
Abstract
Despite decades of study, large parts of the mammalian metabolome remain unexplored1. Mass spectrometry-based metabolomics routinely detects thousands of small molecule-associated peaks in human tissues and biofluids, but typically only a small fraction of these can be identified, and structure elucidation of novel metabolites remains challenging2,3,4. Biochemical language models have transformed the interpretation of DNA, RNA and protein sequences, but have not yet had a comparable impact on understanding small molecule metabolism. Here we present an approach that leverages chemical language models5,6,7 to anticipate the existence of previously uncharacterized metabolites. We introduce DeepMet, a chemical language model that learns from the structures of known metabolites to anticipate the existence of previously unrecognized metabolites. Integration of DeepMet with mass spectrometry-based metabolomics data facilitates metabolite discovery. We harness DeepMet to reveal several dozen structurally diverse mammalian metabolites. Our work demonstrates the potential for language models to advance the mapping of the mammalian metabolome.
Similar content being viewed by others


Main
Mass spectrometry-based metabolomics typically detects thousands of distinct chemical entities in any given biological sample8, but even in human tissues or biofluids, the majority of these are not routinely linked to a chemical structure2,3. This profusion of unidentified chemical entities has been dubbed the chemical ‘dark matter’ of the metabolome4. The existence of this metabolic dark matter suggests that existing metabolic maps are far from complete9,10,11,12. New approaches are needed to illuminate the dark matter of the metabolome in a systematic manner.
Generative models based on deep neural networks have emerged as a powerful approach to study the structure and function of biological macromolecules13. Language models trained on protein sequences are capable of learning the latent evolutionary forces that have shaped extant sequences in order to design new proteins with desired functions, predict the effects of unseen variants, and even forecast protein sequences that are likely to evolve in the future14,15,16,17. Language models can also be trained on the chemical structures of small molecules by leveraging formats that represent these structures as short strings of text, a concept that has been exploited by a large body of work over the past decade5,6,7. So far, however, this paradigm has primarily been applied to explore synthetic chemical space in the setting of drug discovery. Here we introduce DeepMet, a chemical language model trained on the structures of known metabolites that anticipates the existence of previously unrecognized metabolites (Fig. 1a). We develop approaches to integrate DeepMet with mass spectrometry-based metabolomics data that enable de novo identification of metabolites in complex tissues and harness these approaches to reveal several dozen previously unrecognized metabolites.

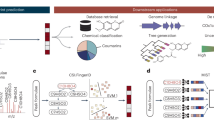
a, Schematic overview of DeepMet. RT, retention time. b, UMAP visualization of the chemical space occupied by known metabolites and generated molecules. Left, known metabolites superimposed over generated molecules. Right, generated molecules superimposed over known metabolites. Known metabolites are coloured by their assigned superclasses in the ClassyFire chemical ontology. c, Receiver operating characteristic (ROC) curve of a random forest classifier trained to distinguish between known metabolites and generated molecules in cross-validation. d, Proportion of enzymatic biotransformations of known metabolites23 recapitulated by DeepMet, shown as a function of the number of rule-based transformations applied sequentially to the original metabolite.
Learning the language of metabolism
Metabolites are synthesized from a small pool of precursors such as amino acids, organic acids, sugars and acetyl-CoA via a limited repertoire of enzymatic transformations. These shared biosynthetic origins result in the overrepresentation of certain physicochemical properties and substructures among metabolites, compared with synthetic compounds made in the laboratory18,19,20. We hypothesized that a chemical language model could learn from the structural features of known metabolites to access previously unrecognized structures from metabolite-like chemical space.
To test this hypothesis, we assembled a training set of 2,046 metabolites that had been experimentally detected in human tissues or biofluids21 and represented these as short strings of text in simplified molecular-input line-entry system (SMILES) notation22. We trained a long short-term memory (LSTM) language model on this dataset of known metabolite structures after first pretraining it on drug-like structures from the ChEMBL database, and used the trained model to generate 500,000 SMILES strings in order to evaluate its understanding of metabolism.
Several lines of evidence indicated that our language model was able to appreciate the structural features of known metabolites and exploit this understanding to generate metabolite-like structures. First, we visualized the chemical space occupied by generated molecules and known metabolites using the nonlinear dimensionality reduction algorithm uniform manifold approximation and projection (UMAP). Generated molecules overlapped extensively with known metabolites (Fig. 1b). Second, we trained a random forest classifier to distinguish the generated molecules from a set of known human metabolites that had been deliberately withheld from the language model during training. We found that this classifier could not accurately separate the two classes of molecules, and instead performed only marginally better than random guessing (Fig. 1c and Extended Data Fig. 1a–c). Third, because many biosynthetic enzymes are known to be promiscuous in the substrates that they accept, we tested whether the generated molecules could be rationalized as enzymatic transformations of known metabolites. We found that the language model recapitulated 77.5% of one-step enzymatic transformations of known metabolites predicted by the rule-based platform BioTransformer23 (Fig. 1d and Extended Data Fig. 1d,e), despite not having been provided any explicit information about enzymatic reactions during training. Our model, however, predicted a much broader spectrum of structures than the rule-based approach, with the vast majority of structures generated by the language model not being predicted by BioTransformer (Extended Data Fig. 1f). Fourth, we found that the generated molecules were more structurally similar to known metabolites than molecules with identical molecular formulas sampled at random from PubChem or ChEMBL (Extended Data Fig. 1g–i).
These results introduce a language model of metabolite-like chemical space, which we named DeepMet.
Anticipating unrecognized metabolites
In the setting of protein biochemistry, language models can be leveraged to predict the functional impacts of unseen mutations and to forecast the evolution of future proteins14,15,16,17. We hypothesized that the same principle could be applied to predict the structures of previously unrecognized metabolites. Unlike nucleotide or protein sequences, however, chemical structures do not have a unique textual representation24, and we observed that DeepMet assigned markedly different likelihoods to different SMILES strings representing the same chemical structure (Extended Data Fig. 2a–c).
In lieu of calculating the likelihoods of individual SMILES strings, we reasoned that chemical structures viewed by DeepMet as more plausible extensions of the training set would be sampled more frequently in aggregate, considering all possible representations. To test this hypothesis, we drew a sample of 1 billion SMILES strings from DeepMet, and then tabulated the frequency with which each unique chemical structure appeared in this sample (Fig. 2a). Whereas the vast majority of these structures appeared at most a handful of times in the language model output, others were generated thousands of times (Fig. 2b).

a, Schematic overview of sampling frequency calculation. b, Distribution of sampling frequencies within a sample of 1 billion SMILES strings. c–f, Properties of molecules generated with progressively increasing frequencies. c, Tanimoto coefficient (Tc) between generated molecules and their nearest neighbours in the training set (n = 50,000 randomly sampled molecules per bin). d, Proportion of generated metabolites recapitulating one-step enzymatic transformations of known metabolites predicted by BioTransformer. e, Jensen–Shannon distances between Murcko scaffolds of generated molecules and known metabolites (n = 10 folds). f, Fréchet ChemNet distances between generated molecules and known metabolites (n = 10 folds). g, Frequencies with which known metabolites withheld from the training set were sampled, compared to all generated molecules (in n = 109 sampled SMILES). h, Proportion of HMDB 5.0 metabolites generated by DeepMet. i, Categorization of the 61 HMDB 5.0 metabolites not generated by DeepMet. j, ROC curve showing prioritization of HMDB 5.0 metabolites on the basis of their sampling frequencies. k, Enrichment of HMDB 5.0 metabolites among the most frequently generated molecules (two-sided χ2 test). l, Proportion of known or predicted/expected metabolites from versions 4.0 or 5.0 of the HMDB within the top-10,000 molecules most frequently generated by DeepMet. m, Examples of metabolites annotated as predicted or expected that are actually well-studied human metabolites, and were generated with frequencies comparable to experimentally detected metabolites despite being withheld from the training set. n–p, Examples of previously unrecognized human metabolites identified in human urine (chemical structures, extracted ion chromatograms (EICs) from chemical standards (Std) and representative urine metabolomes, and mirror plots comparing MS/MS from standards versus experimental spectra). Vertical red lines show times of MS/MS acquisitions. n, N-carbamyl-proline. o, N-succinyl-tryptophan. p, N-lactoyl-glutamine.
We sought to characterize these frequently generated molecules. Molecules sampled more frequently by DeepMet exhibited a higher degree of structural similarity to known metabolites (Fig. 2c and Extended Data Fig. 2d); were disproportionately likely to overlap with plausible enzymatic transformations23 of known metabolites (Fig. 2d and Extended Data Fig. 2e); were more likely to share a chemical scaffold with a known metabolite (Fig. 2e); and, as quantified by the Fréchet ChemNet distance25, were predicted to have a more similar spectrum of biological activities to known metabolites (Fig. 2f). Thus, molecules generated more frequently by DeepMet were disproportionately metabolite-like.
This finding led us to more directly test whether this sampling frequency could be used to prioritize candidate metabolites for discovery. To evaluate this possibility, we withheld known metabolites from the training set in order to simulate the discovery of unknown metabolites. The withheld metabolites were generally among the most frequently generated molecules proposed by the language model (Fig. 2g), such that the sampling frequency alone separated withheld metabolites from other generated molecules with an area under the receiver operating characteristic curve (AUC) of 0.98 (Extended Data Fig. 2f).
We therefore sought to prospectively evaluate the ability of DeepMet to predict future metabolite discoveries. A total of 313 metabolites had been added to version 5.0 of the Human Metabolome Database (HMDB) after our training dataset was finalized26. DeepMet successfully generated 252 of these 313 metabolites (81%; Fig. 2h), and most of the 61 structures that were not successfully generated were not products of endogenous human metabolism, but were instead derived from prescription drugs, food, the microbiome or environmental chemicals (Fig. 2i and Extended Data Fig. 2g,h). Moreover, we again found that the sampling frequency alone separated the HMDB 5.0 metabolites from other generated molecules (AUC = 0.97; Fig. 2j).
HMDB 5.0 metabolites were markedly enriched in the uppermost extremities of the sampling frequency distribution. The top-10,000 most frequently generated molecules, for instance, contained 105 of the 252 generated metabolites, an enrichment of about 1,500-fold over random expectation (Fig. 2k and Supplementary Table 1). Notably, this subset also included 1,888 metabolites annotated as predicted or expected in version 4.0 or 5.0 of the HMDB (Fig. 2l), which had been excluded from the training set. Several of the most frequently sampled predicted or expected metabolites were in fact well-studied metabolites that had been misannotated in the HMDB (Fig. 2m), underscoring the ability of our model to fill gaps in existing metabolic databases.
Among the top-10,000 most frequently sampled metabolites, 6,301 were absent from any version of the HMDB (Fig. 2l). These structures are those considered by DeepMet to represent the most plausible extensions of the known metabolome. We hypothesized that many of these structures were indeed mammalian metabolites.
To test this hypothesis, we obtained or synthesized chemical standards for 80 putative metabolites that ranked in the top-10,000 structures. Each of these standards was profiled by liquid chromatography–tandem mass spectrometry (LC–MS/MS) and then compared against a large bank of urine and blood metabolomics data that had been collected by one laboratory using identical analytical methods. A total of 17 metabolites predicted by DeepMet were identified in human biofluids by the combination of retention time and tandem mass spectrometry (MS/MS), although careful review of the literature revealed a subset of these to be known metabolites missing from the HMDB27,28,29,30 (Fig. 2n–p, Supplementary Fig. 1, and Supplementary Table 2).
Thus, DeepMet can fill the gaps in our understanding of metabolism by predicting the structures of previously unrecognized metabolites.
Prioritizing structures from accurate masses
These experiments introduce a structure-centric approach to metabolite discovery, whereby hypothetical metabolites are prioritized by a chemical language model for synthesis and targeted discovery. We also envisioned, however, that DeepMet could support more conventional approaches to metabolite discovery, whereby metabolites are targeted for structure elucidation on the basis of mass spectrometric data.
We began by asking whether DeepMet could prioritize plausible chemical structures for an unidentified metabolite given a single measurement as input: the metabolite’s exact mass. To test this possibility, we again simulated metabolite discovery by withholding known metabolites from the training set. For each held-out metabolite, we filtered the structures generated by DeepMet to those matching its exact mass (±10 ppm). We then tabulated the total frequency with which each of these structures was generated by DeepMet (Fig. 3a).

a, Schematic overview of the workflow to prioritize metabolite structures given an accurate mass measurement as input. CLM, chemical language model. b, Top-1 accuracy with which the complete chemical structures of held-out metabolites were assigned by DeepMet or two baseline approaches: AddCarbon or searching within the training set. c, Illustrative example demonstrating the use of DeepMet to prioritize candidate metabolite structures based on an accurate mass. A total of n = 27,509 sampled SMILES strings matched the input mass of 176.0950 ± 10 ppm, corresponding to n = 2,818 unique structures. Left, lollipop plot shows the sampling frequencies of the 15 most frequently generated molecules as a proportion of the 27,509 SMILES strings. Right, a subset of the generated molecules is shown, including the four most frequently generated as well as a selection of less frequently generated structures. Structures 1, 2 and 3 are known human metabolites that were not present in the training set. d, As in b, but showing the top-k accuracy curve, for k ≤ 30. e, Tanimoto coefficients between the structures of held-out metabolites and the top-ranked structures prioritized by DeepMet, random structures generated by DeepMet, or two baseline approaches. f, Proportion of held-out metabolites that were ever generated by the language model. g, Tanimoto coefficients between held-out metabolites and their nearest neighbour in the HMDB, for metabolites that were ever versus never generated by the language model. h, Proportion of correct structure assignments for held-out metabolites as a function of the DeepMet confidence score.
Across all withheld metabolites, the most frequently generated structure matched that of the held-out metabolite in 29% of cases (Fig. 3b). For instance, providing the mass of serotonin (176.0950 Da ± 10 ppm) as input yielded 27,509 SMILES strings, representing 2,818 unique chemical structures; of these, the single most frequently sampled structure was that of serotonin itself (Fig. 3c). Because serotonin had been withheld from the training set, this required DeepMet to simultaneously generate the chemical structure of an unseen metabolite, and to prioritize this structure from among thousands of chemically valid candidates.
In cases where the top-ranked structure was not that of the held-out metabolite, the correct structure was often found among a short list of candidates (Fig. 3c,d and Extended Data Fig. 3a). Moreover, when the top-ranked structure was incorrect, it was often structurally similar to the true metabolite (Fig. 3e and Extended Data Fig. 3b–j). Only 10% of held-out metabolites were never reproduced by the language model, and these metabolites tended to demonstrate a low degree of structural similarity to any other metabolite in the training set (Fig. 3f, g).
To contextualize the performance of our language model, we compared DeepMet to the AddCarbon baseline proposed by Renz et al.31 Although this simple approach has frequently outperformed more sophisticated generative models31, we nonetheless found that DeepMet markedly outperformed AddCarbon on all metrics (Fig. 3b,d,e and Extended Data Fig. 3c–e). Structures prioritized by DeepMet also demonstrated a higher degree of structural similarity to the held-out metabolites than isobaric known metabolites, reflecting the ability of the model to generalize beyond the training set into unseen chemical space.
We computed confidence scores for each structure based on the sampling frequencies of all generated molecules matching the query mass, and found that these confidence scores correlated well with the likelihood that any given structure assignment was correct (Fig. 3h and Extended Data Fig. 3k). This observation highlights a particularly useful property of DeepMet: namely, that its most confident predictions are expected to be the best candidates for experimental follow-up.
We then turned again to the 313 metabolites added in version 5.0 of the HMDB, and tested whether DeepMet would demonstrate similar performance in this prospective test set. This is a challenging task, as these metabolites are structurally distinct from those in the training set (Extended Data Fig. 3l). Nonetheless, DeepMet demonstrated comparable performance in this prospective test set (Extended Data Fig. 3m–s).
Together, these experiments establish that DeepMet can simultaneously generate and prioritize candidate structures for unidentified peaks detected by mass spectrometry.
Integration of DeepMet and MS/MS
Because it is impossible to distinguish between isomeric metabolites with the same molecular formula on the basis of accurate mass information alone, most mass spectrometry-based metabolomics workflows rely on MS/MS for metabolite identification. We therefore next sought to integrate our language model with MS/MS data.
A number of existing computational approaches leverage MS/MS data to search databases of known chemical structures32. One such method, CFM-ID33,34, learns from a training dataset of experimental MS/MS spectra and their associated structures to predict MS/MS spectra for unseen compounds. Applying CFM-ID to a database of known chemical structures produces an in silico MS/MS spectral library that can be used to identify compounds by comparing predicted and experimental spectra. We reasoned that this approach could be adapted to search a database of hypothetical metabolites generated by DeepMet, in analogy to approaches that search databases of combinatorially enumerated structures35,36 and in line with an approach proposed, although not implemented, by a previous study35. We further envisioned that both the sampling frequency from DeepMet and the MS/MS spectral match could be integrated for enhanced accuracy (Fig. 4a).

