
Abstract
The bacterial CRISPR–Cas9 (Cas9) nuclease has become a powerful genome manipulation tool for a wide range of organisms1,2,3. However, it has yet to fully leverage the pervasive presence of DNA methylation in genomes4,5,6,7,8,9,10. Here, to fill this gap, we report biochemical, structural and human genome-editing characterizations of a methylation-sensitive Cas9 (ThermoCas9). ThermoCas9 efficiently binds to and cleaves DNA upstream of its protospacer adjacent motif (PAM) 5′-NNNNCGA-3′ or 5′-NNNNCCA-3′ in vitro. Methylation of the fifth cytosine in either PAM sequence (5mCpG or 5mCpC), however, significantly inhibits ThermoCas9 activity. Cryo-electron microscopy structures of ThermoCas9 in pre-cleavage and post-cleavage states at 2.8 Å and 2.2 Å resolution, respectively, reveal the molecular basis for the stringent requirement of the unmethylated cytosine in PAM binding and provide guidance for further enzyme engineering. We demonstrate methylation-sensitive editing by ThermoCas9 in human cell lines with distinct DNA methylation landscapes. Moreover, we demonstrate that a catalytically enhanced ThermoCas9 efficiently targets luminal expression signature genes that are consistently hypomethylated in patients with breast cancer. Owing to its sensitivity to DNA methylation, ThermoCas9 can specifically target cells with disease-related hypomethylation, which adds another layer of precision to genome-editing technologies.
Similar content being viewed by others

Main
Directed by a complementary guide RNA (gRNA), Cas9 proteins catalyse the cleavage of double-stranded DNA (dsDNA)11,12,13 and this activity has been shown to be largely insensitive to DNA methylation14,15. Cytosine methylation (5mC) and its dynamic counterpart, demethylation, are hallmarks of gene regulation in animals and plants, having pivotal roles in cell differentiation, transposon silencing, ageing, pathogenesis of various diseases and development of therapeutics4,5,6,7,8,9,10. To fully harness the power of epigenetic information in genome editing and beyond, the discovery of a 5mC-sensitive Cas9 would substantially extend the reach of Cas9-based tools, including but not limited to site-specific gene editing, base editing, transcription regulation, prime editing and virus eradication16 in a methylation-sensitive manner.
Comprehensive analysis of human cell-type methylomes display well-conserved and cell-type-specific DNA methylation patterns in healthy cells4. Comparison of the methylation patterns in healthy cells with those in diseased or ageing cells unveil disease-specific and location-specific biomarkers for potential diagnosis and therapeutic applications4,17. Many cancer types, for instance, arise from a combination of specific genetic mutations and epigenetic alterations18,19. As a result, methylation profiling has emerged as a powerful tool for disease detection and post-treatment monitoring20,21,22. A methylation-sensitive Cas9 would serve as a simple enzyme-based tool that can both map epigenetic changes and support highly precise gene-editing applications. Although Cas9 has been engineered for site-specific methylation or demethylation in cells through fusion with epigenetic modifiers23,24, which has demonstrated the power of epigenome regulation, these approaches are distinct from methylation-sensitive genome manipulation. Cytosine methylation-sensitive Cas9 systems would allow for the direct response to methylation changes in cells.
We previously characterized two type II-C Cas9 proteins, Acidothermus cellulolyticus (AceCas9) and Geobacillus thermodenitrificans T12 Cas9 (ThermoCas9), that are both controlled by cytosine-containing PAM sequences25,26. We have previously shown that AceCas9 is sensitive to 5mC followed by another cytosine (5mCpC) in its PAM25. However, how ThermoCas9 responds to cytosine methylation in its 5′-NNNNCNR-3′ (R = purine) PAM26, especially in its human cell-editing applications27, remains unknown. Whereas 5mCpC has increasingly been shown to occur in human stem and brain cells28,29,30 and on the mitochondrial genome31, a large majority of cytosine methylation occurs on the CpG sequence as 5mCpG32,33. Therefore, a Cas9 sensitive to 5mCpG would enable much broader epigenetic applications. Here we report and characterize the sensitivity of ThermoCas9 to 5mCpG and 5mCpC both in vitro and in human cells. We determined cryo-electron microscopy (cryo-EM) structures of ThermoCas9 bound to DNA substrates in two distinct functional states, revealing the molecular basis for sensing DNA methylation. More importantly, we demonstrate a proof of concept for ThermoCas9 in performing genome editing in a DNA methylation-sensitive manner. Delivery of an engineered ThermoCas9 ribonucleoprotein (RNP) with enhanced catalytic activity into a breast cancer cell line (MCF-7) and the non-tumorigenic breast epithelial cell line (MCF-10A) enabled specific and efficient targeting of loci consistently hypomethylated in patients with breast cancer.
ThermoCas9 discriminates against 5mCpC and 5mCpG sequences
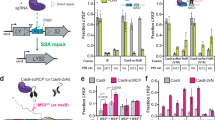
We have previously shown that ThermoCas9 exhibits a broad PAM specificity, with only the fifth position strictly requiring a C–G pair while downstream purines further enhance the activity (optimal PAM, 5′-NNNNCNA-3′ at 37−55 °C and 5′-NNNNCCAA-3′ at 30 °C)26. To test whether ThermoCas9 is sensitive to methylation of the cytosine on the fifth position in its PAM, we programmed a single-guide RNA (sgRNA) to target a 23-base pair (bp) DNA sequence adjacent to either a CpC-containing (5′-NNGGCCA-3′) or a CpG-containing PAM (5′-NNNNCGA-3′) in vitro. We introduced methylation on (1) the 5′-NNGGCCA-3′ PAM by the HaeIII methyltransferase (recognition site 5′-GGCC-3′) to 5′-NNGG5mCCA3′, and (2) the 5′-NNNNCGA-3′ PAM by the M.SssI methyltransferase (recognition site 5′-CG-3′) to 5′-NNNN5mCGA-3′. Although ThermoCas9 cleaved both unmethylated DNA substrates efficiently, it had substantially diminished activity against the DNA associated with either PAM sequence containing 5mC (Fig. 1a and Extended Data Fig. 1a; for gel source data, see Supplementary Fig. 1). To distinguish the effect of 5mC on the non-target strand from that on the target strand, we used synthetic oligo DNA duplexes containing strand-specific methylation, which revealed that ThermoCas9 is more sensitive to methylation on the non-target strand than on the target strand. Methylation on both strands caused the strongest inhibition (Fig. 1b and Extended Data Fig. 1b; for gel source data, see Supplementary Fig. 2). In contrast to the methylation of the PAM cytosine, methylation of cytosines within the protospacer, including four in the seed region, had no notable effect on ThermoCas9 activity (Fig. 1b and Extended Data Fig. 1b; for gel source data, see Supplementary Fig. 2), underscoring the high specificity of PAM methylation in inhibiting ThermoCas9.

For DNA cutting reactions, ThermoCas9 at 1 μM was incubated with 100 nM DNA at 37 °C for 30 min. The experiment was repeated two times independently with similar results. NTS, non-target strand; TS, target strand. a, DNA cleavage results for DNA plasmids analysed on agarose gels. 5mCpGw or 5mCpCw, methylated plasmid by M.SssI or HaeIII methyltransferase. b, DNA cleavage results for DNA oligo substrates analysed on polyacrylamide gels and imaged by fluorescent probes associated with each strand (HEX-NTS and FAM-TS). 5mCpGNTS, oligo substrate with methylation on the non-target strand; 5mCpGTS, oligo substrate with methylation on the target strand; 5mCpGw, oligo substrate with methylation on both the non-target strand and target strand; mNTS, oligo substrate with methylation on three cytosines in the protospacer region of the non-target strand and no methylation on the target strand; mNTS–mTS, oligo substrate with methylation on cytosine in the protospacer region of both the target strand and non-target strand; mTS, oligo substrate with methylation on four cytosines in the protospacer region of the target strand and no methylation on the non-target strand. The asterisk indicates a likely small amount of the unpaired FAM-NTS strand. c, Competition assay results. Cleavage of a pUC19 plasmid containing a protospacer followed by a 5′-NNNNCGA-3′ PAM by ThermoCas9 in the presence of no or increasing concentrations of the DNA oligos as indicated in panel b were analysed by electrophoresis. The fraction of cleavage as a function of competitor DNA concentrations in log scales is plotted. The error bars were obtained from three experimental replicates and are presented as mean ± s.e.m. The inhibition constants for the unmethylated and target strand-methylated oligos are estimated to be 64 ± 9 and 767 ± 250 nm, respectively. d, Native gel mobility shift analysis of ThermoCas9–sgRNA complex binding to the DNA oligos of the same protospacer as indicated in panel b.
We next examined which functional steps of ThermoCas9 are inhibited by DNA methylation. We introduced DNA duplexes assembled from either methylated or unmethylated synthetic oligos into cleavage reactions to compete with unmethylated plasmid substrates (Fig. 1c and Extended Data Fig. 1c; for gel source data, see Supplementary Fig. 1c). Although unmethylated DNA duplexes inhibited plasmid cleavage efficiently, methylated DNA duplexes failed to inhibit the reaction even at a 100-fold molar excess (Fig. 1c and Extended Data Fig. 1c; for gel source data, see Supplementary Fig. 1c). Consistent with the strand-specific methylation sensitivity observed for the oligo cleavage assay (Fig. 1b; for gel source data, see Supplementary Fig. 2), the DNA oligos containing methylation either on the non-target strand alone or on both strands had the weakest competition (Fig. 1c and Extended Data Fig. 1c; for gel source data, see Supplementary Fig. 2). Moreover, a gel mobility shift assay showed reduced binding of the methylated DNA substrates to ThermoCas9 (Fig. 1d; for gel source data, see Supplementary Fig. 2). These results suggest that, in contrast to AceCas9, which is inhibited at steps after DNA binding25, ThermoCas9 is inhibited at the step of DNA binding.
