
Abstract
Cardiolipin (CL) is the signature phospholipid of the inner mitochondrial membrane, where it stabilizes electron transport chain protein complexes1. The final step in CL biosynthesis relates to its remodelling: the exchange of nascent acyl chains with longer, unsaturated chains1. However, the enzyme responsible for cleaving nascent CL (nCL) has remained elusive. Here, we describe ABHD18 as a candidate deacylase in the CL biosynthesis pathway. Accordingly, ABHD18 converts CL into monolysocardiolipin (MLCL) in vitro, and its inactivation in cells and mice results in a shift to nCL in serum and tissues. Notably, ABHD18 deactivation rescues the mitochondrial defects in cells and the morbidity and mortality in mice associated with Barth syndrome. This rare genetic disease is characterized by the build-up of MLCL resulting from inactivating mutations in TAFAZZIN (TAZ), which encodes the final enzyme in the CL-remodelling cascade1. We also identified a selective, covalent, small-molecule inhibitor of ABHD18 that rescues TAZ mutant phenotypes in fibroblasts from human patients and in fish embryos. This study highlights a striking example of genetic suppression of a monogenic disease revealing a canonical enzyme in the CL biosynthesis pathway.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
All the data used to generate the figures can be found in the Supplementary Tables. Raw data for genetic screens and RNA sequencing are available at NCBI GEO (Accession: GSE297454 and PRJNA1301228). The Bio-ID data are available through MassIVE (Accession: PXD064381 (MSV000098022)). Links to the publicly available datasets analysed in this manuscript are as follows: DepMap, https://depmap.org. Tissue expression data (GTEX), https://gtexportal.org; and tissue expression and subcellular localization data, https://www.proteinatlas.org.
Code availability
No new code was generated for this study.
References
Schlame, M. & Xu, Y. The function of tafazzin, a mitochondrial phospholipid-lysophospholipid acyltransferase. J. Mol. Biol. 432, 5043–5051 (2020).
Schlame, M. & Ren, M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 (2006).
Hornby, B. et al. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome. Orphanet J. Rare Dis. 17, 336 (2022).
Kim, A. Y., Vernon, H., Manuel, R., Almuqbil, M. & Hornby, B. Quality of life in Barth syndrome. Ther. Adv. Rare Dis. 3, 26330040221093743 (2022).
Sabbah, H. N., Taylor, C. & Vernon, H. J. Temporal evolution of the heart failure phenotype in Barth syndrome and treatment with elamipretide. Future Cardiol. 19, 211–225 (2023).
Taylor, C. et al. Clinical presentation and natural history of Barth syndrome: an overview. J. Inherit. Metab. Dis. 45, 7–16 (2022).
Wang, S. et al. Genetic modifiers modulate phenotypic expression of tafazzin deficiency in a mouse model of Barth syndrome. Hum. Mol. Genet. 32, 2055–2067 (2023).
He, Q. & Han, X. Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 179, 75–81 (2014).
Zhu, S. et al. Cardiolipin remodeling defects impair mitochondrial architecture and function in a murine model of Barth syndrome cardiomyopathy. Circ. Heart Fail. https://doi.org/10.1161/CIRCHEARTFAILURE.121.008289 (2021).
Whited, K., Baile, M. G., Currier, P. & Claypool, S. M. Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 22, 483–492 (2013).
Anzmann, A. F. et al. Diverse mitochondrial abnormalities in a new cellular model of TAFFAZZIN deficiency are remediated by cardiolipin-interacting small molecules. J. Biol. Chem. 297, 101005 (2021).
Costanzo, M. et al. Global genetic networks and the genotype-to-phenotype relationship. Cell 177, https://doi.org/10.1016/j.cell.2019.01.033 (2019).
Chen, R. et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 34, 531–538 (2016).
Pu, W. T. Experimental models of Barth syndrome. J. Inherit. Metab. Dis. 45, 72–81 (2022).
Aregger, M. et al. Systematic mapping of genetic interactions for de novo fatty acid synthesis identifies C12orf49 as a regulator of lipid metabolism. Nat. Metab. 2, https://doi.org/10.1038/s42255-020-0211-z (2020).
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015).
Mair, B. et al. Essential gene profiles for human pluripotent stem cells identify uncharacterized genes and substrate dependencies. Cell Rep. 27, 599–615 (2019).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576 (2017).