a, Schematic overview of the workflow for metabolite annotation via MS/MS. b, Top-1 accuracy with which the chemical structures of held-out metabolites were assigned by the combination of DeepMet with CFM-ID in positive-mode spectra from the Agilent MS/MS library, compared with a series of baseline approaches, including ranking structures based on the sampling frequency alone, based on the dot-product between predicted and experimental spectra, or the combination of CFM-ID with two baseline approaches, AddCarbon or searching within the training set. c, As in b, but showing the top-k accuracy curve, for k ≤ 30. d, Tanimoto coefficients between the structures of held-out metabolites (n = 558 with positive-mode spectra) and the top-ranked structures prioritized by the combination of CFM-ID with DeepMet as compared to baseline approaches. e,f, As in c,b, but also showing the top-k accuracy when considering prioritized structures with minimum Tanimoto coefficients of 0.4 or 0.675 as matches. g, Number of MS/MS spectra in the human blood metabolome dataset linked to a chemical structure when searching against a database of predicted spectra for known human metabolites only or a combined library containing both known and generated metabolites. h, As in g, but for a minimum cosine similarity of 0.8. i, Cumulative distribution of cosine similarities between predicted and experimental spectra in the human blood metabolome dataset, for generated metabolites binned into deciles by their sampling frequencies. j, Top, structure of N1-methyl-imidazeolelactic acid. Bottom, mirror plot showing the similarity between MS/MS spectra from the human blood metabolome dataset versus a synthetic standard. k, ROC curve showing the separation of patients with sepsis from healthy controls by the abundance of N1-methyl-imidazeolelactic acid in the MTBLS7878 dataset (P value calculated as described in ref. 60).
To test this possibility, we applied CFM-ID to predict MS/MS spectra for 2.4 million structures generated by DeepMet (Extended Data Fig. 4a–c). We again simulated metabolite discovery by withholding known metabolites from the training sets of both models, and found that the combination of DeepMet and CFM-ID correctly assigned the exact chemical structures for 52% and 49% of held-out metabolites in the positive and negative ion modes, respectively (Fig. 4b and Extended Data Fig. 4d). The addition of MS/MS information also robustly increased the number of cases in which the correct structure was ranked among the top-3 or top-10 candidates; the chemical similarity between the predicted and true metabolite structures; and the proportion of spectra for which a close or meaningfully similar match37 was retrieved (Fig. 4c–f and Extended Data Fig. 4e–h). We observed similar performance in a second dataset of MS/MS spectra, or when using alternative machine learning methods for MS/MS prediction38,39 (Extended Data Fig. 4i–n).
The use of auxiliary data such as citation counts or production volumes (collectively referred to as meta-scores) in metabolite annotation has been criticized on the grounds that these features hinder the discovery of novel metabolites35,40. The sampling frequency in DeepMet differs from such meta-scores. Whereas meta-scores bias models towards re-discovery of well-studied metabolites, our approach is explicitly designed to enable discovery of previously unreported structures. Consistent with this objective, whereas meta-scores are by definition only available for known metabolites, DeepMet assigns frequencies to structures that are absent from existing databases (Supplementary Fig. 2a). Moreover, the performance of our approach is not contingent on the use of the sampling frequency, but benefits from it (Supplementary Fig. 2b,c).
Over the past decade, thousands of untargeted metabolomics experiments in human tissues and biofluids have been deposited to public repositories. We reasoned that the combination of DeepMet with MS/MS database search could provide a mechanism to systematically annotate previously unrecognized metabolites within these publicly available data. To explore this possibility, we first assembled a large-scale resource of human blood metabolomics data. We identified a total of 4,510 metabolomic analyses of human blood, from which 29.1 million MS/MS spectra were extracted (Extended Data Fig. 4o–s). We then tested the hypothesis that adding structures generated by DeepMet to an in silico MS/MS spectral library would increase the number of MS/MS spectra that could be putatively annotated. We searched the human blood metabolome data against a library comprising predicted MS/MS spectra for all structures in the HMDB, or a combined library also including DeepMet structures. The combined library markedly increased the number of peaks that could be tentatively matched to a chemical structure at any threshold (Fig. 4g,h), and substantially more matches were observed to predicted MS/MS spectra than to ‘decoy’ spectra created by shuffling fragment ions between predicted spectra with isobaric precursors, indicating that this increase could not be explained solely by chance matches to a larger MS/MS library (Extended Data Fig. 4t and Supplementary Fig. 3). Moreover, structures generated more frequently by DeepMet were disproportionately likely to match to an experimentally collected MS/MS spectrum (Fig. 4i).
We sought to corroborate a subset of these annotations. We initially focused on an unidentified metabolite that DeepMet had annotated as a brominated derivative of nicotinic acid, and which demonstrated an isotopic pattern consistent with the presence of bromine41 (Supplementary Fig. 4a,b). Comparison to a synthetic standard supported the annotation of this peak as 4-bromonicotinic acid, although differences in fragment ion intensity between the synthetic and experimental MS/MS spectra meant that without access to the original sample, this structure remained a leading hypothesis rather than a definitive identification; standards for 12 potential isomers matched less well to the experimental MS/MS (Supplementary Fig. 4c,d). Similarly, comparison to a synthetic standard supported the annotation of an unidentified peak in a metabolomic dataset from patients with sepsis as an N-methylated derivative of imidazolelactic acid, a metabolite that has previously been reported in the literature29 but which was absent from the HMDB42 (Fig. 4j and Supplementary Fig. 4e). The abundance of this metabolite separated patients with sepsis from healthy controls (Fig. 4k and Supplementary Fig. 4f), underscoring the potential to discover metabolic biomarkers by re-interrogating published datasets with DeepMet.
Validation in metabolomics data
High-confidence metabolite annotation requires comparison to data from a reference standard analysed under identical analytical conditions. Accordingly, there are inherent limitations to the confidence with which metabolites can be identified through re-examination of published datasets without access to the original samples. We therefore sought to apply DeepMet to a newly collected metabolomic dataset that would allow for comparison to chemical standards on an identical analytical setup.
We profiled the metabolomes of 23 mouse tissues and biofluids by LC–MS/MS. After an initial round of filtering to discard isotopic peaks, adducts, in-source fragments and other mass spectrometry artefacts with NetID43, a total of 4,814 peaks were detected that represented presumptive metabolites (Extended Data Fig. 5a,b). Of these, 250 (5.2%) could be identified by comparison to an in-house library of metabolite standards, whereas the remaining 94.8% remained unidentified (Extended Data Fig. 5c).
We first leveraged these identifications to benchmark DeepMet in mouse tissues, again simulating metabolite discovery by withholding known metabolites from the training sets of both DeepMet and CFM-ID. The combination of DeepMet and CFM-ID assigned the correct structure to 50% of the known peaks (Supplementary Fig. 5). To further corroborate the performance of DeepMet, we studied a subset of peaks that were annotated as known metabolites by DeepMet, but for which the corresponding standards were absent from our library, and which had been withheld from the training sets of DeepMet and CFM-ID. We obtained standards for 97 of these known metabolites, and experimentally validated 58 of these annotations (60%; Supplementary Table 3 and Extended Data Fig. 5d–u).
Metabolomic profiling collects additional sources of information that are typically not recorded in spectral libraries, including retention times and isotopic patterns at the MS1 level. We hypothesized that these additional data could further increase the accuracy of metabolite discovery. To this end, we trained a random forest classifier in cross-validation to identify correct annotations by integrating multiple sources of information, including the confidence scores emitted by DeepMet, the similarity between predicted and experimental MS/MS spectra, the isotope pattern match at the MS1 level, and the discrepancy between predicted and experimentally measured retention times. This meta-learning approach further increased the accuracy of metabolite annotation to 70% (Fig. 5a). Moreover, annotations that were assigned a higher probability by the meta-learning model were commensurately more likely to be correct (Fig. 5b and Supplementary Fig. 6).

a, Proportion of correct structure assignments for held-out metabolites by a meta-learning model, shown separately for annotations predicted to be correct versus incorrect. b, Proportion of correct structure assignments for held-out metabolites (n = 237) as a function of predicted class probabilities from the meta-learning model. c, Left, MS1 intensity of 3-(methylthio)acryloyl-glycine across 23 mouse tissues. Middle, EICs for the 3-(methylthio)acryloyl-glycine synthetic standard and the peak in mouse urine. Right, mirror plot showing the similarity between MS/MS spectra from 3-(methylthio)acryloyl-glycine synthetic standard versus the experimental spectrum from mouse urine. BAT, brown adipose tissue; diaph, diaphragm; gastrocs, gastrocnemius; gWAT, gonadal white adipose tissue. d, As in c, but for 4,5,6-triaminopyrimidine. Middle, EICs after spiking the standard into urine extract. The peak at 7.6 min in urine extract was confirmed to be 4,5,6-triaminopyrimidine after spiking with the standard at 5 ng ml−1; the peak at 8.0 min is an isomer. e, As in c, but for N-carbamyl-taurine. Middle, EICs after spiking the standard into cecal contents extract (see also Supplementary Fig. 7a). f, As in c, but for 3-hydroxypropane-1-sulfonic acid. g, As in c, but for S-sulfocysteinylglycine.
Metabolite discovery in mouse tissues
We then deployed DeepMet to assign chemical structures to all unidentified peaks in the mouse tissue dataset. To corroborate a subset of the proposed structures, we purchased or synthesized reference standards and profiled these under identical LC–MS/MS conditions. These experiments confirmed the structures of 16 previously unrecognized mammalian metabolites (Fig. 5c–g, Extended Data Fig. 6a–g, Supplementary Fig. 7a and Supplementary Table 2).
These metabolites were structurally diverse. For instance, we identified a series of amino acid conjugates, such as 3-(methylthio)acryloyl-glycine, histamine-C4:0, methionine-C4:0, or (2-(4-hydroxyphenyl)acetyl)-aspartic acid. Other metabolites were nucleotide or nucleoside derivatives, such as methylthioinosine and a triaminopyrimidine that resembled formamidopyrimidines produced by oxidative DNA damage44. A third series included sulfonate-containing metabolites such as N-carbamyl-taurine, 3-hydroxypropane-1-sulfonic acid, and homotaurine. Still other metabolites encompassed carbohydrate derivatives (2-sulfoglycerate, (2-aminoethyl)phosphate-1-hexopyranose, O-sulfo-hexopyranose, and glycerylphosphorylethanol), and nonproteinogenic dipeptides (S-sulfocysteinylglycine and N-acetyl-phenylalanylleucine/isoleucine). Previously unrecognized metabolites were significantly more tissue-specific than known metabolites (P = 1.1 × 10–5, t-test; Extended Data Fig. 6h), an observation that may explain why the former had not been identified previously.
For certain metabolites, we considered the possibility that isomers of the structures assigned by DeepMet could afford similar retention times and MS/MS spectra (Extended Data Fig. 7). DeepMet annotated two metabolites as N-isobutyryl amino acids; however, synthesis of the butyryl analogues established that these afforded comparable or slightly better matches to the retention times of the mouse tissue peaks. Conversely, in the case of 2-sulfoglycerate, the regioisomer 3-sulfoglycerate failed to match the retention time of the queried peak. However, it matched a distinct peak in mouse urine and was thereby identified as another previously unrecognized metabolite.
DeepMet also identified a series of putatively novel metabolites that were revealed after careful review of the literature to be known metabolites that were missing from the HMDB (and, in some cases, even PubChem)27,29,45,46,47,48,49,50,51,52 (Extended Data Fig. 6i–r). That DeepMet recapitulated the existence of metabolites that were not captured in existing maps of the metabolome underscores its ability to fill the gaps in these maps, and raises the possibility that DeepMet could facilitate artificial intelligence-guided curation efforts to more comprehensively catalogue the known metabolome.
A subset of the chemical structures assigned to specific peaks by DeepMet were found to be mismatches on the basis of MS/MS or retention time data acquired from mouse tissues versus synthetic standards. In some cases, the standard afforded a partial match to the MS/MS spectrum acquired in the corresponding tissue, indicating that the predicted structure was likely to resemble that of the true metabolite (Extended Data Fig. 8).
We hypothesized that some of the incorrect predictions might, in fact, represent bona fide metabolites, just not those detected in mouse tissues. Indeed, four of these previously unrecognized metabolites matched peaks in human urine by both MS/MS and LC retention time (Extended Data Fig. 9a–d and Supplementary Table 2).
Motivated by this observation, we searched all of the reference MS/MS spectra acquired in this study against metabolomics data from 35,460 samples from human tissues and cell lines deposited to the MetaboLights and Metabolomics Workbench repositories. This search tentatively identified two additional metabolites, including a glutamyl conjugate of the nucleoside acadesine that was identified in 643 samples, and brought the total number of metabolites discovered in these studies to 36 (Extended Data Fig. 9e–j and Supplementary Fig. 8).
Origins of unrecognized metabolites
We finally sought to further characterize a subset of the previously unrecognized metabolites. To identify metabolites that originate from the diet, we collected metabolomics data from the cecal contents in mice fed standard chow, which is rich in dietary metabolites, or a diet comprising purified macromolecules with few metabolites. To identify metabolites that are produced by the microbiota, we collected metabolomics data from the faeces of mice treated with broad-spectrum antibiotics and untreated controls. Finally, to establish the biosynthetic precursors of these metabolites, we infused mice with 13C-labelled precursors, including glucose, methionine, cysteine and serine.
These experiments situated several of the previously unrecognized metabolites at the nexus of diet, the microbiome and host metabolism (Extended Data Fig. 10 and Supplementary Fig. 7b). For instance, 3-methylthioacrylic acid is known as a metabolite of methionine in soil-dwelling Streptomyces bacteria53. In mice, 3-(methylthio)acryloyl-glycine demonstrated a significant decrease after antibiotic treatment and incorporated 13C-methionine, suggesting that gut microorganisms may encode parallel biosynthetic pathways to those in soil bacteria. N-carbamyl-taurine likewise demonstrated reduced abundance in mice treated with antibiotics, but also decreased in mice fed a purified diet, suggesting contributions from both the diet and the microbiome. This metabolite also incorporated a single carbon from 13C-glucose, suggesting that the carbamyl group itself is derived from glucose metabolism, probably via glucose oxidation to carbon dioxide and subsequent incorporation of bicarbonate into the carbamyl group. By contrast, 4,5,6-triaminopyrimidine was abundant in standard chow but almost completely absent from mice fed a purified diet, did not respond to perturbation of the microbiome, and did not incorporate any isotopically labelled precursors, indicating that this is an exclusively diet-derived metabolite. Finally, S-sulfocysteinylglycine incorporated 13C-cysteine and 13C-serine, and did not respond to perturbations of the diet or microbiota, allowing us to annotate this as an endogenous metabolite.
Discussion
Despite advances in analytical technologies, large parts of the metabolome remain unexplored. Here, we introduce DeepMet, a language model trained on the chemical structures that populate the known metabolome. We demonstrate that DeepMet has learned the metabolic logic embedded within the structures of known metabolites and can leverage this understanding to anticipate the existence of metabolites absent from existing metabolic maps.
DeepMet introduces several approaches to advance the study of metabolism. First, we demonstrate the possibility of computationally anticipating the chemical structures of metabolites that are likely to exist but have not yet been discovered. In turn, we show that this approach can help fill gaps in existing maps of the metabolome, including well-studied metabolites that were absent from or misannotated within the HMDB, and metabolites that were added to the HMDB in a subsequent release26. Whereas prior work has leveraged language models to generate hypothetical natural products54,55, to our knowledge, they have not been applied to expand the chemical space of the mammalian metabolome, nor to prioritize the structures of metabolites that are most likely to be discovered in the future.
Second, we show that by leveraging DeepMet to generate a large database of metabolite-like chemical structures and then filtering this database on the basis of an accurate mass measurement, we can prioritize structures that are most likely to account for a mass spectrometric peak. These prioritizations are well-calibrated and remarkably accurate, even in the absence of any other analytical data. More broadly, this approach transforms scalar accurate mass values into rich distributions over plausible biogenic structures, including those that are absent from existing databases. By design, DeepMet prioritizes structures that are similar to the known metabolites in its training set, allowing it to efficiently navigate a vast chemical space by proposing structures that are likely to have a biogenic origin; however, a drawback of this approach is that DeepMet can only explore a restricted chemical space and is likely to generate incorrect predictions for synthetic compounds.
