and Ts21 HSCs.
Next, we applied a SCENT model using binarized peak accessibility, percent mitochondrial reads, log(number of UMIs) and sample as the covariates, and expression counts as the dependent variable to each peak–gene combination. On the basis of the SCENT paper, we set up an iterative bootstrapping scheme to balance runtime and P value accuracy based on the Poisson regression model P values, where P > 0.1 consisted of 100 bootstraps, P < 0.1 consisted of 1,000 bootstraps and P < 0.01 consisted of 10,000 bootstraps. SCENT was performed on a computing cluster using chunks of 100–500 peak–gene sets. We corrected for multiple testing using FDR, and determined significant peak–gene links at FDR < 0.2. Finally, we repeated the Ts21 analysis after downsampling the 3,784 Ts21 HSCs to match the sample size of 2,431 disomic HSCs to assess the impact of cell population size.
Furthermore, we used SCENT to test whether the effect of peak accessibility on gene expression is modified by trisomy. To do so, we included an interaction term between ATAC peak accessibility and Ts21 status in the SCENT model to address whether the effect of accessibility on expression depends on trisomy. We applied the new SCENT interaction model to a combined Ts21 and disomic HSC dataset. The analysis was performed on all significant peak–gene links in the disomic-only or Ts21-only analyses, and significant interaction terms were assessed at FDR < 0.2.
One reason why Ts21-only peak–gene links were only identified in Ts21 yet have no significant interaction term would be because of a lack of gene expression or peak accessibility in disomic cells. To test whether Ts21-only peak–gene links without a significant interaction term were more accessible and expressed in Ts21 HSCs than in disomic HSCs, we used differential accessibility (from the 10X multiome ATAC) and differential expression results (from the large scRNA-seq analysis). To calculate differential accessibility in the 10X multiome ATAC, we computed pseudobulk profiles and performed limma-voom with trisomy status as the covariate of interest, as similarly described in the large scRNA-seq analysis. We filtered peaks for differential analysis using the filterByExpr() function in edgeR. We performed two separate binomial tests to assess whether the list of (1) Ts21-only peaks or (2) Ts21-only genes were upregulated in Ts21 compared with all other peaks or genes tested in differential analyses. We defined upregulated in Ts21 as nominal P < 0.05 and logFC > 0 in the limma-voom results.
We performed two enrichment tests with regard to our Ts21 peak–gene links and RBC GWAS. We defined SCENT peaks and SCENT genes according to three significance thresholds from the SCENT peak–gene analysis: FDR < 0.1, FDR < 0.2 and nominal P < 0.05, and identified GWAS-related peaks and genes using fine-mapped RBC GWAS SNPs at PIP > 0.2. In the first analysis, we assessed the enrichment of fine-mapped RBC GWAS SNPs (PIP > 0.2) within Ts21 SCENT peaks (also known as active cis-regulatory regions) using a Fisher’s exact test. Here, we are comparing whether peaks with a GWAS variant are more likely to be associated with gene expression than with a background set of peaks without a GWAS variant, which include peaks accessible (in more than 5% of cells) and close to a gene (less than 500 kb) that is expressed (in more than 5% of cells). In the second analysis, we assessed the enrichment of differential expression at target GWAS genes defined by Ts21 SCENT peak–gene links. We defined differential expression as FDR < 0.1 in the differential expression analysis of HSCs using the large scRNA-seq data. We subsetted to significant peak–gene links and calculated a 2 × 2 contingency table reflecting (1) whether the peak contained a fine-mapped variant, and (2) whether the target gene is differentially expressed. A Fisher’s exact test assessed the hypothesis that important enhancers (containing fine-mapped GWAS SNPs) have a role in differential expression within HSCs (affecting their target genes).
Within the results, we report the enrichment P values within the text when using the set of peak–gene links with nominal P < 0.05 (from SCENT). In addition, when reporting peak–gene examples within the results, we use the nominal P values for the SCENT peak–gene test, the differential expression test or the differentially accessibility test. We include the full summary statistics within the Supplementary tables.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The following data have been deposited on ArrayExpress: scRNA-seq FASTQ raw data and CellRanger count matrices (accession number E-MTAB-13067); 10X Visium FASTQ raw data, SpaceRanger count matrices, run summary metrics and spatiality outputs (E-MTAB-13062); and multiome snRNA-seq and snATAC-seq FASTQ raw data, CellRanger ARC count matrices and ATAC fragment files (E-MTAB-13070).