Bachovchin, D. A. & Cravatt, B. F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 11, 52–68 (2012).
Morgenstern, M. et al. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 33, 2464–2483 (2021).
Price, T. R. et al. Lipidomic QTL in Diversity Outbred mice identifies a novel function for α/β hydrolase domain 2 (Abhd2) as an enzyme that metabolizes phosphatidylcholine and cardiolipin. PLoS Genet. 19, e1010713 (2023).
Long, J. Z. & Cravatt, B. F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 111, 6022–6063 (2011).
Brandner, K. et al. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214 (2005).
Le, C. H. et al. Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 295, 12485–12497 (2020).
Seneviratne, A. K. et al. The mitochondrial transacylase, tafazzin, regulates for AML stemness by modulating intracellular levels of phospholipids. Cell Stem Cell 24, 621–636 (2019).
Beranek, A. et al. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 (2009).
Huang, Y. et al. Cardiac metabolic pathways affected in the mouse model of Barth syndrome. PLoS ONE 10, e0128561 (2015).
Kutschka, I. et al. Activation of the integrated stress response rewires cardiac metabolism in Barth syndrome. Basic Res. Cardiol. 118, 47 (2023).
Liu, O., Chinni, B. K., Manlhiot, C. & Vernon, H. J. FGF21 and GDF15 are elevated in Barth syndrome and are correlated to important clinical measures. Mol. Genet. Metab. 140, 107676 (2023).
Tung, C. et al. Elamipretide: a review of its structure, mechanism of action, and therapeutic potential. Int. J. Mol. Sci. 26, https://doi.org/10.3390/ijms26030944 (2025).
Bononi, G., Tuccinardi, T., Rizzolio, F. & Granchi, C. α/β-Hydrolase domain (ABHD) inhibitors as new potential therapeutic options against lipid-related diseases. J. Med. Chem. 64, 9759–9785 (2021).
Ben Ali, Y. et al. Use of an inhibitor to identify members of the hormone-sensitive lipase family. Biochemistry 45, 14183–14191 (2006).
Duncan, A. L. Monolysocardiolipin (MLCL) interactions with mitochondrial membrane proteins. Biochem. Soc. Trans. 48, 993–1004 (2020).
Burkhalter, M. D. et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J. Clin. Invest. 129, 2841–2855 (2019).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Lesurf, R. et al. Whole genome sequencing delineates regulatory, copy number, and cryptic splice variants in early onset cardiomyopathy. npj Genom. Med. 7, 18 (2022).
Carney, O. S. et al. Stem cell models of TAFAZZIN deficiency reveal novel tissue-specific pathologies in Barth syndrome. Hum. Mol. Genet. 34, 101–115 (2024).
Ye, C. et al. Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J. Biol. Chem. 289, 3114–3125 (2014).
Tyurina, Y. Y. et al. Lipidomics characterization of biosynthetic and remodeling pathways of cardiolipins in genetically and nutritionally manipulated yeast cells. ACS Chem. Biol. 12, 265–281 (2017).
Dudek, J. et al. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res 11, 806–819 (2013).
McKenzie, M., Lazarou, M., Thorburn, D. R. & Ryan, M. T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 (2006).
Zong, S. et al. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res 28, 1026–1034 (2018).
Musatov, A. & Robinson, N. C. Bound cardiolipin is essential for cytochrome c oxidase proton translocation. Biochimie 105, https://doi.org/10.1016/j.biochi.2014.07.005 (2014).
Sedlák, E. & Robinson, N. C. Destabilization of the quaternary structure of bovine heart cytochrome c oxidase upon removal of tightly bound cardiolipin. Biochemistry 54, https://doi.org/10.1021/acs.biochem.5b00540 (2015).
Benegiamo, G. et al. COX7A2L genetic variants determine cardiorespiratory fitness in mice and human. Nat. Metab. 4, 1336–1351 (2022).
Pérez-Pérez, R. et al. COX7A2L is a mitochondrial complex III binding protein that stabilizes the III2+IV supercomplex without affecting respirasome formation. Cell Rep. 16, 2387–2398 (2016).
Cogliati, S. et al. Mechanism of super-assembly of respiratory complexes III and IV. Nature 539, 579–582 (2016).
Mair, B., Aregger, M., Tong, A. H. Y., Chan, K. S. K. & Moffat, J. A method to map gene essentiality of human pluripotent stem cells by genome-scale CRISPR screens with inducible Cas9. Methods Mol. Biol. 2377, 1–27 (2022).