Third, we demonstrate the possibility of integrating language model-guided prioritization of hypothetical metabolites with existing approaches that search MS/MS spectra against databases of chemical structures. In contrast to methods that condition structure generation on MS/MS spectra56,57, our approach decouples the generation and prioritization of metabolite-like structures from MS/MS search. We demonstrate that this approach enables annotation of metabolic dark matter in both published and newly collected datasets. DeepMet is agnostic to the specific approach by which chemical structures are matched to MS/MS spectra34,38,39, and other models that leverage MS/MS to search chemical structure databases could be integrated in the future58,59.
Fourth, we introduce a meta-learning approach that integrates the outputs of multiple machine learning models, including predicted MS/MS spectra and retention times, to distinguish correct from incorrect metabolite annotations. This approach provides a principled framework to integrate chemical language models with sources of information that are currently treated in isolation or combined in a heuristic or ad hoc manner.
DeepMet also has limitations. Our metabolite discovery campaign incorporated human oversight in prioritizing structures for synthesis, and we expect that DeepMet will continue to be used in collaboration with chemists, particularly when synthesis is required. A substantial proportion of small molecule-associated peaks represent adducts, in-source fragments, isotopologues or other artefacts. Here we have used NetID to remove such artefacts and limit our discovery efforts to bona fide metabolites, but incorporating co-eluting peaks into the generation and prioritization of candidate structures may further improve performance. De novo structure elucidation from metabolomic data is inherently constrained by the analytical limitations of mass spectrometry, whereby certain isomers—including stereoisomers but also an important fraction of regioisomers—are indistinguishable without dedicated analytical approaches. As a result, even structure assignments supported by chemical standards, including those reported here, may retain some degree of ambiguity. Learning from the structures of known metabolites enables DeepMet to anticipate unexpected connections between known biosynthetic pathways, but implies inherent limitations for its ability to anticipate metabolites that originate from divergent, as of yet undiscovered biosynthetic routes. Our language model was trained and evaluated on the structures of human metabolites, such that evolutionarily distant applications (for instance, to plant or bacterial metabolism) will likely require bespoke models. This limitation is compounded by the fact that the mammalian metabolome encompasses xenobiotic exposures and microbiome-derived metabolites alongside products of endogenous metabolic pathways, only some of which are represented in the HMDB. In the future, scaling chemical language models to encompass all known metabolic pathways may provide a path towards decoding the totality of metabolism in the biosphere.
Methods
Training dataset
A training dataset of known human metabolites was obtained from the HMDB, the largest and most comprehensive database of human metabolism21. Chemical structures were downloaded from the HMDB website in XML format (version 4.0; file ‘hmdb_metabolites.xml’). The HMDB assigns each metabolite to one of four classes: quantified, detected, expected, or predicted. Of the 114,222 metabolites recorded in this XML file, the vast majority fell into the ‘expected’ or ‘predicted’ classes (n = 95,202 and 9,929, respectively), indicating that they had not actually been experimentally detected in human tissues or biofluids. These classes instead include structures identified in cell or tissue cultures or in other species, structures predicted based on rule-based enzymatic derivatizations of known human metabolites, and structures predicted based on combinatorial enumeration (for instance, of lipid head groups and acyl/alkyl chains). To avoid conflating predictions made by our chemical language model with an orthogonal set of predictions based on chemical reaction rules or combinatorial enumeration, we trained our language model exclusively on metabolites annotated as ‘detected’ or ‘quantified.’ Moreover, we found that among the 8,970 detected or quantified metabolites, the vast majority (6,791) of these were lipids. Because comprehensive profiling of lipids generally relies on a distinct set of analytical approaches as compared to efforts to comprehensively profile small (polar) metabolites, and because the preponderance of lipids led language models trained on this dataset to almost exclusively generate structurally simple molecules with long acyl chains, we excluded lipids from the training set. This was achieved by removing structures assigned to the ClassyFire superclass ‘Lipids and lipid-like molecules’61. These filters yielded a training set of 2,046 small molecule metabolites that had been experimentally detected in human tissues or biofluids.
The SMILES strings for these 2,046 metabolites were parsed using the RDKit, and stereochemistry was removed. Salts and solvents were removed by splitting molecules into fragments and retaining only the heaviest fragment containing at least three heavy atoms, using code adapted from the Mol2vec package62. Charged molecules were neutralized using code adapted from the RDKit Cookbook, after which duplicate SMILES (for instance, stereoisomers or alternatively charged forms of the same molecule) were discarded. Molecules with atoms other than Br, C, Cl, F, H, I, N, O, P or S were removed, and molecules were converted to their canonical SMILES representations. The resulting canonical SMILES were then tokenized by splitting the SMILES string into its constituent characters, except for atomic symbols composed of 2 characters (Br, Cl) and environments within square brackets, (such as [nH]), and any SMILES containing tokens found in 10 or fewer structures was removed, on the basis that a language model was unlikely to learn how to use these tokens correctly from such a small number of training examples. Metabolites were subsequently split into ten training folds, each with 10% of the structures withheld, and data augmentation was then performed on each fold by enumeration of 30 non-canonical SMILES for each canonical SMILES string63. This approach takes advantage of the fact that a single chemical structure can be represented by multiple different SMILES strings, and was used here on the basis of previous studies showing that this data augmentation procedure led to more robust chemical language models, particularly when training these models on small datasets64,65.
To prospectively evaluate DeepMet predictions, we obtained the structures of a further 313 experimentally detected metabolites that were added to version 5.0 of the HMDB26. These metabolites were extracted by applying the same filters as described above (except the removal of lipids) to the metabolite XML file from version 5.0, and then removing structures also found in the version 4.0. We additionally removed several thousand exogenous and largely synthetic compounds that had been identified through a text mining approach66.
Language model architecture and training
Our approach to generating metabolite-like chemical structures was based on the use of a language model to generate textual representations of molecules in the SMILES format22, a paradigm that has been extensively explored in the setting of molecular design over the past decade. Although recent efforts have introduced generative models based on transformers67,68, state-space models69, and other architectures70, here, as in previous work5,6,71,72, we trained a recurrent neural network (RNN) on the SMILES strings of the molecules in our training set. SMILES were tokenized as described above, such that the vocabulary consisted of all unique tokens detected in the training data, as well as start-of-string and end-of-string characters that were prepended and appended to each SMILES string, respectively. The language model was then trained in an autoregressive manner to predict the next token in the sequence of tokens for any given SMILES, beginning with the start-of-string token. Language models based on the LSTM architecture were selected on the basis of their excellent performance in previous studies, whereby these were found to outperform both alternative models based on RNNs (e.g., gated recurrent units) as well as models based on the transformer architecture65,68,73. LSTMs were implemented in PyTorch, adapting code from the REINVENT package74. The architecture consisted of a three-layer LSTM with a hidden layer of 1,024 dimensions, an embedding layer of 128 dimensions, and a linear decoder layer. Models were trained to minimize the cross-entropy loss of next-token prediction using the Adam optimizer with default parameters, a batch size of 64, and a learning rate of 0.001. Ten percent of the molecules in the training set were reserved as a validation set and used for early stopping with a patience of 50,000 minibatches.
To further address the data-limited context of the human metabolome, we employed a strategy that we reasoned would first allow our model to learn the syntax of the SMILES representation and subsequently adapt this understanding to the generation of new metabolite-like chemical structures. In particular, we first pretrained the LSTM until convergence on drug-like small molecules from the ChEMBL database, using the same early stopping criteria as above75. ChEMBL (version 28) was obtained from ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/latest/chembl_28_chemreps.txt.gz and processed as described above, except with a single round of non-canonical SMILES enumeration rather than 30. After pretraining using the same stopping criterion as described above, the model was fine-tuned on the HMDB training set, without freezing any layers. This model generated valid SMILES at a rate of 98.9 ± 0.19%, for models trained on each of the ten splits, and novel SMILES at rates of 34.3 ± 7.7% (with respect to the HMDB training set), 49.7 ± 3.0% (with respect to the ChEMBL pretraining set), and 28.0 ± 6.4% (with respect to both sets).
Metabolite likeness of generated molecules
We carried out a series of analyses to first establish that the language model had indeed learned to generate metabolite-like structures. To this end, we first trained a chemical language model as described above on a single training split of the HMDB, then sampled 500,000 SMILES strings from the trained model, and removed those corresponding to known metabolites from the training set. Duplicate structures were likewise removed. No additional filters were imposed on the generated molecules to explicitly remove those falling outside the chemical space of the training set. To visualize the areas of chemical space occupied by generated molecules and known metabolites, we implemented an approach based on nonlinear dimensionality reduction. Briefly, we computed a continuous, 512-dimensional representation of each molecule using the Continuous and Data-Driven Descriptors (CDDD) package76 (available from http://github.com/jrwnter/cddd). These continuous, 512-dimensional descriptors are derived from a machine translation task in which RNNs are used to translate between enumerated and canonical SMILES in a sequence-to-sequence modelling framework, a task that forces the latent space to encode the information required to reconstruct the complete chemical structure of the input molecule. We then sampled CDDD descriptors for an equal number of known metabolites and generated molecules, then embedded the CDDD descriptors for both sets of molecules into two dimensions with UMAP77, using the implementation provided in the R package uwot with the n_neighbors parameter set to 5.
To more quantitatively evaluate the chemical similarity of generated molecules to known metabolites, we used a supervised machine learning approach to test whether the two sets of molecules could be distinguished from one another on the basis of their structures. This was achieved by again sampling an equal number of known metabolites and generated molecules, computing extended-connectivity fingerprints with a diameter of 3 and a length of 1,024 bits, and then splitting the resulting fingerprints into training and test sets in an 80/20 ratio. Known metabolites and duplicate structures were removed from the generated molecules. A random forest classifier was then trained to distinguish between known metabolites and generated molecules, using the implementation in scikit-learn. The performance of the classifier was measured using the area under the receiver operating characteristic curve (AUROC). To ensure that the observed failure of the classifier to separate known metabolites from generated molecules could not be trivially attributed to a poor classifier, an identical model was trained to separate the known metabolites in the training set from an equal number of structures derived from ChEMBL (version 28) containing only the atoms C, H, N, O, P and S, and was found to accurately differentiate these two groups of structures.
Third, we evaluated whether the molecules generated by the language model overlapped with an orthogonal set of enzymatic biotransformations of known metabolites that had been predicted in a rule-based manner by BioTransformer23. BioTransformer comprises a knowledgebase of enzymatic reaction rules that are used to predict generic biotransformation products of endogenous metabolites or xenobiotics based on phase I and II metabolism, promiscuous enzymatic metabolism, and gut microbial metabolism, as well as a machine learning framework to specifically predict human CYP450-catalysed phase I metabolism of xenobiotics78. BioTransformer was applied recursively to the training set in order to generate biotransformation products after one to four steps of enzymatic reactions, and the total fraction of these predictions that were recapitulated by DeepMet was quantified. The inverse (that is, the total fraction of structures generated by DeepMet that were also generated by BioTransformer) was also quantified, both before and after excluding structures also present in ChEMBL from the output of BioTransformer. For this analysis, we used the sample of 1 billion SMILES from all ten models described in detail below, rather than 500,000 from a single split, again removing duplicate structures.
Fourth, we computed the Tanimoto coefficient (Tc) between each generated molecule and its nearest neighbour in the training set, again using 1,024-bit Morgan fingerprints of radius 3 to calculate the Tc and removing duplicate structures from the language model output. As a negative control, for each generated molecule, we drew at random a molecule with the same molecular formula from PubChem. The nearest-neighbour Tc was then computed for molecules sampled from PubChem in order to provide a baseline against which the enrichment for metabolite-like chemical structures within the language model output could be compared.
Sampling frequencies of generated molecules
We initially sought to leverage the trained language model for metabolite discovery by identifying the generated molecules that it viewed as the most plausible extensions of the training set, in analogy to the use of protein language models to forecast the emergence of new protein sequences. Because the use of non-canonical SMILES enumeration implied that multiple SMILES strings could be generated for any given structure, and because we found that different SMILES representations of the same metabolite were often sampled with very different losses, we drew a very large sample of SMILES strings from the trained model and tabulated the frequency with which each unique chemical structure appeared in this output. This was achieved by drawing samples of 100 million SMILES strings from language models trained on each of the ten training folds, for a total of 1 billion SMILES. The sampled molecules were then parsed with the RDKit, invalid outputs were discarded, and the frequency with which each canonical SMILES appeared in the model output was tabulated. Sampling frequencies were then averaged across the outputs of all ten models, removing molecules in the training set from the language model output for each fold before calculating the average such that all of the analyses described below excluded molecules reproduced from the training set.
To evaluate the relationships between the sampling frequency and metabolite-likeness, generated molecules were then divided into six bins on the basis of sampling frequency, and a series of metrics were calculated that quantified the similarity between the non-redundant set of generated molecules in this bin and the molecules in the training set. First, we calculated the nearest-neighbour Tc between each generated molecule and the training set, as described above, and tested for a significant trend with increasing sampling frequency using the Jonckheere-Terpstra test. Second, we again quantified the overlap between generated molecules and rule-based enzymatic transformations predicted by BioTransformer within each sampling frequency bin. Third, we measured the chemical similarity between the generated molecules and the training set as quantified by the Fréchet ChemNet distance25. This metric is calculated from the hidden representations of molecules learned by a neural network trained to predict biological activities in thousands of biological assays recorded in ChEMBL, ZINC, and PubChem, and therefore captures both structural properties as well as inferred biological activity; it was previously found to be among the most reliable metrics for evaluating generative models of small molecules65 and is included in multiple benchmark suites79,80. Fourth, we determined the Murcko scaffolds of generated molecules and the training set81, and then calculated the Jensen–Shannon distance between the scaffold distributions of the training set and generated molecules in each frequency bin65.
Anticipation of previously unrecognized metabolites
To test whether the frequency with which molecules were generated could be used to prioritize previously unrecognized metabolites, we again withheld 10% of the training set at a time to simulate the appearance of unknown metabolites. We then quantified the extent to which sampling frequency alone would separate the held-out metabolites from the background of all generated molecules, using ROC curve analysis and excluding metabolites reproduced from the training set of each model as described above. The same analysis was repeated in a prospective setting for the metabolites newly added in version 5.0 of the HMDB, excluding all version 4.0 metabolites. In addition to ROC analysis, we calculated the fold enrichment of HMDB 5.0 metabolites within the top-10 to 100,000 most frequently sampled molecules over random expectation, and evaluated statistical significance using a χ2 test.
Structure-centric discovery of predicted metabolites
To experimentally confirm the existence of metabolites prioritized by DeepMet, we leveraged a large-scale resource of deidentified human metabolomics data collected by the Provincial Toxicology Centre at the British Columbia Centre for Disease Control as part of its routine operations. Clinical urine and forensic blood samples were subjected to full-scan mass spectrometry as part of routine drug screening. Samples were received in sterile urine containers or vacutainers. Samples were identified by anonymized identifiers for all analyses described here and no identifying information or clinical data was retrieved. The study was approved by the UBC Clinical Research Ethics Board (H22-02722 and H25-00702).
Urine and blood samples were analysed by liquid chromatography–high-resolution mass spectrometry. Urine samples were hydrolysed using IMCSzyme genetically modified β-glucuronidase at 60 °C for 1 h and filtered using a Biotage Isolute PPT+ protein precipitation plate. After cooling, acetonitrile was added to wash the filter. The acetonitrile was evaporated and the extract reconstituted using methanol:type I water (1:1, v:v). One microlitre was injected on a Thermo Scientific Vanquish LC coupled to a Q Exactive Hybrid Quadrupole Orbitrap mass spectrometer (Waltham, MA, USA). Chromatographic separation was achieved using a Thermo Scientific Accucore Phenyl-Hexyl Column (2.1 × 100 mm, 2.6 Å) using a gradient elution. Mobile phase A was 2 mM ammonium formate with 0.1% formic acid in type I water. Mobile phase B was 2 mM ammonium formate with 0.1% formic acid in a 1:1 (v:v) mixture of acetonitrile and methanol. The flow rate was 0.5 ml min−1. The total run time was 12.5 min. The column and the autosampler temperatures were set at 40 °C and 10 °C, respectively. Full scan with targeted data-dependent MS2 (full MS/dd-MS2) was performed in the positive electrospray ionization mode with an inclusion list containing over 200 drugs. The top eight most intense precursors were selected for fragmentation, unless one or more masses from the inclusion list was detected, in which case those masses were prioritized for fragmentation. The sheath gas flow rate and the auxiliary gas flow rate were set at 60 and 20 a.u., respectively. The spray voltage was set at 3,000 V. The capillary and the auxiliary gas heater temperatures were set at 380 °C and 375 °C, respectively. The S-lens RF was set to 60 V.
