
Abstract
The earliest morphologically identifiable dogs are from Europe and date to at least 14,000 years ago1,2,3,4,5, although early remains are also found in other regions. The origin of early dogs in Europe, and their relationships to other dogs, has remained elusive in the absence of genome-wide data. Similarly, although dogs were the only domestic animal to predate agriculture, little is known about how the arrival of Neolithic farmers from Southwest Asia affected the dogs living with European Mesolithic hunter-gatherers. Here we analysed 216 canid remains, including 181 from Palaeolithic and Mesolithic Europe. We developed a genome-wide capture approach that enriched endogenous DNA by 10–100-fold and could distinguish dog from wolf ancestry for 141 of 216 remains. The oldest dog data that we recovered are from a 14,200-year-old dog from the Kesslerloch site in Switzerland, and we find that it shares ancestry with later worldwide dogs—inconsistent with the hypothesis that European Upper Palaeolithic dogs derived wholly from a separate domestication process. The Kesslerloch dog already displays more affinity to Mesolithic, Neolithic and present-day European dogs than to Asian dogs, demonstrating that dog genetic diversification had started well before 14,200 years ago. We find a Neolithic influx of Southwest Asian ancestry into Europe, but this seems to have been of smaller magnitude than in humans, suggesting that Mesolithic dogs contributed substantially to Neolithic, and, ultimately, probably also modern, European dogs.
Similar content being viewed by others


Main
Dogs (Canis lupus familiaris) were domesticated from grey wolves (Canis lupus) towards the end of the last Ice Age, and were the first animals to enter into a domestic relationship with humans. Where in the world this happened, and which human group or groups were involved in the process of domestication, remains unknown6,7,8,9,10,11,12,13,14. The earliest known canid remains with probable dog morphology have been found in Europe, and include remains from the Bonn–Oberkassel2, Kesslerloch1, Le Morin3, Errala4 and Paglicci5 sites, all from the period 14,000–17,000 years ago (14–17 ka); however, putative dog remains within a few thousand years younger than these have also been found in East Asia15, the Levant16 and the Americas17, such that the archaeological record does not clearly point to any particular geographical region as the centre of domestication. Some much earlier remains have also been hypothesized to be dogs, but many morphological assignments remain contested in the absence of genetic data18.
Despite Europe having the earliest archaeological evidence of dogs, European wolves that lived during the Late Glacial period (~23–12 ka), when dogs probably arose, have not been found to have contributed detectably to dog ancestry14. Instead, dogs overall show stronger genetic affinities to Late Glacial wolves from eastern Eurasia14. But further to this, dogs exhibit some degree of variation in their genetic relationships to ancient wolves, implying a ‘dual ancestry’ model in which at least two distinct wolf populations must have contributed ancestry to dogs14. In particular, dogs in Southwest Asia and Africa show an affinity to wolves in Southwest Asia, suggesting a second source from there14. The identities of the wolf source populations, whether they imply a single or multiple domestication processes, and their relationships to the earliest dogs in Europe, remain undetermined.
With the exception of Mesolithic dogs from Karelia in northeastern Europe13,19, previous genome-wide analyses of European dogs have been restricted to the Neolithic period and later8,12,13. DNA analyses of older putative dogs have been restricted to mitochondrial DNA, which does not always reliably distinguish wolves from dogs10. Neolithic and later European dogs have been found to be genetically intermediate between East Eurasian dogs on the one hand, and Southwest Asian and African dogs on the other13. The genomic ancestry of dogs in Europe predating the Neolithic, in the Upper Palaeolithic and Mesolithic periods—including the probable dog remains dating to 14–17 ka—remains unknown. It is also unknown how the Neolithic transition, which involved large-scale migrations of people and domestic animals into Europe from Southwest Asia, affected the dog populations already present in Europe during the Mesolithic.
Targeted genome-wide DNA enrichment
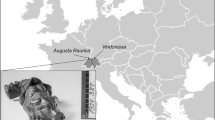
We extracted and analysed DNA from 216 canid skeletal remains (Supplementary Data 1), at least 181 of which come from pre-Neolithic contexts in Europe (Fig. 1a). Some of these remains might derive from the same biological individual (Supplementary Data 5). Our sampling included remains from the Kesslerloch site, Switzerland (14.2 ka, Magdalenian culture, n = 104), the Gnirshöhle site, Germany (~15 ka, Magdalenian, n = 35), Goyet and various other cave sites in Belgium (46–2 ka, n = 12), the Aghitu-3 cave in Armenia (31–30 ka, Upper Palaeolithic, n = 3), the Boncuklu Höyük site in Turkey (11.4 ka, Neolithic, n = 1), a canid found close to the Senckenberg Natural History Museum, Germany (11.1 ka, no cultural context, n = 1), the site of Hardinxveld-Giessendam de Bruin, Netherlands (~7 ka, Mesolithic, n = 7), seven sites in southern Sweden (10–5 ka, Mesolithic Kongemose and Ertebølle, and Neolithic Pitted Ware cultures, n = 9), 20 sites in Denmark (9–5 ka, Mesolithic Kongemose and Ertebølle, and Neolithic, n = 32) and three sites in Scotland (5 ka, Neolithic, n = 4) (Supplementary Data 1). We obtained eleven new radiocarbon dates (Supplementary Data 2).

a, Geographical origins of canid remains and sites analysed in this study, adapted from a Natural Earth base map (http://naturalearthdata.com). b, Endogenous content before and after hybridization capture. c, Testing for dog ancestry by computing the outgroup f3-statistic f3(X, Basenji; Wolf35Xinjiang), which quantifies the amount of genetic drift shared with the modern Basenji dog. A value above the dotted line confidently indicates that an individual is a dog. Error bars are ±2 standard errors. The background colour indicates the most comprehensive type of data generated for a given sample.
We screened remains for DNA preservation and found generally low levels of endogenous DNA in pre-Neolithic remains: 95% of remains from the Upper Palaeolithic and Mesolithic periods had less than 5% endogenous DNA, and 79% of remains had less than 1% endogenous DNA. To overcome this, we turned to hybridization capture of single-nucleotide variants (SNVs), an approach that has proven effective in improving DNA recovery in human studies20.
Following the expectation that ascertainment in an outgroup should enable largely unbiased ancestry inference21, we identified about 311,000 transversion SNVs that are heterozygous in a present-day coyote and also polymorphic among present-day grey wolves. We also included approximately 178,000 SNVs from commonly used dog genotyping arrays, to ensure overlap with existing datasets. The final array design included 486,547 variants, and we applied the capture to 138 remains (Methods). Most remains saw a 10–100-fold enrichment in endogenous content after capture (Fig. 1b), and we see clear enrichment of DNA at the targeted sites (Extended Data Fig. 1), and evidence of some degree of DNA preservation in most if not all remains (Supplementary Data 1). We combined the generated data with previously published modern and ancient canid genomes (Supplementary Data 3).
For seven individuals, we performed deeper whole-genome sequencing with at least one billion sequences each: these were from the Mesolithic sites of Bökeberg (13× coverage) and Tågerup (4×, 0.1×) in Sweden, the Neolithic sites of Tulach an t’Sionnaich (18×), Tulloch Of Assery (0.4×) and Cuween (0.8×) in Scotland, the Late Glacial La Fru rock shelter in France (0.04×) and the Neolithic Boncuklu site in Turkey (0.8×). We found that capture data produce ancestry results that are highly similar to those obtained using shotgun data (Extended Data Fig. 2), although it is possible that there are still batch effects to some degree between the data types22. Although environmental sources of microbial DNA are hard to exclude, we also detected microbial DNA from opportunistic pathogens known to infect dogs in several samples (Extended Data Table 1 and Supplementary Data 6). Among these, Erysipelothrix rusiopathiae has been linked to endocarditis23, Tannerella forsynthia to periodontal disease24 and Clostridium perfringens to acute diarrhoea in dogs25.
Genetic identification of early dogs
We tested whether the genetic ancestry of each sample is consistent with that of a dog or wolf by quantifying the amount of genetic drift shared with a present-day dog (the African Basenji). We found that this test clearly separates known dogs and wolves, and that data on only about 500 out of the captured 486,547 variants are sufficient to distinguish dogs from wolves. We successfully obtained this resolution, and a dog or wolf identification for 141 out of the 216 analysed remains (133 out of 138 for remains subjected to capture) (Fig. 1c). Strictly speaking, genetic data cannot rule out that any of the individuals we classify as wolves had been domesticated independently and thus could also be considered dogs in some sense, but in this work we only intend the word ‘dog’ to refer to members of the known C. lupus familiaris population.