Structural basis for activation of ThermoCas9
To elucidate the molecular basis of the sensitivity of ThermoCas9 to DNA methylation and its effect on catalytic efficiency, we determined cryo-EM structures of active ThermoCas9 bound to DNA substrates containing either a 5′-NNNNCCAA-3′ PAM or a 5′-NNNNCGAA-3′ PAM (Extended Data Table 1 and Extended Data Figs. 2–4). We also assembled active ThermoCas9 with the same DNA substrate containing a 5′-NNNN5mCCAA-3′ PAM (Extended Data Fig. 5). The unmethylated DNA with the 5′-NNNNCCAA-3′ PAM-assembled complex resulted in three reconstructed structures corresponding to three functional states: the post-cleavage state at 2.2 Å resolution, the pre-cleavage state at 2.8 Å resolution and a target DNA strand-only state at 2.5 Å (Figs. 2a–g, Extended Data Table 1 and Extended Data Figs. 2–4). Consistent with a weaker interaction of ThermoCas9 with dsDNA containing a methylated PAM, attempts to obtain such a complex resulted predominantly in assemblies bound to only the single target DNA strand, despite a high molar excess of dsDNA over protein (Extended Data Fig. 5). As this state closely resembles the same complex obtained from the unmethylated DNA samples, it does not offer additional insights and was not pursued further. The unmethylated DNA containing the 5′-NNNNCGAA-3′ PAM resulted in a reduced number of assembled particles and a single reconstructed structure mimicking the post-cleavage state at 2.6 Å resolution (Extended Data Table 1 and Extended Data Fig. 5). We describe most structural features below based on the higher-quality structures obtained with the DNA containing the 5′-NNNNCCAA-3′ PAM.

a, Domain architecture and sequence features of ThermoCas9 with domain boundaries indicated. b, Secondary structures of the observed R-loop structure and nucleotide labelling. The dashed lines indicate unmodelled regions. Asterisks indicate numbering on the non-target strand. PK, pseudoknot. c, Cartoon representation of the observed R-loop structure of the post-cleavage conformation. The dashed lines indicate unmodelled regions. d, Cryo-EM density of the pre-cleavage conformation of ThermoCas9. e, Cartoon representation of the pre-cleavage conformation of ThermoCas9. f, Cryo-EM density of the post-cleavage conformation of ThermoCas9. g, Cartoon representation of the post-cleavage conformation of ThermoCas9. Panels d–g are colour coded as in panel a.
The overall architecture of ThermoCas9 resembles that of other Cas9 nucleases, with the typical nucleic acid recognition (REC) and the nuclease (NUC) lobes (Fig. 2a). Of the available Cas9 structures, ThermoCas9 in its pre-cleavage state most closely resembles another type II-C (Extended Data Fig. 6a), the Cas9 of Geobacillus stearothermophilus (GeoCas9; Protein Data Bank (PDB) 8UZA; root mean square deviation of 1.25 Å for 845 Cα atoms)34, with which it shares approximately 88% amino acid sequence identity. Its post-cleavage-state structure has less similarity to the Cas9 of Neisseria meningitidis (Nme1Cas9; PDB 6JDV; root mean square deviation of 1.62 Å for 787 Cα atoms)35 (Extended Data Fig. 6b), with which it shares approximately 39% amino acid sequence identity and identical size (1,082 amino acids). The trans-activating CRISPR RNA of both ThermoCas9 and GeoCas9 fold similarly in the 3′ terminal region (Extended Data Fig. 4b). Residues 89–105 and 128–132 form a stable pseudoknot coaxially with stem loop III (133–145; Fig. 2b,c and Extended Data Fig. 4b). The extended pseudoknot and stem-loop III lie along the C-terminal extension of ThermoCas9 (residues 1048–1070). In a previous study, we showed that deletion of trans-activating CRISPR RNA nucleotides 104–144, which would disrupt the pseudoknot, severely reduced DNA cleavage activity at high temperatures in vitro26, strongly suggesting a role in the thermostability of ThermoCas9. An analogous pseudoknot was also observed in stabilizing the trans-activating CRISPR RNA scaffold of the Campylobacter jejuni Cas9 (ref. 36). In addition, the extended stem I (repeat–anti-repeat duplex + tetraloop) interacts with the insertion elements (residues 822–908) of its PAM-interaction domain (PID) that are unique to ThermoCas9 and its two close homologues (Extended Data Figs. 4b and 6a,b). Like other type II-C Cas9 nucleases, but unlike type II-A Cas9 nucleases, ThermoCas9 prefers arginine over lysine as RNA-binding residues (Extended Data Fig. 4a).
Studies of other Cas9 nucleases have revealed the importance of domain movements, from an open to a closed conformation, in commencing its catalytic activities37. After recognition of the correct PAM sequence by Cas9, the target DNA is slightly bent to position the target strand for base pairing with the gRNA38. The subsequent formation of the guide–target heteroduplex, from the PAM-adjacent seed region to the PAM-distal end, gradually positions the REC domains along the heteroduplex until it is secured against the RuvC domain39. This process coincides with a large swing of the HNH domain from the inactive (open) to the active (closed) configuration, which also adjusts the RuvC domain towards a cleavage-competent state40. The captured pre-cleavage and post-cleavage conformations of ThermoCas9 support a similar activation process (Fig. 2d–g and Supplementary Video 1) while highlighting distinct structural rearrangements.
In the open state, the target strand remains intact (Extended Data Fig. 3), consistent with the catalytic sites of both the HNH domain and the RuvC domain being in an inactive state. The HNH nuclease domain is rested in a position approximately 60 Å away from its cleavage site on the target DNA strand, whereas the REC2 domain loosely engages the guide–target heteroduplex (Figs. 2d,e and 3a and Supplementary Video 1). The region distal to the PAM of the non-target DNA strand is disordered and thus not placed into the RuvC active site (Fig. 2d,e and Extended Data Fig. 3).

a, Cartoon representation of the pre-cleavage (open) and the post-cleavage (closed) conformation highlighting the two active sites and DNA. The HNH active site residues are coloured in green and those for RuvC are in blue. Magnesium ions are shown as orange spheres. Dashed lines indicate disordered regions. b, Close-up views of the two cleavage sites. Aligned sequences of select Cas9 effectors in regions that comprise the two nuclease centres, HNH and RuvC, respectively (top). Residues participating in metal coordination and additional catalytic functions are highlighted by different colours. Cryo-EM density and stick models of the two active sites are shown around the cleaved nucleotides, the magnesium ion, the coordinated water molecules and protein residues (bottom). The dashed lines indicate close contact distances. Scissile phosphate oxygen atoms are labelled as OSp for pro-Sp, ORp for pro-Rp and ONUC for the nucleophilic oxygen. Water molecules are labelled as W1, W2 and so on, and the protein residues are also labelled. c, Superimposed RuvC active site structures between the pre-cleavage (tan) and post-cleavage (blue) conformation. Magnesium ions are shown as orange spheres. The black dashed lines indicate the contacts that undergo changes from the pre-cleavage-to-post-cleavage transition. d, Bacterial cell survival assay results of the WT, Lys711 to alanine (K711A) and Asp723 to alanine (D723A) of ThermoCas9. The survival rate is calculated by dividing the colony-forming unit (CFU) on the arabinose-plus (inducing ccdB toxic protein) plate by that on the arabinose-minus plate. n = 3 biologically independent survival assay experiments. Data are presented as mean ± s.e.m.
Transition from the open to the closed conformation of ThermoCas9 requires an approximately 180° rotation of the HNH domain. In the closed state, the HNH domain has attacked the target strand, resulting in cleavage of the phosphodiester bond between nucleotides C3 and C4 (Figs. 2f,g and 3a,b). Similar to other Cas9 variants39,40,41, the transition to the closed state results in a cleavage-competent HNH site in which the conserved catalytic residues Asp581 and Asn605 coordinate with the catalytic magnesium ion (Fig. 3b). The leaving 3′-hydroxyl oxygen would detach from the scissile phosphodiester following cleavage, but in the obtained structure, it remains coordinated with the magnesium at a distance of 2.2 Å (Fig. 3b). Together with the pro-Sp oxygen of the scissile phosphate and two water molecules, the six coordination ligands make a perfect octahedral geometry with the catalytic magnesium (Fig. 3b). In addition, Nδ of His582 (equivalent to His840 of SpyCas9) maintains a close distance to the oxygen probably from the nucleophilic water (2.35 Å), consistent with its role in activating the water molecule42. The well-conserved Lys608 (equivalent to Lys866 of SpyCas9; Fig. 3b), computationally predicted to activate His582 (ref. 42), has a constant conformation throughout the open-to-closed transition process (Extended Data Fig. 6c), unlike Lys866 of SpyCas9 that undergoes a significant rearrangement39, suggesting a different regulation process of HNH catalysis between the two enzymes.
Likewise, in the open state, the RuvC active site lacks necessary metal ions and the non-target DNA strand. After transition to the closed state, however, it forms a cleavage-competent configuration. In the obtained structure, the phosphodiester bond between nucleotides G(-4*) and G(-3*) of the non-target strand has been cleaved (Fig. 3b). The RuvC active centre captures two magnesium ions that are coordinated with the pro-Sp oxygen of the scissile phosphate, the side chains of Asp8, Glu500 and His720 as well as four water molecules (Fig. 3b), underscoring the essential role of these universally conserved residues in catalysis. In addition, three conserved residues, Asp723, Arg713 and Lys711 (corresponding to Asp986, Arg976 and Lys974 in SpyCas9, respectively), undergo pronounced rearrangements to further shape the active site through the open-to-closed transition (Fig. 3b,c and Supplementary Video 2). This dynamic behaviour contrasts with the relatively stationary positioning of their counterparts in other Cas9 variants39,40,41. Consistently, mutation of either Asp723 or Lys711 to alanine severely impaired ThermoCas9 activities in bacterial cells (Fig. 3d). To the best of our knowledge, these are the first demonstrated effects of these conserved residues that regulate the Cas9 catalytic activity.
Structural basis for DNA methylation sensitivity
For both 5′-NNNNCCAA-3′ PAM and the 5′-NNNNCGAA-3′ PAM DNA, ThermoCas9 primarily recognizes the fifth base pair C(5*)–G(-5) while imposing additional restrictions on the sixth to eighth base pairs (Fig. 4a,b). G(-5) is recognized by Arg1035 through a pair of hydrogen bonds. C(5*) is simultaneously recognized by Asp1017 and Ser1019 through its major groove edge, leaving little space for additional functional groups such as a C5 methyl (Fig. 4b). The extensive interactions between ThermoCas9 and the C(5*)–G(-5) pair explain the critical role of C(5*) in PAM recognition and why its methylation impairs ThermoCas9 binding. In addition, the base pair A(7*)–T(-7) is recognized by a pair of asparagine residues, Asn961 and Asn1020, supporting a preference for an A–T pair at this position26. The presence of only single contacts between ThermoCas9 and the base pairs at positions 6 and 8 (Fig. 4b) is consistent with the low specificity observed at these positions26.