To prioritize generated metabolites for discovery, we cross-referenced the 6,301 structures that did not appear in any version of the HMDB within the top-10,000 most frequently sampled molecules with catalogues of commercially available compounds. A total of 106 standards were acquired from Mcule, and standards for two additional predicted metabolites that were not available from commercial suppliers were selected for custom synthesis on the basis of manual review (N-lactoyl-glutamine and N-lactoyl-serine, as described in ‘Chemical synthesis’).
The compounds were diluted with methanol to a final stock concentration of 1 mg ml−1. These stock solutions were further diluted to concentrations of 100 ng ml−1 or 1 µg ml−1 with methanol:water 1:1, v:v. Each of the standards was then analysed individually using the same chromatographic and mass spectrometric methods that were used to profile clinical samples. The resulting data files were then manually inspected to determine retention times and extract reference MS/MS spectra; 26 standards did not afford high-quality MS/MS spectra (at least two fragments with intensities greater than 1% of the base peak) and were discarded at this stage. The resulting library of 80 reference spectra and their retention times was then queried against the mass spectrometric data from all urine and blood samples. Initial identification of the predicted metabolite standards was performed on the the basis of a dot-product of 0.75 or greater between reference and experimental MS/MS spectra and a retention time difference of less than 15 s, which was followed by manual inspection to corroborate these matches.
Prioritization of metabolite structures from accurate masses
The finding that the sampling frequency of any given generated structure was correlated with its metabolite-likeness led us to further hypothesize that we could leverage the sampling frequency to suggest chemical structures for unannotated signals in an untargeted metabolomics experiment. Specifically, we posited that given some experimental measurement as input, such as an accurate mass, we could filter the language model output to a subset of molecules matching this measurement, and rank this subset of generated molecules in descending order by sampling frequency in order to produce a ranked list of candidates. We tested this possibility by filtering the language model output based on the exact mass of each held-out metabolite, allowing for a mass error of up to 10 ppm, and ranking the resulting structures by the frequency with which they were generated.
To evaluate the accuracy of this approach, we computed the fraction of held-out HMDB version 4.0 metabolites for which the correct structure was found within the top-k candidates, systematically varying the value of k between 1 and 30. In addition, we calculated the Tc between the top-ranked candidate and the held-out molecule; whereas Morgan fingerprints were used for all other analyses in the study, here RDKit fingerprints were used because these had previously been calibrated based on a user study of expert chemists to define quantitative thresholds that approximated these chemists’ subjective judgements of ‘meaningful similarity’ or a ‘close match’ between true and predicted structures37. The use of chemical similarity thresholds allowed us to also identify cases in which the language model nominated a structure closely related to the ground truth (for instance, where the correct scaffold of the held-out metabolite was predicted, but a single functional group was misplaced). As a secondary measure of chemical similarity, we computed the Euclidean distance between CDDD descriptors76. We additionally hypothesized that held-out metabolites that the model failed to ever generate would tend to occupy more distinct regions of chemical space with few similar structures in the training set; we tested this hypothesis by calculating the nearest-neighbour Tc between each metabolite in version 4.0 of the HMDB and the remainder of the training set. Each of the above analyses was then repeated for the metabolites added to version 5.0 of the HMDB.
We sought to place the performance of DeepMet in context by comparing our model to simple baseline approaches. First, to assess the model’s ability to generalize beyond the chemical space of the training set, we searched by accurate mass in the training set itself, with the recognition that this would yield a top-k accuracy of 0% by definition, but with the goal of comparing the Tanimoto coefficients between the true molecule and structures prioritized by the language model to plausible matches from the training set. A substantial fraction of held-out metabolites had no molecules with matching masses in the training set; these were omitted from the evaluation. Second, we applied the AddCarbon approach that has been advocated as a simple and universal baseline for more complex generative models31. This model inserts a carbon atom (‘C’) at random positions within the SMILES representation of a molecule from the training set. If the insertion of the carbon atom produces a valid SMILES string and the corresponding molecule is not itself in the training set, then the modified SMILES string is retained. Surprisingly, this trivial baseline was found to outperform numerous more complex approaches to molecule generation on the distribution learning tasks proposed in one widely used benchmark suite79. We adapted the Python source code available from https://github.com/ml-jku/mgenerators-failure-modes to exhaustively enumerate all possible ‘AddCarbon’ derivatives of the training set metabolites. Invalid SMILES were removed, the remaining SMILES were converted to their canonical forms, and derivatives that were also found in the training set were removed. For both baselines, the same 10 ppm error window was used as for the language model, and when more than one candidate structure matched, the candidates were ordered at random.
We additionally tested whether this prioritization was robust to the presence of multiple positively or negatively charged adducts. This was achieved by computing the protonated or deprotonated mass of the held-out metabolite in the positive or negative modes, respectively, and then searching in the language model output as described above but here considering three adduct types in each mode, including [M + H]+, [M + NH4]+, and [M+Na]+ in the positive mode and [M-H]–, [M + Cl]–, and [M + FA-H]– in the negative mode.
We further assessed the calibration of confidence scores emitted by DeepMet for any given accurate mass input. The confidence score for a candidate molecule m is calculated as its relative frequency within the set of candidate molecules for a given query (for example, a single monoisotopic mass, or a m/z value and a list of adducts). This score is formalized as follows:
where:
-
Frequency(m) is the number of times that a SMILES string representing molecule m was sampled by DeepMet.
-
The denominator is the sum of the frequencies of all candidate molecules i for a given query.
These scores were then divided into ten bins of equal widths, and the proportion of correct matches within each bin was determined. Because these confidence scores reflect the relative frequencies with which structures were generated by DeepMet, they are independent of analytical data that could be used to differentiate structural isomers (for instance, MS/MS or retention time) and do not capture structures that were never generated by the language model. Consequently, although they can contribute to structure annotation, they should not be interpreted as a probabilistic measure that any given annotation is correct.
Integration of DeepMet and MS/MS
We next sought to integrate DeepMet with MS/MS data as a means to experimentally differentiate between isobaric metabolites, which cannot be distinguished by accurate mass measurements alone. Our efforts to this end began by applying CFM-ID34,82 to create an in silico MS/MS library for metabolites generated by DeepMet. CFM-ID is a machine learning method that is trained on a reference library of MS/MS spectra for known small molecules, and learns to predict MS/MS spectra for unseen chemical structures on the basis of the information within this dataset. During the training phase, for each input molecule, CFM-ID first employs a combinatorial bond cleavage approach to enumerate all theoretically possible fragments. The output of this procedure is a molecular fragmentation graph, in which each node represents a theoretically possible fragment from the parent molecule with one bond cleavage, and each edge (also known as transition) between nodes encodes the chance that one fragment directly produces another fragment through a fragmentation event. The probability of each transition is estimated by parameters that CFM-ID learns from its training dataset of known molecules and their associated MS/MS spectra. These parameters are learned by minimizing a negative log-likelihood loss within a training dataset of known molecule-MS/MS spectrum pairs using expectation maximization (EM). Finally, CFM-ID uses the fragmentation graph and associated transition probability estimates for each molecular fragment to reconstruct the corresponding MS/MS spectrum for the input molecule. CFM-ID predicts MS/MS spectra at three different collision energies (at 10 eV, 20 eV and 40 eV) and in both positive and negative ionization modes, functionality which differentiates it from many alternative machine-learning methods for MS/MS spectrum prediction from chemical structures.
To balance performance with the computational requirements necessary to predict MS/MS spectra for tens of millions of generated structures, we limited these predictions to a subset of 2.4 million molecules that were generated at least five times by DeepMet. This threshold was selected by removing molecules sampled less than 2, 3, 4, 5 or 10 times and repeating the analyses of metabolite prioritization based on exact mass information described above, which indicated that the ability of DeepMet to prioritize metabolites from exact masses was minimally affected by removing molecules sampled less than 5 times.
To evaluate the performance of the combination of DeepMet and CFM-ID in the context of metabolite discovery, we again simulated de novo structure elucidation by withholding the structures and MS/MS spectra of known metabolites from both of these models to simulate the emergence of a metabolite not found within the training set. CFM-ID was trained on ESI-QTOF MS/MS spectra from the Agilent MassHunter METLIN Metabolite reference spectral library. These models were used to predict MS/MS spectra for metabolites in the held-out test set for each fold. An 11th model was trained on the entire Agilent MS/MS library and used to predict MS/MS spectra for metabolites without reference spectra in the training set. Spectra predicted at multiple collision energies were merged to produce a single predicted MS/MS spectrum per generated structure. For each reference MS/MS spectrum in the test set, candidates were retrieved from the database of generated metabolites produced by DeepMet (again with molecules from the training set removed as described above), and a final score was assigned to each candidate structure by multiplying the confidence scores assigned by the language model on the basis of the precursor m/z by the dot-product between the predicted and experimental MS/MS spectra. This combined score (referred to in the figures as DeepMet + CFM-ID, or DeepMet + MS/MS for alternative MS/MS prediction models) for a candidate molecule m is formalized as follows:
where:
-
CDeepMet(m) is the DeepMet confidence score for molecule m (as defined above).
-
CosineSimilarity(m) is the cosine similarity between the experimental MS/MS spectrum and the predicted spectrum for candidate m.
Performance was then evaluated using the same metrics as described above—that is, the top-k accuracy for values of k between 1 and 30; the Tc between the top-ranked candidate and the held-out molecule, using RDKit fingerprints; and the top-k accuracy when considering predicted metabolites with a Tc ≥ 0.675 (close match) or ≥ 0.40 (meaningfully similar) as matches37. In addition, we applied CFM-ID to structures proposed by the same two baseline methods as above, AddCarbon and searching within the training set, and compared the performance of these approaches to the combination of CFM-ID with DeepMet. To further place the performance of the combined approach in context, we computed the top-k accuracy and Tc between top-ranked candidate and the held-out molecule when ranking structures solely on the basis of the dot-product between predicted and experimental MS/MS (CFM-ID alone), or solely on the basis of the DeepMet confidence score, discarding the MS/MS spectra altogether.
To demonstrate the robustness of our approach to metabolite identification by combining DeepMet with MS/MS prediction, we carried out several additional experiments. First, we retrained CFM-ID on a second library of MS/MS reference spectra, obtained from the HMDB, and again evaluated performance as described above. Second, we benchmarked alternative models for MS/MS prediction from chemical structures. CFM-ID is one of numerous methods that have been introduced to predict MS/MS from chemical structures. We initially selected CFM-ID because of its permissive open-source license, its widespread use in metabolomics, and the distribution of code required to re-train models in structure-disjoint cross-validation. We also leveraged two alternative models, FraGNNet38 and NEIMS39, to predict MS/MS spectra for generated metabolites, here employing five rather than ten folds. The goal of this evaluation was to demonstrate that DeepMet is not intrinsically tied to CFM-ID but rather could be integrated with a range of different computational methods to interpret MS/MS spectra; future work could also evaluate methods that predict chemical fingerprints from MS/MS spectra, rather than predicting high-resolution MS/MS spectra from chemical structures. Each of these models employs a different approach to MS/MS prediction. CFM-ID models fragmentation as a Markov decision process, and is trained to predict the probability of each fragmentation (that is, bond cleavage) event. FraGNNet applies a graph neural network (GNN) to a combinatorial fragmentation graph in order to model mass spectra as distributions over molecule fragments. NEIMS performs MS/MS prediction via vector regression, taking molecular fingerprints as input and passing these through a multilayer perceptron (MLP) to predict binned MS/MS spectra. NEIMS was modified to predict high-resolution MS/MS spectra at a resolution of 0.01 m/z, rather than 1 m/z as originally described by the authors, a modification which required replacing the fully connected MLP output layer with a low-rank layer to fit the high-resolution model into memory. Both FraGNNet and NEIMS were additionally modified to condition MS/MS prediction on adduct type and collision energy as input, in order to match their output MS/MS spectra with those predicted by CFM-ID. Notably, each of the three models has limitations that precluded the prediction of MS/MS spectra for some generated metabolites. CFM-ID and FraGNNet cannot predict MS/MS spectra for structures with a formal charge. FraGNNet additionally cannot predict MS/MS spectra for structures with more than 60 heavy atoms. NEIMS cannot predict MS/MS spectra for structures with a precursor m/z greater than 1,500 Da. An empty spectrum was assigned to structures that violated these constraints, which comprised 5%, 8%, and 1% of the generated metabolites for CFM-ID, FraGNNet, and NEIMS, respectively.
Previous generations of tools that sought to apply rule-based biochemical transformations to a ‘seed’ population of known metabolites23,83,84 implicitly assign a binary ‘metabolite-likeness’ score to candidate structures, insofar as structures accessed by rule-based transformations can be used to annotate a given MS/MS spectrum, whereas structures that cannot be accessed by these transformations will never be considered as candidates. The sampling frequency provides a quantitative metric that correlates with ‘metabolite-likeness’ (Fig. 2b–g) rather than implicitly performing a binary classification of metabolite-like versus non-metabolite-like structures, but with the same underlying premise (that is, that unrecognized metabolites are likely to structurally resemble known metabolites). To demonstrate that our approach benefits from, but is not contingent on, the use of the sampling frequency as a quantitative metric, we drew progressively smaller samples of SMILES strings from the language models trained on each split (Supplementary Fig. 2b). The analyses of the Agilent MS/MS library described above were then repeated with the resulting generated structures and their sampling frequencies. Generated structures were additionally ranked by the dot-product between experimental and predicted MS/MS spectra alone, and the performances of the weighted and unweighted dot-products were found to converge when limiting the degree of SMILES sampling to generate smaller databases of metabolite-like chemical structures (Supplementary Fig. 2c).
Meta-analysis of the human blood metabolome
To showcase the potential for DeepMet to enable metabolite discovery in published metabolomics data at the scale of thousands of experiments, we carried out a meta-analysis of the human blood metabolome, as shown in Fig. 4. Data was obtained from the MetaboLights database85, as this resource requires extensive metadata annotation for each deposited sample, including the species and tissue of origin. An XML record of all studies deposited to MetaboLights was obtained (file ‘eb-eye_metabolights_studies.xml’) and filtered to only mass spectrometry-based metabolomics studies that included at least one sample from human serum, plasma, or whole blood. Complete data depositions for this subset of studies were then downloaded from MetaboLights. The assay-level metadata (‘a_*’ files) were parsed to obtain a complete list of all mass spectrometric runs for all of the human blood metabolome studies and to exclude GC-MS, imaging MS, and targeted MS experiments, inspecting the relevant MTBLS pages and the corresponding publications as necessary to ensure that no LC–MS metabolomics studies were inadvertently removed and manually correct any filenames that were discordant between the assay-level metadata and the deposited raw files. Compressed archives (.tar, .gz, .zip) were decompressed, and vendor-specific file formats (.d, .raw, and .wiff) were converted to mzML format using the msconvert utility bundled with ProteoWizard86. MS/MS spectra from each run were then extracted and written to MGF files after ensuring the following quality control (QC) criteria were met: at least 50 unique precursor m/z values; at least 100 non-empty MS/MS spectra; both precursor m/z and fragment m/z recorded to at least four decimal places. LC–MS/MS files that did not meet one or more of these filters were manually inspected to ascertain why they did not pass these QC criteria and, ultimately, were all discarded. A handful of duplicate files, representing cases where the same mass spectrometry run was uploaded as part of more than one accession, were identified by their checksums and removed. Finally, sample-level metadata files (‘s_*’), study protocols, and/or the original publications were manually reviewed for each of the files that passed all of the above steps in order to confirm that the run in question was indeed a human blood sample. In total, these steps afforded a resource comprising 29.1 million MS/MS spectra from 4,510 mass spectrometry runs. The complete list of accession numbers and raw data files included in this analysis is provided in Supplementary Table 4a.
All 29.1 million MS/MS spectra were then searched against the resources of spectra predicted by CFM-ID for both known metabolites and molecules generated by DeepMet at least 5 times, merging predicted spectra across collision energies for each known or generated structure. We first calculated the total number of human blood MS/MS spectra with at least 1 match to a predicted MS/MS spectrum above any given cosine similarity threshold between 0 and 1, when considering known metabolites alone or when combining known metabolites with molecules generated by DeepMet.