In Mesolithic Sweden, out of eight samples with sufficient data, we find that all are dogs. At Mesolithic and Neolithic sites in Denmark, we identify 21 dogs and five wolves, the latter being from the Ertebølle, Ramløse and Halleby Å sites and previously morphologically identified as wolves according to museum records. At the Mesolithic site of Hardinxveld in the Netherlands, we identify four dogs and one wolf. We confirm that a canid found close to the Senckenberg Museum in Frankfurt, Germany—which we radiocarbon date to 11.1 ka—is a dog. All four analysed Scottish Neolithic samples are dogs. In the cave sites in Belgium, France and Armenia, we only identify wolves. The presence of wolves alongside dogs in several Mesolithic human settlements in northwestern Europe is consistent with zooarchaeological records rich in wolves and other carnivores in this period26, with hunting being the most parsimonious explanation26.
We find that a canid from the Goyet cave in Belgium, directly dated to 13.7 ka and previously suggested to be a dog on the basis of its small size in combination with traces of human modification and red-coloured stains27, has fully wolf-like ancestry. This finding highlights the importance of genetic confirmation of hypothesized dog remains, but we also note that genetic data cannot rule out that this, or other wolves, could have interacted with humans in a variety of ways (for example, taming).
We identified eight remains displaying dog ancestry from the sites of Předmostí (Czechia), Goyet (Belgium) and Gnirshöhle (Germany) that all hold Upper Palaeolithic material culture; however, in every case, radiocarbon dating or analysis of the genetic data demonstrates either later deposition of dog bones at these sites in the past few thousand years, or DNA contamination from modern dogs (Methods). Similarly, some of the identified dogs from the Dutch and Danish Mesolithic and Neolithic sites display ancestry typical of more recent dogs, suggesting later deposition. We also directly radiocarbon dated a dog from the Gökhem site in Sweden, previously thought to be of Neolithic age but displaying ancestry more similar to recent European dogs13, and found it dates to the Late Iron Age (1.1 ka).
Among the analysable remains from the Kesslerloch site (some of which might derive from the same individual), we find that 62 have wolf ancestry and one is from a dog. The specimen we identify as a dog genetically was previously proposed to be a dog on the basis of its morphology1, and has been radiocarbon dated to 14.2 ka. Our genetic results thus confirm that this individual is a dog, thereby pushing back the record of dogs confirmed by genome-wide genetic analyses to the Upper Palaeolithic.
Shared origin of European and other dogs
The fact that early European dogs are identifiable through their shared genetic drift with present-day members of the C. lupus familiaris dog lineage (Fig. 1c) reveals that they cannot derive from an independent source population of wolves. If they shared no ancestry with other dogs, they would look no different from wolves in these analyses. As further evidence for a shared origin, in a principal component analysis (PCA) based on projecting dogs into a space defined by modern wolf diversity (Supplementary Data 4), Upper Palaeolithic and Mesolithic European dogs cluster among previously analysed dogs, implying their relationship to wolves is similar to that of other dogs (Fig. 2a).

a, A PCA constructed using modern wolf genomes, with dogs then projected into the resulting space. The right-hand image magnifies the dog cluster. Dogs fall on a west-to-east cline, with European pre-Neolithic dogs falling on the eastern side of that cline. b, Results from qpAdm, testing whether various dog targets are compatible with fully eastern progenitor ancestry (using the Siberian Zhokhov as a representative) or require a contribution from a western progenitor (using a present-day Syrian wolf as proxy). c, Heterozygosity in dog genomes, estimated by sampling two reads per site. Error bars represent ±2 standard errors. PWC, Pitted Ware Culture.
Under the previously proposed dual ancestry model, dogs derive ancestry from two distinct wolf sources: an eastern progenitor that probably lived somewhere in eastern Eurasia, and a western progenitor that probably lived somewhere in western Eurasia—perhaps in Southwest Asia14. All dog genomes analysed so far have some amount of eastern progenitor ancestry, whereas only some dogs seem to have western progenitor ancestry. The largest proportion of western progenitor ancestry observed so far is in Neolithic dogs in Southwest Asia, which trace approximately half of their ancestry to this source, with smaller proportions in Neolithic and later European dogs14. The east–west cline in the proportions of eastern and western progenitor ancestry carried by different dogs is visible in their projection onto the wolf PCA (Fig. 2a). In this cline, we find that pre-Neolithic European dogs tend to fall on the eastern side—similarly to East Asian and Arctic dogs—rather than together with Southwest Asian and African dogs. Consistent with this, f4-statistics demonstrate that pre-Neolithic European dogs—like dogs generally14—have stronger affinities to Late Pleistocene Siberian wolves than to the European wolves they were contemporary with (Extended Data Fig. 3). In contrast to nearby dogs in the Levant and Iran, the Neolithic Anatolian Boncuklu dog does not fall towards the western extreme of the wolf PCA cline, and instead projects alongside early European dogs (Fig. 2a).
We next used the qpAdm framework28 to more formally model how different dogs fit into the dual ancestry model. We used a 9,500-year-old dog from Zhokhov island in the East Siberian Sea29 to represent a baseline of likely fully eastern progenitor ancestry. We asked whether a single-source eastern progenitor model can explain the ancestry of a given target, or whether a contribution from a second, western Eurasian wolf is needed (we use a present-day Syrian wolf as a proxy for this western progenitor). We find that the single-source Zhokhov model fits or nearly fits for all the pre-Neolithic European dogs analysed here, including the 14,200-year-old Kesslerloch dog (Fig. 2b and Supplementary Data 7). This implies that the ancestry of these early dogs in Europe is consistent with deriving from the same eastern progenitor source as dogs in Siberia, East Asia and Australasia, and that no additional contributions from western Eurasian wolf sources are required to explain the data. Surprisingly, given that dogs in the nearby Levant 7 ka had the largest western progenitor contribution observed so far14, the Boncuklu dog in Anatolia is also consistent with having entirely eastern dog progenitor ancestry, suggesting that the western progenitor ancestry present in Neolithic Levant and Iran might not have been present in Anatolia 11.4 ka.
The Kesslerloch dog, and especially the Boncuklu dog, do display a slight westward shift in the exploratory analyses (Fig. 2a and Extended Data Fig. 3), raising the possibility that they could have a small contribution of western wolf ancestry that is outside the detection power of qpAdm. Regardless, we can rule out a substantial western wolf contribution, and conclude that most of the ancestry of these Palaeolithic European and early Neolithic Anatolian dogs derives from the same wolf source as that of dogs in eastern Eurasia. The finding that the Kesslerloch dog shares ancestry with other worldwide dogs also allows us to reject the hypothesis that the Palaeolithic dogs in Europe derived wholly from a separate domestication process, independent from dogs in Asia8.
Dogs overall show a reduction in genetic diversity levels of about one-third relative to Late Glacial wolves14, but it is not known whether this loss of diversity occurred early during the domestication process itself, or during later episodes of dog population history. We quantified conditional heterozygosity at captured SNVs, and find that the lower diversity levels typical of dogs was already in place in Upper Palaeolithic and Mesolithic European dogs, including in the Kesslerloch dog 14.2 ka (Fig. 2c). This observation strengthens the hypothesis that the domestication process itself was responsible for the diversity loss.
Genetic affinities of pre-Neolithic dogs
To explore the genetic relationships of pre-Neolithic dogs in Europe to later and present-day European and worldwide dogs, we ran the model-based clustering software ADMIXTURE30 (Fig. 3a and Extended Data Fig. 4). This results in what we refer to as ancestry components, which should not be viewed as population genetic estimates of ancestry, but rather as exploratory evidence of affinities between individuals. We find that all Upper Palaeolithic and Mesolithic European dogs share an ancestry component (shown in red in Fig. 3a) that is not assigned to any dogs elsewhere in the world. This red component is gradually replaced in Neolithic and later European dogs, initially by a component maximized in ancient Southwest Asia, and eventually completely by a component that is maximized in present-day European dogs. These results suggest a largely shared ancestry of pre-Neolithic dogs in Europe, at least in the regions of northwestern Europe covered by our dataset, as well as some degree of continuity with later Neolithic dogs.

a, Results of clustering with ADMIXTURE (k = 7), with each colour representing a modelled ancestry component. Bolded samples are new to this study. b, Contrasting the affinities of global dogs to a 7,200-year-old dog from the Levant (Tel Hreiz) versus a 9,500-year-old dog from Siberia (Zhokhov) reveals a cline. c, Contrasting the affinities of global dogs to the 14,200-year-old Kesslerloch dogs versus a 7,800-year-old Mesolithic Scandinavian dog reveals a largely linear relationship, but Southwest Asian dogs depart from this relationship. Error bars for ancient genomes denote ±2 standard errors (See also Extended Data Fig. 5).