a, Cartoon representations of ThermoCas9 structures highlighting the PAM recognition region for the 5′-NNNNCCA-3′ (CC PAM) and the 5′-NNNNCGA-3′ (CG PAM) DNA (centre). Close-up views of the PAM nucleotides as stick models and the interacting protein residues (within 3.5 Å) are shown for the CC PAM (left) and the CG PAM (right) structures. Asterisks indicate numbering on the non-target strand. b, Recognition of each PAM base pair as stick models is overlaid with the cryo-EM density for the CC PAM (top row) and the CG PAM (bottom row) structures. The dashed lines indicate close contacts between the protein residues and the DNA bases. The black arrows indicate the predicted 5-methyl group location if the CC PAM (top row) or the CG PAM (bottom row) were methylated. The dashed box highlights the different base pairs of the two DNA substrates at position 6. Asterisks indicate numbering on the non-target strand. c, Cell survival assay (top row) and in vitro DNA cleavage (bottom row) results of WT, Asp1017 to alanine (D1017A) and Ser1019 to alanine (S1019A) of ThermoCas9. The survival rate is calculated by dividing the CFU on the arabinose-plus (inducing ccdB toxic protein) plate by that on the arabinose-minus plate. CpG indicates unmethylated, whereas 5mCpGw indicates fully methylated DNA plasmid substrates. Percent survival columns show mean ± s.e.m. (n = 3 biological replicates per group).
The anticipated critical roles of Asp1017 and Ser1019 in C(5*) base recognition were investigated using an activity assay in bacterial cells and an in vitro cleavage assay (Fig. 4c; for gel source data, see Supplementary Fig. 1d). Mutation of Asp1017 to alanine virtually abolished DNA cleavage activity, whereas substitution of Ser1019 to alanine retained partial activity in bacteria cells. Unlike the purified Asp1017 to alanine mutant, the Ser1019 to alanine mutant retained the ability of ThermoCas9 to discriminate methylated DNA in the cleavage assay (Fig. 4c; for gel source data, see Supplementary Fig. 1d). This indicates that Asp1017 is crucial for PAM recognition and probably influences methylation sensitivity, with other residues potentially contributing as well.
Whereas ThermoCas9 does not bind to DNA containing a methylated PAM (Fig. 1c and Extended Data Fig. 1), AceCas9 indeed forms a stable complex with the PAM-methylated DNA25. We thus determined a cryo-EM structure of AceCas9 bound with a methylated DNA at 3.0 Å resolution (Extended Data Table 1 and Extended Data Fig. 7). A large majority of particles formed from the active AceCas9 incubated with methylated DNA resulted in the pre-cleavage structure of AceCas9 where its HNH domain is positioned far from the target strand cleavage site (Extended Data Fig. 8). This result strongly suggests that methylation in PAM inhibits the open-to-closed transition, a step critical to Cas9 activation39,40,41. Comparison of structures of AceCas9 bound with dsDNA in the presence (this work) or in the absence (PDB 8DKL) of PAM methylation revealed no substantial structural changes, except for an increased mobility of the key PAM-interacting residues Asp1044 and Arg1088, as indicated by weak density (Extended Data Fig. 8b).
This suggests that methylation may perturb the PAM-interacting residues of AceCas9 in a way that impedes the conformational transition necessary for activation. Consistent with this model, mutation of the phosphate lock residues that have been previously shown to overcome the weaker 5′-NNNAC-3′ PAM40 alleviated inhibition by methylation (Extended Data Fig. 8c).
Methylation-sensitive genome editing in human cells by ThermoCas9
Human genomes undergo dynamic (hyper or hypo) methylation changes to allow for desired differentiation in healthy individuals. In case of disease-associated disruption of methylation regulation, this may lead to a wide range of undesired alterations in gene expression. The programmability and methylation sensitivity of ThermoCas9 offer the prospect of differential gene editing in human cells with distinct methylation landscapes.
We tested the utility of ThermoCas9 for methylation-sensitive editing in cells that differ in methylation profile. On the basis of in silico analysis of reduced-representation bisulfite sequencing data from the Encyclopedia of DNA Elements (ENCODE) database, we identified three DNA target sites with a 5′-NNNNCGAA-3′ PAM that show various methylation status in human embryonic kidney (HEK293T) and human colorectal carcinoma (HCT116) cells (Extended Data Fig. 9a). These include the EMX1 gene (target 4 (T4)), the PRDX4 gene (T5), and locus 1 (T3) on the VEGFA gene. We further performed bisulfite sequencing of these sites (Extended Data Fig. 9b). T4 and T5 showed methylation patterns consistent with those identified in ENCODE, whereas T3, surprisingly, exhibited methylation in both cell types (Extended Data Fig. 9b). We also selected a non-methylated site, locus 2 on the VEGFA gene (T9), with a 5′-NNNNCCAA-3′ PAM previously shown to be an effective target for ThermoCas9 (ref. 27).
We subsequently performed genome-editing experiments using ThermoCas9 programmed to target T3, T4, T5 and T9 both in HEK293T and in HCT116 cells and quantified average indel (small insertions and/or deletions) formation at each target site (Fig. 5a and Supplementary Figs. 3–8). As expected, ThermoCas9 successfully targeted the unmethylated VEGFA T9 site with mean indel frequencies up to 33% in HEK293T and 16% in HCT116 cells, respectively. On the contrary, ThermoCas9 was unable to edit VEGFA T3 of which the PAM is methylated in both cell lines, resulting in a null mean indel frequency for both HEK293T and HCT116 cells (Fig. 5a and Supplementary Figs. 3–8). Because the T3 and T9 sites are in close proximity (145 bp), the observed editing in both cell lines most likely resulted from the methylation sensitivity of ThermoCas9 rather than their differences in chromatin accessibility (Supplementary Fig. 9). Consistently, the commonly used SpyCas9 was able to edit T3–T5 efficiently in HEK293T cells regardless of their methylation status (Fig. 5a,b, Extended Data Fig. 9 and Supplementary Fig. 3). At the two differentially methylated sites, EMX1 T4 and PRDX4 T5, we again observed methylation-sensitive editing across the two cell lines. As expected, ThermoCas9 efficiently edited the unmethylated EMX1 T4 in HEK293T cells, with mean indel frequencies reaching 18%, but failed to edit the same site in HCT116 cells, where it is methylated, resulting in a mean null indel frequency (Fig. 5a and Supplementary Figs. 5–8). Similarly, at the PRDX4 T5 site, ThermoCas9 failed to edit the methylated T5 in HEK293T cells, whereas it efficiently edited the same site in HCT116 cells, where the PAM is unmethylated, with a mean indel frequency of 22% (Fig. 5a and Supplementary Figs. 5–8). As expected, the commonly used SpyCas9 was able to edit the EMX1 T4 and PRDX4 T5 sites in HEK293T regardless of their methylation status (Fig. 5a, Extended Data Fig. 9 and Supplementary Fig. 3). The observed methylation-sensitive editing of these two sites is also independent of the chromatin accessibility at these two sites between the two cell lines (Supplementary Fig. 9).

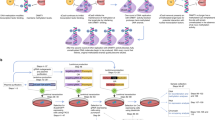
The colour keys indicate the specific Cas9, delivery method and the targeted cells. The filled circles indicate methylated sites, whereas open circles indicate unmethylated sites based on ENCODE reduced-representation bisulfite sequencing or EPIC data. The error bars are calculated from the indel percent of n ≥ 2 biologically independent samples. Data are presented as mean ± s.e.m. a, Methylation-sensitive editing of ThermoCas9 (plasmid delivery) in HEK293T cells (grey bars) and HCT116 cells (white bars). ICE, Inference of CRISPR Edits. b, Comparison of ThermoCas9 and SpyCas9 editing in HEK293T cells for six target sites. The protospacers chosen for both Cas9 effectors are either overlapping or within 10 bp. A cartoon summary of observed methylation sensitivity between Thermocas9 and SpyCas9 from panels a,b is also shown (bottom). c, Methylation-sensitive editing of ThermoCas9 (mRNA delivery) in HEK293T cells (grey bars) and HCT116 cells (white bars). d, Heatmap of DNA methylation based on β values measured by Illumina human methylation EPIC probes with greater than 20% difference for the target genes and surrounding regions between the MCF-10A (top) and MCF-7 (bottom) cells, respectively. EPIC probes are shown as equal-width columns arranged in the 5′-to-3′ direction of each gene and coloured according to their measured β values, ranging from hypomethylated (green) to hypermethylated (red), as indicated by the scale bar. The targeted genes and surrounding regions are displayed below the probe columns. Genome coordinates of the target sites (t; green arrows) and the nearest probe (p) and the mean β values of the nearest EPIC probes for each target are indicated. e, Methylation-sensitive editing of catalytically enhanced (CE) ThermoCas9 in MCF-7 and MCF-10A cells (mRNA or RNP delivery). The black-filled circles indicate sites within the PAM sequence of ThermoCas9 with estimated β > 0.90, whereas the grey gradient-filled circles indicate sites with estimated β values of 0.3–0.8.
To further substantiate these results, we utilized a PCR-based procedure to directly observe methylation-sensitive cutting of genomic DNA in vitro. We exposed the isolated genomic DNA from HEK293T or HCT116 cells to ThermoCas9 RNP complexes programmed with appropriate RNA guides for one of the target sites (T3, T4 or T5). Following ThermoCas9 RNP exposure, we PCR amplified the DNA fragments flanking the three sites along with that of a control site (Supplementary Fig. 10). For the targets whose PAM sequences contain 5mCpG, ThermoCas9 would fail to cut, resulting in distinct post-cutting PCR products. Conversely, for those whose PAMs lack CpG methylation, ThermoCas9 would cleave the sites, yielding no or weak PCR products. As expected, clear PCR products were observed for the VEGFA T3 in both HEK293T and HCT116, EMX1 T4 in HCT116 and PRDX4 T5 in HEK293T genomic DNA (Supplementary Fig. 10). However, no or weak products were detected for EMX1 T4 in HEK293T and PRDX4 T5 in HCT116 genomic DNA, whereas the untreated control had a clear PCR product (Supplementary Fig. 10). These results support our hypothesis that ThermoCas9 can be repurposed for methylation-sensitive editing and screening.