Brockmann, M. et al. Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nature 546, 307–311 (2017).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, https://doi.org/10.14806/ej.17.1.200 (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Hart, T. et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015).
Herzog, K. et al. Lipidomic analysis of fibroblasts from Zellweger spectrum disorder patients identifies disease-specific phospholipid ratios. J. Lipid Res. 57, 1447–1454 (2016).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221 (2007).
Pinault, M. et al. A 1D high performance thin layer chromatography method validated to quantify phospholipids including cardiolipin and monolysocardiolipin from biological samples. Eur. J. Lipid Sci. Technol. 122, 1900240 (2020).
Plekhanov, A. Y. Rapid staining of lipids on thin-layer chromatograms with amido black 10B and other water-soluble stains. Anal. Biochem. 271, 186–187 (1999).
Jha, P., Wang, X. & Auwerx, J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 6, 1–14 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Aken, B. L. et al. Ensembl 2017. Nucleic Acids Res. 45, D635–D642 (2017).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Fonslow, B. R. et al. Digestion and depletion of abundant proteins improves proteomic coverage. Nat. Methods 10, 54–56 (2013).
Washburn, M. P., Wolters, D. & Yates, J. R. III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001).
He, L., Diedrich, J., Chu, Y. Y. & Yates, J. R. 3rd Extracting accurate precursor information for tandem mass spectra by RawConverter. Anal. Chem. 87, 11361–11367 (2015).
Xu, T. et al. ProLuCID: an improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 (2015).
Tabb, D. L., McDonald, W. H. & Yates, J. R. III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 (2002).
Gao, J. et al. CIMAGE2.0: an expanded tool for quantitative analysis of activity-based protein profiling (ABPP) data. J. Proteome Res. 20, 4893–4900 (2021).
Weerapana, E. et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010).
Wang, S. et al. AAV gene therapy prevents and reverses heart failure in a murine knockout model of Barth syndrome. Circ. Res. 126, 1024–1039 (2020).
Ren, M. et al. Extramitochondrial cardiolipin suggests a novel function of mitochondria in spermatogenesis. J. Cell Biol. 218, 1491–1502 (2019).
Molenaars, M. et al. Metabolomics and lipidomics in Caenorhabditis elegans using a single-sample preparation. Dis. Model. Mech. 14, https://doi.org/10.1242/dmm.047746 (2021).
Kulik, W. et al. Bloodspot assay using HPLC–tandem mass spectrometry for detection of Barth syndrome. Clin. Chem. 54, 371–378 (2008).
van der Sande, M. et al. Seq2science: an end-to-end workflow for functional genomics analysis. PeerJ 11, e16380 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Frölich, S., van der Sande, M., Schäfers, T. & van Heeringen, S. J. genomepy: genes and genomes at your fingertips. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btad119 (2023).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Sayols, S., Scherzinger, D. & Klein, H. dupRadar: a Bioconductor package for the assessment of PCR artifacts in RNA-Seq data. BMC Bioinformatics 17, 428 (2016).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Gertsenstein, M. & Nutter, L. M. J. Production of knockout mouse lines with Cas9. Methods 191, 32–43 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Acknowledgements
We thank J. C. Bozelli Jr; the late R. Epand for characterizing HAP1 TAZ clonal cell lines for altered mCL levels using NMR spectroscopy; the proteomics team at SPARC BioCentre at the Hospital for Sick Children for isolating biotinylated preys with KingFisher Flex, mass spectrometry data generation and analysis, and use of Agilent Seahorse equipment; Cayman Chemicals for lipidomics sample preparation and mass spectrometry data generation and analysis; the Imaging Facility and Nanoscale Biomedical Imaging Facility at the Hospital for Sick Children for confocal microscope use; A. Darbandi for TEM sample preparation and instrument use; J. Burgess, A. Zalazar, S. Prokop, M. Carpenter and T. Cunningham for animal care and