The 14,200-year-old Kesslerloch dog, the earliest in our dataset, is the only pre-Neolithic dog in Europe that is modelled as sharing ancestry with ancient dogs from Southwest Asia—about half of its ancestry is assigned to a component maximized in Neolithic and later dogs in the Levant and Iran. Meanwhile, the 11,400-year-old Boncuklu dog from Anatolia is the only Southwest Asian dog that is assigned a considerable amount of the Mesolithic European component, displaying an overall ADMIXTURE profile similar to that of the Kesslerloch dog. This hints at an early connection between dogs in Southwest Asia and Upper Palaeolithic Europe that, by 7 ka, is no longer visible; however, these ADMIXTURE results do not inform on the directionality or temporal depth of any genetic connectivity that would have given rise to this pattern.
For a few pre-Neolithic European dogs, the ADMIXTURE software assigns parts of their ancestry to other components. Dogs from the Mesolithic Veretye culture in Karelia13,19, dated to 10.9 ka, are assigned one-third of their ancestry to a component found in Siberian and North American dogs. This component is absent in the Swedish and Danish Mesolithic dogs, demonstrating a greater eastern affinity in the Karelian dogs, which would mirror differences in human ancestry between Scandinavian and eastern hunter-gatherers28,31.
Dogs associated with the Neolithic Pitted Ware hunter-gatherer culture in Scandinavia (Ajvide and Möllehusen sites) are the most recent dogs in our dataset that are assigned all of their ancestry to the Mesolithic ancestry component. Our results demonstrate over three millennia of ancestry continuity in hunter-gatherer Scandinavia, between our earliest Mesolithic Scandinavian dogs at around 8 ka and the most recent Pitted Ware Culture dogs 4.8 ka. Like the humans that they lived alongside32,33, these Pitted Ware Culture dogs probably represented some of the last populations with ancestry profiles typical of pre-agricultural Europe.
Deep genetic structure in Europe
The 14,200-year-old Kesslerloch dog is several thousand years older than other dog genomes published so far, and therefore of particular interest for understanding early dog history. Global dog diversity has been found to be empirically well-described by a cline stretching between eastern Eurasian dogs on one end, and African and Southwest Asian dogs on the other, with European dogs intermediate13. We find that the Mesolithic Scandinavian dogs fall towards the eastern side of this cline, but that the Kesslerloch dog behaves very differently (Fig. 3b). The Kesslerloch individual is the only dog to fall off the cline, displaying low amounts of shared drift with both the eastern Eurasian and the Southwest Asian dog populations.
We next directly contrasted Kesslerloch with a Mesolithic Scandinavian dog, and asked how much genetic drift other worldwide dogs share with each of these (Fig. 3c). This reveals a largely linear relationship, but almost all dogs are heavily shifted off-diagonally towards the Scandinavian dog. This is even true of dogs geographically distant from Europe, including dingoes and North American dogs, both of which are closer to Mesolithic Scandinavia than to Kesslerloch. This again indicates a divergent ancestry in Kesslerloch, and implies either that there has been some amount of shared ancestry connecting virtually all dogs in the world after the divergence of Kesslerloch from other dog ancestors, or that Kesslerloch received a portion of ancestry by admixture from a lineage basal to other dogs. The Anatolian Boncuklu dog (11.4 ka) displays a similarly divergent profile, although less pronounced than in Kesslerloch (Extended Data Fig. 5a).
There are two exceptions to the linear relationship in the Kesslerloch–Scandinavia contrast (Fig. 3c). First, dogs in Southwest Asia and Africa depart from the trend line and display excess affinity to Kesslerloch. Second, the early Neolithic Anatolian Boncuklu dog also falls off the trend line, but shows much higher affinity to Kesslerloch than other Southwest Asian dogs do. These Southwest Asian, African and Neolithic Anatolian dogs are the only dogs that are almost equally similar to Kesslerloch as to Mesolithic Scandinavia. This mirrors the connection between Kesslerloch and ancient Southwest Asia, and in particular between Kesslerloch and Boncuklu, observed in the ADMIXTURE clustering results (Fig. 3a).
One possibility is that populations related to Kesslerloch were a partial source of ancestry for dogs in Southwest Asia, and another possibility is that earlier dogs in Southwest Asia were a source of ancestry for Kesslerloch—our data do not allow us to dissect the directionality of this affinity. But it does set Kesslerloch apart from later dogs in Europe, as by the Neolithic, this connection to Southwest Asia is not as pronounced. One hypothesis that could explain both the basal position of Kesslerloch (Fig. 3b) and the weakened Southwest Asian affinity in Mesolithic Europe (Fig. 3c) is that some more easterly ancestry, perhaps first observed in the Karelian Veretye dogs, came into Europe after 14.2 ka and mixed with Kesslerloch-like dogs to produce the ancestry that characterizes the Mesolithic at around 8–10 ka. Filling the current gap in genomic sampling between 14 and 10 ka will be needed to better understand this change in European dog ancestry.
Although the Kesslerloch dog displays a divergent ancestry profile, our results also show that it is not a strict outgroup to later dog diversity. First, Kesslerloch shares more genetic drift with Boncuklu than with any other dog. Second, Kesslerloch clearly shares more drift with later dogs in Europe than with dogs in Asia (Fig. 3c). This can also be seen in f4-statistics directly contrasting affinities to European versus eastern dogs living today, for example, f4(Andean Fox, Kesslerloch; German Shepherd, New Guinea Singing Dog) = −0.014, Z = −15.6. Our results thus demonstrate that the differentiation of European dog ancestry had already occurred by 14.2 ka, and that there has been at least some degree of ancestry continuity in European dogs since then to the present day.
Effects of the Neolithic transition
In humans, ancient DNA studies have shown that the onset of the Neolithic transition and the arrival of farming in Europe was driven by large-scale migration of people from Southwest Asia28,34,35,36. Dogs were the only domestic animal that existed in Europe before the Neolithic, but the extent to which dogs experienced a similarly large-scale ancestry replacement is not known. An influx of Southwest Asian dog ancestry has been suggested on the basis of mitochondrial DNA37 and ancestry clines among Neolithic dogs13, but direct genome-wide data to address this question have not been previously available. The ADMIXTURE results show a gradual replacement of the Mesolithic ancestry component, starting in the Neolithic with a component that peaks in Southwest Asian dogs (Fig. 3a). This could be consistent with a history similar to that of humans, but the exploratory nature of this method means it is difficult to draw firm conclusions about an influx of ancestry.
To test for an influx of Neolithic ancestry more formally, we used qpAdm to model the ancestry of European dogs post-dating the arrival of Neolithic groups as a combination of sources drawn from pre-Neolithic Europe (Ertebølle, Denmark; Bökeberg, Sweden; Veretye, Karelia; Kesslerloch, Switzerland) and Neolithic Southwest Asia (Tel Hreiz, Levant; Boncuklu, Anatolia), with individuals from Siberia, North America and Oceania as reference populations. We find that a Southwest Asian contribution is needed as a source to fit the ancestry of nearly all available Neolithic European dogs (Fig. 4a and Supplementary Data 8), but with the sizes of those contributions varying greatly (Fig. 4b and Extended Data Fig. 6). A 7,000-year-old dog from the Herxheim site fits equally well without as with a 13% contribution. The estimates of Southwest Asian ancestry are 21–25% for three Scottish Neolithic dogs, and 33–34% for three Croatian and Serbian Neolithic and Eneolithic dogs. The highest values are obtained for Neolithic dogs in southern Europe—66% in a dog from Greece and 40% in a dog from Spain—and a Late Neolithic (Corded Ware) dog from Germany with 53%. Overall, these Neolithic ancestry proportions are substantially lower than in humans, where early Neolithic groups in Europe typically had at least 70–80% Southwest Asian ancestry38.

a, qpAdm results for dog targets living around or after the arrival of the Neolithic in Europe. Each dot is a model with different combinations of sources, and a low P value indicates poor model fit. If any model with n sources fits with P > 0.01, models with >n models are not plotted. b, Ancestry proportions inferred by qpAdm, for the best-fitting single-source model or a fixed, two-source model using Ertebølle (Denmark) and Tel Hreiz (Israel) as representatives of Mesolithic European and Neolithic Southwest Asian source populations, respectively. Error bars represent ±2 standard errors (refer to Extended Data Fig. 6 for analyses of the robustness of the estimated proportions to choices of sources and genetic variant sets).