Code availability
The code is available at https://github.com/drewmard/t21-proj, https://github.com/drewmard/t21_multiome and https://gitlab.com/cvejic-group/downsyndrome/.
References
Belson, M., Kingsley, B. & Holmes, A. Risk factors for acute leukemia in children: a review. Environ. Health Perspect. 115, 138–145 (2007).
Hasle, H., Clemmensen, I. H. & Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 355, 165–169 (2000).
Choi, J. K. Hematopoietic disorders in Down syndrome. Int. J. Clin. Exp. Pathol. 1, 387–395 (2008).
Hasaart, K. A. L. et al. Mutation accumulation and developmental lineages in normal and Down syndrome human fetal haematopoiesis. Sci. Rep. 10, 12991 (2020).
Cabelof, D. C. et al. Mutational spectrum at GATA1 provides insights into mutagenesis and leukemogenesis in Down syndrome. Blood 114, 2753–2763 (2009).
Hitzler, J. K., Cheung, J., Li, Y., Scherer, S. W. & Zipursky, A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101, 4301–4304 (2003).
Wagenblast, E. et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science 373, eabf6202 (2021).
Laurent, A. P., Kotecha, R. S. & Malinge, S. Gain of chromosome 21 in hematological malignancies: lessons from studying leukemia in children with Down syndrome. Leukemia 34, 1984–1999 (2020).
Muskens, I. S. et al. The genome-wide impact of trisomy 21 on DNA methylation and its implications for hematopoiesis. Nat. Commun. 12, 821 (2021).
Antonarakis, S. E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18, 147–163 (2017).
Korenberg, J. R. et al. Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc. Natl Acad. Sci. USA 91, 4997–5001 (1994).
Akdemir, K. C. et al. Somatic mutation distributions in cancer genomes vary with three-dimensional chromatin structure. Nat. Genet. 52, 1178–1188 (2020).
Polak, P. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518, 360–364 (2015).
Meharena, H. S. et al. Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors. Cell Stem Cell 29, 116–130.e7 (2022).
Roy, A. et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc. Natl Acad. Sci. USA 109, 17579–17584 (2012).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Popescu, D.-M. et al. Decoding human fetal liver haematopoiesis. Nature 574, 365–371 (2019).
Ranzoni, A. M. et al. Integrative single-cell RNA-seq and ATAC-seq analysis of human developmental hematopoiesis. Cell Stem Cell 28, 472–487.e7 (2021).
Ito, K. et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451 (2006).
Tothova, Z. et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339 (2007).
Guo, P. et al. SFPQ is involved in regulating arsenic-induced oxidative stress by interacting with the miRNA-induced silencing complexes. Environ. Pollut. 261, 114160 (2020).
Liu, Y. et al. Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J. Neurosci. 34, 2813–2821 (2014).
Kokoszka, J. E., Coskun, P., Esposito, L. A. & Wallace, D. C. Increased mitochondrial oxidative stress in the Sod2+/− mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl Acad. Sci. USA 98, 2278–2283 (2001).
Kornblum, C. et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 45, 214–219 (2013).
Ronchi, D. et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am. J. Hum. Genet. 92, 293–300 (2013).
Peter, B. et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet. 27, 1743–1753 (2018).
Fang, D., Xie, H., Hu, T., Shan, H. & Li, M. Binding features and functions of ATG3. Front. Cell Dev. Biol. 9, 685625 (2021).
Kim, J. et al. VDAC oligomers form mitochondrial pores that release small mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536 (2019).
Papadopoulos, C. et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36, 135–150 (2017).
Qin, Y. et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 10, e1004358 (2014).
Starokadomskyy, P. et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat. Immunol. 17, 495–504 (2016).
Kondo, T. et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl Acad. Sci. USA 110, 2969–2974 (2013).