zebrafish-facility maintenance at the Hospital for Sick Children; the SickKids Heart Centre Biobank Registry for access to patient samples; the Temerty Faculty of the Medicine Flow Cytometry Facility at the University of Toronto for instrument use; and the core staff in the BSU, Histology and Molecular Technology facilities at the CRUK Scotland Institute. S.N.M. was the recipient of Mito2i graduate fellowships. This work was partly supported by the Azrieli PCHP Catalyst Program to I.C.S., the German Research Foundation (DFG; TRR259_397484323) to M.B., a BTHS IDEA grant to J.M., Cancer Research UK to the CRUK Scotland Institute (A31287), the Canadian Institutes for Health Research (PJT-GMX-463531 to J.M.), the Ontario Research Fund (RE11 to C.B., J.M. and B.J.A.). J.M. is the GlaxoSmithKline chair in Genetics and Genome Biology at the Hospital for Sick Children.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.N.M., A. Srivastava, P.M., S.M.B.N., V.A.B. and J.M. Methodology: S.N.M., A. Srivastava, V.S.E., P.M., I.C.S., M.J.N. and D.S. Data acquisition: S.N.M., A. Srivastava, V.S.E., P.M., E.A., J.W., D.T.T., A.G.F., O.S.C., L.C., J.v.A., F.N., L.M., C.S., L.W.-G., N.N., R.M.S., F.M.V., B.E., R.L., S.M., H.V., D.S., M.J.N., B.M.M. and L.M.J.N. Analysis and interpretation: S.N.M., A. Srivastava, V.S.E., P.M., L.V.B., N.M., A. Shaw, S.v.H., B.M.M., S.P., L.M.J.N., K.R.B., R.L., M.B., M.J.N. and D.S. Visualization: S.N.M., P.M., A. Srivastava, L.V.B., K.R.B., S.M.B.N., V.A.B. and J.M. Drafting of the manuscript: S.N.M., P.M., A. Srivastava, S.M.B.N., V.A.B. and J.M., with input from all authors. Technical support: K.C., A.H.Y.T., T.P., O.S., A.H., L.N., M.C. and M.L. Funding and supervision: B.J.A., C.L.M., T.R.B., C.B., I.C.S., S.M.B.N., V.A.B. and J.M.
Corresponding authors
Ethics declarations
Competing interests
S.M. serves on the Advisory Committee of Bristol Myers Squibb, Tenaya Therapeutics and Rocket Pharmaceuticals. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Quality control and validation of CRISPR and gene-trap screens.
(a) Precision-recall curves for five biological replicate CRISPR screens in HAP1 TAZ KO cells using the reference core essential gene set (CEG2) defined in Hart et al. 2017. (b) Scatterplots representing TAZ qGIs derived from all possible pairwise combinations of five replicate screens. Pearson’s correlation based on comparison of qGI scores calculated on all grey points (|qGI | >0.5, FDR < 0.5) is reported in all cases, with R and p-value (TAZ_051 vs. TAZ_186 P = 3.6667×10−91, TAZ_203 vs. TAZ_208 P = 9.31965×10−210, TAZ_203 vs. TAZ_264 P = 1.68248×10−163, TAZ_208 vs. TAZ_264 P = 3.79×10−83) determined using the stat_cor() method in the ggpubr R package. Significant points in both screens highlighted (red). (c) Fitness effect (LFC) distributions for reference core essential (CEG2) and non-essential gene sets across five TAZ KO query screens, the ABHD18-c1 screen and the TAZ;ABHD18-c2 screen respectively. (d) Enrichment for Gene Ontology (GO), molecular function, GO bioprocesses and Reactome terms among genes that exhibited a significant negative GI with TAZ (significant in at least 2 TAZ replicates, |qGI | >0.5, FDR < 0.5) (top), and positive GI with TAZ (bottom). Number of genes overlapping a particular term and term size are indicated. P-values determined by Fisher’s one-tailed test using gProfileR. (e) Mutagenesis-based viability screens in HAP1 WT, and two independent TAZ cell lines. X-axis: log10-transformed mean number of insertions (± strand). Y-axis: ratio, defined as # sense reads/ (# sense + # antisense reads). Significant genes (P adj<1e-3, as compared to four WT controls) are dark grey. Selected genes are marked in red. (f) Overview of unique insertions in the ABHD18 gene in both TAZ KO (s1 and s2 clones) gene-trap screens. Gene transcript (top), sense integrations (middle), and antisense insertions (bottom). (g) Competitive growth assays. Co-cultures of TAZ;ABHD18 double KO cells with either TAZ KO cells or WT HAP1 cells followed over time by flow cytometry and normalised to day 0. (h) Relative proliferation of WT, TAZ KO, ABHD18 KO and ABHD18;TAZ double-KO