Most of the dogs from Mesolithic and early Neolithic sites in Denmark are consistent with lacking Neolithic Southwest Asian ancestry, consistent with a relatively late Neolithic ancestry turnover in humans in Denmark39. Both dogs from the Neolithic–Mesolithic transitional site of Syltholm were consistent with having little or no Neolithic Southwest Asian ancestry, mirroring human DNA from chewed birch pitch at the site showing completely Mesolithic ancestry40. The only Danish dogs with confidently detectable Neolithic ancestry are from the Middle Neolithic site of Bundsø (estimated contributions of 18–19%), demonstrating an arrival of that ancestry to Denmark at least by about 5,000 years before present (bp; although we caution that these Bundsø dogs are not directly dated). The Swedish Neolithic Pitted Ware Culture dogs are consistent with entirely Mesolithic ancestry, or a small Neolithic contribution (8.5% in one individual).
In humans, the Neolithic transition in Europe was associated with an increase in genetic diversity, with heterozygosity levels at around 20–40% higher in Neolithic farmers than in Mesolithic hunter-gatherers32,35. In dogs, we do not find the same pattern, as pre-Neolithic dogs in Europe generally have similar heterozygosity levels to Neolithic dogs (Fig. 2c). Only in the Late Neolithic and later periods do European dogs display higher heterozygosity levels. We find a very low level of diversity in the dog from the early Neolithic Anatolian Boncuklu site, not mirroring human Anatolian farmers who generally had higher diversity levels than European hunter-gatherers41, but we caution that with just one genome we cannot say how representative this is of Neolithic Anatolian dogs.
Overall, our results demonstrate that dogs qualitatively mirror the demographic processes seen in humans in that the Neolithic period brought an influx of ancestry from Southwest Asia, but that the overall effect of this was of a smaller magnitude in dogs. This suggests that dogs from local hunter-gatherer groups contributed substantially to the dog populations that lived with Neolithic farmer communities in Europe. The legacy of Mesolithic European dogs is substantial, and has seemingly been maintained until the present day. Modern European dogs still largely fall within the diversity established during the Neolithic13, and in the dog ancestry cline they fall roughly halfway between Mesolithic European and Neolithic Southwest Asian dogs (Fig. 3b), implying that they might trace approximately half of their ancestry to the pre-agricultural dogs of Europe.
Conclusion
Our understanding of dog history before the spread of agriculture primarily relies on morphological assessments, but these are often contentious. Furthermore, even if a morphology distinct from wolves can be established for a given individual, it is still difficult to confidently conclude that it belongs to the same lineage as the domestic dogs that we know today (C. lupus familiaris). Our results demonstrate the value of genome-wide nuclear DNA analysis in distinguishing members of the domestic dog lineage from wolves. By making use of capture technology, we were able to make a dog versus wolf ancestry call for two-thirds of the tested specimens, and to confirm dog ancestry for at least 14 new individuals from European hunter-gatherer contexts.
The observation that the Kesslerloch dog already shows higher affinity to later dogs in Europe than those in other regions demonstrates that population structure in dogs is at least 14,200 years old. This pushes back the time depth of dog genetic diversification supported by genome-wide ancient DNA past the 10.9 ka previously demonstrated13. Although this result does not impose any strong constraints on the timing of dog domestication, it seems reasonable to assume that domestication would have occurred several millennia before 14.2 ka, to allow time for this structure to emerge. Beyond this, our results cannot resolve where in the world, and how many times, dogs were domesticated.
How dogs spread across Europe and the rest of the world, and how this fits into the human history of the Upper Palaeolithic, remains unclear. The Kesslerloch site is associated with the Magdalenian technocomplex, as are several other sites with putative early dog remains in Europe1,2,3,4. The period around 14 ka is one of substantial change in human populations, with large-scale and rapid replacement following the spread of the so-called Villabruna hunter-gatherer ancestry42,43. The expanding Villabruna ancestry had an elevated affinity to populations in Southwest Asia44,45, in that sense mirroring our findings on the Kesslerloch dog. Coincidentally, the earliest observation of the Villabruna ancestry north of the Alps is in individuals from the Bonn-Oberkassel site in Germany 14 ka (ref. 43), where they were buried alongside one of the earliest known canids with clear dog morphology2 (although nuclear genetic data are not available from this probable dog). A tempting hypothesis is thus that dogs spread through Europe with, or just ahead of, the Villabruna expansion; however, this hypothesis would not fully align with remains suggested to be dogs on morphological grounds in Europe as early as 17 ka (ref. 4), and leaves question marks in terms of where people in Europe ultimately obtained dogs.
Dogs were the only domestic animal that existed in Europe before the Neolithic, meaning their history can provide unique insights into how farming societies from Southwest Asia spread across the continent. Our results show that rather than replacing local Mesolithic dogs, incoming farmers incorporated them to a large extent. This contrasts with other dramatic expansions in human history, such as the arrival of Europeans to the Americas in the colonial era, in which European dogs rapidly and almost completely replaced native American dogs13,46. Our results suggest a less sudden and violent process in Neolithic Europe, consistent with admixture and interactions between farmers and hunter-gatherers ongoing for several centuries in many parts of the continent38. The smaller Neolithic effect on dogs is also reflected in how they do not mirror the genetic diversity increase seen in humans. The persistence of Mesolithic dog ancestry in the Neolithic might imply that hunter-gatherers and farmers used dogs for at least partially overlapping purposes, but we stress that genetic data cannot reveal the social and economic processes underlying the admixture between Mesolithic and Neolithic dogs. In any case, the modern European dog population probably traces a substantial portion of their ancestry to the dogs that lived in Europe before the advent of agriculture.
Methods
Radiocarbon and contextual dating
We generated eleven new radiocarbon dates (Supplementary Data 2). Dates were calibrated using the IntCal20 calibration curve, using the OxCal v.4.4 software. For samples where δ¹³C values were obtained independently of accelerator mass spectrometry using high-precision stable-isotope mass spectrometry—providing dietary information—no high values indicative of potential marine diets were observed, and therefore no corrections for marine reservoir effect were applied. The previously published C88 dog from the site of Frälsegården, Gökhem in southwestern Sweden had been attributed to a Neolithic cultural context and an approximate age of 5,000 years bp13, but the new radiocarbon date of 1,154 calibrated years bp instead places it in the Late Iron Age or Viking Age.
For remains that did not have radiocarbon dates, we assigned dates usable in analyses on the basis of available archaeological context. The ‘Analysis date’ column in Supplementary Data 3 holds the best available single number date estimate, with a value of ‘NA’ if we were not able to confidently assign a date.
Sampling, DNA extractions and library preparation
When sampling skeletal remains for DNA, in most instances we aimed to take one sample per biological individual. In some instances, samples were obtained from different remains that possibly could derive from the same individual, based on the zooarchaeological context (in particular this was the case for the Kesslerloch site). In a few cases in which it was unambiguous that data from multiple remains came from the same individual, we merged those data. But in most cases, we treated data from different remains separately, to err on the side of caution and not incorrectly merge data. The true number of sampled individuals is thus not precisely known, but we provide a table listing the groups of remains where archaeological information suggest they might derive from the same individual (Supplementary Data 5).
University of Tübingen
A number of samples had already been processed in the cleanroom facilities of the University of Tübingen and analysed for DNA preservation, as previously described47. In brief, samples were subjected to UV radiation for 30 min, followed by DNA extraction48 and double-stranded library preparation49. For the current project, these double-stranded libraries were transferred to the Max Planck Institute for Evolutionary Anthropology, where they were enriched for targeted canid single nucleotide polymorphisms using an in-solution capture as described below.
Max Planck Institute
Material from the Kesslerloch site was sampled and processed into powder at the cleanroom facilities of the Institute of Evolutionary Medicine, University of Zurich. All bone and teeth were decontaminated via UV irradiation for 30 min in a cross-linker on at least two sides before drilling. For some specimens, the external surface was lightly ablated by hand with a dentist drill to access more sterile tissue. Samples were then drilled using a dentist drill with sterilized bits to produce powder. The powders were then transferred to the Max Planck Institute for Evolutionary Anthropology for further processing. Three samples of the Senckenberg dog (a vertebra, a foot phalanx and a rib), and 25 new specimens from Gnirshöhle, were sampled in the designated clean room laboratories at the Max Planck Institute for Evolutionary Anthropology, in which they were decontaminated under ultraviolet irradiation for 30 min on each side and drilled with a sterile dentist drill to produce bone powder. DNA extractions for all of the aforementioned samples were then performed at the Max Planck Institute.