We separately sought to quantify the number of MS/MS matches that would be expected by random chance when searching a spectral database of equivalent size. To this end, we constructed a decoy database of MS/MS spectra by shuffling fragment ions between predicted MS/MS spectra for isobaric structures. For each query, we first select a set of predicted spectra from the predicted MS/MS library for DeepMet spectra library for which the mass-to-charge ratios of the precursor are within 10 ppm of the query spectrum. All peaks from these selected spectra were aggregated into a pool of candidate peaks, retaining duplicate m/z entries to preserve the peak distribution. To generate a shuffled candidate spectrum, the number of peaks k was uniformly sampled from the integers in the range [1, 20]. Next, k unique peaks were randomly sampled from the pool of candidate peaks. Finally, if duplicate m/z values were present within the sampled peaks, only the peak with the highest intensity was kept to ensure unique m/z entries in the final shuffled spectrum. The 29.1 million MS/MS spectra from human blood were then searched against the resulting library of shuffled MS/MS spectra as described above.
To corroborate the quality of the shuffled ‘decoy’ spectra generated by the approach described above, we tested the assumption that incorrect matches are equally likely to involve decoy and experimental spectra87. Because testing this assumption requires reference spectra for which the true structure is known, we generated shuffled ‘decoy’ spectra for all of the MS/MS in the Agilent PCDL library, and then compared the distribution of dot-product similarities for incorrect matches to other reference MS/MS spectra and to shuffled decoy MS/MS spectra.
We then tested whether metabolites generated more frequently demonstrated a greater propensity to match to experimentally collected MS/MS spectra. To address this question, we iterated over each experimentally collected MS/MS spectrum and annotated this spectrum on the basis of the combination of cosine similarity and DeepMet sampling frequency, as described above, excluding spectra without at least one match to a predicted spectrum with a dot-product greater than zero. We then binned these annotated metabolites by their sampling frequencies into deciles, here again considering only structures generated at least five times, and computed the proportion of annotations within each decile where the predicted and experimental MS/MS spectra matched with a cosine similarity score exceeding a given threshold between 0 and 1.
We last sought to experimentally corroborate a subset of the annotations made by the combination of DeepMet and CFM-ID by acquiring MS/MS spectra from synthetic standards. We initially focused on a peak detected in MTBLS70041 (sample ns94, RT 3.25 min, precursor m/z 201.9492 in positive mode) that was annotated as 6-bromonicotinic acid both when ranking candidate structures by the sampling frequency alone or when integrating this score with CFM-ID, and for which the experimental MS1 data supported the presence of bromine. Reference spectra for 2-, 4-, 5- and 6-bromonicotinic acid were acquired as described in ‘Metabolite standards’. In addition, reference spectra were acquired for all possible brominated isomers of picolinic and isonicotinic acid, as well as all regioisomers of bromonitrobenzene. Catalogue numbers were as follows: 2-bromonicotinic acid, A111216 (AmBeed); 4-bromonicotinic acid, A291101 (AmBeed); 5-bromonicotinic acid, 211390100 (Thermo Fisher Scientific); 6-bromonicotinic acid, A169647 (AmBeed); 3-bromopicolinic acid, A115820 (AmBeed); 4-bromopicolinic acid, A113480 (AmBeed); 5-bromopicolinic acid, A635338 (AmBeed); 6-bromopicolinic acid, CS-W009049 (ChemScene); 2-bromoisonicotinic acid, A139877 (AmBeed); 3-bromoisonicotinic acid, A258659 (AmBeed); 1-bromo-2-nitrobenzene, 002709 (Oakwood); 1-bromo-3-nitrobenzene, 143070 (Beantown); and 1-bromo-4-nitrobenzene, 078591 (Oakwood). To visualize the match between the experimental spectrum from MTBLS700 and the synthetic standard, the former was preprocessed to remove MS2 fragments that were uncorrelated with the MS1 precursor, as described in more detail below, with a minimum Pearson correlation coefficient of 0.95. We also sought to corroborate an annotation of a peak in MTBLS787842 (sample neg_C13, RT 1.76 min, precursor m/z 169.0596 in negative mode). Synthetic 2-hydroxy-3-(1-methyl-1H-imidazol-5-yl)propanoic acid was obtained from Enamine (Z8914008850) and a reference MS/MS spectrum was acquired as described ‘Metabolite standards’. We also considered 2-hydroxy-3-(1-methylimidazol-4-yl)propanoic acid as a possible regioisomer (Enamine, EN300-314547). To evaluate the relationship between the quantitative abundance of this metabolite and disease status in this study, the dataset was re-processed with xcms88 and the intensity of this peak was compared between patients with sepsis and healthy controls using ROC curve analysis as implemented in the R package AUC.
Mouse sample collection
Animal studies adhered to protocols approved by the Princeton University Institutional Animal Care and Use Committee (IACUC). Male C57BL/6 mice (Charles River), aged 10–12 weeks, were maintained on standard mouse chow. On the sample collection day, urine was collected after a 6 h fast. Blood samples were taken via tail snip, kept on ice for up to 60 min, and then centrifuged at 10,000 rcf for 10 min at 4 °C. The plasma was transferred to another tube and stored at –80 °C. The mice were then euthanized by cervical dislocation, and tissues were dissected, wrapped in foil, clamped with a Wollenberger clamp precooled in liquid nitrogen, and subsequently immersed in liquid nitrogen. All samples were stored in a –80 °C freezer. A total of 23 tissue and fluid samples were collected, including the brain, liver, kidney, spleen, pancreas, gWAT, stomach, small intestine, lung, heart, quadriceps, BAT, soleus, diaphragm, gastrocnemius, colon, skin (ear), testicles, bladder, cecal content, eye, serum and urine.
Metabolite extraction
Frozen solid tissue samples were first weighed to aliquot approximately 40 mg of each tissue and then transferred to 2.0 ml Eppendorf tubes on dry ice. Samples were then ground into powder with a cryomill machine (Retsch) maintained at cold temperature using liquid nitrogen. Thereafter, for every 30 mg tissue, 1 ml 40:40:20 acetonitrile:methanol:water with 0.5% formic acid was added to the tube, vortexed, and allowed to sit on ice for 10 min and 85 µl 15% NH4HCO3 (w:v) was added and vortexed to neutralize the samples89. The samples were incubated on ice for another 10 min and then centrifuged at 14,000 rpm for 25 min at 4 °C. The supernatants were transferred to another Eppendorf tube and centrifuged at 14,000 rpm again for 25 min at 4 °C with supernatant collected for analysis. For metabolite extraction from serum and urine, frozen samples were allowed to thaw on ice. For 10 µl urine or serum, 200 µl methanol was added and vortexed for 10 s, and centrifuged for 25 min. The supernatant was collected, then dried down under N2 stream, and re-dissolved into 200 µl 40:40:20 acetonitrile:methanol:water for analysis.
Liquid chromatography–mass spectrometry
LC–MS analysis was performed on a Vanquish UHPLC system coupled with an Orbitrap Exploris 480 mass spectrometer. LC separation was achieved using a Waters XBridge BEH Amide column (2.1 × 150 mm, 2.5 µm particle size, 186006724), with column oven temperature at 25 °C and injection volume of 5 µl. The method has a running time of 25 min at a flow rate of 150 µl min−1. Solvent A is 95:5 water:acetonitrile with 20 mM ammonium hydroxide and 20 mM ammonium acetate, pH 9.4. Solvent B is acetonitrile. The gradient is, 0 min, 90% B; 2 min, 90% B; 3 min, 75%; 7 min, 75% B; 8 min, 70%, 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 14 min, 25% B; 16 min, 0% B, 20.5 min, 0% B; 21 min, 90% B; 25 min, 90% B (ref. 90). The Exploris 480 mass spectrometer was operated in full-scan mode at MS1 level for the 23 metabolite extracts. This allows the relative quantitation of the individual metabolite across all tissues and fluids by ion counts. In addition, the 23 extracts were mixed to generate a ‘mixture’ sample and analysed using the same LC–MS method. Peak picking was performed from the mixture sample for further analysis. Full scan parameters are: resolution, 120,000; scan range, m/z 70–1,000 (negative mode); AGC target, 107; ITmax, 200 ms. Other instrument parameters are: spray voltage 3,000 V, sheath gas 35 (Arb), aux gas 10 (Arb), sweep gas 0.5 (Arb), ion transfer tube temperature 300 °C, vaporizer temperature 35 °C, internal mass calibration on, RF lens 60.
Peak picking and annotation
Thermo Raw data files were converted to mzXML format using the msconvert utility bundled with ProteoWizard86. Peak picking for the mixture sample was then performed using El-MAVEN version 12.091 with the following parameters, mass domain resolution 10 ppm, time domain resolution 20 scans, minimum peak intensity 10,000, minimum quality 0.5, minimum signal/blank ratio 3.0, minimum signal/baseline ratio 2.0, minimum peak width 10 scans. The analysis resulted in 17,386 peaks (features) in negative mode defined by their m/z and RT. EIC curves for each peak were then retrieved, plotted and saved as grey-scale images of the same format (.png, 700 by 525 pixels). A computer vision algorithm implemented in Matlab was then used to classify these peaks as reliable versus spurious. The classifier comprised a convolutional neural network (CNN), with similar architecture as previously described92, and was trained on the dataset provided by the authors of EVA. The CNN flagged 960 poor-quality peaks, which were removed from the dataset, along with 3,012 duplicate peaks, yielding a table of 14,374 peaks for further annotation.
This peak table, along with the intensities of each peak in each mixture sample, was provided to NetID for metabolite annotation43, along with a database of 114,014 known metabolites from the HMDB; a table containing the m/z and retention times of 500 metabolite standards; and a transformation rule table describing the formula and mass difference of 84 transformations, as previously described43. The penalty for 1 ppm m/z difference between annotated formula and measured m/z was set at –0.5. The propagation and recording thresholds were set at 10 and 5 ppm, respectively. All other parameters were set at their default values. NetID annotated 7,015 peaks as artefactual (including isotopes, adducts, fragments, and ringing artifacts), 2,369 peaks as known metabolites, 2,305 peaks as putative derivatives of known metabolites, and 2,685 peaks as putative unknowns.
Annotated peaks were then further filtered on the basis of their intensities. The intensities of each peak across all tissues were retrieved, and the most abundant tissue along with its intensity (Imax) was recorded for each peak. Among known metabolites and their putative derivatives, 2,285 and 1,972 peaks with Imax > 105 were selected, respectively. Among putative unknown peaks, 557 peaks with log10(Imax) > 6.5 were selected. These filters yielded a total of 4,814 peaks for which MS/MS spectra were acquired in a targeted manner, as described in the section below. Each peak was subsequently annotated with DeepMet, and the top-ranked structures (as determined by the combined DeepMet + MS/MS score) for a subset of peaks were selected to be synthesized or purchased. The DeepMet confidence scores, cosine similarities between predicted and experimental spectra, and combined DeepMet + MS/MS scores are provided for the previously unrecognized metabolites identified in mouse tissues in Supplementary Table 2. The same scores are provided for all of the candidate structures generated by DeepMet for the 25 peaks identified in mouse tissues (Fig. 5 and Extended Data Figs. 6 and 7) in Supplementary Table 5. For four of these metabolites (homotaurine, N-acetyl-phenylalanylleucine/isoleucine, 3-hydroxypropane-1-sulfonic acid, and N1-methyl-imidazolelactic acid), the chemical standard did not match to the original tissue peak, but instead matched a peak with a distinct MS/MS spectrum and/or a retention time difference that could not be explained by chromatographic drift (see also ‘Success rate of metabolite discovery’).
Targeted MS/MS analysis
For each of the 4,814 peaks of interest, signal intensity was retrieved from the full-scan data of the 23 tissue extracts to identify the tissue with the highest signal intensity, and MS/MS was performed for the corresponding tissue extract. Samples were analysed with a full scan, followed by targeted MS2 scans using an inclusion list in the same LC–MS run. Full scan parameters were: resolution 60,000, range m/z 70–1,000, AGC target 107, ITmax 200 ms. MS/MS parameters were: isolation window 1.7 m/z, collision energies 15, 30, 50 eV, resolution 15,000, AGC target 1e6, ITmax 300 ms, RT window 3 min.
In complex biological samples, the presence of chimeric MS/MS spectra containing fragments from multiple precursor ions within the isolation window can hinder metabolite identification93,94. To deconvolve fragment ions from co-isolated precursors, we implemented a procedure based on the Pearson correlation between MS1 and MS2 ions, with the assumption that only fragment ions whose intensities are correlated with that of the precursor ion originated from this precursor. Each precursor ion yielded multiple MS2 scans spanning a RT window of up to 3 min. The scan associated with the highest MS1 intensity was used to obtain the MS2 spectrum for that precursor ion, from which the m/z and intensity for individual fragment ions were retrieved. For each fragment peak, its EIC curve at the MS2 level was constructed and correlated with the EIC of the precursor ion in MS1, after alignment of the scan times by interpolation and filtering to a RT window of 0.3 min around the scan with the highest MS1 intensity. Fragment ions with a Pearson correlation coefficient less than 0.8 were discarded.
Meta-learning
The availability of additional sources of information in the mouse tissue dataset to support metabolite annotation, including retention times and isotopic patterns at the MS1 level, suggested an avenue to improve the accuracy of metabolite annotation relative to that which had been achieved using MS/MS alone. To explore this possibility, we devised a meta-learning framework to combine DeepMet confidence scores and predicted MS/MS spectra from CFM-ID with retention time and isotope patterns. We first used the combination of DeepMet and CFM-ID to annotate the MS/MS spectra for all 246 identified metabolites with MS/MS spectra in the mouse tissue dataset, using the weighted combination of normalized sampling frequency and cosine similarities between predicted and experimental MS/MS spectra as described above. A 5 ppm window of error was used to identify candidates for each precursor m/z, searching for only [M-H]- adducts; as above, any DeepMet or CFM-ID predictions were made by models trained with 10% of metabolites withheld at a time to avoid data leakage between training and test sets. We then calculated a series of features for each annotation that were provided as input to a random forest classifier. The features were as follows: (1) the DeepMet confidence score, as described above; (2) the frequency with which the annotated structure was generated; (3) the rank of the annotated structure by sampling frequency among all candidates generated by DeepMet; (4) the cosine similarity between the experimental MS/MS spectrum and that predicted by CFM-ID for each candidate; (5) the number of fragment ions matching between the predicted and experimental spectra; (6) the mass error between the experimental and theoretical m/z, in parts per million; (7) the cosine similarity between the theoretical and observed isotope patterns at the MS1 level; and (8) the difference between experimental and predicted retention times. The random forest classifier was then trained in tenfold cross-validation on the set of known metabolites to predict whether a given annotation was correct or incorrect. Calibration was assessed as described above by binning annotations by the probability assigned by the meta-learning model into deciles, and calculating the proportions of correct annotations within each bin.
Prediction of unmeasured retention times for structures generated by DeepMet was achieved using a structure-based retention time prediction model based on a GNN. The GNN model was implemented in PyTorch and the Deep Graph Library (DGL) and comprised four GraphSAGE layers95 with a LSTM feature aggregator and 4 dense layers, each with a hidden dimension of 256. The RT prediction model was trained in tenfold cross-validation on an in-house library of metabolite standards and their retention times to minimize a mean absolute error loss for 1,000 epochs using the Adam optimizer, a batch size of 512, and a learning rate of 0.001.
To assess the possibility that the random forest classifier was overfit to a relatively small training dataset, we additionally fit a logistic regression model to the same dataset and inspected the coefficients associated with each feature (Supplementary Fig. 6b,c). The direction of the coefficients was generally consistent with expert interpretation (for instance, higher dot-product between predicted and experimental MS/MS is indicative of a better annotation), with the major exception being the cosine similarity between theoretical and experimental isotope patterns at the MS1 level, which was counterintuitively assigned a negative coefficient. In the future, enforcing directionality of certain features35 might further reduce overfitting. It is important to emphasize that the classifier presented in this manuscript is trained on features that are specific to our particular analytical setup, particularly because chromatographic methods vary widely across laboratories.
Metabolite standards
Synthetic standards for putative chemical structures assigned by DeepMet were obtained from commercial suppliers (Mcule, Enamine) or via chemical synthesis. Catalogue numbers for commercially available compounds are provided in Supplementary Table 2. Protocols for chemical synthesis are described in ‘Chemical synthesis’.
Standards were dissolved into 50:50 methanol:H2O at 1 mg ml−1. The stock solution was further diluted into 40:40:20 acetonitrile:methanol:H2O at 2 µg ml−1 and analysed by full-scan LC–MS to determine the retention time on the 25 min HILIC method. The 23 tissue extracts were then re-analysed side-by-side with the synthesized compounds using full-scan followed by targeted MS2 scans using an inclusion list. Full scan parameters were: resolution 60,000, range m/z 70–1,000, AGC target 107, ITmax 200 ms. MS2 parameters are, isolation window 1.7 m/z, collision energies 15, 30, 50 eV or 15, 20, 30 eV, resolution 15,000, AGC target 106, ITmax 300 ms.