RPE1 pools, presented as means of n = 2 biological replicates). (i) Relative proliferation of WT, TAZ KO, ABHD18 KO and ABHD18;TAZ double-KO H1-iCas9 pools (left; WT n = 5, TAZ-KO1 n = 4, ABHD18-KO1 n = 3, ABHD18-KO1: TAZ KO-1 n = 4 biological replicate assays) and proportion of pluripotent cells in selected pools (right; WT n = 4, TAZ-KO1 n = 4, ABHD18-KO1 n = 3, ABHD18-KO1: TAZ-KO1 n = 3 biological replicate assays). Data presented as means ± s.d. with significance calculated using one-way ANOVA followed by Dunnett’s post-hoc test. (j) Representative flow cytometric analysis of OCT4 & SOX2 expression in WT, TAZ, ABHD18 and ABHD18;TAZ H1 hPSC pools. (k) Bar plot showing relative fitness effects when overexpressing either TAZ (TAZ OE) or ABHD18 (ABHD18 OE) across double mutant backgrounds. OE of either TAZ [clone 2 P = 0, clone 8 P = 0.0007] or ABHD18 [clone 2 P = 0, clone 8 P = 0.0001] reduces fitness in TAZ;ABHD18 double-KO cells. Data presented are means ± s.d. (n = 3 biological replicates) with significance determined by one-way ANOVA followed by Tukey’s multiple comparisons test. (l) Volcano plot showing qGI scores and associated significance (-log10(P value)) for TAZ;ABHD18-c2 CRISPR screen. (m) Scatterplot of the overlap between TAZ;ABHD18-c2 and ABHD18-c1 qGIs showing GI overlap. Yellow: Shared positive GIs, blue: shared negative GIs (qGI < −0.5, >0.5; FDR < 0.5) in both screens. (n) Heat map displaying genes (y axis) with significant interactions with TAZ across five replicate screens and at least one additional screened query (x-axis) (|qGI | >0.5, FDR < 0.5). Positive and negative qGI scores are indicated by yellow and blue, respectively. (o) Bar plot illustrating relative cell proliferation of partial LOF mutant TAZ;ABHD18-c8 (partial LOF of ABHD18) compared with WT HAP1. Data presented as means ± s.d. (n = 3 biological replicates) with significance determined by one-way ANOVA followed by Dunnett’s post-hoc test. (p) Scatterplot displaying profile similarity of ABHD18 across genome-wide DepMap CRISPR/Cas9 screens. Similarity was quantified by taking all pairwise gene-gene Pearson correlation coefficients of CERES score profiles across 563 screens (19Q2 DepMap data release). Distribution of 17,633 CERES profile similarity is plotted as a quantile-quantile plot (right). Pathway analysis of ABHD18 profile similarity for all 17,634 genes represented in DepMap were mean summarized by pathway as defined in the HumanCyc standard (left). Pathway similarity and dissimilarity with ABHD18 were tested using a two-sided Wilcoxon rank-sum test with multiple-hypothesis correction using the Benjamini and Hochberg procedure.
Extended Data Fig. 2 BioID enrichments support mitochondrial localization of ABHD18.
(a) Confocal images of ABHD18-FLAG HEK293 Flp-In T-Rex cells stained for FLAG (green) and HSP60 (red). Intensity measurements across the indicated white line are indicated in the bottom right panel. At least 30 cells from 2 biological replicate experiments were analyzed and a representative image shown. (b) GO biological process (BP), molecular function (MF), and cellular compartment (CC) enrichments for BioID preys captured by both N- and C-term miniTurbo-tagged ABHD18 in either glucose or galactose growth conditions. Enrichment terms for significantly enriched gene sets (p < 0.05, maximum term size 105) are indicated and bars depict -log10 (FDR) determined by p-values calculated using gProfileR. (c) Scatterplots of interaction specificity of N- and C-term tagged ABHD18 detected preys captured with BioID in either glucose or galactose growth conditions. Average spectral counts of preys captured in proximity to either N- or C-term miniTurbo BirA-tagged ABHD18 are plotted against their specificity across hundreds of baits from the Human Cell Map BioID dataset. Avg Spec was calculated using SAINTexpress v3.6.1. Data are representative of three biologically independent experiments per condition. (d) Representative TEM images of HAP1 parental, TAZ KO, ABHD18 KO, TAZ;ABHD18 double-KO cells, green: mitochondria. Scale bars, 1 μm. Representative of n = 4 biological replicate cell pellets per genotype.