Tun-Kyi, A. et al. Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 12, 733–741 (2011).
Izzo, A. et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 26, 1056–1069 (2017).
Valenti, D., Manente, G. A., Moro, L., Marra, E. & Vacca, R. A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochem. J. 435, 679–688 (2011).
Vannini, N. et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell 24, 405–418.e7 (2019).
Pendergrass, W., Wolf, N. & Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 61, 162–169 (2004).
Mansell, E. et al. Mitochondrial potentiation ameliorates age-related heterogeneity in hematopoietic stem cell function. Cell Stem Cell 28, 241–256.e6 (2021).
Snoeck, H.-W. Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol. 49, 91–98 (2017).
Novotna, B., Bagryantseva, Y., Siskova, M. & Neuwirtova, R. Oxidative DNA damage in bone marrow cells of patients with low-risk myelodysplastic syndrome. Leuk. Res. 33, 340–343 (2009).
Lynch, A. W. et al. MIRA: joint regulatory modeling of multimodal expression and chromatin accessibility in single cells. Nat. Methods 19, 1097–1108 (2022).
Dreos, R., Ambrosini, G., Groux, R., Cavin Périer, R. & Bucher, P. The Eukaryotic Promoter Database in its 30th year: focus on non-vertebrate organisms. Nucleic Acids Res. 45, D51–D55 (2017).
Isakova, A. et al. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat. Methods 14, 316–322 (2017).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Makova, K. D. & Hardison, R. C. The effects of chromatin organization on variation in mutation rates in the genome. Nat. Rev. Genet. 16, 213–223 (2015).
Richard, C. & Verdier, F. Transferrin receptors in erythropoiesis. Int. J. Mol. Sci. 21, 9713 (2020).
Jardine, L. et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature 598, 327–331 (2021).
Moore, J. E. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Chapman, M. S. et al. Lineage tracing of human development through somatic mutations. Nature 595, 85–90 (2021).
Velten, L. et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 19, 271–281 (2017).
Bagchi, A. et al. Erythroid lineage-specific lentiviral RNAi vectors suitable for molecular functional studies and therapeutic applications. Sci. Rep. 12, 14033 (2022).
Lun, A. T. L. et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Li, B. et al. Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 17, 793–798 (2020).
Lange, M. et al. CellRank for directed single-cell fate mapping. Nat. Methods 19, 159–170 (2022).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Chen, Y., Lun, A. T. L. & Smyth, G. K. From reads to genes to pathways: differential expression analysis of RNA-seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. https://doi.org/10.12688/f1000research.8987.2 (2016).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 (2022).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Machlab, D. et al. monaLisa: an R/Bioconductor package for identifying regulatory motifs. Bioinformatics 38, 2624–2625 (2022).
Yu, F. et al. Variant to function mapping at single-cell resolution through network propagation. Nat. Biotechnol. 40, 1644–1653 (2022).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231.e11 (2020).
Ulirsch, J. C. et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 51, 683–693 (2019).
Sakaue, S. et al. Tissue-specific enhancer–gene maps from multimodal single-cell data identify causal disease alleles. Nat. Genet. 56, 615–626 (2024).
Acknowledgements
The human embryonic and fetal material was provided by the Joint MRC–Wellcome Trust (grant number MR/006237/1) Human Developmental Biology Resource (http://www.hdbr.org). We thank the Cambridge NIHR BRC Cell Phenotyping Hub for their advice and support in cell sorting; D. Nachun for discussions regarding somatic mutations found in Ts21 and statistical modelling of mitochondrial mass data; J. Xu, C. Nicu, A. M. Ranzoni, B. Myers and E. Panada for sample collection and processing; M. Nelson for computational support with initial clustering of scRNA-seq; the Cancer Research UK Cambridge Institute (CRUK CI; grant number CTRQQR-2021\100012) Genomics Core Facility for library preparation and sequencing services; the Wellcome Sanger Institute (WSI) DNA pipelines for their contribution to sequencing the data; S. Leonard from the New Pipeline Group (NPG) for pre-processing of sequencing data; R. Somasundaram from the BRIC FACS Core Facility for support with the flow cytometry experiments; and P. Rainer and U. Tiemann for critical reading of the manuscript. This study was supported by the European Research Council (CONTEXT 101043559). However, the views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. This study was also supported by the Novo Nordisk Foundation (grant number 0066260); and core support grants from both the Wellcome Trust and the MRC to the Wellcome Trust-MRC Cambridge Stem Cell Institute (203151/Z/16/Z). A.R.M. was funded by the Stanford Dean’s Fellowship. Montgomery lab members were funded by R01MH12524404 (S.B.M., A.R.M. and Y.X.), U01HG01206904 (S.B.M. and A.R.M.) and U01HG011762 (S.B.M., A.R.M. and E.M.P.). J.B. was funded by the Stanford Graduate Fellowship. T.H.H.C. is the recipient of an EMBO long-term fellowship (ALTF 172-2022). The figures were created using BioRender (https://biorender.com).