We extracted DNA from between 6 mg and 104 mg of the powdered sample applying a silica-based method optimized to recover short DNA fragments48,50. In brief, lysates were prepared by adding 1 ml lysis buffer (0.45 M EDTA at pH 8.0, 0.25 mg ml–1 proteinase K, 0.05% Tween-20) to the sample material in a sterile 2.0 ml BioPure Eppendorf tube, which was rotated at 37 °C overnight. DNA was purified from 125 µl of the lysate using silica-coated magnetic beads and the binding buffer D (ref. 50) on an automated liquid-handling system Bravo NGS Workstation B (Agilent Technologies). Purified DNA extracts were eluted in 30 µl Tris–EDTA–Tween buffer (TET). Extraction blanks without sample material were processed with the samples.
Single-stranded DNA libraries were built using a protocol adapted for automated library preparation51,52 on the Bravo-B NGS Workstation (Agilent Technologies). In short, all DNA molecules in the complete volume of the DNA extract (30 μl) were dephosphorylated at the 5′ and 3′ ends and denatured into single strands by heating. A 3′-biotinylated synthetic DNA adaptor was attached to the 3′ ends of the DNA fragments using T4 DNA ligase and a splinter oligonucleotide carrying a sequence of six random nucleotides. Ligation products were then immobilized on streptavidin-coated magnetic beads and splinter oligonucleotides were removed by washing beads at elevated temperature. The second DNA strand was synthesized using the Klenow fragment of Escherichia coli DNA polymerase I. The unincorporated primers were removed during a bead wash at elevated temperature. After the blunt-end ligation of the second DNA library adaptor, the final synthesized strand was released from the beads by heat denaturation. Extraction blanks were turned into single-stranded DNA libraries alongside the samples. DNA libraries were diluted tenfold and quantified via qPCR in the LightCycler 96 (Roche).
DNA fragments in each library were extended via PCR with a sample-unique pair of indices in 100 μl reactions using AccuPrime Pfx DNA Polymerase (Thermo Fisher Scientific) and amplified to plateau to enable simultaneous sequencing of multiple libraries later on. Indexed libraries were purified using the solid-phase reversible immobilization method with magnetic beads, eluted in 20 µl Tris–Tween buffer (EBT) and quantified using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific). Half of the indexed purified library was amplified again for 15 cycles in a 100 µl reaction using Herculase II Fusion DNA Polymerase (Agilent Technologies) to increase the concentration and were purified with the SPRI bead approach. Before sequencing, DNA libraries were normalized to the final concentration of 200 ng μl–1, reconditioned with a single PCR cycle and pooled in equal volumes. Library pool was purified over silica columns using the MinElute PCR Purification Kit (QIAGEN N.V.), and eluted in a 30 µl EBT. The pool was quantified using the Agilent TapeStation System 4200 (Agilent Technologies). Equimolar pools of libraries and of extraction blanks were shotgun-sequenced on the Illumina HiSeq 4000. Five million reads (single-end, 75 cycles) were generated for each library and analysed to obtain basic quality metrics.
The Francis Crick Institute
For material processed at the Francis Crick Institute, powders were obtained using an Emax EVOlution fine dentistry drill in a cleanroom. Cementum samples were taken by removing a thin layer of powder from the surface of the tooth root, while the dentine was accessed by drilling a small hole directly into the root to access and hollow out the pulp cavity. Petrous bones were sampled by drilling a hole directly into the bone to access the cochlea, and the cortical bone was sampled on postcranial material. The initial lysis extraction protocol was performed manually, where extraction buffer (HPLC water, 0.5 M EDTA pH 8.0, Tween-20, 0.25 mg ml–1 Proteinase K; ref. 51) was added to the powders based on weight: 300 µl for <10 mg of powder, 500 µl for 10–15 mg, 700 µl for 15–25 mg and 1,000 µl for >25 mg. These were incubated and rotated for 24 h at 37 °C before being centrifuged for 2 min at 13,200 r.p.m. Then, 140 µl of the supernatant was transferred into LVL tubes with 10 µl EBT (HPLC water, 1 M Tris–HCL pH 8.0, Tween 20; ref. 51) where automated extraction, indexing and library preparation on an Agilent Bravo Workstation was then undertaken.
For the samples from Denmark, sampling and DNA extraction was performed in the ancient DNA facility of the PalaeoBARN laboratory at the University of Oxford. Sampling was performed by removing a small area of the outer surface layer, cutting off a small piece and then pulverizing it with a mixer mill to produce 50–70 mg of bone powder. DNA extraction was performed following the Dabney protocol48 but with the addition of 30 min pre-digestion. The DNA extracts were transferred to the Francis Crick Institute for further processing.
All samples, including those extracted in Oxford, were prepared as single-stranded libraries and amplified with unique double index combinations50,53. A qPCR quality control step was undertaken in between library preparation and indexing, using a QuantStudio 7 Flex machine. The sample plates were processed with negative extraction controls and positive and negative library controls. All samples underwent paired-end sequencing with a 2 × 100 paired-end read configuration on Illumina HiSeq4000, NovaSeq 6000 and NovaSeqX platforms.
Capture design
Single-nucleotide variants from the nuclear genome to include on the capture were taken from three sets:
-
1)
SNVs ascertained for population genetics. We first identified transversions SNVs that are heterozygous in a single high-coverage coyote genome (Coyote01 from California, BioSample: SAMN02921301). Ascertainment in an outgroup should ensure unbiased population genetic inferences21,54. To reduce the number of variants that are monomorphic in dogs, we further ascertained the list by requiring a minor allele count of at least 2 in a set of 44 Eurasian grey wolves. This set had 311,037 SNVs.
-
2)
SNVs found on commonly used dog genotyping arrays. We downloaded the list of variants used by the Embark company for a dog genome-wide association study55 and excluded variants that cannot be unambiguously strand-flipped (C/G and A/T variants). This set had 178,357 SNVs.
-
3)
A small number of SNVs hand-picked due to potentially being associated with trait variation in dogs, taken from previous studies56,57. This set had 273 SNVs.
We merged the above three sets, which overlapped to some degree, and then designed probes. For each targeted SNV, we designed four probes, each of 52 base pair (bp) length, by extracting sequences from the canFam3.1 reference genome. Two probes of 52 bp centre on the SNV, but carry the two different alleles at the SNV. The other two probes end 1 bp upstream and start 1 bp downstream of the SNV, respectively. We excluded SNVs where any of the four probes contained a 15-mer that was found more than 100 times in the reference genome, or contained N’s. The final set contained 486,547 SNVs. An additional 8-bp linker sequence (CACTGCGG) was added to each probe, and the complete set of probes was ordered on two Agilent SureSelect DNA Capture Arrays, which were turned into an in-solution DNA capture library as previously described20.
In-solution capture
Capture was performed at the Max Planck Institute for Evolutionary Anthropology. Targeted single nucleotide variants (SNVs) were enriched from the total DNA in a library by applying an in-solution capture technology that is based on synthetic modified, immortalized probe sequences58,59. DNA (1,000 ng) was hybridized with the single-stranded capture probes for 24 h under stringent conditions in the thermocycler at 65 °C with a heated lid. Hybridization reactions were subjected to multiple automated washing steps on magnetic beads using the Bravo-B NGS Workstation. Purified enriched libraries were eluted in 30 μl TT and 1 μl was quantified via qPCR in the LightCycler 96 (Roche). The complete remaining eluate was amplified to the plateau using Herculase II Fusion DNA Polymerase (Agilent Technologies), purified with the SPRI method and eluted in 20 μl EBT. Purified enriched libraries were quantified on the NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific) and normalized to 100 ng μl–1. Normalized libraries were reconditioned and prepared for sequencing following the same protocol as for shotgun sequencing. Enriched libraries were sequenced for 20 million reads per library single-read on the Illumina HiSeq 4000 with 75 cycles.
Decisions on which libraries to subject to capture were made slightly differently for different sets of samples. For libraries built at the University of Tübingen and the Francis Crick Institute, all libraries were captured regardless of what the screening metrics looked like. An exception to this was the set of libraries built from Danish samples at the Francis Crick Institute, seven of which showed poor screening metrics and were not captured. Three samples (two La Fru, Boncuklu) were processed later in the project and not captured for that reason. For libraries built at the Max Planck Institute, capture decisions were informed by screening metrics with generally any library showing at least 0.1% endogenous DNA being captured. An exception to this was the final batch of samples from the Gnirshöhle site, which were processed late in the project and were not considered for capture.