To rule out the possibility that other genome processes (such as DNA repair or heterochromatin compaction that may impact Cas9 editing43) might have contributed to the observed methylation-sensitive editing, we compared gene editing by either ThermoCas9 or SpyCas9 at three additional unmethylated (T10–T12) and three methylated (T13–T15) sites in HEK293T cells. We also performed in silico analysis of the accessibility of these and the previously targeted sites (Supplementary Fig. 9). Whereas ThermoCas9 exhibited notable gene-editing efficacy at T10–T12, no editing activity was observed at T13–T15 (Fig. 5b and Supplementary Figs. 11–13). By contrast, SpyCas9 displayed editing activities at all six sites regardless of their methylation states (Fig. 5b and Supplementary Figs. 14–17). We did not observe a notable difference in indel distributions between the methylated and unmethylated sites, suggesting a similar DNA repair process following dsDNA break at these sites. The combined editing activities of ThermoCas9 at all sites show that ThermoCas9 consistently discriminates methylated sites with a minimal impact by the chromatin accessibility or other genomic processes.
To enhance the efficiency of methylation-sensitive editing by ThermoCas9, we performed protein-directed evolution following the strategy previously used for AceCas9 (refs. 40,44,45). In brief, we screened a library of ThermoCas9 with varied HNH hinge (linker II, between HNH and RuvC-III; Supplementary Fig. 18a) and selected for catalytically enhanced variants. A single variant emerged that contained two mutations, Glu655 to Gly and Asn696 to Ile, that we termed catalytically enhanced ThermoCas9 (Supplementary Fig. 18).
In addition to enhancing the catalytic efficiency of ThermoCas9, we also used mRNA delivery for gene editing. Compared with delivery as DNA (plasmid or viral vectors), transfection of mRNA enables rapid expression, reduces immunogenicity through chemical modifications, avoids genomic integration by ensuring transient expression, and is compatible with lipid nanoparticles for in vivo therapeutic applications46. When the wild-type (WT) ThermoCas9-mRNA was paired with gRNAs targeting T9, T3, T4 and a new target T6, we observed significantly improved editing efficiency compared with plasmid delivery (Fig. 5c and Supplementary Figs. 19–23). Catalytically enhanced ThermoCas9-mRNA produced higher editing levels than WT ThermoCas9-mRNA without compromising its methylation sensitivity for both targets with 5′-NNNNCGAA-3′ PAM, although the magnitude of improvement varied depending on the target site (Supplementary Fig. 24).
ThermoCas9 targets hypomethylated genes in MCF-7 breast cancer cells
To explore the therapeutic potential of the 5mCpG sensitivity of ThermoCas9, we assessed its ability to target hypomethylated genes associated with breast cancer. The luminal expression signature genes, such as ESR1 and GATA3, are among the most frequently mutated in luminal/oestrogen receptor-positive (ER+) breast cancers and are often overexpressed in patients, largely due to loss of DNA methylation47,48. Targeting overexpressed ESR1 is a cornerstone of treatment in ER+ breast cancers49. However, treatment often drives the emergence of ESR1 mutations, which confer oestrogen-independent receptor activation and are associated with reduced overall survival, indicative of a more aggressive clinical phenotype50. Specific modulation of ESR1 and GATA3 within lesion cell populations could enable new intervention strategies.
To confirm that DNA methylation changes in our model cells reflect those widely observed in breast cancer genomes and corresponding normal tissues48, we performed an Infinium Methylation EPIC array on genomic DNA isolated from healthy MCF-10A and cancer-derived MCF-7 cells (Fig. 5d). The EPIC assay quantifies DNA methylation at more than 900,000 CpG sites including regions of ESR1 and GATA3. By considering DNA methylation levels and the available 5′-NNNNCGAA-3′ PAM sites, we selected targets for ThermoCas9 in enhancer or promoter regions of ESR1, GATA3 and the gene body of a control gene EGFLAM (Fig. 5d). We first transfected MCF-7 cells with either the WT or catalytically enhanced ThermoCas9-mRNA targeting these sites (T11 on the control EGFLAM gene, T17 on ESR1 gene and T18 on GATA3 gene) and observed moderate efficiency with modified read frequencies ranging from 2% to 13% in the bulk population (Fig. 5e and Supplementary Figs. 25 and 26). In addition, the improvement in efficiencies with the catalytically enhanced over the WT ThermoCas9-mRNA was not significant in MCF-7 cells (Supplementary Fig. 27a).
To further improve editing efficiency, we purified WT and catalytically enhanced ThermoCas9 proteins containing three nuclear localization signals (Supplementary Fig. 27b) and evaluated an alternative delivery method using nucleofection of RNPs (Fig. 5e and Supplementary Figs. 28–30). Of note, catalytically enhanced ThermoCas9 RNP outformed mRNA delivery and the WT RNP substantially for two of the three targets (Supplementary Fig. 27c), yielding 25% modified reads at ESR1 (T17) and up to 78% at GATA3 (T18; Fig. 5e and Supplementary Figs. 28–30). Motivated by the substantial improvement observed with catalytically enhanced ThermoCas9 RNPs, we next targeted the same sites in MCF-10A cells with the catalytically enhanced ThermoCas9 RNPs and observed editing efficiencies ranging from 14% at EGFLAM to 28% at GATA3 with no editing at ESR1 (Fig. 5e and Supplementary Figs. 31–33). The levels of editing in MCF-10A at the three sites reflect their estimated methylation levels (Fig. 5d), supporting the target selectivity of the catalytically enhanced ThermoCas9 variant in breast cell lines.
The notable targeting of the hypomethylated GATA3 by catalytically enhanced ThermoCas9 is significant, considering its coordinated role with ESR1 in driving oestrogen-responsive transcription. In MCF-7, GATA3 contains a frameshift mutation in exon 6, leading to a truncated protein that is overexpressed and believed to contribute to the pathogenesis51,52. In other breast cancer cell models, GATA3 truncation mutations, which contribute to 50% of all GATA3 mutations in luminal/ER+ cases, cause dominant-negative effects that impair the normal transcriptional functions of WT GATA3 (ref. 53), contributing to disrupted differentiation programs and poor prognosis54. In addition, overexpression of GATA3 in breast cancer is correlated with the loss of DNA methylation in its associated enhancer regions55. Along with ESR1, successful targeting of GATA3 in MCF-7 by ThermoCas9 highlights its potential for therapeutic targeting of hypomethylated sites in breast cancer.
Discussion
Here we report a unique feature of ThermoCas9 in that it recognizes its DNA target using a 5′-N4CGAA-3′ PAM sequence, and that methylation of the corresponding cytosine (5mC) prevents target cleavage by ThermoCas9 both in vitro and in human cells. We further report the structural characterization of two methylation-sensitive Cas9 nucleases: ThermoCas9 when bound with a non-methylated DNA target and the previously characterized AceCas9 when bound with methylated DNA. Whereas both nucleases are inhibited by 5mC in their PAM sequences, the molecular basis of the inhibitory effect differs between them. Whereas the nuclease activity of AceCas9 is disturbed after target binding, the methylated cytosine abolished ThermoCas9 binding to a dsDNA target. Our discoveries provide a potential opportunity to repurpose both Cas9 nucleases for epigenetic genome detection and manipulation.
The demonstrated methylation-sensitive gene editing in human cells by ThermoCas9 expands the precision of CRISPR–Cas9 technologies by allowing for discrimination of different natural epigenetic states. Recent studies have shown that unrepaired dsDNA breaks or nicks, such as those generated by CRISPR–Cas9 nickases, can lead to unintended mutations, indels and genomic instability56,57,58. ThermoCas9-based technology, including base and prime editing, can reduce these adverse outcomes by restricting editing to genomic sites that have lost 5mCpG methylation. This selectivity is particularly important when hypomethylated alleles are the intended targets.
The PAM sequences, 5′-NNNNCCAA-3′ or 5′-NNNNCGAA-3′, used by ThermoCas9 in the human genome-editing experiments described in this study were based on those identified from previous in vitro studies26. For unrestricted epigenetic applications, minimal dependence on nucleotides flanking CpG or CpC is desirable. In vitro DNA cleavage results and the structural data indicate that the adenosine immediately following CpG may be relevant, whereas the last adenosine is less important. If this dependence is conserved in human genome editing, additional engineering of ThermoCas9 could be a way to lessen or eliminate it. Owing to its reliance on a long guide–protospacer heteroduplex (23 bp), relaxing the PAM requirement for ThermoCas9 is unlikely to compromise its demonstrated high specificity26.
Similar to other type II-C Cas9 variants, ThermoCas9 has lower DNA cleavage activities than those of type II-A Cas9 effectors such as SpyCas9 (ref. 59). Apart from the shifted temperature optimum of nucleases from thermophilic bacteria, this is believed to stem from the inherently weaker DNA-unwinding activity of type II-C relative to type II-A Cas9 nucleases. Although type II-C Cas9 effectors may offer improved editing fidelity60, which could be linked to their weaker catalytic efficiencies, efforts have been made in successfully improving catalytic efficiencies through enzyme engineering34,45. Here we have demonstrated that combining protein engineering with optimized delivery methods enables robust editing in traditionally difficult-to-edit cells, substantially broadening the utility of type II-C Cas9 effectors in genome editing. More importantly, we have shown that ThermoCas9 can selectively edit therapeutic target genes based on their methylation status.
ThermoCas9 offers the potential for a novel type of gene therapy and unlocks a new generation of methylation-sensitive tools beyond DNA cleavage, thereby contributing to the spectacular progress of the CRISPR–Cas9 technology. The three-dimensional (3D) structures of the active ThermoCas9 provide a crucial foundation for further engineering of improved and safer variants. Once developed, these variants could enable innovative therapeutic strategies.