Author information
Authors and Affiliations
Contributions
A.C. designed the study and oversaw all experiments. A.R.M., S.B.M. and A.C. led and oversaw all analyses. M.D.Z., S.W. and R.M. performed the experiments and created the figures. H.X. analysed the spatial transcriptomics under supervision of A.C. and A.R.M. J.B. led the application of MIRA to the multiome data. T.H.H.C. performed the somatic mutation calling in scRNA-seq samples. A.R.M., H.X. and J.B. provided computational support. E.M.P. performed transcription factor motif analysis. Y.X. performed the ABC enhancer analysis. A.C. and A.R.M. wrote the manuscript, and all authors edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.R.M. consults for Third Rock Ventures, Inc. S.B.M. advises BioMarin, MyOme and Tenaya Therapeutics. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Rosario Isasi, Nathan Salomonis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Differentiation trajectory within the BM niche.
a, PAGA graphs overlaid on the diffusion maps, computed from cell types representing the BM niche population and their connectivity (top-disomic, bottom-Ts21). b, Left: trajectory with the directionality of the differentiation process from CAR cells to osteoprogenitors and osteoblasts in disomic and Ts21 samples. Stream plot overlaid on Force-directed (FLE) graph of diffusion map of CAR cells, LepR + CAR cells, osteoprogenitors and osteoblasts. Right: scatter plot of the Pearson correlation between gene expression and CellRank’s terminal state absorption probabilities in disomic and Ts21. Significantly correlated genes (FDR < 0.05) were plotted. Colours indicate associated GO terms. c. Left - stream plot overlaid on FLE graph of diffusion map of arterial endothelial cells, transitioning endothelial cells and osteoblasts. Right - scatter plot of GO terms estimated from top 500 genes most positively/negatively correlated with the absorption probabilities.
Extended Data Fig. 2 Impact of chromosome 21 on differential expression analysis.
In liver (a) and femur (b), differential expression analysis was performed between Ts21 and disomic cells using limma-voom and sample-level pseudobulks. The median log2-fold change (LFC) was calculated for chr21 genes (blue) and all other genes (Not chr21, red). c, Percentage of differentially expressed genes (DEG), (FDR < 0.05) on chromosome “N” versus other chromosomes. Each point represents a cell type. n = 1,107,552 cells and k = 18 foetuses, represented by Ts21 liver (n = 780,299, k = 15), Ts21 femur (n = 162,775, k = 12), disomic liver (n = 110,671, k = 3) and disomic femur (n = 53,807, k = 3). d, Left, median log-fold changes for genes on each chromosome between fetuses #15633 and #15781. Right, median log-fold changes for genes on each chromosome between fetuses #15633 and #15781, after increasing mapped reads to genes on chromosome 21 by 50% in two #15781 pseudobulk samples. e, Comparison of log-fold changes for all genes between the two analyses. Orange points represent the genes on chromosome 21, which have increased expression within the two #15781 pseudobulk samples in the spiked chr21 analysis (y-axis). The log-fold changes of the underlying real data is along the x-axis. f, Average normalised gene expression of statistically significant Environment-driven DEGs and Ts21-induced HSC/MPPs DEGs within each dataset. Normalised expression was calculated by first transforming a gene’s expression to have mean = 0 and variance = 1, and then averaging transformed expression across all genes. Mean normalised expression across cells is shown, with vertical bars representing 2 standard errors around the mean. g, Comparison of the percentage of cells cycling within disomic samples (x-axis) and Ts21 samples (y-axis). Each dot represents a different cell type. Asterisk (*) signifies statistically significant difference in cycling of cells in Ts21 vs disomic by performing a Mann-Whitney U test on sample-level pseudobulk proportions. Dashed line represents y = x.