Extended Data Fig. 3 ABHD18 regulates mitochondrial respiratory chain assembly and generates MLCL from nCL.
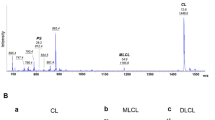
(a) BN-PAGE immunoblots (total OxPHOS, CII, CIII, CIV, CV) and in-gel activity assays (CI, CIV) from isolated mitochondria of HAP1 mutant cell lines. Blots probed for individual complexes and supercomplexes. Total OXPHOS (n = 5 biological replicates) obtained using antibody cocktail targeting CI, CII, CIII, CIV, and CV. CIII and CIV blots (n = 3 biological replicates), and CIV activity are representative (n = 2 biological replicates). (b) Bar plots of Seahorse Cell Mito Stress Test for respiratory parameters: basal respiration, maximal respiration, mitochondrial ATP generation, and glycolytic ATP generation across mutant HAP1 cell lines. Data presented are means ± s.d. of n = 4 technical replicates. Scatterplots of OCR and ECAR measurements taken during seahorse assays (right). (c) Dot plots of lipidomics analysis of CL and MLCL abundance in HAP1 WT and mutant cell lines (left and middle panel respectively). Relative abundance shown as bar plot above dot plot for each genotype. Bar plot quantifying MLCL/CL ratio changes across HAP1 cell lines using total CL and MLCL species abundance (right). (d) Dot plots showing lipidomics analysis of CL and MLCL abundance in an additional set of HAP1 mutant cell lines (n = 3 clonal mutant cell lines, n = 4 WT cell lines). (e) Thin Layer Chromatography (TLC) showing wild-type ABHD18[WT], but not catalytic dead ABHD18[S199A] recombinant protein deacylates different cardiolipin species to yield MLCL and DLCL, and deacylates MLCL to yield DLCL. Negative control does not contain any ABHD18 protein present, only buffer.
Extended Data Fig. 4 Taz−/YAbhd18−/− mice are fertile and demonstrate normal heart function.
(a) Representative CL spectrum of control wildtype mice (n = 3 animals, left), and Abhd18 knockout mice (n = 3 animals, right) obtained by blood spot assay. Blue: summation of carbon atoms in the fatty acid side chains. Red: CL peaks for each carbon group along with number of double bonds. (b) qRT-PCR analysis of Taz and Abhd18 expression levels in mice heart tissue. Expression was normalized to Gapdh. Data are presented as means ± s.e.m. from three animals. (c) Schematic showing sterility test cross between Taz−/Y:Abhd18−/− double KO male mice and Taz+/−Abhd18−/− female mice. (d) Representative ultrasound images of mouse hearts recorded in M mode (WT n = 8, Taz−/Y n = 3, Abhd18−/− n = 7, Abhd18−/−Taz−/Y n = 6 mice). Six measurements were taken for each image using ImageJ: 1. Anterior Wall Thicknesss (AWT) – diastole (d), 2. Left Ventricle End Diastole Diameter (LVEDD), 3. Posterior Wall Thickness (PWT) – diastole (d), 4. AWT – systole (s), 5. Left Ventricle End Systole Diameter (LVESD) and 6. PWT – systole (s).
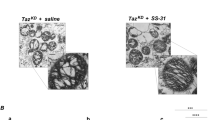
Extended Data Fig. 5 ABD646 treatment mimics ABHD18 LOF phenotypes.