Read data processing
Genomic data were processed through the nf-core/eager v.2 pipeline60. Adaptors were trimmed, overlapping paired-end reads collapsed and bases with qualities below 20 trimmed using AdapterRemoval v.2 (ref. 61) (--trimn --trimqualities --collapse --minadapteroverlap 1 --preserve5p). Collapsed reads with lengths of ≥35 bp were mapped to the canFam3.1 reference genome (NCBI assembly accession no. GCF_000002285.3) using bwa aln v.0.7.17 (ref. 62) with permissive parameters (-l 16500 -n 0.01). Pseudohaploid genotypes were called for all ancient genomes by sampling a single allele per position using htsbox pileup r345 (ref. 63), requiring a minimum read length of 35 bp, a mapping quality of 20 and a base quality of 30. For samples where we had both shotgun and capture data, we added the suffix ‘c1’ to the capture data sample ID in genotype files. We inferred biological sex for samples with greater than 0.01× coverage on the captured sites, assigning male if the chrX-to-autosome coverage ratio was less than 0.7, and female if the ratio was greater than 0.9.
As the starting point for population genetic analyses, we used a VCF file containing diploid genotypes across 722 modern canids (NCBI BioProject accession no. PRJNA448733)64, augmented with diploid genotypes from additional modern dogs and wolves65,66,67,68 as described previously14, and pseudohaploid calls from ancient canids8,12,13,14,19,46,69,70, at 67.8 million biallelic SNVs. We then added the pseudohaploid genotypes from the newly generated ancient genomes onto this VCF, and subsetted to sites targeted in the capture, resulting in a dataset of 470,024 SNVs (336,297 transversions, 133,727 transitions). Metadata on the previously published genomes used are provided in Supplementary Data 3.
Metagenomic screening and authentication
Reads not aligning to the dog reference genome were screened for microbial DNA by competitive alignment to a curated pathogen reference panel using BWA aln62, with parameters identical to those for host mapping. This panel was manually curated to include clinically relevant species with bloodstream invasion potential, while minimizing within-genus redundancy by selecting single representatives with less than 10% mutual genome coverage, or restricting to specific chromosomes or organelles (Supplementary Data 6). Duplicate reads were removed using DeDup v.0.12.8 (ref. 71) and only alignments with a mapping quality ≥30 and sequence identity ≥80% were retained. A taxon was considered as authentically present if it met four authentication criteria: (1) a minimum of 30 unique reads; (2) a 5′ terminal C-to-T substitution rate of ≥0.1, estimated using PMDtools v.0.60 (--first)72; (3) a monotonically decreasing mismatch distribution (freq(zero mismatches) > freq(one mismatch) > freq(two mismatches)), assessed after excluding characteristic C-to-T and G-to-A damage-related substitutions; and (4) an observed genome coverage of at least 80% of the value expected under a Poisson distribution.
Ancestry analyses
Clustering was performed using ADMIXTURE v.1.3.0 (ref. 30), running on all 470,012 captured SNVs, on samples having data on at least 4,000 SNVs. To perform PCA in the context of a larger number of modern wolves, we merged our data with a previously compiled dataset of array (CanineHD BeadChip) genotypes73, which includes data from numerous past studies74,75,76,77,78 (Supplementary Data 4). After merging, 91,280 SNVs remained. PCA was performed using EIGENSOFT v.7.2.1 (ref. 79), using the poplistname parameter to specify 211 modern Eurasian wolves to use for inferring principal components, with dogs instead being projected. The options ‘lsqproject: YES’ and ‘shrinkmode: YES’ were used.
To assess stability of ADMIXTURE to the choice of random seed, we reran ADMIXTURE (at k = 7 as in Fig. 3a) 100 times with different random seeds, and used the CLUMPAK ‘Compare’ program80 to compare these results with our primary results shown in Fig. 3a. CLUMPAK identified a cluster of solutions that 82/100 of the replicate runs converged on, and this solution had a correlation coefficient with our primary results of 99.92%. There is thus minimal variation between runs with different random seeds, and the primary results we present in Fig. 3a represent the solution most commonly found by ADMIXTURE.
Conditional heterozygosity was calculated on the 343,875 captured transversion SNVs, by randomly sampling two reads per site and per sample and calculating the fraction of sites at which those two reads display mismatching alleles (ignoring any alleles other than those targeted by the capture). Standard errors were obtained through block jackknifing across the 38 chromosomes. Samples with data from 1,525 or fewer sites covered by two reads were excluded from the figure. For the sample KSL101 we had five separate libraries, and we noticed that heterozygosity estimates obtained when merging these were higher than the per-library estimates, potentially owing to between-library batch effects—we therefore counted matches and mismatches only using reads from the same library, aggregated these per chromosome and performed the block jackknifing across chromosomes as above.
qpAdm analyses and f3 and f4 statistics were performed using ADMIXTOOLS2 v.2.0.0 (ref. 81), with statistics being calculated directly from genotype files, using all 38 chromosomes (auto_only = FALSE), and the allsnps = TRUE argument. Outgroup f3 statistics were calculated using a modern wolf (Wolf35Xinjiang) as the outgroup, using all 470,012 captured SNVs for wolf-versus-dog ancestry testing to maximize power, and 343,875 transversion SNVs for pairwise dog comparisons. qpAdm was run on all 470,012 captured SNVs to maximize power. We ranked qpAdm models by favouring models with fewer sources (except if a fewer-source model had a P value of <0.01). The two different qpAdm analyses were set up as follows:
-
1)
Testing for dual wolf ancestry: we tested one- and two-source models involving a 9,500-year old dog from Zhokhov island in Siberia and a modern Syrian wolf as proxies for eastern dog and Southwest Asian wolf ancestry, respectively14. We used a fixed set of reference populations consisting of pre-Last-Glacial-Maximum wolves from Siberia (LOW008, CGG29, IN18_016, VAL_005, LOW002, CGG23, VAL_008), the Yukon (SC19.MCJ010), the Altai (AL2744), Europe (JK2175), the Caucasus (JK1557) and an ancient dhole (HOV4). In one-source models, the other of the two candidate sources was also added to the reference list. All model test results are provided in Supplementary Data 7.
-
2)
Testing for Mesolithic versus Neolithic ancestry in Europe: we tested all possible one-, two- and three-source models across the following set of dogs as candidate sources. European pre-Neolithic: Kesslerloch (KSL101), Veretye (OL4061), Bökeberg (C10604), Ertebølle (C11744); Southwest Asian Neolithic: Tel Hreiz (THRZ02), Boncuklu (C16498); eastern dogs: NewGuineaSingingDog, Zhokhov (CGG6), Port au Choix (AL3194), Baikal (OL4223) and an ancient dhole (HOV4). We rotated fully across populations, meaning any population not used as a source in a given model was placed in the reference list. We considered models with ancestry proportions outside [−0.1,1.1] as failed. All model test results are provided in Supplementary Data 8.
Identification of recent dogs at old sites
We identified a number of dogs from sites with early cultural context (Upper Palaeolithic, Mesolithic or Neolithic), but where the dogs display evidence of being more recent. We therefore excluded these dogs from analyses of dog population history in Neolithic or earlier time periods.
At the sites of Goyet, Belgium and Předmostí, Czechia, two dogs have direct radiocarbon dates of 2.3 ka (sample TU96) and 2.9 ka (sample JK1560), respectively. At Hardinxveld in the Netherlands, the sample C11478 is assigned fully to the modern European dog component in ADMIXTURE analyses (Fig. 3a). Similarly, for four samples from Denmark (C11741, C11748, C11754, C11727), 50–100% of their ancestry are assigned to the modern component. At Gnirshöhle, Germany, we identified several individuals with dog ancestry typical of modern dogs, despite direct radiocarbon dates of around 15.4 ka, consistent with the chronology of the site47. These individuals with dog ancestry also displayed much lower rates of ancient DNA damage (C-to-T substitution at 1–8% of terminal C’s) than individuals displaying wolf ancestry from the same site (31–40%). Furthermore, we found that these samples share high amounts of genetic drift with a specific modern breed, the Bernese Mountain Dog. Given the probably recent emergence of such a modern breed, a specific affinity such as this would be very unlikely unless the DNA is recent. Given that the radiocarbon dates from these samples are consistently Late Pleistocene in age, a probable explanation for the recent dog DNA in the Gnirshöhle samples might be DNA contamination at some stage. Consistent with these samples being wolves with modern dog DNA contamination, a previous study of their mitochondrial DNA filtered reads based on ancient DNA damage patterns, and then recovered mitochondrial sequences consistent with Late Pleistocene wolf diversity47.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomic data generated in this project are available at the European Nucleotide Archive under project accession no. PRJEB90148. We archive all genomic data generated, regardless of endogenous content or other data properties. This includes different combinations of shotgun screening, genome-wide capture and deep shotgun data for different samples. We make the data available in two forms: all reads in FASTQ format, and analysis-ready BAM files containing the reads as mapped and filtered for our analyses. The SNPs targeted by capture and the probe sequences are available via Zenodo at https://zenodo.org/records/17080078.