Where appropriate, two orthogonal approaches were used to further confirm matches between synthetic standards and metabolites identified in tissue extracts. The first such approach involved differentiating chromatographic drift from slight differences in retention time by spiking the synthetic standard into the corresponding tissue extract to establish whether the two features (retention time, MS1 and MS2) merged into a single peak, as expected. These spike-in experiments were performed for N-carbamyl-taurine, glycerylphosphorylethanol, and 4,5,6-triaminopyrimidine (in the positive ionization mode). Spike-in EICs were visualized in Thermo Xcalibur with nine-point Gaussian smoothing. The second such approach involved re-acquiring data from both the synthetic standard and from the tissue extract in positive mode. Data acquired in positive mode is shown in the manuscript for the following metabolites: diacetylputrescine, O-methyl-5-methyluridine, 4,5,6-triaminopyrimidine, and histamine-C4:0.
Thermo raw files were then analysed by the Xcalibur QualBrowser to determine the retention time for each synthetic standard and visualize the MS2 spectra for the standards and corresponding metabolite peaks in the tissue samples. Raw files were then converted to mzML files using ProteoWizard, and contaminating fragment ions from co-isolated precursors were identified as those that showed low correlation with the precursor m/z and removed, as described in ‘Targeted MS/MS analysis’ but with the following modifications: (1) internal mass calibration ions were removed; (2) fragment ions with an absolute difference of less than 1 m/z to the precursor ion were removed as these could not be explained by a neutral loss; (3) the minimum correlation between MS1 and MS2 ions was manually adjusted in a data-adaptive manner as a function of retention time and MS1 signal intensity; and (4) fragment ions with an absolute intensity greater than 120% of the precursor ion in MS1 were removed.
Success rate of metabolite discovery
An estimate of the overall success rate of our metabolite discovery campaign can be derived by comparing the numbers of standards that matched (n = 25; Fig. 5 and Extended Data Figs. 6 and 7) or did not match (n = 42) to both MS/MS and retention times of mouse tissue peaks (25/67, 37%). If excluding the four cases where the chemical standard matched a peak other than that originally targeted for structure elucidation (‘Peak picking and annotation’), this rate would drop to 21/67 (31%). For the purpose of this comparison, an annotation was considered to be correct if it was compatible with the level of structural annotation described in the manuscript; for instance, N-isobutyryl-histamine and N-isobutyryl-methionine were considered correct predictions for histamine-C4:0 and methionine-C4:0, respectively. The total number of correct predictions includes the metabolites that were thought to be novel at the time of chemical standard acquisition or synthesis, but which were later found to be known (Extended Data Fig. 6i–r), but excludes 3-sulfoglycerate, which was synthesized in the course of our attempts to confirm the regiochemistry of 2-sulfoglycerate (Extended Data Fig. 7c) rather than on the basis of a DeepMet prediction. Metabolites that did not match to mouse tissue peaks, but which were later identified in human samples (Extended Data Fig. 9), were treated as failures in this comparison, as were metabolites that afforded partial but imperfect matches to the mouse tissue data (Extended Data Fig. 8). In the latter regard, it is important to emphasize that experimental outcomes can be influenced by factors beyond the accuracy of structure annotation, including metabolite instability or degradation in tissue, low endogenous concentrations leading to low-quality MS/MS spectra, and matrix effects in biological samples. For example, biological samples may contain multiple structural isomers that co-elute, producing MS2 spectra from a single chromatographic peak that differ subtly from spectra of a pure synthetic standard; in cases such as these, we considered the predicted structure to be incorrect for the purpose of this comparison. Last, some of the incorrectly annotated peaks may represent mass spectrometry artifacts not detected by NetID (for instance, unusual adducts or multimers) and therefore could not possibly have been correctly annotated by our workflow, which predicted structures for deprotonated ions. For the above reasons, this methodology provides a conservative and context-dependent estimate of the success rate.
Metabolomics of antibiotics-treated mice
Wild type C57BL/6NCrl mice (strain no. 027) were obtained at 8 weeks of age from Charles River Laboratories and used for experiments at age 8–15 weeks. Antibiotics were provided in the drinking water for 14 days as a mixture containing ampicillin (1 g/L), neomycin (1 g l−1), metronidazole (1 g l−1) and vancomycin (0.5 g l−1) (all from Sigma-Aldrich). To improve the taste of the drinking water, 0.5% aspartame (from Bulk Supplements) was added to the antibiotic solution, and drinking water with 0.5% aspartame was used as control. To collect faecal samples, mice were restrained and gently massaged on the belly to induce defecation, and the faecal pellets were immediately frozen on dry ice. All samples were stored at –80 °C until further analysis.
Metabolomics of dietary perturbations
To assess the difference between chow and purified diets, 8-week-old male C57BL/6NCrl mice were fed either PicoLab Rodent Diet 20 (5053, n = 4) or a standard casein protein based purified diet (Research Diets, D11112201i, n = 4). After 10 days on the respective diets, faeces were collected at 07:00. The faecal samples were collected fresh and immediately flash frozen. For extraction, faeces were ground at liquid nitrogen temperature with a cryomill (Restch). The resulting powder was extracted with 40:40:20 methanol: acetonitrile: water (40 μl extraction solvent per 1 mg tissue) for 10 min on ice and centrifuged at 15,000g for 10 min.
Isotope tracing
Infusions were performed in conscious, free-moving mice which had been catheterized in the jugular vein at least five days prior. Specifically, male C57BL/6 mice, housed in a normal light cycle and aged 12–16 weeks, were brought to a procedure room at 09:00. Mice (n = 2 per tracer) were placed in a new cage and the infusion line was connected. For the U13C-glucose infusion, animals were provided food in their new cage, but other animals were not provided food and remained fasting to the end of the infusion. Immediately after connecting the infusion line, it was primed with 14 μl of infusate to replace the dead volume of saline. At 11:00, a sample of blood was collected by tail snip, and then the infusion started. Infusion rates and concentrations were designed to target 50% enrichment based on previously published measurements of Fcirc (circulatory flux, also known as rate of appearance; 13C3-serine, 141.667 mM, 3 µl min−1, 2 h; 13C5-methionine, 33.333 mM, 3 µl min−1, 2 h; 13C6-glucose, 1,875 mM, 4 µl min−1, 6 h; 13C3-cysteine, 30 mM, 2.5 µl min−1, 6 h)96,97. Urine was collected from the animal if it urinated when scruffed at the end of the infusion. The mice were euthanized by cervical dislocation, and tissues were quickly dissected and snap frozen in liquid nitrogen using a Wollenberger clamp. If urine had not already been collected, then urine was withdrawn from the bladder using an insulin syringe. In addition, a set of mice were not infused with anything but handled in parallel to the infused mice as controls.
Chemical synthesis
Synthetic methods and NMR spectra are provided in Supplementary Note 1. Because stereoisomers are not expected to be distinguishable by our LC–MS/MS metabolomics approach, structures are drawn with undefined stereochemistry in both the main text and Supplementary Note 1 except for chiral building blocks in the latter.
Searching reference spectra against metabolomic repositories
The observation that several chemical structures assigned by DeepMet yielded poor matches to the corresponding peaks in mouse tissues, but that these putative metabolites could nonetheless be identified in human biofluids via LC–MS/MS analysis of synthetic standards, motivated us to more comprehensively search the reference MS/MS spectra acquired in this study against published human metabolomics data, as shown in Extended Data Fig. 9. To this end, we assembled a compendium of untargeted metabolomic runs from human tissues from the MetaboLights and Metabolomics Workbench repositories. Human files were identified by a combination of automated metadata search followed by extensive manual review to remove non-human samples and blanks. For MetaboLights, study metadata was downloaded in XML format and filtered as described in ‘Meta-analysis of the human blood metabolome’ to include only liquid chromatography-mass spectrometry datasets, but here including all tissues rather than limiting this analysis to blood. For Metabolomics Workbench, the REST API was queried first to retrieve all LC–MS studies (endpoint ‘/rest/study/study_id/ST/summary’) and then manually curated in order to subset these to experiments in human tissues. These files were then downloaded, archives (.tar, .gz and .zip) were decompressed, and vendor-specific formats (for example, .raw, .d and .wiff) were converted to mzML, after which MS/MS spectra from each run were then extracted and written to MGF files after quality control. MS/MS spectra from each run were then extracted and written to MGF files after ensuring the following QC criteria were met: at least 50 unique precursor m/z values; at least 100 non-empty MS/MS spectra; both precursor m/z and fragment m/z recorded to at least 4 decimal places. Files that failed one or more of these criteria were discarded. Duplicate files, representing cases where the same mass spectrometry run was uploaded as part of more than one accession, were identified by their checksums and removed. In total, these steps afforded a resource comprising 356.3 million MS/MS spectra from 35,460 mass spectrometry runs. The complete list of accession numbers and raw data files included in this analysis is provided in Supplementary Table 4b. The reference MS/MS spectra acquired from chemical standards were then used to search the entire compendium of published human metabolomics data, using the implementation of the normalized dot-product in the Spectra R package98, with a precursor m/z tolerance of 20 ppm, a fragment m/z tolerance of 50 ppm, and considering only peaks in the reference spectrum that were within the scan limit of the experimental spectrum. Matches with a normalized dot-product greater than 0.8 and at least three matching peaks greater than 1% relative intensity in both spectra were retained and manually inspected to discard unreliable matches.
Citation counts
A table linking PubChem identifiers to PMIDs referencing the corresponding compounds was obtained from the PubChem FTP site (https://ftp.ncbi.nlm.nih.gov/pubchem/Compound/Extras/CID-PMID.gz). The corresponding SMILES strings were obtained from the same FTP site (file CID-SMILES.gz). Structures were preprocessed in RDKit by removing stereochemistry and standardizing tautomers, and citation counts associated with stereoisomers or tautomers of the same compound (as identified by the InChI key) were summed. In parallel, a dataset comprising 718,097 biomolecular structures obtained from the union of 14 molecular structure databases99, including metabolites, drugs, toxins and other small molecules of biological interest, was obtained from https://github.com/boecker-lab/myopic-mces-data. The distribution of citation counts was then retrieved for the metabolites comprising the training set of DeepMet, a random subset of 1 million generated structures, a random subset of 1 million structures from PubChem, and the biomolecules database.
Impact of pretraining
The presence of numerous known metabolites from our HMDB training set in the ChEMBL dataset on which the language model was pretrained raised the possibility that the performance of DeepMet might be attributable at least in part to the presence of these metabolites in ChEMBL. To evaluate this possibility, we retrained a series of identical LSTM models on the same dataset and splits as DeepMet, but without any pretraining on ChEMBL, and then sampled a total of 1 billion SMILES strings (100 million from each training fold) from the non-pretrained models. These SMILES strings were preprocessed and the sampling frequency computed for each unique chemical structure in an identical fashion to those sampled from DeepMet itself. We then repeated a number of the analyses described above for DeepMet for the structures generated by the non-pretrained models. We found that, as we had observed for DeepMet, molecules generated more frequently by the non-pretrained model were disproportionately metabolite-like; withheld metabolites were among the most frequently generated molecules proposed by the non-pretrained language model; the non-pretrained model generated the majority of the metabolites added to version 5.0 of the HMDB; and these HMDB 5.0 metabolites were generated with significantly higher sampling frequencies than other generated molecules (Supplementary Fig. 9a–h). We then reproduced the evaluations shown in Fig. 3, in which we had shown that DeepMet can prioritize plausible chemical structures for a metabolite withheld from the training set, given only its exact mass as input. We observed that the performance of the non-pretrained model was essentially identical to that reported for DeepMet in the original manuscript, in terms of the top-1 and top-k accuracies, the Tanimoto coefficient between the prioritized and true structures, and the proportion of withheld metabolites that were ever generated by the non-pretrained model (Supplementary Fig. 9i–l). Finally, we validated that overlap between ChEMBL and the HMDB did not compromise our evaluation of the integration of DeepMet with computational methods for MS/MS annotation, such as CFM-ID. We re-analysed the database of MS/MS spectra that had been predicted for structures generated by DeepMet itself, but here omitted structures from the HMDB that were also part of the ChEMBL pretraining set from our evaluation. We continued to observe excellent performance of DeepMet when removing all metabolites also found in ChEMBL (Supplementary Fig. 9m–q).
Terminology
Throughout the manuscript, we use the terminology ‘previously unrecognized metabolite’ to refer to small molecules that, to the best of our knowledge, had not previously been recognized as mammalian metabolites. None of these metabolites are present in the HMDB, so their identification with DeepMet is consistent with the goal of enabling de novo metabolite identification without relying on existing metabolite databases. However, because the HMDB is an incomplete catalogue, it is more challenging to assert that a molecule is unknown in the broader context of mammalian metabolism. We employed a multi-tiered process involving extensive manual review to support this categorization. This review was undertaken for all metabolites reported in the manuscript. First, we removed any structures present in any version of the HMDB, with any annotation status (quantified, detected, expected or predicted). Second, if the structure was present in PubChem or CAS SciFinder, we manually reviewed all of the associated literature references to establish whether any of these reported its detection in mammals. Third, we formulated potential common names or synonyms that we could envision describing the compound in question, and performed literature searches using Google Scholar and PubMed. Fourth, we searched for potential isomers on PubChem and SciFinder that could be envisioned to afford similar MS/MS spectra to identify whether these were known to be mammalian metabolites. This multi-tiered review procedure led us to identify that certain structures presented in Extended Data Fig. 6i–r, which we had targeted on the belief that these were previously unrecognized metabolites, were in fact known to be mammalian metabolites. In Supplementary Note 2, we provide additional context for each of the previously unrecognized metabolites, including reports of their detection in non-mammalian species. Despite our best efforts, the possibility that this review of the literature may have been incomplete for certain compounds must be acknowledged, in part because of both false positives and false negatives in databases that attempt to automatically link chemical structures to their appearance in the literature.
Visualization
Throughout the paper, box plots show the median (horizontal line), interquartile range (hinges) and smallest and largest values no more than 1.5 times the interquartile range (whiskers).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data, including all of the generated structures, the subset of generated structures not found in the HMDB, and MS/MS spectra for each structure predicted by CFM-ID, can be accessed via an interactive web application (see Code availability) or directly via Zenodo (https://doi.org/10.5281/zenodo.16813151 (ref. 100)). All mass spectrometry-based metabolomics data acquired in this study has been deposited to MassIVE (https://massive.ucsd.edu/) with accession number MSV000097536, with the exception of the data from clinical or forensic samples; investigators interested in accessing this data should contact A.M.S. (aaron.shapiro1@phsa.ca). Reference MS/MS spectra are provided as Supplementary Files 1 and 2. Accessions and filenames for published metabolomic experiments re-analysed in this study are provided in Supplementary Table 4. ChEMBL (version 28) is available from https://doi.org/10.6019/CHEMBL.database.28. HMDB is freely available at https://hmdb.ca/downloads. Source data are provided with this paper.
Code availability
An interactive web application, available at http://deepmet.org, allows users to explore the structures of generated molecules and prioritize metabolite structures based on accurate masses, inferred molecular formulas, or MS/MS spectra. Users can provide masses, formulas or MS/MS spectra for features of interest (as identified using software such as MZmine101 or xcms88) and removal of degenerate features such as isotopologues, multimers and adducts prior to structure annotation is recommended; the DeepMet web application is not designed to process raw mass spectrometry data files or identify degenerate features. We have also released a Snakemake pipeline that allows investigators to rapidly train, sample from and evaluate chemical language models, allowing the paradigm described here to be extended to other classes of metabolites (available from GitHub at https://github.com/skinniderlab/clm or from Zenodo at https://doi.org/10.5281/zenodo.14917571 (ref. 102)).
References
El Abiead, Y. et al. Discovery of metabolites prevails amid in-source fragmentation. Nat. Metab. 7, 435–437 (2025).
Aksenov, A. A., da Silva, R., Knight, R., Lopes, N. P. & Dorrestein, P. C. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 1, 0054 (2017).
Dias, D. A. et al. Current and future perspectives on the structural identification of small molecules in biological systems. Metabolites 6, 46 (2016).
da Silva, R. R., Dorrestein, P. C. & Quinn, R. A. Illuminating the dark matter in metabolomics. Proc. Natl Acad. Sci. USA 112, 12549–12550 (2015).
Segler, M. H. S., Kogej, T., Tyrchan, C. & Waller, M. P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131 (2018).
Olivecrona, M., Blaschke, T., Engkvist, O. & Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminformatics 9, 48 (2017).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Alseekh, S. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021).
Gizzi, A. S. et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558, 610–614 (2018).