Extended Data Fig. 3 CellRank and MIRA differentiation analysis.
a, Left, force directed layout (FLE)(ForceAtlas2) of cells belonging to the myeloid lineage. Harmony batch corrected and PAGA initialised. HSC cluster set as the root for pseudotime calculation. Black arrows indicate the projected transition matrix (CellRank). Right, PAGA representation. b, Five terminal states were automatically identified using CellRank. Normalised expression of genes with highest correlation with each terminal state plotted in FLE space (CellRank lineage drivers) (IRF8:pDCs, ITGA2B:Megakaryocytes, TFRC:Late erythroid, LMO4:Mast cells, SAMHD1: monocytes). PROM1 was added to illustrate the location of HSCs. c, MIRA topic analysis performed on data including both disomic and Ts21 HSCs. Topic analysis was performed separately on RNA and ATAC raw data. Cross-correlation matrix between RNA topics and ATAC topics. Left, Ts21 HSCs. Right, disomic HSCs. d, Each cell is associated with a single RNA topic according to the topic with the highest cumulative expression in that cell. Bar graph illustrating the composition of topics each MIRA identified branch, as measured by topics assigned to each cell of that branch. RNA topic 9 has the highest frequency in branch 2 and topic 8 has the highest frequency in branch 3. e, Left, gene set enrichment analysis (GSEA) of branch 2 specific topic 9 from querying “MSigDB_Hallmark_2020” (enrichr). GSEA analysis of branch 3 specific topic 8 from querying “HDSigDB_Human_2021” (enrichr). f. UMAP plot representing the joint RNA and ATAC MIRA latent space of HSCs. Expression of cell-cycling gene MKI67 (raw counts).
Extended Data Fig. 4 Trans-regulated genes in HSC multiome.
a, Compositional STREAM graph indicating topic abundance proportion within cells ordered along pseudotime for each branch. Left, RNA expression topics. Right, ATAC accessibility topics. b, Left, scatter plot comparing the TFs regulating expression of genes turned on in branch 2 or 3. P-values calculated using a Wilcoxon rank sum test over the association scores. Right, top genes regulated by labelled branch 2 TFs. The expression (blue) and accessibility (green) indicate Ts21 HSC specificity. Data is normalised by row to illustrate preference between disomic and Ts21. Analyses include Ts21 liver (n = 35,633 cells, k = 3) and disomic liver (n = 21,257, k = 3) samples; n = 3,784 Ts21 and n = 2,431 disomic cells are HSCs. c, The cumulative gene NITE score (MIRA) across all HSCs separated by differentially expressed genes (DEGs) (pseudobulk DESeq2) between disomic and Ts21. Significance of P < 0.001 was determined using a two-sided Wilcoxon rank sum with Benjamini-Hochberg correction. d, Distribution of the cumulative gene NITE score across all modelled genes (n = 4,446). Black, top genes regulated by branch 2 transcription factors. Blue, top genes with highest NITE score. e, UMAP plots (MIRA’s joint RNA and ATAC topic model latent representation) indicating the raw expression of genes with the highest cumulative NITE score. f, Representation of the gene, TFR2, with the highest NITE score. UMAP plots from left to right illustrate raw expression, LITE modelling prediction, NITE modelling prediction, and the chromatin differential (difference between LITE and NITE). Scatter plots (right) indicate the correlation between regulatory potential modelling prediction and the normalised expression of the gene. The NITE model that incorporates both cis and trans accessibility greater than doubles the predictive efficiency of TFR2 expression (LITE: R2 = 0.3, NITE: R2 = 0.64). g, Gene set enrichment analysis (GSEA) (enrichr) results from the top 500 genes according to the cumulative NITE score. Left, GSEA results from querying “BioPlanet_2019”. Right, GSEA results from querying “RNA-Seq_Disease_Gene_and_Drug_Signatures_from_GEO”. Colouring indicates adjusted p-value (-log 10).