Methods
Cloning, protein expression and purification
The DNA encoding ThermoCas9 with a C-terminal His6 tag was integrated into the pML-1B vector and expressed in the Escherichia coli NiCo21(DE3) strain. Cells were grown in Luria–Bertani (LB) medium with 0.2% d-(+)-glucose at 37 °C until optical density at 600 nm reached 0.8, at which point addition, isopropyl-β-d-thiogalactopyranoside was added to 0.5 mM concentration. Cells were grown for an additional 16–18 h at 20 °C and harvested by centrifugation and stored in −80 °C. Previously frozen cells were lysed via sonication in a lysis buffer (500 mM NaCl, 50 mM phosphate buffer pH 8.0 (sodium phosphate dibasic and sodium phosphate monobasic), 5 mM imidazole and 1 mM β-mercaptoethanol) containing 1 tablet of cOmplete Mini Protease Inhibitor Cocktail (Sigma-Aldrich) per 100 ml. The lysate was centrifuged at a speed of 16,000 rpm for 60 min at 4 °C, after which the supernatant was loaded on a pre-equilibrated 5-ml HisTrap HP His tag protein purification column (Cytiva Life Sciences). The resin was subsequently washed with 200 ml wash buffer (500 mM NaCl, 50 mM phosphate buffer pH 8.0, 30 mM imidazole and 1 mM β-mercaptoethanol), before being eluted with elution buffer (500 mM NaCl, 50 mM phosphate buffer pH 8.0, 250 mM imidazole and 1 mM β-mercaptoethanol). The resultant eluate was transferred onto a pre-equilibrated HiTrap Heparin HP affinity column (Cytiva Life Sciences) and eluted with a 100 mM to 2 M NaCl gradient. The purified protein was then concentrated and stored at −80 °C until further use.
For purification of ThermoCas9 used in human gene-editing experiments, the DNA encoding 3×-nuclear localization sequence (2× SV40 NLS and 1× nucleoplasm NLS) fused with ThermoCas9 with a C-terminal His6 tag was integrated into the pML-1B vector and expressed in E. coli Rosetta (DE3) cells. The same purification method was used with the exception that the gel-filtration buffer was made with cytotoxin-free water.
In vitro RNA transcription
We used the T7 in vitro transcription method to produce the sgRNA for both ThermoCas9 and AceCas9. The sgRNA templates containing a T7 promotor were purchased from Eurofins Genomics. A 149 nt sgRNA for ThermoCas9 and a 106 nt sgRNA for AceCas9 (Supplementary Table 1), respectively, were transcribed by T7 RNA polymerase in a transcription buffer (5 mM NTPs, 50 mM Tris-HCl pH 7.5, 15 mM MgCl2, 5 mM dithiothreitol and 2 mM spermidine) and purified via the Monarch RNA Cleanup Kits (New England Biolabs). The DNA used in cryo-EM and biochemical assays was purchased from Eurofins Genomics.
Cryo-EM sample preparation, data collection and 3D reconstruction
The heparin-purified protein was incubated with sgRNA at a 1:1.5 molar ratio at 37 °C for 30 min, and the resulting RNP was further purified via size-exclusion chromatography with a Superdex 200 10/300 GL column (Cytiva Life Sciences) in gel-filtration buffer (300 mM NaCl, 30 mM HEPES pH 7.5 and 1 mM dithiothreitol). The Cas9–RNA–DNA ternary complex was assembled by adding pre-annealed substrate dsDNA into the RNP at a 2:1 molar ratio with the presence of 10 mM magnesium chloride. The reactive ternary complex was incubated at 37–50 °C for 15–30 min. Of the sample, 4 µl was added to glow-discharged Gold 300 mesh R1.2/1.3 grids, which was then allowed to adsorb for 30 s before blotting for 2.5 s under conditions of 20 °C and 100% humidity. These grids were rapidly frozen in liquid nitrogen cooled ethane within Vitrobot Mark IV.
Raw micrographs of ThermoCas9 bound with DNA containing 5′-NNNNCCA-3′ PAM and AceCas9 bound with DNA containing 5′-NNN5mCC-3′ PAM were collected at the Laboratory for Biomolecular Structure of the Brookhaven National Laboratory using a Titan Krios G3i cryo transmission electron microscope equipped with a Gatan K3 direct electron detector. Raw micrographs of ThermoCas9 bound with DNA containing 5′-NNNNCGA-3′ PAM were collected at the Pacific Northwester Center for Cryo-EM using a Titan Krios Electron Microscope equipped with a Gatan K3 direct electron detector (Thermo Fisher Scientific). Movies were recorded at a nominal magnification of 105,000 in a super-resolution mode with an energy filter of 15 eV, corresponding to a corrected physical pixel size of 0.82 Å per pixel. A total dose of 50–60 e− Å−2 was spread over 60 frames with random defocus set to −0.8 to −2.5 µm. Motion correction was executed in bin 2 via MotionCorr2 and contrast transfer function (CTF) estimation was carried out with Gctf61. A total of 6,080 micrographs were collected and 2,516,939 particles were picked using Topaz62, followed by multiple rounds of 2D classification using cryoSPARC63, resulting in 2,015,088 good particles for 3D classification. After heterogenous refinement in cryoSPARC, the dataset was classified into five classes. Several rounds of 3D refinement and 3D classification were then performed using Relion 4.0 (ref. 64) to obtain high-quality particles. Finally, several rounds of non-uniform refinement65 were performed using cryoSPARC to reach the final 3D structures. Structural models were built in COOT66 and refined in PHENIX67 to satisfactory stereochemistry and real-space map correlation parameters. Note that water molecules were only modelled based on both density and interaction chemistry in the two high-resolution structures.
Bacterial survival assay
The survival assay in bacterial cells followed a previously outlined procedure44 with minor modifications. In brief, electrocompetent E. coli BW25141 cells, harbouring the modified p11-LacY-wtx1 plasmid encoding toxic ccdB protein, were transformed with 60 ng of WT or mutant ThermoCas9 plasmids. Afterwards, the cells were recovered in LB for 30 min with shaking at 37 °C. Subsequently, 0.05 mM isopropyl-β-d-thiogalactopyranoside was introduced, and the recovery process continued for an additional 60 min. The recovered cells were then plated on LB agar plates containing either chloramphenicol (15 mg ml−1) or a combination of chloramphenicol and 10 mM arabinose. The plates were incubated at 37 °C for 16–20 h. Manual counting of colonies was performed on both plates, and survival rates were determined by dividing the CFUs on arabinose-containing plates by those on chloramphenicol-only plates. For directed evolution of ThermoCas9, a library of ThermoCas9 linker II variants were transformed into BW25141 cells harbouring a modified p11-LacY-wtx1 plasmid containing a PAM-distal truncated protospacer of 17 nucleotides (17-mer) in the same manner as stated above. CFUs that grew on arabinose in the 17-mer cells were selected for Sanger sequencing.
In vitro DNA cleavage assay and competition assay
ThermoCas9 was combined with sgRNA at a 1:2 ratio and incubated at 37 °C for 30 min to form the RNP. The target plasmid at 6 nM was then added to the RNP at 1 µM and allowed to incubate for varying lengths of time. The reactions were stopped by adding a 5× stop buffer (25 mM Tris pH 7.5, 250 mM EDTA pH 8.0, 1% SDS, 0.05% w/v bromophenol blue and 30% glycerol). The reaction products were separated on a 0.8% agarose gel and stained by ethidium bromide.
Fluorescently labelled oligonucleotides were also used to prepare DNA substrates. Six-carboxyfluorescein (FAM)-labelled non-target strand DNA was annealed with an unlabelled target strand DNA at a 1:1 molar ratio. Separately, hexachlorofluorescein (HEX)-labelled target strand DNA was annealed with unlabelled non-target strand DNA at a 1:19 molar ratio. Annealing was performed by heating the DNA mixtures to 75 °C for 5 min, followed by a gradual cooling to room temperature. Pre-annealed dsDNA substrates were prepared at concentrations of 100–200 nM for the labelled strand. These substrates were then added to a ThermoCas9 RNP solution at 1 µM to initiate the cutting reaction. Divalent metal ions, specifically 10 mM of MgCl2, were also included in each reaction. The reaction mixtures were incubated at 37–50 °C for 1 h before adding 2× RNA loading buffer (97% formamide, 0.02% SDS and 1 mM EDTA). The reaction products were resolved using a 7 M urea 20% polyacrylamide denaturing gel. Gel electrophoresis was performed under denaturing conditions to ensure the separation of DNA fragments based on size. Following electrophoresis, the gel was visualized using a Bio-Rad ChemiDoc gel imaging system. Fluorescent labels were detected using excitation wavelengths of 488 nm for FAM and 580 nm for HEX.
For competition assays, ThermoCas9 RNP at 1 μM was mixed with the target plasmid at 10 nM, and a competing oligo DNA substrate at concentrations of 50 nM to 1 μM. The reactions were incubated at 50 °C for 15 min and stopped by adding the 5× stop buffer. The reaction products were separated on a 0.8% agarose gel and stained by ethidium bromide. The fraction of cleavage versus oligo concentration plots were fitted to a competitive one-site binding model in GraphPad to yield the estimated binding constant of each competing oligo (Ki).
Native gel-binding assay
FAM-labelled non-target strand DNA was annealed with an unlabelled target strand DNA at a 1:1 molar ratio. dsDNA (100 nM) was mixed with 1 μM ThermoCas9 RNP in the reaction buffer without MgCl2 for 1 h. The reaction product was then mixed with 6X purple gel loading dye (New England Biolabs) and loaded onto a 10% TBE gel (Invitrogen) for electrophoresis.
In vitro methylation screening
Genomic DNA from HEK293T and HCT116 cells was extracted with QuickDNA microprep kit (Zymo Research). The extracted genomic DNA was then incubated with 125–250 nM ThermoCas9 RNP in the reaction buffer containing 5 mM MgCl2 at 37 °C for 30–45 min. The reaction product was treated with E.Z.N.A. Plasmid DNA Mini Kit Solution I (Omega Bio-tek) for 10 min at a 1:1 volume ratio. DNA was subsequently cleaned up using the Monarch PCR & DNA Cleanup Kit (New England Biolabs). For PCR amplification, 1 µl of the reaction product was mixed with 0.25–1 µM primers and Q5 High-Fidelity 2X Master Mix (New England Biolabs). The PCR product was then mixed with 6X blue gel loading dye (New England Biolabs) and loaded onto a 2% agarose gel with a 100-bp DNA ladder (New England Biolabs) for electrophoresis.