Li, V. L. et al. An exercise-inducible metabolite that suppresses feeding and obesity. Nature 606, 785–790 (2022).
Mohanty, I. et al. The underappreciated diversity of bile acid modifications. Cell 187, 1801–1818.e20 (2024).
Gentry, E. C. et al. Reverse metabolomics for the discovery of chemical structures from humans. Nature 626, 419–426 (2024).
Bepler, T. & Berger, B. Learning the protein language: evolution, structure, and function. Cell Syst. 12, 654–669.e3 (2021).
Hie, B., Zhong, E. D., Berger, B. & Bryson, B. Learning the language of viral evolution and escape. Science 371, 284–288 (2021).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape. Nature 622, 818–825 (2023).
Hie, B. L., Yang, K. K. & Kim, P. S. Evolutionary velocity with protein language models predicts evolutionary dynamics of diverse proteins. Cell Syst. 13, 274–285.e6 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Gupta, S. & Aires-de-Sousa, J. Comparing the chemical spaces of metabolites and available chemicals: models of metabolite-likeness. Mol. Divers. 11, 23–36 (2007).
Hert, J., Irwin, J. J., Laggner, C., Keiser, M. J. & Shoichet, B. K. Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 5, 479–483 (2009).
Peironcely, J. E., Reijmers, T., Coulier, L., Bender, A. & Hankemeier, T. Understanding and classifying metabolite space and metabolite-likeness. PLoS ONE 6, e28966 (2011).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 28, 31–36 (1988).
Djoumbou-Feunang, Y. et al. BioTransformer: a comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminformormatics 11, 2 (2019).
O’Boyle, N. M. Towards a Universal SMILES representation - A standard method to generate canonical SMILES based on the InChI. J. Cheminformatics 4, 22 (2012).
Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S. & Klambauer, G. Fréchet chemnet distance: A metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 58, 1736–1741 (2018).
Wishart, D. S. et al. HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res. 50, D622–D631 (2022).
Nissinen, S. I. et al. Discrimination between pancreatic cancer, pancreatitis and healthy controls using urinary polyamine panel. Cancer Control 28, 10732748211039762 (2021).
Ariaey-Nejad, M. R. & Pearson, W. N. 4-Methylthiazole-5-acetic acid–a urinary metabolite of thiamine. J. Nutr. 96, 445–449 (1968).
Fernandes Silva, L., Ravi, R., Vangipurapu, J. & Laakso, M. Metabolite signature of simvastatin treatment involves multiple metabolic pathways. Metabolites 12, 753 (2022).
Lee-Sarwar, K. et al. Intestinal microbial-derived sphingolipids are inversely associated with childhood food allergy. J. Allergy Clin. Immunol. 142, 335–338.e9 (2018).
Renz, P., Van Rompaey, D., Wegner, J. K., Hochreiter, S. & Klambauer, G. On failure modes in molecule generation and optimization. Drug Discov. Today Technol. 32–33, 55–63 (2019).
Krettler, C. A. & Thallinger, G. G. A map of mass spectrometry-based in silico fragmentation prediction and compound identification in metabolomics. Brief. Bioinformatics 22, bbab073 (2021).
Allen, F., Greiner, R. & Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 11, 98–110 (2015).
Wang, F. et al. CFM-ID 4.0: More accurate ESI-MS/MS spectral prediction and compound identification. Anal. Chem. 93, 11692–11700 (2021).
Hoffmann, M. A. et al. High-confidence structural annotation of metabolites absent from spectral libraries. Nat. Biotechnol. 40, 411–421 (2022).
Peironcely, J. E. et al. OMG: open molecule generator. J. Cheminform. 4, 21 (2012).
Butler, T. et al. MS2Mol: A transformer model for illuminating dark chemical space from mass spectra. Preprint at https://doi.org/10.26434/chemrxiv-2023-vsmpx-v4 (2023).
Young, A. et al. FraGNNet: a deep probabilistic model for tandem mass spectrum prediction. Trans. Machine Learning. Res https://openreview.net/pdf?id=UsqeHx9Mbx (2025).
Wei, J. N., Belanger, D., Adams, R. P. & Sculley, D. Rapid prediction of electron-ionization mass spectrometry using neural networks. ACS Cent. Sci. 5, 700–708 (2019).
Hoffmann, M. A., Kretschmer, F., Ludwig, M. & Böcker, S. MAD HATTER correctly annotates 98% of small molecule tandem mass spectra searching in pubchem. Metabolites 13, 314 (2023).
Wang, X. et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 69, 2131–2142 (2020).
Wang, Z., Qi, Y., Wang, F., Zhang, B. & Jianguo, T. Circulating sepsis-related metabolite sphinganine could protect against intestinal damage during sepsis. Front. Immunol. 14, 1151728 (2023).
Chen, L. et al. Metabolite discovery through global annotation of untargeted metabolomics data. Nat. Methods 18, 1377–1385 (2021).
Dizdaroglu, M., Kirkali, G. & Jaruga, P. Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects. Free Radic. Biol. Med. 45, 1610–1621 (2008).
Kožich, V. et al. Human ultrarare genetic disorders of sulfur metabolism demonstrate redundancies in H2S homeostasis. Redox Biol. 58, 102517 (2022).
Pan, G., Ham, Y.-H., Chan, H. W., Yao, J. & Chan, W. LC-MS/MS coupled with a stable-isotope dilution method for the quantitation of thioproline-glycine: a novel metabolite in formaldehyde- and oxidative stress-exposed cells. Chem. Res. Toxicol. 33, 1989–1996 (2020).
Hey, J. A. et al. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs 32, 849–861 (2018).
Madhu, C., Gregus, Z., Cheng, C. C. & Klaassen, C. D. Identification of the mixed disulfide of glutathione and cysteinylglycine in bile: dependence on gamma-glutamyl transferase and responsiveness to oxidative stress. J. Pharmacol. Exp. Ther. 262, 896–900 (1992).
Hofmann, U., Eichelbaum, M., Seefried, S. & Meese, C. O. Identification of thiodiglycolic acid, thiodiglycolic acid sulfoxide, and (3-carboxymethylthio)lactic acid as major human biotransformation products of S-carboxymethyl-L-cysteine. Drug Metab. Dispos. 19, 222–226 (1991).
Waring, R. H. Variation in human metabolism of S-carboxymethylcysteine. Eur. J. Drug Metab. Pharmacokinet. 5, 49–52 (1980).
He, L. et al. Metabolic analysis of nucleosides/bases in the urine and serum of patients with alcohol-associated liver disease. Metabolites 12, 1187 (2022).
Rocchiccioli, F. et al. Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr. Res. 28, 657–662 (1990).
Surette, R. & Vining, L. C. Formation of 3-methylthioacrylic acid from methionine by Streptomyces lincolnensis. Isolation of a peroxidase. J. Antibiot. 29, 646–652 (1976).
Tay, D. W. P., Yeo, N. Z. X., Adaikkappan, K., Lim, Y. H. & Ang, S. J. 67 million natural product-like compound database generated via molecular language processing. Sci. Data 10, 296 (2023).
Shen, X., Zeng, T., Chen, N., Li, J. & Wu, R. NIMO: a natural product-inspired molecular generative model based on conditional transformer. Molecules 29, 1867 (2024).
Stravs, M. A., Dührkop, K., Böcker, S. & Zamboni, N. MSNovelist: de novo structure generation from mass spectra. Nat. Methods 19, 865–870 (2022).
Bohde, M., Manjrekar, M., Wang, R., Ji, S. & Coley, C. W. DiffMS: diffusion generation of molecules conditioned on mass spectra. In Proc. 42nd Int. Conf. Machine Learning https://openreview.net/pdf?id=EvILcv2v8L (Vancouver, Canada, 2025).
Dührkop, K. et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019).
Goldman, S. et al. Annotating metabolite mass spectra with domain-inspired chemical formula transformers. Nat. Mach. Intell. 5, 965–979 (2023).
Mason, S. J. & Graham, N. E. Areas beneath the relative operating characteristics (ROC) and relative operating levels (ROL) curves: statistical significance and interpretation. Q. J. R. Met. Soc. 128, 2145–2166 (2002).
Djoumbou Feunang, Y. et al. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminformatics 8, 61 (2016).
Jaeger, S., Fulle, S. & Turk, S. Mol2vec: unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 58, 27–35 (2018).
Bjerrum, E. J. SMILES enumeration as data augmentation for neural network modeling of molecules. Preprint at https://doi.org/10.48550/arXiv.1703.07076 (2017).
Arús-Pous, J. et al. Randomized SMILES strings improve the quality of molecular generative models. J. Cheminformatics 11, 71 (2019).
Skinnider, M. A., Stacey, R. G., Wishart, D. S. & Foster, L. J. Chemical language models enable navigation in sparsely populated chemical space. Nat. Mach. Intell. 3, 759–770 (2021).
Barupal, D. K. & Fiehn, O. Generating the blood exposome database using a comprehensive text mining and database fusion approach. Environ. Health Perspect. 127, 97008 (2019).
Bagal, V., Aggarwal, R., Vinod, P. K. & Priyakumar, U. D. MolGPT: molecular generation using a transformer–decoder model. J. Chem. Inf. Model. 62, 2064–2076 (2022).
Chen, Y. et al. Molecular language models: RNNs or transformer?. Brief. Funct. Genomics 22, 392–400 (2023).
Özçelik, R., de Ruiter, S., Criscuolo, E. & Grisoni, F. Chemical language modeling with structured state space sequence models. Nat. Commun. 15, 6176 (2024).
Özçelik, R., Brinkmann, H., Criscuolo, E. & Grisoni, F. Generative deep learning for de novo drug design─a chemical space odyssey. J. Chem. Inf. Model. 65, 7352–7372 (2025).
Arús-Pous, J. et al. Exploring the GDB-13 chemical space using deep generative models. J. Cheminform. 11, 20 (2019).
Moret, M., Friedrich, L., Grisoni, F., Merk, D. & Schneider, G. Generative molecular design in low data regimes. Nat. Mach. Intell. 2, 171–180 (2020).
Skinnider, M. A. Invalid SMILES are beneficial rather than detrimental to chemical language models. Nat. Mach. Intell. 6, 437–448 (2024).
Blaschke, T. et al. REINVENT 2.0: an AI tool for de novo drug design. J. Chem. Inf. Model. 60, 5918–5922 (2020).
Mendez, D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940 (2019).
Winter, R., Montanari, F., Noé, F. & Clevert, D.-A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 10, 1692–1701 (2019).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://doi.org/10.48550/arXiv.1802.03426 (2018).
Tian, S., Djoumbou-Feunang, Y., Greiner, R. & Wishart, D. S. Cypreact: A software tool for in silico reactant prediction for human cytochrome P450 enzymes. J. Chem. Inf. Model. 58, 1282–1291 (2018).
Brown, N., Fiscato, M., Segler, M. H. S. & Vaucher, A. C. Guacamol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108 (2019).
Polykovskiy, D. et al. Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 11, 565644 (2020).
Bemis, G. W. & Murcko, M. A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 39, 2887–2893 (1996).
Allen, F., Pon, A., Wilson, M., Greiner, R. & Wishart, D. CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 42, W94–W99 (2014).
Menikarachchi, L. C., Hill, D. W., Hamdalla, M. A., Mandoiu, I. I. & Grant, D. F. In silico enzymatic synthesis of a 400,000 compound biochemical database for nontargeted metabolomics. J. Chem. Inf. Model. 53, 2483–2492 (2013).
Jeffryes, J. G. et al. MINEs: open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminformatics 7, 44 (2015).
Haug, K. et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444 (2020).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Lu, W., Wang, L., Chen, L., Hui, S. & Rabinowitz, J. D. Extraction and quantitation of nicotinamide adenine dinucleotide redox cofactors. Antioxid. Redox Signal. 28, 167–179 (2018).
Wang, L. et al. Peak annotation and verification engine for untargeted LC–MS metabolomics. Anal. Chem. 91, 1838–1846 (2019).
Agrawal, S. et al. El-MAVEN: a fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol. Biol. 1978, 301–321 (2019).
Guo, J. et al. EVA: evaluation of metabolic feature fidelity using a deep learning model trained with over 25000 extracted ion chromatograms. Anal. Chem. 93, 12181–12186 (2021).
Xing, S. et al. Recognizing contamination fragment ions in liquid chromatography–tandem mass spectrometry data. J. Am. Soc. Mass. Spectrom. 32, 2296–2305 (2021).
Stancliffe, E., Schwaiger-Haber, M., Sindelar, M. & Patti, G. J. DecoID improves identification rates in metabolomics through database-assisted MS/MS deconvolution. Nat. Methods 18, 779–787 (2021).
Hamilton, W. L., Ying, R. & Leskovec, J. Inductive representation learning on large graphs. In Proc. 31st Int. Conf. Neural Inf. Processing 1025–1035 (2017).
Hui, S. et al. Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688.e4 (2020).
Lee, W. D. et al. Impact of acute stress on murine metabolomics and metabolic flux. Proc. Natl. Acad. Sci. USA 120, e2301215120 (2023).
Rainer, J. et al. A modular and expandable ecosystem for metabolomics data annotation in R. Metabolites 12, 173 (2022).
Kretschmer, F., Seipp, J., Ludwig, M., Klau, G. W. & Böcker, S. Coverage bias in small molecule machine learning. Nat. Commun. 16, 554 (2025).
Skinnider, M. Predicted metabolites and their MS/MS spectra [Data set]. Zenodo https://doi.org/10.5281/zenodo.16813151 (2025).
Schmid, R. et al. Integrative analysis of multimodal mass spectrometry data in MZmine 3. Nat. Biotechnol. 41, 447–449 (2023).
Bansal, V., Acharya, A., Skinnider, M. & Chaloo, A. skinniderlab/CLM: v0.0.1 (v0.0.1). Zenodo https://doi.org/10.5281/zenodo.14917576 (2025).
Acknowledgements
This work was supported by Ludwig Cancer Research; the National Institutes of Health (DP5OD036960 to M.A.S., R50CA211437 to W.L., P30DK019525 to J.D.R., P30CA072720 to J.D.R.); Genome Canada, Genome British Columbia, and Genome Alberta (project 284MBO); the Canada Foundation for Innovation (CFI MSIF 42495); Canada Research Chairs (CRC TIER1 100628); the Princeton Language Institute; NSERC; AMII; and CIFAR. We thank M. Belan for assistance with the illustrations.
Author information
Authors and Affiliations
Contributions
H.Q., F.W., W.L., X.X., H.K., S.A.M.M., L.B.A., J.E.A., A.R., M.N., R.A.C., W.D.L., V. Gupta, S.L.N., M.H.-G. and A.Y. conducted experiments. H.Q., F.W., W.L., X.X., L.B.A., E.H., V. Gupta, S.L.N., A.Y., S.T., L.C., C.W.J. and M.A.S. analysed the data. E.O. and V. Gautam developed the web server. B.W., H.R., J.B., R.G., L.J.F., A.M.S., D.S.W., J.D.R. and M.A.S. supervised the study. M.A.S. wrote the manuscript, and all authors contributed to its editing.
Corresponding author
Ethics declarations
Competing interests
J.D.R. is a member of the Rutgers Cancer Institute of New Jersey (RCINJ) and the University of Pennsylvania Diabetes Research Center (U Penn DRC); a director of the U Penn DRC-Princeton inter-institutional metabolomics core and RCINJ metabolomics core; an advisor and stockholder of Colorado Research Partners, Bantam Pharmaceuticals, Barer Institute, Rafael Pharmaceuticals, Empress Therapeutics and Marea Therapeutics; a founder, director, and stockholder of Farber Partners, Raze Therapeutics and Sofro Pharmaceuticals; and a founder, advisor, and stockholder of Marea Therapeutics and Fargo Biotechnologies. H.K. is a member of the Rutgers Cancer Institute of New Jersey (RCINJ); a co-founder, director, stockholder and consultant for Crescenta Biosciences; a co-founder, director, stockholder for Farber Partners; a co-founder and shareholder of Chiromics; a shareholder and scientific advisor to Colorado Research Partners and Integrated Biosciences. M.A.S. is a member of the Rutgers Cancer Institute of New Jersey (RCINJ). H.K. and J.D.R. are inventors on patents held by Princeton University. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Sebastian Böcker, Pieter Dorrestein, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 A language model of the human metabolome.
a, Schematic overview of the random forest classifier trained in cross-validation to distinguish generated molecules from a held-out set of human metabolites, withheld from the language model during training. b, Receiver operating characteristic (ROC) curve of a random forest classifier trained to distinguish between known metabolites and structures from ChEMBL containing only the atoms C, H, N, O, P, and S in cross-validation. Inset text shows the area under the ROC curve (AUC). c, Confusion matrix showing predictions by the random forest classifier versus true classes. d, Proportion of one-step biotransformations of known human metabolites recapitulated by DeepMet, shown across individual enzymatic reactions. e, Examples of enzymatic biotransformations recapitulated by DeepMet in a rule-free manner. f, As in Fig. 1d, but showing the proportion of molecules generated by DeepMet that are also BioTransformer products, as a function of the number of rule-based transformations applied sequentially to the original metabolite and with or without the removal of structures also present in ChEMBL. g, Nearest-neighbour Tanimoto coefficients to a known metabolite for generated molecules versus molecules with identical molecular formulas sampled randomly from PubChem. h, Nearest-neighbour Tanimoto coefficients to a known metabolite for generated molecules versus molecules with identical molecular formulas sampled randomly from ChEMBL. i, Nearest-neighbour Tc to a known human metabolite for generated molecules versus molecules with identical molecular formulas sampled randomly from PubChem, shown separately for each superclass in the ClassyFire chemical ontology.