Extended Data Fig. 5 Transcription factor motif enrichment in Ts21 and disomic HSCs.
a, Enrichment of selected motifs by monaLisa in peaks with greater accessibility in Ts21 (left) and reduced accessibility in disomy (right). b, Barplots describing the number and proportion of (i) EPD promoters that overlap with peaks accessible in HSCs, (ii) expressed genes with at least one EPD promoter, and (iii) expressed genes with at least one EPD promoter that overlaps with peaks accessible in HSCs. c, The association between differentially accessible EPD promoters and differentially expressed (DE) linked genes (odds ratios with standard errors). Left used DE genes at FDR < 0.05 (adjusted) and right used DE genes at nominal P < 0.05. d, Gene set enrichment analysis of genes linked to differentially accessible (DA) promoters with greater accessibility in Ts21. e-f, Gene set enrichment analysis results for (e) genes linked to CRE-containing promoters with greater accessibility in trisomy and (f) genes linked to TRE-containing promoters with greater accessibility in disomy.
Extended Data Fig. 6 Transcription factor motif enrichment in enhancers in Ts21 and disomic HSCs.
a-c, Histogram of the number of ABC enhancers per gene (a), differentially accessibility ABC enhancers per gene (b), and association between differentially accessible ABC enhancers and differential expression of their target genes (c). d, The absolute value of log-fold changes for differentially accessible peaks, stratified by peaks with significantly higher accessibility in disomy (left) and Ts21 (right), and ABC enhancers (blue) or EPD promoters (red). e, Proportion of variance explained in the differential accessibility of trisomy 21 HSCs for each TF motif instance. f, The differential accessibility, estimated by |logFC| between Ts21 and disomic HSCs, between different peak sets and stratified by GATA-containing elements or not.
Extended Data Fig. 7 Gene regulation and somatic mutation accrual in HSCs.
a, Distribution of significant peak-gene links identified in Ts21 (top) or disomic HSCs (bottom) (FDR < 0.2). The y-axis represents the proportion of links that are on that chromosome versus genome-wide. b-c, The enrichment of Ts21 somatics relative to disomic somatics within CREs of differentially expressed genes from (b) Ts21 vs Disomy HSCs and (c) Ts21 cycling vs less-cycling HSCs, across different P-value thresholds.
Extended Data Fig. 8 Enrichment of RBC traits GWAS SNPs in multiome peaks.
a, aggregated accessibility and expression of HSCs from disomic and Ts21 HSCs for TFR2 and TSPAN32 using 10X multiome data. b, Pseudobulk expression within HSC populations using the scRNA-seq data. Each point represents a different sample. c, CD235a expression in Ctrl HUDEP-2 and in the population of highly expressing TFR2 cells. *** p-value < 0.001 (two-sided Mann-Whitney U test). Boxplots represent 25th, 50th and 75th percentiles, whiskers represent 1.5× interquartile range.
Supplementary information
Supplementary Information
Supplementary Notes 1–20, Supplementary Figs. 1–21, full descriptions for Supplementary Tables 1–21 (Tables supplied separately) and Supplementary References.
Supplementary Tables
Supplementary Tables 1–21 – see Supplementary Information file for full descriptions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Marderstein, A.R., De Zuani, M., Moeller, R. et al. Single-cell multi-omics map of human fetal blood in Down syndrome. Nature 634, 104–112 (2024). https://doi.org/10.1038/s41586-024-07946-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-07946-4
This article is cited by
-
Charting the future: current and future directions in translational research for individuals with Down syndrome
Journal of Neurodevelopmental Disorders (2025)
-
Perspectives on the origin and therapeutic opportunities in Down syndrome-associated leukemia
Cancer and Metastasis Reviews (2025)
-
Childhood leukaemia in Down’s syndrome primed by blood-cell bias
Nature (2024)