(a) Dot plots showing lipidomic analysis of CL and MLCL abundance in HAP1 and TAZ-c1 KO cells treated with either DMSO or 1μM of ABD646 for 5 days (left and middle panel respectively). Bar plot quantifying MLCL/CL ratio changes across HAP1 cell lines using total CL and MLCL species abundance (right). (b) Bar plots illustrating the proliferative effect of 10 μm ABD646 on HAP1 and TAZ KO cells. All values normalized to HAP1 DMSO (n = 3 biological replicates), with significance determined by one-way ANOVA followed by Dunnett’s post-hoc test. (c) TEM images of HAP1 TAZ KO cells treated with DMSO or ABD646 for five days. Green: mitochondria. Scale bars, 1 μm (top) and 200 nm (bottom). (d) Schematic illustrating mode of action of three substrate oxidation pathway inhibitors for glucose/pyruvate (UK5099), long chain fatty acids (LCFA, Etomoxir), and glutamine oxidation (BPTES). (e) Bar plots of seahorse substrate oxidation tests using Etomoxir, BPTES, and UK5099 on HAP1 WT and mutant cells, with ABD646 or DMSO treatment. Data are maximal respiration values (OCR pmol/min, n = 6 technical replicates). (f) Representative TEM images showing partial restoration of mitochondrial morphologies observed in BTHS fibroblasts treated with 1μM of ABD646 for 5 days. Green: mitochondria. Scale bars: 1 μm, and 200 nm. n = 2 biological replicate cell pellets per treatment. (g) Flow cytometry analysis of mitochondrial mass (Mitotracker Green FM) and membrane potential (Mitotracker CMXROS) in fibroblasts from patients with BTHS treated with vehicle or 5μM ABD646 for five days. (h) BN-PAGE immunoblots and in-gel activity assays for mitochondrial complexes in fibroblasts from patients with BTHS treated with vehicle or 5μM ABD646 for five days. (i) Dot plots showing lipidomics analysis of CL and MLCL abundance in BTHS fibroblasts treated with either DMSO or 1μM of ABD646 for 5 days (left and middle panel respectively). Bar plot quantifying MLCL/CL ratio changes across HAP1 cell lines using total CL and MLCL species abundance (right). (j) Representative images at day 5 of zebrafish embryos injected with increasing concentrations of TAZ MOs (2.5ng-10.0 ng) (left; scale bars, 200 μm). Bar plot quantifying observed phenotypes across doses of TAZ MOs (right). (k) 24 h acute treatment of ABD646 has chronic benefit for TAZ MO zebrafish. Representative images of zebrafish embryos following either a 24-h treatment with ABD646 on WT (n = 45), and TAZ MO treated (n = 40) embryos. Embryos were washed once a day for 5 consecutive days with imaging and quantification on day 5 (left; scale bars, 200 μm). Right, quantification of embryos demonstrating “rescue” or near WT phenotypes per wash day. (l) Box plot of ABHD18 gene expression across HAP1 and TAZ mutant clones in nutrient rich (IMDM) or nutrient limiting (DMEM) media conditions. Data are median read counts ± interquartile range (IQR), n = 3 biological replicates per media condition. Significance determined by one-way ANOVA followed by Dunnett’s post hoc test. (m) Bar plot of spearman correlations between TAZ and ABHD18 gene expression across tissue samples. Data from the Genotype-Tissue Expression (GTEx) Project.
Supplementary information
Supplementary Information (download PDF )
This file contains the methods for the chemical synthesis of ABD646, Supplementary Figs. 1 and 2, Supplementary Methods Tables 1–4 and full descriptions for Supplementary Videos 1–4 and Supplementary Tables 1–4 (supplied separately).
Supplementary Table 1 (download XLSX )
Genetic screens and related data analyses.
Supplementary Table 2 (download XLSX )
ABHD18 Bio-ID interaction data.
Supplementary Table 3 (download XLSX )
Lipidomics data.
Supplementary Table 4 (download XLSX )
RNA-seq data from mouse hearts.
Supplementary Video 1 (download MOV )
Video of a wild-type GFP-labelled fish heart showing cardiac contractions following treatment with drug vehicle (see Methods for details).
Supplementary Video 2 (download MOV )
Video of a wild-type GFP-labelled fish heart treated for 5 days with ABD646 showing cardiac contractions (see Methods for details).
Supplementary Video 3 (download MOV )
Video of a GFP-labelled fish heart showing cardiac contractions following treatment with Taz morpholinos and subsequently with drug vehicle (see Methods for details).
Supplementary Video 4 (download MOV )
Video of a GFP-labelled fish heart showing cardiac contractions following treatment with Taz morpholinos and subsequently with ABD646 for 5 days (see Methods for details).
Supplementary Information (download PPTX )
Supplementary Video Summary Slide.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Masud, S.N., Srivastava, A., Mero, P. et al. Genetic suppression features ABHD18 as a Barth syndrome therapeutic target. Nature 645, 1029–1038 (2025). https://doi.org/10.1038/s41586-025-09373-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09373-5
This article is cited by
-
Rebalancing cardiolipin biosynthesis to treat cardiomyopathy
Nature Reviews Drug Discovery (2025)