In silico analysis of differentially methylated sites in human cells
Reduced representation bisulfite sequencing (RRBS) data were collected from the ENCODE functional genomics database for various cell lines68. We downloaded the call sets (bed files) from the ENCODE portal69 (https://www.encodeproject.org/) with the following identifiers: ENCFF001TMR, ENCFF001TMQ, ENCFF001TMS and ENCFF001TMT for the HEK293T cell line and ENCFF001TMM and ENCFF001TMN for the HCT116 cell line. An in-house program was used to compare the methylation profiles based on the methylation scores. The RRBS methylation profiles across various genetic loci in different cell lines were visualized using the Integrative Genomics Viewer70. An in-house program based on Python scripts and bed utilities was used to identify genes that are differentially methylated in different cell lines.
Transfections and gene editing in HEK293T and HCT116 cells using plasmid DNA
Human-codon-optimized thermocas9-sv40nls gene and its sgRNA module were expressed under the control of the constitutive cytomegalovirus (PCMV) and U6 RNA polymerase III (PU6) promoters, respectively (Supplementary Table 1). We co-expressed the EGFP reporter gene under the constitutive elongation factor 1α promoter (PEF1α) to allow for sorting of successfully transfected cells, as previously described27. We designed four spacers that target protospacers in the chromosomal genes VEGFA, EMX1 and PRDX4. All differentially methylated protospacers were flanked by a PAM of (5′-NNNNCGAA-3′) thus representing a potential CpG methylated PAM. The targeting spacers of EMX1 and PRDX4 are differentially methylated in the PAM sequence between HEK293T and HCT116; the negative and positive control targets are located on the VEGFA gene. HCT116 cells were maintained in McCoy’s 5A media and HEK293T cells were maintained in DMEM media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C with 5% CO2. Both HEK293T and HCT116 cells were seeded on physically surface-treated 24-well plates (Corning/Falcon) at a seeding density of 1.0 × 105 cells per well. After 24 h of incubation, 0.5 μg of genome-editing plasmid was transfected into the HEK293T and HCT116 cells using Lipofectamine 3000 Transfection reagent (L3000015, Thermo Fisher). For each well on the plate, transfection plasmids were combined with OptiMEM Reduced Serum Medium (31985062, Thermo Fisher) to a total volume of 25 µl and mixed with 1 µl P3000 reagent. Separately, 25 µl OptiMEM was combined with 1.1 µl Lipofectamine 3000 reagent. Plasmid and Lipofectamine solutions were then combined, incubated at room temperature for 10 min and pipetted on to cells. The transfected cells were cultured 72 h and further evaluated for the presence of GFP using fluorescence-activated cell sorting (FACS). For SpyCas9 gene-editing experiments, HEK293T cells were transfected with 0.5 μg of plasmid co-expressing SpyCas9 and sgRNA (Addgene #42230). The transfection methods were consistent with ThermoCas9, except cells were harvested 48 h post-transfection for genomic DNA isolation without FACS sorting.
FACS
After 72 h of incubation, the transfected HEK293T and HCT116 cells were harvested, centrifuged at 1,000 rpm for 5 min, resuspended in 250 μl DMEM (10% FBS and 1% penicillin–streptomycin), and filtered through Nylon Mesh 52 micron, 32% open area filter (Component Supply Co.). GFP+ fluorescent cells were bulk sorted using the BD FACSAria III cell sorter device (BD; 488 nm laser, FITC detection channel for GFP fluorescence). The cells were gated for ‘high-green’ to reduce the signal to noise of auto-fluorescent cells (Supplementary Fig. 4). Cells were transferred to a 96-well nucleon plate and centrifuged at 200 rpm for 2 min and cultured for approximately 1–2 weeks (37 °C; 5% CO2). When approximately 75% confluency was reached, the propagated cells of each experiment were steadily passaged to 24-well plates and further screened for indels.
ThermoCas9-mRNA production and nucleofection
In vitro transcription reactions for ThermoCas9-mRNA (Supplementary Table 1) were assembled with T7 buffer (NEB), 100 mM ATP (NEB), 100 mM GTP (NEB), 100 mM CTP (NEB), 100 mM pseudo-UTP (Trilink), CleanCap AG (Trilink), human-codon-optimized ThermoCas9-NLS (Gene Fragment with Adapters Twist Bioscience; 108612) and T7 RNA Polymerase (NEB) and incubated at 37 °C overnight. The following day, the reaction was further treated with DNase enzyme (NEB) followed by Monarch spin RNA cleanup kit (500 µg column) before transfection.
For ThermoCas9-mRNA delivery, all transfections were performed with a 4D Lonza nucleofector. Before the addition of nucleofection buffers, cells were detached with TrypLE and washed with PBS pH 7.2 1X (Gibco) to remove potential RNases. The ThermoCas9-mRNA nucleofection conditions were as follows: 16.4 µl SF or SE nucleofection buffer supplemented with 3.6 µl Supplement 1; 1.0 × 105 cells; 1 µl of 100 µM µl−1 custom sgRNA (SC1518-CRISPR Oligo, Genscript) and 3.8 µg µl−1 CleanCap ThermoCas9 mRNA. Pulse codes for nucleofections were DS-150, EN-113, DS-137 and EN-150 for HEK293T (CRL-3216, American Type Culture Collection (ATCC)), HCT116 (CCL-247, ATCC), MCF-7 (HTB-22, ATCC) and MCF-10A (CRL-10317, ATCC), respectively. MCF-7 cells were maintained in EMEM media supplemented with a final concentration of 2 mM L-glutamine, 0.01 mg ml−1 insulin and 10% fetal bovine serum. MCF-10A cells were maintained in Lonza MEGM Mammary Epithelial cell Growth Medium BulletKit supplemented with 100 ng ml−1 cholera toxin and grown at 37 °C with 5% CO2. For RNP, all conditions are the same as above but RNP conditions were 1 µl of 100 µM µl−1 custom sgRNA (SC1518-CRISPR Oligo, Genscript), and 1 µl of 3 mg ml−1 ThermoCas9 protein. All nucleofections were conducted with a 16-well nucleocuvette strip within the 4D-Nucelofector X Unit. After applying the electroporation pulse, cells were allowed to rest within the nucleocuvette strip for approximately 10 min before adding 80 µl of respective media to transfer to a 24-well plate.
Screening for genome editing
HEK293T and HCT116 genomic DNA was isolated from the bulk population of propagated cells grown approximately 2–3 weeks post-FACS sorting. Genomic DNA was extracted using the Zymo Research Quick-DNA MicroPrep kit. Genomic target regions (VEGFA, EMX1 and PRDX4) were PCR amplified with Q5 High-Fidelity 2X Master Mix (New England Biolabs). The PCR products were verified on a 2% DNA agarose gel, and they were subsequently gel purified with the E.Z.N.A. gel extraction kit (Omega-BioTek). To detect indel formation, the gel-purified PCR products were subjected to Sanger sequencing (FSU sequencing facility). The sequencing results of the genome-editing assays were analysed using the Inference of CRISPR Edits (ICE) tool70 (EditCo) (Supplementary Figs. 7, 8 and 12–17). For ThermoCas9-mRNA and RNP editing experiments: HEK293T, HCT116, MCF-7 and MCF-10A genomic DNA was isolated from cells 72 h post-nucleofection. All downstream sample processing is the same as mentioned above. To detect modified reads from mRNA-treated or RNP-treated samples, the gel-purified PCR products were subjected to premium PCR sequencing by Plasmidsaurus using Oxford Nanopore Technology with custom analysis and annotation. The ThermoCas9-mRNA and RNP genome-editing sequence analysis was performed using CRISPResso2 by uploading FASTQ files as single-end reads and using the standard settings for Cas9.
Bisulfite sequencing
Genomic DNA of both HEK293T and HCT116 were bisulfite treated via the EpiJET Bisulfite Conversion Kit (K1461, Thermo Scientific) following the manufacturer’s instructions. The MethPrimer online tool was utilized to design primers to amplify bisulfite-converted samples flanking the regions of gene-editing targets followed by Sanger sequencing (FSU sequencing facility).
5mC interrogation by Infinium Methylation EPIC array
DNA was quantified by Qubit fluorometry (Promega) and 250 ng of DNA from each sample was bisulfite converted using the Zymo EZ DNA Methylation Kit (Zymo Research) following the manufacturer’s protocol using the specified modifications for the Illumina Infinium methylation assay. After conversion, all bisulfite reactions were cleaned using the Zymo-Spin binding columns and eluted in 12 µl of Tris buffer. Following elution, bisulfite-converted DNA was processed through the Infinium Methylation EPIC array v2.0 protocol (Illumina). The EPIC array v2.0 contains more than 930,000 probes querying methylation sites including CpG islands and non-island regions, RefSeq genes, ENCODE open chromatin, ENCODE transcription factor-binding sites and FANTOM5 enhancers. To perform the assay, 4 µl of converted DNA was denatured with 4 µl 0.1 N sodium hydroxide. DNA was then amplified, hybridized to the EPIC bead chip, and an extension reaction was performed using fluorophore-labelled nucleotides per the manufacturer’s protocol. Array beadchips were scanned on the Illumina iScan platform and probe-specific calls were made using Illumina Genome Studio software. ThermoCas9 target sites with contrasting methylation scores between the MCF-7 and the MCF-10 cells were identified from the processed EPIC array data using an in-house script.
Data processing for Infinium Methylation EPIC array
The R package SeSAMe71 was used to process Illumina microarray platform files in IDAT format generated from the EPIC v2.0 array, followed by downstream differential methylation locus (DML) and region analyses. The ‘openSesame’ function from SeSAMe was used to convert the files into DNA methylation level (β value) matrices in R. For DML detection, SeSAMe applies linear models to identify DMLs between two groups in a contrast. For differential methylation region analysis, neighbouring CpGs that show consistent methylation variation were merged into differentially methylated regions, and adjusted P values were calculated using the Benjamini–Hochberg procedure. Methylation sites were annotated using SesameData71, and additional annotation regarding genomic context and proximity to nearby genes was obtained from Noguera-Castells et al.72.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Uncropped and unedited gel data that support this study are included in this published article (and its Supplementary Information files). Reported cryo-EM structures and associated maps have been deposited to the PDB and Electron Microscopy Data Bank, respectively, with 9AR4 and 43769 for the CpC DNA–ThermoCas9 post-cleavage complex, 9AR6 and 43771 for the CpC DNA–ThermoCas9 pre-cleavage complex, 9AR7 and 43772 for the CpC DNA target-only complex, 9AR5 and 43770 for the 5mCpC AceCas9 complex, and 9BS6 and 44859 for the CpG DNA–ThermoCas9 post-cleavage complex. Amplicon nanopore sequencing data have been deposited to the NCBI Sequence Read Archive database under accession PRJNA1426056. Infinium Methylation EPIC array data for MCF-10A and MCF-7 cells have been deposited to the Gene Expression Omnibus repository under accession GSE322563. Plasmids generated in this study have been deposited to Addgene (254684 and 254685).