References
Napierala, H. & Uerpmann, H.-P. A. ‘New’ palaeolithic dog from central Europe. Int. J. Osteoarchaeol. 22, 127–137 (2012).
Janssens, L. et al. A new look at an old dog: Bonn–Oberkassel reconsidered. J. Archaeol. Sci. 92, 126–138 (2018).
Boudadi-Maligne, M., Mallye, J.-B., Langlais, M. & Barshay-Szmidt, C. Magdalenian dog remains from Le Morin rock-shelter (Gironde, France). Socio-economic implications of a zootechnical innovation. Paléo 39, 54 (2012).
Hervella, M. et al. The domestic dog that lived ∼17,000 years ago in the Lower Magdalenian of Erralla site (Basque Country): a radiometric and genetic analysis. J. Archaeol. Sci. Rep. 46, 103706 (2022).
Boschin, F. et al. The first evidence for Late Pleistocene dogs in Italy. Sci. Rep. 10, 13313 (2020).
Savolainen, P., Zhang, Y.-P., Luo, J., Lundeberg, J. & Leitner, T. Genetic evidence for an East Asian origin of domestic dogs. Science 298, 1610–1613 (2002).
Wang, G.-D. et al. Out of southern East Asia: the natural history of domestic dogs across the world. Cell Res. 26, 21–33 (2016).
Frantz, L. A. F. et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 352, 1228–1231 (2016).
Shannon, L. M. et al. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc. Natl Acad. Sci. USA 112, 13639–13644 (2015).
Thalmann, O. et al. Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs. Science 342, 871–874 (2013).
Vonholdt, B. M. et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902 (2010).
Botigué, L. R. et al. Ancient European dog genomes reveal continuity since the Early Neolithic. Nat. Commun. 8, 16082 (2017).
Bergström, A. et al. Origins and genetic legacy of prehistoric dogs. Science 370, 557–564 (2020).
Bergström, A. et al. Grey wolf genomic history reveals a dual ancestry of dogs. Nature 607, 313–320 (2022).
Zhang, M. et al. Ancient mitogenomes reveal the maternal genetic history of East Asian dogs. Mol. Biol. Evol. 41, msae062 (2024).
Tchernov, E. & Valla, F. F. Two new dogs, and other Natufian dogs, from the southern Levant. J. Archaeol. Sci. 24, 65–95 (1997).
Perri, A. et al. New evidence of the earliest domestic dogs in the Americas. Am. Antiq. 84, 68–87 (2019).
Perri, A. A wolf in dog’s clothing: initial dog domestication and Pleistocene wolf variation. J. Archaeol. Sci. 68, 1–4 (2016).
Feuerborn, T. R. et al. Modern Siberian dog ancestry was shaped by several thousand years of Eurasian-wide trade and human dispersal. Proc. Natl Acad. Sci. USA 118, e2100338118 (2021).
Fu, Q. et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl Acad. Sci. 110, 2223–2227 (2013).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Rohland, N. et al. Three assays for in-solution enrichment of ancient human DNA at more than a million SNPs. Genome Res 32, 2068–2078 (2022).
Cabrera-García, A. I., Müller, F., Rödler, F. S., Traub, F. & Heilmann, R. M. Infective endocarditis due to Erysipelothrix rhusiopathiae in a dog—a case report. BMC Vet. Res. 16, 328 (2020).
Di Bello, A. et al. Periodontal disease associated with red complex bacteria in dogs. J. Small Anim. Pract. 55, 160–163 (2014).
Marks, S. L., Rankin, S. C., Byrne, B. A. & Weese, J. S. Enteropathogenic bacteria in dogs and cats: diagnosis, epidemiology, treatment, and control. J. Vet. Intern. Med. 25, 1195–1208 (2011).
Magnell, O. in Bear and Human, Vol. 1 (ed. Grimm, O.) 209–234 (Brepols, 2023).
Germonpré, M., Jimenez, E.-L. & Boudin, M. in The Beef Behind all Possible Pasts (eds Sabine, G.-W. & Olaf, J.) 505–520 (Romisch-Germanisches Zentralmuseum, 2021).
Haak, W. et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207 (2015).
Sinding, M.-H. S. et al. Arctic-adapted dogs emerged at the Pleistocene–Holocene transition. Science 368, 1495–1499 (2020).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Günther, T. et al. Population genomics of Mesolithic Scandinavia: investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol. 16, e2003703 (2018).
Skoglund, P. et al. Genomic diversity and admixture differs for Stone-Age Scandinavian foragers and farmers. Science 344, 747–750 (2014).
Coutinho, A. et al. The Neolithic Pitted Ware culture foragers were culturally but not genetically influenced by the Battle Axe culture herders. Am. J. Phys. Anthropol. 172, 638–649 (2020).
Skoglund, P. et al. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science 336, 466–469 (2012).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014).
Hofmanová, Z. et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc. Natl Acad. Sci. 113, 6886–6891 (2016).
Ollivier, M. et al. Dogs accompanied humans during the Neolithic expansion into Europe. Biol. Lett. 14, 20180286 (2018).
Lipson, M. et al. Parallel palaeogenomic transects reveal complex genetic history of early European farmers. Nature 551, 368–372 (2017).
Allentoft, M. E. et al. 100 ancient genomes show repeated population turnovers in Neolithic Denmark. Nature 625, 329–337 (2024).
Jensen, T. Z. T. et al. A 5700 year-old human genome and oral microbiome from chewed birch pitch. Nat. Commun. 10, 5520 (2019).
Marchi, N. et al. The genomic origins of the world’s first farmers. Cell 185, 1842–1859.e18 (2022).
Charlton, S. et al. Dual ancestries and ecologies of the Late Glacial Palaeolithic in Britain. Nat. Ecol. Evol. 6, 1658–1668 (2022).
Posth, C. et al. Palaeogenomics of Upper Palaeolithic to Neolithic European hunter-gatherers. Nature 615, 117–126 (2023).
Fu, Q. et al. The genetic history of Ice Age Europe. Nature 534, 200–205 (2016).
Feldman, M. et al. Late Pleistocene human genome suggests a local origin for the first farmers of central Anatolia. Nat. Commun. 10, 1218 (2019).
Ní Leathlobhair, M. et al. The evolutionary history of dogs in the Americas. Science 361, 81–85 (2018).
Baumann, C. et al. A refined proposal for the origin of dogs: the case study of Gnirshöhle, a Magdalenian cave site. Sci. Rep. 11, 5137 (2021).
Dabney, J. et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA 110, 15758–15763 (2013).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, db.prot5448 (2010).
Rohland, N., Glocke, I., Aximu-Petri, A. & Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 13, 2447–2461 (2018).
Gansauge, M.-T. & Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 8, 737–748 (2013).
Gansauge, M.-T. & Meyer, M. in Ancient DNA (eds Shapiro, B. et al.) 75–83 (Springer, 2019).
Gansauge, M.-T., Aximu-Petri, A., Nagel, S. & Meyer, M. Manual and automated preparation of single-stranded DNA libraries for the sequencing of DNA from ancient biological remains and other sources of highly degraded DNA. Nat. Protoc. 15, 2279–2300 (2020).
Wang, Y. & Nielsen, R. Estimating population divergence time and phylogeny from single-nucleotide polymorphisms data with outgroup ascertainment bias: estimating phylogeny from ascertained data. Mol. Ecol. 21, 974–986 (2012).
Deane-Coe, P. E., Chu, E. T., Slavney, A., Boyko, A. R. & Sams, A. J. Direct-to-consumer DNA testing of 6,000 dogs reveals 98.6-kb duplication associated with blue eyes and heterochromia in Siberian Huskies. PLoS Genet. 14, e1007648 (2018).
Mikkola, L. I. et al. Novel protective and risk loci in hip dysplasia in German Shepherds. PLoS Genet. 15, e1008197 (2019).
Mikkola, L. et al. An across-breed validation study of 46 genetic markers in canine hip dysplasia. BMC Genom. 22, 68 (2021).
Gnirke, A. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189 (2009).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015).