Extended Data Fig. 2 DeepMet anticipates metabolites absent from the training set.
a, Number of distinct SMILES strings generated for each held-out metabolite in the test set, within a sample of 10 million SMILES. b, Density plot showing the distribution of negative log-likelihoods for all sampled SMILES, with red ticks showing likelihoods for all sampled SMILES corresponding to the structure of suberylglycine, a held-out metabolite from the test set. Right, structure of suberylglycine. c, As in b, but showing another held-out metabolite, isatin. d, Tc between generated molecules and their nearest neighbour in the training set of known metabolites for molecules generated with progressively increasing frequencies, shown separately for each superclass in the ClassyFire chemical ontology. e, Heatmap showing the proportion of generated metabolites recapitulating one- to four-step enzymatic transformations of human metabolites predicted by BioTransformer, for molecules generated with progressively increasing frequencies. f, ROC curve showing the prioritization of held-out human metabolites from HMDB 4.0 on the basis of sampling frequency, as compared to the background distribution of all generated molecules. g, Examples of structures added to version 5.0 of the HMDB that were not generated by DeepMet, with synthetic or biosynthetic origins outside of endogenous mammalian metabolism. h, Examples of bona fide human metabolites added to version 5.0 of the HMDB that were not generated by DeepMet.
Extended Data Fig. 3 Prioritization of metabolite structures from accurate mass measurements.
a, Illustrative example demonstrating the use of DeepMet to prioritize candidate metabolite structures in which the correct structure is ranked second. A total of 23,664 sampled SMILES strings matched the input mass of 253.0950 ± 10 ppm, corresponding to 8,624 unique structures. Top, lollipop plot shows the sampling frequencies of the 15 most frequently generated molecules as a proportion of the 23,664 SMILES strings. Bottom, a subset of the generated molecules is shown, including the four most frequently generated as well as two less frequently generated structures. Here, the held-out metabolite was N-lactoyl-tyrosine (structure 2); however, structure 1, 1-carboxyethyltyrosine, is also a known metabolite. b, Histogram showing the distribution of Tanimoto coefficients between random pairs of training set metabolites from version 4.0 of the HMDB. Arrow, mean Tc between held-out metabolites and structures prioritized by DeepMet. c, Tc between the structures of held-out metabolites and the top-ranked structures prioritized by DeepMet, random structures generated by DeepMet, or two baseline approaches, as shown in Fig. 3e, but here with the Tc represented as a quantile of the empirical distribution of Tanimoto coefficients between random pairs of training set metabolites. d, Euclidean distance between CDDD embeddings for held-out metabolites and the top-ranked structures prioritized by DeepMet, random structures generated by DeepMet, or two baseline approaches. e, Top-1 accuracy with which the complete chemical structures of held-out metabolites were assigned by DeepMet or two baseline approaches when considering prioritized structures with minimum Tc of 0.4 or 0.675 as matches. f, As in a, but showing an example where the top-ranked structure is incorrect but demonstrates a high degree of chemical similarity to the true held-out metabolite. Left, lollipop plot showing sampling frequencies; middle, top-ranked structure; right, true structure of 11-β-hydroxyandrosterone-3-glucuronide. g, Top-k accuracy with which the complete chemical structures of held-out metabolites were assigned by DeepMet when considering prioritized structures with minimum Tc of 0.4 or 0.675 as matches. h, Top-1 accuracy with which the complete chemical structures of held-out metabolites were assigned by DeepMet when considering multiple positively or negatively charged adducts. i, Tc between the structures of held-out metabolites and the top-ranked structures prioritized by DeepMet when considering multiple positively or negatively charged adducts. j, As in h, but showing the top-k accuracy. k, Distribution of confidence scores assigned to top-ranked structures for each held-out metabolite, given the exact mass of that metabolite as input. l, ROC curve showing the performance of a random forest classifier trained in cross-validation to separate metabolites from versions 4.0 and 5.0 of the HMDB. m, Top-1 accuracy with which the complete chemical structures of metabolites added in version 5.0 of the HMDB were assigned by DeepMet or two baseline approaches. n, As in l, but showing the top-k accuracy curve. o, Tc between the structures of metabolites added in version 5.0 of the HMDB and the top-ranked structures prioritized by DeepMet, random structures generated by DeepMet, or two baseline approaches. p, As in n, but showing the top-k accuracy curves when considering prioritized structures with minimum Tc of 0.4 or 0.675 as matches. q-s, Illustrative examples of metabolites added in version 5.0 of the HMDB whose two-dimensional structures would have been correctly prioritized by DeepMet, given an accurate mass as input (but note that orthogonal analytical data would be needed to confirm these annotations in real data). Top, lollipop plots showing sampling frequencies; bottom, structures of the metabolites.
Extended Data Fig. 4 Metabolite annotation via MS/MS.
a, Top-1 accuracy with which the complete chemical structures of held-out metabolites were assigned by DeepMet when considering only structures generated at least n times, for n between 1 and 10,000. b, As in a, but showing the top-k accuracy curve, for k ≤ 30. c, Tanimoto coefficients (Tc) between the structures of held-out metabolites and the top-ranked structures prioritized by DeepMet when considering only structures generated at least n times, for n between 1 and 10,000. d, Top-1 accuracy with which the complete chemical structures of held-out metabolites were assigned by the combination of DeepMet with CFM-ID in the Agilent MS/MS library for negative ion mode spectra, as compared to a series of baseline approaches, including ranking structures based on the sampling frequency alone (“DeepMet alone”), based on the dot-product between predicted and experimental spectra (“CFM-ID alone”), or the combination of CFM-ID with two baseline approaches, AddCarbon or searching within the training set. e, As in d, but showing the top-k accuracy curve, for k ≤ 30. f, Tc between the structures of held-out metabolites and the top-ranked structures prioritized by the combination of CFM-ID with DeepMet as compared to baseline approaches. g, As in e, but also showing the top-k accuracy when considering prioritized structures with minimum Tc of 0.4 or 0.675 as matches. h, As in d, but also showing the top-1 accuracy when considering prioritized structures with minimum Tc of 0.4 or 0.675 as matches. i, As in d, but in the HMDB MS/MS library for positive and negative ion mode spectra. j, As in e, but in the HMDB MS/MS library for positive and negative ion mode spectra. k, As in f, but in the HMDB MS/MS library for positive and negative ion mode spectra. l, As in g, but in the HMDB MS/MS library for positive and negative ion mode spectra. m, As in e, but when using FraGNNet to predict MS/MS spectra in the positive ion mode. n, As in e, but when using NEIMS to predict MS/MS spectra in the positive ion mode. o, Number of mass spectrometry runs, left, and number of MS/MS spectra per run, right, in the human blood metabolome meta-analysis dataset. p, Total number of experiments comprising the human blood metabolome meta-analysis dataset, grouped by polarity. q, Total number of MS/MS spectra comprising the human blood metabolome meta-analysis dataset, shown by polarity. r, Distribution of precursor m/z’s in the human blood metabolome meta-analysis dataset. s, Number of fragment ions per MS/MS spectrum in the human blood metabolome meta-analysis dataset. t, Left, number of MS/MS spectra in the human blood metabolome dataset linked to a chemical structure when searching against databases of MS/MS spectra predicted by CFM-ID, or ‘decoy’ MS/MS spectra generated by shuffling fragment ions between isobaric predicted spectra, at four dot-product cutoffs. Right, enrichment of matches to predicted versus shuffled spectra, at four dot-product cutoffs.
Extended Data Fig. 5 Overview of the mouse tissue dataset and DeepMet-guided identification of simulated unknown metabolites.
a-b, Visualization of the 4,814 peaks in the mouse tissue metabolomics data, coloured by maximum precursor intensity across 23 tissues, left, or the tissue with the highest intensity, right. c, Proportion of the 4,814 peaks identified by a match to the m/z and retention time of a chemical standard. d-u, Representative examples of known metabolites absent from our in-house library of chemical standards but which were correctly identified by the combination of DeepMet and CFM-ID despite being withheld from the training sets of both models. Left, chemical structure of the metabolite (top) and MS1 intensity across 23 mouse tissues (bottom), coloured as in b. Right, mirror plot showing the similarity between MS/MS spectra from the synthetic standard versus the experimental spectrum in mouse tissues. d, APGPR enterostatin; e, UDP glucuronic acid; f, deoxycytidine; g, 4-ethylphenylsulfate; h, acadesine; i, GDP glucose; j, galactosylglycerol; k, 7-methylguanine; l, isoleucyltyrosine; m, indoxyl glucuronide; n, phenylglucuronide; o, phenol sulfate; p, tryptophanamide; q, amylose; r, N-acetylhistamine; s, peracitol; t, glycylserine; u, 3-hydroxytetradecanedioic acid.
Extended Data Fig. 6 Additional mouse metabolites.
a, Left, structure of (2-(4-hydroxyphenyl)acetyl)-aspartic acid (top) and MS1 intensity across 23 mouse tissues (bottom). Right, mirror plot showing the similarity between MS/MS spectra from the synthetic standard versus the experimental spectrum from mouse urine. b, As in a, but for methylthioinosine. c, As in a, but for homotaurine. d, As in a, but for (2-aminoethyl)phosphate-hexopyranose. e, As in a, but for N-acetyl-phenylalanylleucine/isoleucine. f, As in a, but for glycerylphosphorylethanol. g, As in a, but for O-sulfo-hexopyranose. h, Tissue specificity of previously unrecognized metabolites versus known metabolites identified by comparison to an in-house library of synthetic standards, as quantified by the number of tissues in which the metabolite in question was identified with a MS1 intensity of 105 or greater. i, As in a, but for thioprolylglycine. j, As in a, but for S-sulfohomocysteine. k, As in a, but for 3-carboxymethyl-thiolactic acid. l, As in a, but for 3-sulfopropanoic acid. m, As in a, but for glutathione-cysteinylglycine mixed disulfide. n, As in a, but for N-acetyl-S-carboxymethyl-cysteine. o, As in a, but for diacetylputrescine. p, As in a, but for O-methyl-5-methyluridine. q, As in a, but for hydroxyazelaic acid. r, As in a, but for N1-methyl-imidazolelactic acid.
Extended Data Fig. 7 Isomers of annotated metabolites.
a, Methionine-C4:0. DeepMet annotated the peak as N-isobutyryl-methionine; N-butyryl-methionine was also synthesized. Left, extracted ion chromatograms (EICs) for both synthetic standards and mouse cecal contents. Middle, MS1 intensity of the corresponding peak across 23 mouse tissues and mirror plot showing the similarity between MS/MS spectra from N-isobutyryl-methinione versus the experimental spectrum from mouse cecal contents. Right, mirror plot showing the similarity between MS/MS spectra from N-butyryl-methinione versus the experimental spectrum from mouse cecal contents. b, As in a, but for histamine-C4:0. c, As in a, but for 2-sulfoglycerate versus 3-sulfoglycerate.
Extended Data Fig. 8 Examples of incorrect predictions.
From left to right, panels show: (1) putative structure assigned by DeepMet (top) and MS1 intensity of the corresponding peak across 23 mouse tissues (bottom); (2) lollipop plot showing the sampling frequencies of the top-15 most frequently generated molecules as a proportion of all sampled SMILES strings within ± 5 ppm of the query mass; (3) mirror plot showing the similarity between the MS/MS spectrum predicted by CFM-ID (top) versus the experimental spectrum from mouse tissues (bottom); (4) distribution of dot-products among all generated structures within ± 5 ppm of the query mass, with the dot-product for the structure in question marked by a black point; (5) extracted ion chromatograms (EICs) for the chemical standards, top, and in the corresponding peak in mouse tissues, bottom; and (6) mirror plot showing the similarity between MS/MS spectra from the chemical standards versus mouse tissues. a, The standard affords a partial match by MS/MS, but a key fragment at m/z 86.06 is missing, and there is a discrepancy in retention times. b, The standard matches the retention time of the mouse tissue peak, and all of the MS/MS fragment ions observed in the spleen are matched to the standard, but with different relative intensities. c, All major fragment ions match between the standard and mouse tissue MS/MS, but with different relative intensities, and there is a discrepancy in retention times. d, The standard matches by MS/MS to the mouse tissue peak, but there is a discrepancy in retention times. A spike-in experiment confirmed the presence of two distinct compounds. e, The standard matches by MS/MS to the mouse tissue peak, but there is a large discrepancy in retention times. f, Not a convincing match by either MS/MS or retention time. g, Not a convincing match by MS/MS.
Extended Data Fig. 9 Additional metabolite discoveries.
a-d, Additional previously unrecognized human metabolites predicted to exist by DeepMet, and assigned to peaks in the mouse tissue dataset, that were subsequently experimentally identified in human urine. Left, extracted ion chromatograms from the chemical standard and a representative urine metabolome; middle, mirror plots showing the similarity between MS/MS spectra from the standard versus the experimental spectrum from human urine; right, chemical structures of the predicted metabolites. Vertical red lines show the times of the MS/MS acquisitions. a, N-glutarylvanillylamine; b, β-carboline-propionic acid; c, N-lactoylmethoxytyrosine; d, N-benzoylmethionine. e, Overview of metabolites tentatively identified in public repositories by comparisons to reference spectra acquired in this study. Points show matches between reference spectra and experimental MS/MS spectra from human metabolomics experiments in MetaboLights or Metabolomics Workbench, as quantified by the dot-product. Left, metabolites also identified through comparisons to chemical standards under identical LC-MS/MS conditions in human or mouse; right, metabolites tentatively identified only by MS/MS search in public repositories. f-j, Mirror plots showing similarities between MS/MS spectra from chemical standards and experimental spectra from public repositories. f, Glutamyl-acadesine; g, desmethoxy-secoisolariciresinol; h, S-sulfocysteinylglycine; l, N-lactoyl-glutamine; j, N-succinyl-tryptophan.
Extended Data Fig. 10 Origins of selected metabolites.
Far left, inferred origins of each metabolite. Middle left, metabolite structures and positional labelling from the indicated infused 13C-labelled precursors. Positional labelling is manually inferred from the structure and experimental isotope labeling patterns in Supplementary Fig. 7b. Middle right, metabolite intensities in cecal contents from mice fed chow versus purified (casein) diets. Far right, metabolite intensities in the feces of mice treated with broad-spectrum antibiotics versus untreated controls. None, no significant difference (p < 0.05, two-sided t-test, and two-fold change).
Supplementary information
Supplementary information
This file contains Supplementary Figs. 1–9, Supplementary Note 2 and references.
Supplementary Note 1
Synthetic methods and NMR spectra
Supplementary File 1
Reference MS/MS spectra for previously unrecognized metabolites
Supplementary File 2
Reference MS/MS spectra for known metabolites
Supplementary Tables 1-5
Supplementary Table 1. Top-10,000 most frequently generated structures. Supplementary Table 2. Previously unrecognized metabolites. Supplementary Table 3. Known metabolites. a, Known metabolites purchased as part of the experiments shown in Extended Data Fig. 5. b, Metabolites revealed on review of the literature to be known metabolites missing from the HMDB. Supplementary Table 4. Accession numbers and raw data files for published metabolomic datasets re-analysed in this study. a, 4,510 runs from human blood; b, 35,460 runs across human tissues. Supplementary Table 5. All candidate structures generated by DeepMet that were considered as potential matches to the 25 peaks identified in mouse tissues, and the DeepMet confidence scores, cosine similarities between experimental spectra and spectra predicted by CFM-ID, and combined DeepMet + MS/MS scores for each
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiang, H., Wang, F., Lu, W. et al. Language model-guided anticipation and discovery of mammalian metabolites. Nature (2026). https://doi.org/10.1038/s41586-025-09969-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41586-025-09969-x