Change history
23 April 2026
In the version of this article initially published, the Gene Expression Omnibus accession GSE322563 was listed incorrectly in the Data availability section and is now amended in the HTML and PDF versions of the article.
References
Wang, J. Y. & Doudna, J. A. CRISPR technology: a decade of genome editing is only the beginning. Science 379, eadd8643 (2023).
Zhang, F. Development of CRISPR-Cas systems for genome editing and beyond. Q. Rev. Biophys. 52, e6 (2019).
Charpentier, E. CRISPR-Cas9: how research on a bacterial RNA-guided mechanism opened new perspectives in biotechnology and biomedicine. EMBO Mol. Med. 7, 363–365 (2015).
Loyfer, N. et al. A DNA methylation atlas of normal human cell types. Nature 613, 355–364 (2023).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Ziller, M. J. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013).
Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002).
Greenberg, M. V. C. & Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607 (2019).
Krepelova, A. & Neri, F. DNA methylation controls hematopoietic stem cell aging. Nat. Aging 3, 1320–1322 (2023).
Jones, P. A., Issa, J. P. & Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641 (2016).
Tsui, T. K. & Li, H. Structure principles of CRISPR-Cas surveillance and effector complexes. Annu. Rev. Biophys. 44, 229–255 (2015).
Nunez, J. K., Harrington, L. B. & Doudna, J. A. Chemical and biophysical modulation of Cas9 for tunable genome engineering. ACS Chem. Biol. 11, 681–688 (2016).
Garcia-Doval, C. & Jinek, M. Molecular architectures and mechanisms of class 2 CRISPR-associated nucleases. Curr. Opin. Struct. Biol. 47, 157–166 (2017).
Yaung, S. J., Esvelt, K. M. & Church, G. M. CRISPR/Cas9-mediated phage resistance is not impeded by the DNA modifications of phage T4. PLoS ONE 9, e98811 (2014).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020).
Yousefi, P. D. et al. DNA methylation-based predictors of health: applications and statistical considerations. Nat. Rev. Genet. 23, 369–383 (2022).
Baylin, S. B. & Jones, P. A. A decade of exploring the cancer epigenome — biological and translational implications. Nat. Rev. Cancer 11, 726–734 (2011).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Chemi, F. et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. Cancer 3, 1260–1270 (2022).
Nuzzo, P. V. et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 26, 1041–1043 (2020).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018).
Liu, X. S. et al. Editing DNA methylation in the mammalian genome. Cell 167, 233–247.e17 (2016).
Nakamura, M., Gao, Y., Dominguez, A. A. & Qi, L. S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23, 11–22 (2021).
Das, A. et al. The molecular basis for recognition of 5′-NNNCC-3′ PAM and its methylation state by Acidothermus cellulolyticus Cas9. Nat. Commun. 11, 6346 (2020).
Mougiakos, I. et al. Characterizing a thermostable Cas9 for bacterial genome editing and silencing. Nat. Commun. 8, 1647 (2017).
Trasanidou, D. et al. Efficient genome and base editing in human cells using ThermoCas9. CRISPR J. 6, 278–288 (2023).
Pinney, S. E. Mammalian non-CpG methylation: stem cells and beyond. Biology 3, 739–751 (2014).
Guo, J. U. et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222 (2014).
Yu, B. et al. Genome-wide, single-cell DNA methylomics reveals increased non-CpG methylation during human oocyte maturation. Stem Cell Rep. 9, 397–407 (2017).
Patil, V. et al. Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res. 47, 10072–10085 (2019).
Schmitz, R. J., Lewis, Z. A. & Goll, M. G. DNA Methylation: shared and divergent features across eukaryotes. Trends Genet. 35, 818–827 (2019).
de Mendoza, A., Lister, R. & Bogdanovic, O. Evolution of DNA methylome diversity in eukaryotes. J. Mol. Biol. 432, 1687–1705 (2019).
Eggers, A. R. et al. Rapid DNA unwinding accelerates genome editing by engineered CRISPR-Cas9. Cell 187, 3249–3261.e14 (2024).
Sun, W. et al. Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol. Cell 76, 938–952.e5 (2019).
Yamada, M. et al. Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol. Cell 65, 1109–1121.e3 (2017).
Jiang, F. & Doudna, J. A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529 (2017).
Cofsky, J. C., Soczek, K. M., Knott, G. J., Nogales, E. & Doudna, J. A. CRISPR-Cas9 bends and twists DNA to read its sequence. Nat. Struct. Mol. Biol. 29, 395–402 (2022).
Pacesa, M. et al. R-loop formation and conformational activation mechanisms of Cas9. Nature 609, 191–196 (2022).
Das, A. et al. Coupled catalytic states and the role of metal coordination in Cas9. Nat. Catal. 6, 969–977 (2023).
Bravo, J. P. K. et al. Structural basis for mismatch surveillance by CRISPR-Cas9. Nature 603, 343–347 (2022).
Nierzwicki, L. et al. Principles of target DNA cleavage and the role of Mg2+ in the catalysis of CRISPR-Cas9. Nat. Catal. 5, 912–922 (2022).
Přibylová, A., Fischer, L., Pyott, D. E., Bassett, A. & Molnar, A. DNA methylation can alter CRISPR/Cas9 editing frequency and DNA repair outcome in a target-specific manner. New Phytol. 235, 2285–2299 (2022).
Hand, T. H., Das, A. & Li, H. Directed evolution studies of a thermophilic Type II-C Cas9. Methods Enzymol. 616, 265–288 (2019).
Hand, T. H. et al. Catalytically enhanced cas9 through directed protein evolution. CRISPR J. 4, 223–232 (2021).
Paunovska, K., Loughrey, D. & Dahlman, J. E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 23, 265–280 (2022).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Dennis, S. R. et al. DNA methylation patterns in breast cancer, paired benign tissue from ipsilateral and contralateral breast, and healthy controls. Breast Cancer Res. 27, 103 (2025).
Burstein, H. J. Systemic therapy for estrogen receptor-positive, HER2-negative breast cancer. Reply. N. Engl. J. Med. 384, 1176–1177 (2021).
Turner, N. C. et al. ESR1 mutations and overall survival on fulvestrant versus exemestane in advanced hormone receptor-positive breast cancer: a combined analysis of the phase III SoFEA and EFECT trials. Clin. Cancer Res. 26, 5172–5177 (2020).
Takaku, M., Grimm, S. A. & Wade, P. A. GATA3 in breast cancer: tumor suppressor or oncogene? Gene Expr. 16, 163–168 (2015).
Usary, J. et al. Mutation of GATA3 in human breast tumors. Oncogene 23, 7669–7678 (2004).
Takaku, M., Grimm, S. A., De Kumar, B., Bennett, B. D. & Wade, P. A. Cancer-specific mutation of GATA3 disrupts the transcriptional regulatory network governed by estrogen receptor alpha, FOXA1 and GATA3. Nucleic Acids Res. 48, 4756–4768 (2020).
Takaku, M. et al. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat. Commun. 9, 1059 (2018).
Detilleux, D., Spill, Y. G., Balaramane, D., Weber, M. & Bardet, A. F. Pan-cancer predictions of transcription factors mediating aberrant DNA methylation. Epigenetics Chromatin 15, 10 (2022).
Fiumara, M. et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat. Biotechnol. 42, 877–891 (2024).
Kimble, M. T. et al. Repair of replication-dependent double-strand breaks differs between the leading and lagging strands. Mol. Cell 85, 61–77.e6 (2025).
Chauhan, V. P., Sharp, P. A. & Langer, R. Engineered prime editors with minimal genomic errors. Nature 646, 1254–1260 (2025).
Ma, E., Harrington, L. B., O’Connell, M. R., Zhou, K. & Doudna, J. A. Single-stranded DNA cleavage by divergent CRISPR-Cas9 enzymes. Mol. Cell 60, 398–407 (2015).
Mir, A., Edraki, A., Lee, J. & Sontheimer, E. J. Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chem. Biol. 13, 357–365 (2018).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. eLife 11, e83724 (2022)
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Zhou, W., Triche, T. J. Jr, Laird, P. W. & Shen, H. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 46, e123 (2018).
Noguera-Castells, A., Garcia-Prieto, C. A., Alvarez-Errico, D. & Esteller, M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics 18, 2185742 (2023).
Acknowledgements
We acknowledge the use of instruments at the Biological Science Imaging Resource supported by Florida State University, the Laboratory for Biomolecular Structure (LBMS) and the Pacific Northwest Center for Cryo-EM (PNCC). We thank G. Seo of the Institute of Molecular Biophysics Protein Expression Facility for providing facilities and resources for human tissue culture; B. Alexander of the FSU Flow Cytometry Laboratory for cell sorting; S. Miller of the FSU Sequencing facility for assistance with Sanger sequencing of genomic DNA PCR products; R. Hoffman of IMRA America for scripts used in the ENCODE data analysis; the staff at the LBMS, especially G. Hu, and at the PNCC, especially N. Meyer and M. Miletto, for assistance with data collection; the Van Andel Institute Genomics Core (RRID: SCR_022913), especially T. Avequin and D. Fu, for their assistance with the Infinium Methylation EPIC array; and M. Myers and Y. Zhang at the University of Michigan for thoughtful discussion and the protocol for mRNA synthesis and delivery. This work was supported by US National Institutes of Health (NIH) grant R35 GM152081 to H.L., and by Dutch Research Council (NWO) Spinoza grant SPI 93-537, European Research Council (ERC) Advanced Grant (834279), University Fund Wageningen (UFW) grant, and Dutch Ministry of Economic Affairs (Groeifonds, NXTGEN HighTech) grant to J.v.d.O. The Titan microscope was funded from NIH grant S10 RR025080. The BioQuantum/K3 was funded from NIH grant U24 GM116788. The Vitrobot Mk IV was funded from NIH grant S10 RR024564. The Solaris Plasma Cleaner was funded from NIH grant S10 RR024564. The DE-64 was funded from NIH grant U24 GM116788. The LBMS is supported by the DOE Office of Biological and Environmental Research (KP160711). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. Research reported in this publication was supported by the National Cancer Institute of the NIH under award number T32CA251066 to M.O.R. at the Van Andel Institute.