Fellows Yates, J. A. et al. Reproducible, portable, and efficient ancient genome reconstruction with nf-core/eager. PeerJ 9, e10947 (2021).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
lh3/htsbox (GitHub, 2025); https://github.com/lh3/htsbox.
Plassais, J. et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 10, 1489 (2019).
Kardos, M. et al. Genomic consequences of intensive inbreeding in an isolated wolf population. Nat. Ecol. Evol. 2, 124–131 (2018).
Liu, Y.-H. et al. Whole-genome sequencing of African dogs provides insights into adaptations against tropical parasites. Mol. Biol. Evol. 35, 287–298 (2018).
Sinding, M.-H. S. et al. Population genomics of grey wolves and wolf-like canids in North America. PLoS Genet. 14, e1007745 (2018).
Gopalakrishnan, S. et al. Interspecific gene flow shaped the evolution of the genus Canis. Curr. Biol. 28, 3441–3449.e5 (2018).
Skoglund, P., Ersmark, E., Palkopoulou, E. & Dalén, L. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol. 25, 1515–1519 (2015).
Ramos Madrigal, J. et al. Genomes of extinct Pleistocene Siberian wolves provide insights into the origin of present-day wolves. Curr. Biol. 31, 1–9 (2021).
Peltzer, A. et al. EAGER: efficient ancient genome reconstruction. Genome Biol. 17, 60 (2016).
Skoglund, P. et al. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl Acad. Sci. USA 111, 2229–2234 (2014).
Pilot, M. et al. Global phylogeographic and admixture patterns in grey wolves and genetic legacy of an ancient Siberian lineage. Sci. Rep. 9, 17328 (2019).
Vaysse, A. et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 7, e1002316 (2011).
Stronen, A. V. et al. Genome-wide analyses suggest parallel selection for universal traits may eclipse local environmental selection in a highly mobile carnivore. Ecol. Evol. 5, 4410–4425 (2015).
Cronin, M. A., Cánovas, A., Bannasch, D. L., Oberbauer, A. M. & Medrano, J. F. Single nucleotide polymorphism (SNP) variation of wolves (Canis lupus) in Southeast Alaska and comparison with wolves, dogs, and coyotes in North America. J. Hered. 106, 26–36 (2015).
Fitak, R. R., Rinkevich, S. E. & Culver, M. Genome-wide analysis of SNPs is consistent with no domestic dog ancestry in the endangered Mexican wolf (Canis lupus baileyi). J. Hered. 109, 372–383 (2018).
Pilot, M. et al. On the origin of mongrels: evolutionary history of free-breeding dogs in Eurasia. Proc. Biol. Sci. 282, 20152189 (2015).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A. & Mayrose, I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191 (2015).
Maier, R. et al. On the limits of fitting complex models of population history to f-statistics. eLife 12, e85492 (2023).
Acknowledgements
A. Bergström was supported by the Leverhulme Trust (grant no. PLP-2023-281). P.S. was supported by a UKRI Horizon guarantee/ERC Consolidator award (grant no. UKRI338), the European Molecular Biology Organisation, the Vallee Foundation, the European Research Council (grant no. 852558), the Wellcome Trust (grant no. 217223/Z/19/Z) and Francis Crick Institute core funding (grant no. FC001595) from Cancer Research UK, the UK Medical Research Council and the Wellcome Trust. We thank K. M. Gregersen, the Langelands Museum, Museum Sydøstdanmark and Provincial Archaeological Depots of Zuid-Holland (I. Riemersma) for facilitating sample access.
Author information
Authors and Affiliations
Contributions
V.J.S., J.K. and P.S. supervised the study. E. Rosengren, C. Baumann, H.A.-W., D.B., A.B.G., H.B., A. Bridault, R.B., D.G.D., A.S.F., L.F., B.G., L.G., M.G., L.J., A.W.K., K.K., M.L.G., D.L., O.M., L. Martin, S.C.M., G.M., B.M., A.P., F.P., M.R., A.R.-B., Ö.S., K.S., J.A.S., K-G.S., J.S., L.V.S., Y.T., F.T-N., O.T. and G.L. excavated or curated samples. A.F., S.J., E. Rosengren, A. Breidenstein, T.B., J.B.M., J.P., M.W., M.K., F.T., C. Baumann, R.R., K.A., I.G., S.P., J.G., T.R.F., E. Reiter, A.L., S.C. and V.J.S. generated data through sample preparation and/or laboratory work. A. Bergström, C. Barrington, A.G., M.S., A. Brativnyk, A.H., K.P., F.R. and L. Mikkola analysed and/or curated genomic data. A. Bergström and P.S. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Mikkel-Holger Sinding, François Lanoe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Enrichment of DNA at sites targeted by capture.
For selected samples, the number of mapped reads as a function of distance from the single-nucleotide variant targeted by the capture is plotted as histograms. There is clear enrichment at the target sites. Note that each panel has a different vertical scale. Shotgun data from three previously published wolf samples14 are included for comparison.
Extended Data Fig. 2 Comparing results obtained from shotgun and capture data.
For a dog from the Swedish Mesolithic site of Bökeberg we have both shotgun (13× coverage) and capture data. The results of outgroup f3-statistics measuring the amount of genetic drift shared with other dogs obtained with each of these datasets is plotted against each other. Error bars for ancient genomes denote ±2 standard errors. All values line up along the diagonal, showing that highly comparable results are obtained with the two approaches.
Extended Data Fig. 3 f4-statistics relating ancient dogs to wolves.
Only transversion SNVs were used, requiring data on at least 10,000 SNVs for a given statistic. Bars denote ±3 standard errors. a) Contrasting each ancient dog to an ancient Siberian wolf (Belaya Gora, 18.1 ka) versus an ancient European wolf (Hohle Fels, 13.2 ka). b) Using a 9,500- year-old Siberian dog from Zhokhov island in Siberia as a baseline, to test other dogs for any excess affinity to an ancient European wolf (Hohle Fels, 13.2 ka). c) Using a 9,500-year-old Siberian dog from Zhokhov island in Siberia as a baseline, to test other dogs for any excess affinity to a present-day wolf from Syria.
Extended Data Fig. 4 ADMIXTURE clustering results for different values of k.
Results are shown for k = 2-10. Each colour represents a modelled ancestry component. White lines delimit sample groups in the same way as in Fig. 3a.
Extended Data Fig. 5 Contrasting the genetic affinities of pairs of early dogs.
Scatter plots of outgroup f3 statistics, using a present-day wolf from Xinjiang, China, as the outgroup. Error bars for ancient genomes denote ±2 standard errors. a) Neolithic Anatolia against Mesolithic Sweden. b) Neolithic Anatolia against Siberia 9.5 ka. c) Neolithic Levant against Neolithic Anatolia. d) Upper Palaeolithic Kesslerloch, Switzerland against Neolithic Anatolia.
Extended Data Fig. 6 Robustness of Mesolithic-Neolithic admixture estimates.
Estimates of the Neolithic Southwest Asian ancestry contributions in European dog targets, from the same qpAdm analyses presented in Fig. 4b but varying the choice of Neolithic and Mesolithic sources, and using either all sites or only transversions. Single-source Neolithic models were excluded, as high amounts of genotype missingness for some targets sometimes leads to insufficient power to reject such models (especially when using the source THRZ02, which has a sequencing coverage of only 0.1x).
Supplementary information
Supplementary Information (download PDF )
Supplementary notes with descriptions of the sites and specimens analysed in the study, and additional references
Supplementary Data (download XLSX )
Supplementary Data 1: ancient genome metadata (metadata for all ancient canid remains sampled in this study). Supplementary Data 2: new radiocarbon dates (results from new radiocarbon dates obtained in this study). Supplementary Data 3: previously published genomes (metadata for previously published modern and ancient canid genomes used in analyses). Supplementary Data 4: modern wolf data used for PCA (list of modern wolf individuals used for PCA; Fig. 2a). Supplementary Data 5: remains possibly same ind. (sets of sampled remains that potentially could derive from the same biological individual, one set per row). Supplementary Data 6: pathogen reference panel (list of microbial genomes used as the pathogen reference panel). Supplementary Data 7: dual ancestry qpAdm (results of all qpAdm models addressing ‘dual ancestry’ contributions from one or two wolf sources to dogs). Supplementary Data 8: Mesolithic-Neolithic qpAdm (results of all qpAdm models addressing Mesolithic and Neolithic contributions to European dogs).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bergström, A., Furtwängler, A., Johnston, S. et al. Genomic history of early dogs in Europe. Nature 651, 986–994 (2026). https://doi.org/10.1038/s41586-026-10112-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-026-10112-7