Author information
Authors and Affiliations
Contributions
M.O.R., Y.S., Y.Z., D.T., R.D.H., J.v.d.O. and H.L. designed the experiments. M.O.R., D.T. and R.D.H. designed the gene-editing sites with the assistance of N.T. M.O.R. and R.D.H. performed the gene-editing experiments. M.O.R., M.L.M. and M.K.G. performed the ThermoCas9 engineering experiments. Y.S. performed the in vitro assays and prepared the cryo-EM samples. Y.Z. performed the cryo-EM experiments of ThermoCas9 with the assistant of Y.S. and H.N.G. A.D. and J.R. performed the cryo-EM studies of AceCas9 with the assistance of B.W. M.Z. and R.D.H. wrote the data analysis scripts for DNA methylation analysis. D.T., C.S. and E.B. provided the expression vectors. Y.S. purified the ThermoCas9 used in human cell gene editing. M.O.R., Y.S., Y.Z., R.D.H., J.v.d.O. and H.L. analysed the data and wrote the manuscript. All authors contributed to discussions, finalizing figures, and reviewing and editing the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
A patent application on methylation-sensitive gene editing using ThermoCas9 and its variants has been filed related to this work with the following information: Florida State University Research Foundation, INC, Wageningen University are the patent applicants under PCT/US2025/014770 (published as US2025/0250641 on 8 July 2025), with H.L., M.O.R., Y.S., Y.Z., R.D.H., J.v.d.O. and D.T. named as inventors. The application is pending. J.v.d.O. is an advisor of NTrans Technologies, Scope Biosciences and Hudson River Biotechnology. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Jun-Jie (Gogo) Liu, Krishanu Saha and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sample preparation and DNA binding analysis results.
a. Purification of ThermoCas9. Left, elution profile of ThermoCas9, following Ni-NTA affinity purification on a Heparin column. The shaded fractions are analyzed on an SDS-PAGE gel and pooled before used in biochemical analysis or cryo-EM sample preparation. Right, elution profile of ThermoCas9, following incubation with the in vitro transcribed single guide RNA at 37 °C for 30 min, on a S200i gel filtration column. The shaded fractions were analyzed on an SDS-PAGE gel and were used immediately for cryo-EM analysis. b. DNA oligo substrates used for in vitro cleavage assay and native gel analysis. Hexachlorofluorescein (HEX) and 6-Carboxyfluorescein (FAM) fluorescent dye labels are indicated. c. DNA binding competition assays with ThermoCas9. Gel images illustrate the cleavage results of a DNA plasmid by ThermoCas9 in the presence of increasing amount of four different competing double-stranded DNA oligos containing 5′-NNNNCGA-3′ (CpG), 5′-NNNN5mCGA-3′ on nontarget strand (5mCpGNTS), 5′-NNNN5mCGA-3′ on target strand (5mCpGTS) and 5′-NNNN5mCGA-3′ on both target and nontarget strand (5mCpGW) PAM, respectively. Each experiment was repeated with independent samples three times.
Extended Data Fig. 2 Cryo-EM image collection, analysis and 3D reconstruction results of the ThermoCas9 bound with a cognate DNA substrate.
a. Example micrograph and 2D class averages (scale bar 50 nm). b. Data collection, particle selection, and reconstruction flowchart. All classes reconstructed and fitted with atomic models are indicated with reported resolutions. c. Upper, the final map used for building the model of the post-cleavage and dsDNA-bound state (CLOSED) and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core. Lower, the Fourier Shell Correlation (FSC) curves of the refined model. 0.143 FSC cutoff was used for resolution estimation. d. Upper, the final map used for building the model of the post-cleavage and target strand-bound state (CLOSED) and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core. Lower, the Fourier Shell Correlation (FSC) curves of the refined model. 0.143 FSC cutoff was used for resolution estimation. e. Upper, the final map used for building the model of the pre-cleavage state (OPEN) and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core. Lower, the Fourier Shell Correlation (FSC) curves of the refined model. 0.143 FSC cutoff was used for resolution estimation.
Extended Data Fig. 3 Cryo-EM density maps for ThermoCas9 observed at the post-cleavage (CLOSED) and pre-cleavage (OPEN) states shown for the protein- (top) and nucleic acids- (middle) in two orientations.
Each functional domain or nucleic acid element is labeled. The bottom row shows close-up views of the density around the target strand cleavage site of the CLOSED and OPEN states, respectively.
Extended Data Fig. 4 Detailed interactions between ThermoCas9 and the single guide RNA.
a. Observed percent of five charged amino acids involved in contacting guide RNA for ThermoCas9 and four other Cas9 variants. The coordinates used for analysis are 8DLK for AceCas9, 6JDJ for Nme1Cas9, and 7S4X for SpyCas9. b. Depiction of the secondary structure of the single guide RNA and the contacts with ThermoCas9 for the CLOSED state.
Extended Data Fig. 5 Cryo-EM image collection, analysis and 3D reconstruction results of the ThermoCas9 bound with a methylated DNA substrate or with DNA substrate containing 5′-NNNCGA-3′ PAM.
a. Example micrograph and 2D class averages (scale bar 100 nm) of the ThermoCas9-gRNA-(5mCpG)dsDNA complex. b. Micrograph cellection, particle selection, and reconstruction flowchart. Due to redundance, no model was refined to the final stage. c. Left, the final map is indicated with a reported resolution and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core. Right, the Fourier Shell Correlation (FSC) curves of the refined model. 0.143 FSC cutoff was used for resolution estimation. d. Example micrograph and 2D class averages (scale bar 100 nm) of the ThermoCas9-gRNA-(CpG)dsDNA complex. e. Micrograph collection, particle selection, and reconstruction flowchart. f. Left, the final map is indicated with a reported resolution and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core. Right, the Fourier Shell Correlation (FSC) curves of the refined model. 0.143 FSC cutoff was used for resolution estimation.
Extended Data Fig. 6 ThermoCas9 structural comparison.
a. Superimposed structures of ThermoCas9 (colored) and GeoCas9 (gray, PDB 8UZA). b. Superimposed structures of ThermoCas9 (colored) and Nme1Cas9 (gray, PDB 6JDV). c. Superimposed HNH domain between the pre-cleavage (OPEN) and the post-cleavage (CLOSED) state, indicating minor adjustment of the catalytic residues despite a large domain rotation between the two different enzyme conformations. Metal ion and water molecules are models from the CLOSED state.
Extended Data Fig. 7 Cryo-EM image collection, analysis and 3D reconstruction results of the AceCas9 bound with a methylated DNA substrate.
a. Micrograph collection, particle selection, and reconstruction flowchart. All classes reconstructed and fitted with atomic models are indicated with reported resolutions. b. Example micrograph and 2D class averages (scale bar 100 nm). c. The Fourier Shell Correlation (FSC) curves of the three refined models. 0.143 FSC cutoff was used for resolution estimation. d. The final map used for building the model of the uncleaved state 1 and analyzed by Resmap. Resolutions are color-coded according to a scale bar, showing the comparably high-resolution inner core.
Extended Data Fig. 8 Structural features of AceCas9 bound with a methylated DNA.
a. The schematic of AceCas9 protein and the nucleic acids used in cryoEM structure study. b. The structure of AceCas9 bound with its guide RNA (sgRNA) and methylated DNA. Top, cryoEM density with each domain and nucleic acids color coded as in panel a. and labeled. Bottom, cartoon representation of AceCas9 bound with the methylated DNA color coded as in panel a. and labeled. Insets show close-up views of the target strand cleavage site and the PAM interactions. “Me” indicates the methyl group on the methylated cytosine. Dash lines indicate close contacts between the DNA bases and protein residues. c. Left, stick models showing the contacts of the two phosphate lock residues, Glu839 and Glu840, with the kinked target strand. Arrows indicate the Glu839 to arginine and Glu840 to tyrosine or leucine (RY or RL), respectively. Right, gel analysis of phosphate lock residue mutants RY and RL cleaving unmethylated and methylated DNA plasmid. Methylated DNA was obtained with treatment with HaeIII methyltransferase. HaeIII restriction endonuclease (HaeIII endo) is included as a control.
Extended Data Fig. 9 DNA methylation at the targeted sites in HEK293T and HCT116 cells.
a. Heat map of DNA methylation based on β values obtained from reduced-representation bisulfite sequencing (RRBS) data of the Encyclopedia of DNA Elements (ENCODE) database for HEK293T (top) and HCT116 (bottom) cells, respectively. CpG sites are shown as equal-width columns arranged in the 5′ to 3′ direction of each gene and colored according to their β-values, ranging from hypomethylated (green) to hypermethylated (red), as indicated by the scale bar. The targeted sites by ThermoCas9 and SpyCas9 are indicated. Genome coordinates of the target sites (t, black arrows) and the nearest CpG site (p) for ThermoCas9 are listed along with the mean β-values of the nearest CpG. b. Bisulfite sequencing results for EMX1 T4, VEGFA T3, and PRDX4 T5 sites. Colored traces represent Sanger sequencing results at indicated genomic sites from bisulfite-coverted genomes of the HEK293T and HCT116 cells, respectively. Detected sequences are shown above the traces with converted cytosine nucleotides in parentheses and unconverted (methylated) marked by solid circles. Asterisks indicate possible cytosine nucleotides with incomplete bisulfite conversion.
Supplementary information
Supplementary Video 1 (download MP4 )
Overview of structural transitions from the OPEN to CLOSED conformation of ThermoCas9. Domains and nucleic acids are colored as in Figure 3
Supplementary Video 2 (download MP4 )
Structural transitions from the OPEN to CLOSED conformation of the RuvC active site of ThermoCas9. Residues and magnesium ions are labeled
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Roth, M.O., Shu, Y., Zhao, Y. et al. Molecular basis for methylation-sensitive editing by Cas9. Nature 653, 1229–1239 (2026). https://doi.org/10.1038/s41586-026-10384-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-026-10384-z
